

Abstract
Alzheimer's disease (AD) is characterized by the extracellular deposition of amyloid-β (Aβ), neurofibrillary tangle formation, and a microglial-driven inflammatory response. Chronic inflammatory activation compromises microglial clearance functions. Because peroxisome proliferator-activated receptor γ (PPARγ) agonists suppress inflammatory gene expression, we tested whether activation of PPARγ would also result in improved microglial Aβ phagocytosis. The PPARγ agonist pioglitazone and a novel selective PPARα/γ modulator, DSP-8658, currently in clinical development for the treatment of type 2 diabetes, enhanced the microglial uptake of Aβ in a PPARγ-dependent manner. This PPARγ-stimulated increase of Aβ phagocytosis was mediated by the upregulation of scavenger receptor CD36 expression. In addition, combined treatment with agonists for the heterodimeric binding partners of PPARγ, the retinoid X receptors (RXRs), showed additive enhancement of the Aβ uptake that was mediated by RXRα activation. Evaluation of DSP-8658 in the amyloid precursor protein/presenilin 1 mouse model confirmed an increased microglial Aβ phagocytosis in vivo, which subsequently resulted in a reduction of cortical and hippocampal Aβ levels. Furthermore, DSP-8658-treated mice showed improved spatial memory performance. Therefore, stimulation of microglial clearance by simultaneous activation of the PPARγ/RXRα heterodimer may prove beneficial in prevention of AD.
Introduction
Cerebral inflammation occurs through the activation of microglia, the brain innate immune system in response to misfolded or aggregated proteins and neuronal debris. In addition to its classical hallmarks, amyloid-β (Aβ) deposition and neurofibrillary tangle formation, Alzheimer's disease (AD) is characterized by a neuroinflammatory component (Lucin and Wyss-Coray, 2009). In AD, activated microglia are found in close vicinity to Aβ deposits and produce a wide range of cytokines and chemokines (Rogers and Lue, 2001). Because microglia are critical for Aβ phagocytosis, the microglial presence at the Aβ plaque can be interpreted as an attempt to clear this pathological deposit. Evidence for a important role of microglial phagocytosis comes from studies that show that restricting microglial Aβ uptake leads to increased cerebral Aβ levels (El Khoury et al., 2007; Lee and Landreth, 2010). Microglial expression of receptors that promote the clearance and phagocytosis of Aβ, such as CD36, TLR4, and TLR6 (Lee and Landreth, 2010), are mandatory for restricting amyloid plaque formation. However, as AD progresses, microglia acquire a chronically activated phenotype through unremitting stimulation by cytokines and Aβ itself, which compromises their clearance function (Bamberger et al., 2003; Hickman et al., 2008; Heneka et al., 2010a) .
Because impaired Aβ clearance is likely to account for the majority of sporadic AD cases (Mawuenyega et al., 2010), identification of factors the regulate microglial Aβ uptake seems to be of upmost importance.
The recognition that a robust inflammatory response contributes to AD also fuelled the discovery that long-term treatment with non-steroidal anti-inflammatory drugs (NSAIDs) reduced the risk for AD, delayed disease onset, and slowed cognitive decline (McGeer et al., 1996; Stewart et al., 1997; in t' in t'Veld et al., 2001). Of note, a subset of NSAIDs directly regulates gene expression through binding and activating the peroxisome proliferator-activated receptor-γ (PPARγ) (Lehmann et al., 1997) that belongs to the nuclear receptor superfamily and forms a heterodimer with the retinoid X receptor (RXR) before binding to PPAR response elements in the promoter region of target genes. Currently marketed PPARγ ligands modulate insulin sensitivity but also cause considerable side effects, including body weight gain and fluid retention. DSP-8658 is a novel selective PPARα/γ modulator that is currently developed for type 2 diabetes, because the described side effects are absent in relevant preclinical models.
In microglia and macrophages, PPARγ activation inhibits proinflammatory gene expression (Jiang et al., 1998; Ricote et al., 1998). In the brain, PPARγ agonists reduce Aβ- and cytokine-mediated neuroinflammation and neurotoxicity in vitro (Heneka et al., 1999; Combs et al., 2000) and in vivo (for review, see Heneka and Landreth, 2007).
Here we demonstrate the potential of the PPARγ ligand pioglitazone and the PPARα/γ modulator DSP-8658 to modulate microglial Aβ phagocytosis in vitro and in vivo.
Materials and Methods
Materials.
DSP-8658 was provided from Dainippon Sumitomo Pharma. Pioglitazone was obtained by Takeda Pharmaceuticals. 9-cis-Retinoic acid, actinomycin D, and cycloheximide were obtained from Sigma-Aldrich. Bexarotene was purchased from LC Laboratories. Rat PPARγ siRNA (L-080081-01-0010), rat RXRα siRNA (L-089934-01-0010), rat RXRβ siRNA (L-094287-01-0010), and nontargeting control siRNA (D-001810-10-05) were purchased from Thermo Fisher Scientific.
Primary microglial cell culture.
Primary microglial cell cultures were prepared as described previously in detail (Terwel et al., 2011). Briefly, mixed glial cultures were prepared from newborn rat or mouse cultured in DMEM supplemented with 10% FCS and 100 U/ml penicillin/streptomycin. Microglial cells were used after 10–14 d of primary cultivation. They were harvested by shake off, seeded, and allowed to attach to the substrate for 30–60 min.
Transactivation assay.
Monkey kidney COS-1 cells were maintained in standard culture conditions (DMEM) supplemented with 10% FCS, 1% sodium pyruvate, 1% essential amino acids, and 1% streptomycin/penicillin at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every other day. Cells were transfected with GAL4–UAS–luc plasmid and the plasmid coding the nuclear receptor of interest (GAL4 DNA–BD/PPARα–LBD, GAL4 DNA–BD/PPARγ–LBD). Transfected cells were incubated for 24 h at 37°C in medium containing the compound tested. The luciferase activity was measured with the Steady-Glo Luciferase Assay System (Promega). The reference compounds for PPARα and PPARγ were fenofibrate and rosiglitazone, respectively.
Phagocytosis of 6-carboxyfluorescein-labeled Aβ1–42 by microglia.
Microglial phagocytosis of 6-carboxyfluorescein (FAM)-labeled Aβ1–42 (FAM-Aβ) (Peptide Specialty Laboratories) was measured by plate-based assay as described previously (Floden and Combs, 2006). In brief, cells were plated at a density of 50,000 per well. Cells were incubated with 500 nm Aβ for up to 4 h, starting 1 h after plating. The Aβ-containing medium was removed, and extracellular Aβ was quenched with 100 μl of 0.2% trypan blue in PBS, pH 4.4, for 1 min. After aspiration, fluorescence was measured at 485 nm excitation/535 nm emission using an Infinity 200 reader (Tecan). To normalize for cell numbers, cells were incubated with 100 μl of 50 μg/ml H33342 [2′-(4-ethoxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5′-bi-1H-benzimidazole trihydrochloride] in PBS and 0.1% Triton X-100 for 15 min, and fluorescence was measured at 360 nm excitation/465 nm emission. For analysis of intracellular Aβ degradation, cells were incubated for 30 min with pioglitazone or DSP-8658. Afterward, FAM-Aβ1–42 was added for 30 min, and the cell culture media were replaced. After 4 h of incubation, fluorescence was determined as described above. For intracellular localization of Aβ mouse primary microglia, cells were incubated with 150 nm Aβ1–42. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and blocked with 20% goat serum in PBS. Subsequently, cells were coincubated with antibodies IC16 against Aβ (Jäger et al., 2009) and MCA711 (AbD Serotec) against CD11b. Fluorescence microscopy was done on an BX61 equipped with a disc-spinning unit (Olympus). Microglial cell death in response to drug or Aβ exposure was excluded by LDH assay.
Nucleofection of siRNA.
All electroporations were performed using Nucleofector II (Lonza) and the mouse macrophage nucleofection kit (Amaxa Biosystems) according to the instructions of the manufacturer.
Western blotting.
Protein concentration in the RIPA-soluble or SDS-soluble fraction was determined using the BCA Protein Assay kit (Pierce). Protein samples (10–50 μg) were separated by 4–12% NuPAGE (Invitrogen) using MES buffer and transferred to nitrocellulose membranes. Protein levels of PPARγ [anti-PPARγ (H-100); Santa Cruz Biotechnology], RXRα [anti-RXRα (PP-K8508); Perseus Proteomics], RXRβ [anti-RXRβ (PP-H7341); Perseus Proteomics] (1:500), CD36 [anti-CD36 (ab36977); Abcam], and tubulin [E7; Developmental Studies Hybridoma Bank] were assessed. Primary antibody incubation was followed by incubation with appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactivity was detected by enhanced chemiluminescence reaction (Millipore), and luminescence intensities were analyzed using Chemidoc XRS documentation system (Bio-Rad).
RNA preparation and real-time reverse transcription-polymerase chain reaction.
Total RNA was isolated from microglia using the RNeasy Mini kit (Qiagen) and quantified spectrophotometrically and reverse transcribed using the RevertAid First Strand cDNA Synthesis kit (Fermentas) according to the instructions of the manufacturer. Predesigned PPARγ, CD36, and GAPDH primers were purchased from Applied Biosystems. The real-time PCR reactions were performed using the StepOnePlus PCR System (Applied Biosystems). mRNA expression values were normalized to the level of GAPDH expression.
Animals.
Amyloid precursor protein/presenilin 1 (APP/PS1) transgenic animals expressing the mouse APP containing the human Aβ domain as well as the Swedish mutation and the PS1 Δexon 9 mutation both under the control of the prion promoter were used (Jankowsky et al., 2001). Three-month-old wild-type and APP/PS1 transgenic mice were divided into control and DSP-8658-treated groups and fed with a standard food not containing substance or containing 0.1% DSP-8658 for 3 months. The amounts of food and body weight were assessed weekly. The daily food intake was calculated from the following equation: daily food intake (g/d) = was total food intake during the treatment period/treatment days. The mean dose administered to the DSP-8658-treated group was estimated from the following equation: estimated administered dose (mg · kg−1 · d−1) = daily food intake × 1000 × 0.1% (content of DSP-8658 in the diet)/100/mean body weight during the dosing period. Mice were housed in groups under standard conditions at a temperature of 22 ± 1°C and a 12 h light/dark cycle with access to standard food (Altromin) and water ad libitum. Animal care and handling were performed according to the Declaration of Helsinki and approved by local ethical committees. Mice were deeply anesthetized with isoflurane and killed, and brains were removed and processed for neurochemical and immunochemical determinations.
Morris water maze.
The test was conducted in a pool consisting of a circular tank (1 m diameter) filled with opacified water at 24°C. The water basin was dimly lit (20–30 lux) and surrounded by a white curtain. The maze was virtually divided into four segments (quadrants). In one of the quadrants, a hidden platform (15 × 15 cm) was present 1.5 cm below the water surface. Mice were trained to find the platform, orientating by means of three intermaze cues placed asymmetrically as spatial references. They were let into the water in a quasi-random manner to prevent strategy learning. Mice were allowed to search for the platform for 40 s. If the mice did not reach the platform in the allotted time, they were placed manually on it. The mice were allowed to stay on the platform for 15 s before the next trial was started. After four trials were completed, mice were dried and placed back in their home cage. Mice received four training trials per day for 8 consecutive days. Movements of the mice were recorded by a computerized tracking system that calculated distances moved and latencies (Noldus).
Extraction of brain lysates.
Snap-frozen forebrain hemispheres were homogenized in PBS with protease inhibitor mixture (Sigma). Protein was extracted in RIPA (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% sodium desoxycholate, 1% NP-40, and 0.1% SDS) for 30 min on ice. After centrifugation at 100,000 × g for 30 min at 4°C, the resulting supernatant (RIPA-soluble fraction) was saved, and the pellet was sonified in 25 mm Tris-HCl, pH 7.5, and 2% SDS (RIPA-insoluble fraction).
Aβ determination by sandwich ELISA.
Human (6E10) Aβ1–40 or Aβ1–42 ultrasensitive kits were used (Meso Scale Discovery). Signals were measured on a SECTOR Imager 2400 reader (Meso Scale Discovery).
In vivo phagocytosis assay.
Six-month-old wild-type and APP/PS1 transgenic mice were intraperitoneally injected 3 h before the animals were killed with 10 mg/kg methoxy-X04 (kindly provided by Dr. Alfons Verbruggen, Catholic University of Leuven, Leuven, Belgium) in 50% DMSO/50% NaCl (0.9%), pH 12 (Bolmont et al., 2008). Mice were perfused with ice-cold PBS, the brain was removed, and one hemisphere was chopped into pieces using scalpels. Brain pieces were homogenized in HBSS, 0.6% glucose, and 15 mm HEPES, pH 7, using a Potter-Elvehjem tissue homogenizer. Additional homogenization was achieved by gently up and down pipetting using Pasteur pipettes with decreasing opening diameter. The resulting homogenate was filtered through a cell strainer (70 μm) and centrifuged at 155 × g (Beckman Allegra) at 4°C for 10 min without brake. The pellet was resuspended in 9 ml of 75% Percoll in PBS and underlayered with ice-cold 10 ml of 25% Percoll in PBS and overlayered with 6 ml ice-cold PBS. The gradient was centrifuged at 800 × g at 4°C for 25 min (Beckman Allegra) without brake. Microglial cells are recovered from the 25/75% Percoll interphase, diluted with 3 vol of PBS, and centrifuged at 880 × g at 4°C for 25 min (Beckman Allegra) without brake. The pellet containing the microglial cells was resuspended in 200 μl of PBS.
Flow cytometry analysis of methoxy-X04 containing microglia.
Binding of antibodies to Fc receptors was prevented by adding Fc block (Merck) diluted 1:100 for 10 min on ice. Fifty microliters of cells were diluted with 0.5 ml of HBSS and centrifuged at 1000 rpm for 5 min at 4°C. Cells were taken up in 50 μl of HBSS containing biotinylated TLR4 antibody (1:100), incubated 30 min on ice, and centrifuged at 1000 rpm for 5 min at 4°C. Cells were taken up in 50 μl of HBSS containing streptomycin–phycoerythrin (PE)–Cy7 (1:200), incubated 30 min on ice, and centrifuged at 1000 rpm for 5 min at 4°C. Finally, cells were incubated with antibody mix [CD11b–adenomatous polyposis coli (APC), 1:100; CD45–FITC, 1:100] and incubated for 30 min on ice. Cells were centrifuged at 1000 rpm for 5 min at 4°C and resuspended in 200 μl of HBSS. For control and compensation, we used corresponding isotype control antibodies. Samples were analyzed on an FACS Canto II flow cytometer (BD Bioscience). Data evaluation of the CD11b-positive (CD11b+)/CD45+ population concerning methoxy-X04 incorporation was performed. Flow cytometry analysis of microglia was performed using antibodies CD11b–APC (catalog #101212; BioLegend), CD45–FITC (catalog #11-0451; eBioscience), and CD36–PE (catalog #12-0361; eBioscience).
Immunohistochemistry.
Free-floating 40-μm-thick serial sections were cut on a vibratome (Leica). Sections obtained were stored in 0.1% NaN3 and PBS in a cold room. For immunohistochemistry, sections were treated with 50% methanol for 15 min. Then, sections were washed three times for 5 min in PBS and blocked in 3% BSA, 0.1% Triton X-100, and PBS (blocking buffer) for 30 min, followed by overnight incubation with the primary antibody in blocking buffer. Next, sections were washed three times in 0.1% Triton X-100 and PBS and incubated with Alexa Fluor 488-conjugated or Alexa Fluor 594-conjugated secondary antibodies (1:500; Invitrogen) for 90 min, washed three times with 0.1% Triton X-100 and PBS for 5 min. Finally, the sections were mounted on glasses in tap water and embedded. The following primary antibodies were used with respective concentrations: rabbit polyclonal 2964 against Aβ (1:400, generous gift from Dr. Jochen Walter) and rat monoclonal anti-mouse CD11b (1:200, Serotec). In thioflavin S staining, slices were rinsed in water, incubated in 0.01% thioflavin S in 50% ethanol, and differentiated in 50% ethanol. Fluorescence microscopy was done on an BX61 equipped with a disc-spinning unit (Olympus) or an A1-MP (Nikon) laser-scanning microscope, and images were processed in Cell-P (Olympus) or NIS elements (Nikon).
Statistical analyses.
Data were analyzed by using Student's t test, one-way ANOVA followed by Tukey's post hoc test, or Dunnett's multiple comparison test using Prism 5.03 (GraphPad Software).
Results
Effects of PPARγ agonist on Aβ phagocytosis in rat primary microglia
To test whether PPARγ agonists affect Aβ phagocytosis, rat primary microglia were incubated with increasing concentrations of DSP-8658, a PPARα/γ agonist with an EC50 of ∼1 μm for both isoforms (Table 1), or pioglitazone in the presence of FAM-Aβ (0.5 μm). After 4 h exposure to Aβ, intracellular levels of Aβ were determined. Both DSP-8658 and pioglitazone enhanced the uptake of Aβ in rat primary microglia in a concentration-dependent manner (Fig. 1A,B). Intracellular presence of Aβ in primary microglia was confirmed by immunocytochemistry using antibody IC16 against Aβ and antibody MCA711 against the cell-surface antigen CD11b (Fig. 1C). Time-dependent intracellular degradation was analyzed as described previously (Fleisher-Berkovich et al., 2010) over the time course of 4 h and found to be similar between substances (Fig. 1D,E). Therefore, these results indicate that DSP-8658 and pioglitazone induce the phagocytic uptake of Aβ by microglia. To firmly establish the contribution of PPARγ in the regulation of Aβ phagocytosis and to exclude off-target drug effects, PPARγ gene expression was silenced by siRNA. Quantitative PCR analysis indicated that the siRNA transfection suppressed PPARγ gene expression by 50% (Fig. 1F), resulting in a substantial reduction of PPARγ protein levels when compared with the nontarget control siRNA (Fig. 1G). Although DSP-8658 and pioglitazone enhanced Aβ uptake at 3 μm in nontarget control siRNA-transfected primary microglia, the same treatments failed to increase Aβ phagocytosis in PPARγ siRNA-transfected primary microglia (Fig. 1H,I). Comparing these results with Figure 1, A and B, it seemed that the phagocytic response during drug stimulation was reduced in the siRNA transfection experiments. Although cell death was excluded as the source of this phenomenon, siRNA transfection may have led to microglial prestimulation, resulting in an overall decreased phagocytic response. Together, these results suggest that microglial Aβ clearance can be increased by PPARγ activation.
Table 1.
Activation of human PPARs by DSP-8658
| Compound | Human, EC50 |
|
|---|---|---|
| α | γ | |
| DSP-8658 | 1.08 μm | 1.01 μm (76%) |
| Pioglitazone | >10 μm | 0.52 μm (100%) |
| Rosiglitazone | 0.084 μm (104%) | |
| Fenofibric acid | 16.9 μm | |
Activation of PPARα and PPARγ was determined by transactivation assay in COS-1 cells after incubation with compounds for 24 h. The reference compounds for PPARα and PPARγ were fenofibrate and rosiglitazone, respectively.
Figure 1.
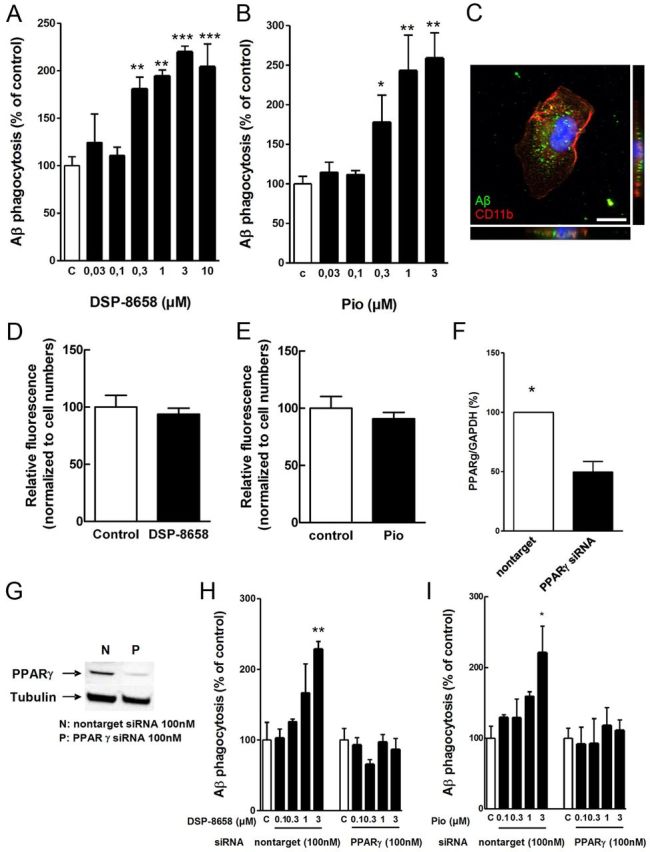
Effect of PPARγ agonist on Aβ phagocytosis in primary microglia. Rat primary microglia were incubated with increasing concentrations of DSP-8658 or pioglitazone in the presence of FAM-Aβ (0.5 μm). After 4 h exposure to Aβ, intracellular levels of Aβ were determined. Both DSP-8658 (A) and pioglitazone (B) enhanced the uptake of Aβ in rat primary microglia in a concentration-dependent manner (mean ± SEM of n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA, Tukey's post hoc test). C, Intracellular uptake of Aβ1–42 was confirmed by immunocytochemical staining of Aβ using antibody IC16 and the cell-surface antigen CD11b using antibody MCA711. Scale bar, 10 μm. Rat primary microglia were incubated for 30 min with FAM-Aβ in the presence of 0.3 μm DSP-8658 (D) or 0.3 μm pioglitazone (pio) (E) (mean ± SEM of n = 3). PPARγ mRNA (F) and protein levels (G) in primary microglia transfected with siRNA for PPARγ (mean ± SEM of n = 3, *p < 0.05, Student's t test). Primary microglia transfected with siRNA for PPARγ or nontarget were incubated with DSP-8658 (H) or pioglitazone (I) in the presence of FAM-Aβ (0.5 μm). DSP-8658 and pioglitazone enhanced Aβ uptake at 3 μm in nontarget control siRNA-treated primary microglia, although the same treatments failed to increase Aβ phagocytosis in PPARγ siRNA-treated primary microglia (mean ± SEM of n = 3, *p < 0.05, **p < 0.01, one-way ANOVA, Tukey's post hoc test).
Effects of PPARγ agonist on CD36 expression in primary microglia
To determine whether PPARγ-mediated Aβ uptake requires de novo synthesis of mRNA and protein, rat primary microglia were coincubated with inhibitors of transcription (actinomycin D, 5 μm) or translation (cycloheximide, 10 μm). Interestingly, the increasing effect of DSP-8658 and pioglitazone was abolished once either actinomycin D or cycloheximide were coincubated, suggesting that de novo mRNA/protein synthesis is required (Fig. 2A,B).
Figure 2.
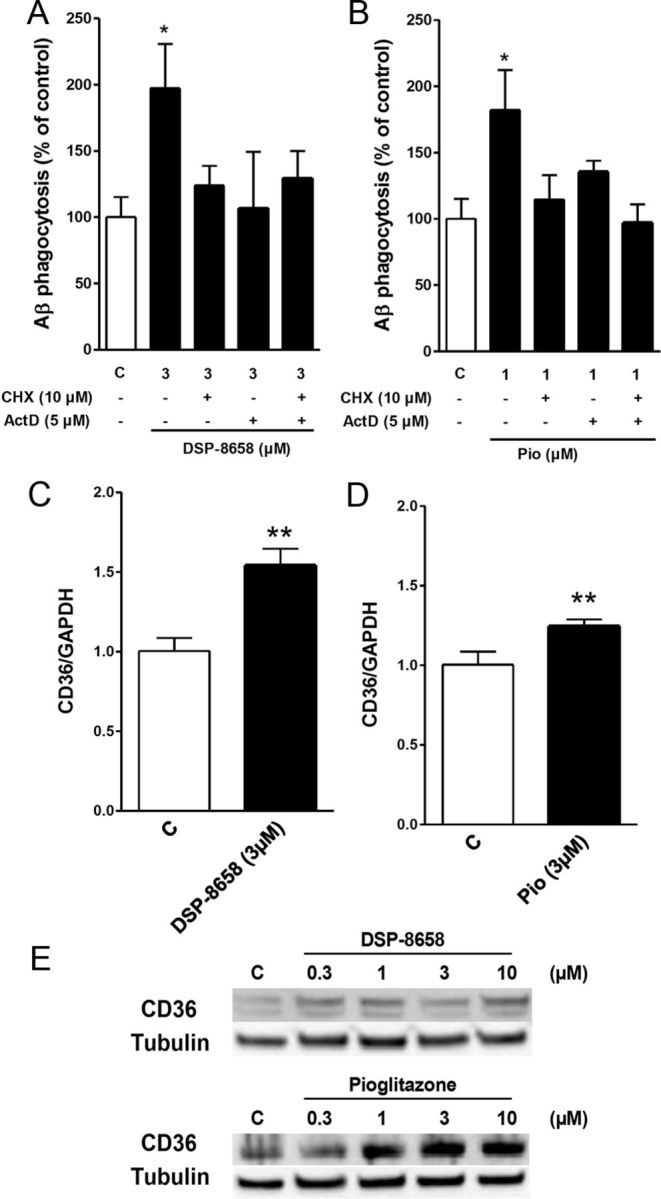
Effects of PPARγ agonist on CD36 expression in primary microglia. Primary microglia was coincubated with inhibitors of transcription [actinomycin D (ActD), 5 μm] or translation [cycloheximide (CHX), 10 μm] in the presence of FAM-Aβ (0.5 μm). After 4 h, intracellular levels of Aβ were determined. Aβ phagocytosis by DSP-8658 (A) or pioglitazone (B) was abolished by coincubation with the respective inhibitors. The data show the intracellular Aβ uptake as percentage of control (mean ± SEM of n = 3, *p < 0.05, one-way ANOVA, Tukey's post hoc test). C–E, Primary microglia was incubated with DSP-8658 or pioglitazone for 6 h, and CD36 mRNA and protein levels were detected. CD36 mRNA was upregulated in response to DSP-8658 (C) or pioglitazone (D), followed by an increase of the respective protein levels in a concentration-dependent manner (E) (mean ± SEM of n = 3, **p < 0.01, Student's t test).
To further address the mechanism underlying the stimulation of microglial Aβ phagocytosis by PPARγ agonists, we focused on the search for a cell-surface marker that is regulated by PPARγ and is involved in Aβ phagocytosis. Indeed, the expression of the scavenger receptor CD36 is controlled by PPARγ (Tontonoz et al., 1998) and is involved in the uptake of Aβ. To substantiate this hypothesis, primary microglia were incubated with DSP-8658 or pioglitazone for 6 h, and CD36 mRNA and protein levels were measured. CD36 mRNA was upregulated in response to DSP-8658 or pioglitazone, followed by an increase of the respective protein levels in a concentration-dependent manner (Fig. 2C,D). Restricting the action of CD36 by either an inhibitory antibody against CD36 or siRNA knockdown of CD36 resulted in the abolishment of the stimulatory effect of DSP-8658 or pioglitazone on Aβ phagocytosis prevented the increase of Aβ uptake during incubation with DSP-8658 or pioglitazone (Fig. 3). These results identify CD36 as a key mediator of PPARγ agonist-induced microglia Aβ phagocytosis.
Figure 3.

CD36 mediates the PPARγ-stimulated increase in microglial Aβ uptake. Primary microglia was incubated for 4 h with either DSP-8658 (A) or pioglitazone (B; Pio) in the presence of CD36 antibody or isotype control. After 4 h, intracellular Aβ was determined. PPARγ agonist-stimulated Aβ phagocytosis was suppressed by coincubation with CD36 antibody (mean ± SEM of n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA, Tukey's post hoc test). C, Protein levels of CD36 in primary microglia transfected with siRNA for CD36. D, E, Primary microglia transfected with siRNA for CD36 or nontarget were incubated with DSP-8658 or pioglitazone in the presence of FAM-labeled Aβ1–42 (0.5 μm). After 4 h, intracellular level of Aβ was determined. Transfection of cell with CD36 siRNA blocked the PPARγ agonist-stimulated uptake of Aβ. The data show the intracellular Aβ uptake as percentage of control (mean ± SEM of n = 3, **p < 0.01, one-way ANOVA, Tukey's post hoc test).
Additive effects of combined activation of PPARγ and RXR in primary microglia
Because all PPARs heterodimerize with RXRs and subsequently bind to DNA responsive elements, thereby regulating the expression of target genes, we analyzed whether a combined activation of the PPARγ/RXR heterodimer would lead to an increase in Aβ uptake. Two RXR agonists, retinoic acid and bexarotene, were tested for their effect on Aβ phagocytosis. Both RXR agonists enhanced Aβ phagocytosis in primary microglia in a concentration-dependent manner (Fig. 4A,B). Primary microglia was then incubated with DSP-8658 or pioglitazone and the respective RXR agonists, and intracellular levels of Aβ were determined after 4 h of incubation. Interestingly, the combination of pioglitazone or DSP-8658 with RXR agonists revealed an additive effect on Aβ uptake (Fig. 4C–F). Because both retinoic acid and bexarotene are not isoform specific and may activate both RXRα and RXRβ, we determined which isoform is involved in the agonist-induced increase of Aβ phagocytosis by siRNA knockdown of either RXRα or RXRβ. Transfection of primary rat and mouse microglia with siRNA effectively decreased RXRα expression (Fig. 5A,C), whereas RXRβ expression was not (rat) or only to a minor level (mouse) detected in primary microglia after transfection of both nontarget control or RXRβ siRNA. In contrast and as a positive control, RXRβ expression was found in BV2 cells (Fig. 5A), indicating that RXRβ is either not expressed or at levels below detection by Western blot. Importantly, nontarget siRNA and RXRβ siRNA did not affect the uptake of Aβ in response to bexarotene (Fig. 5B,D). In contrast, suppression of RXRα by siRNA inhibited the bexarotene-induced increase of Aβ uptake (Fig. 5B,D). These results indicated that RXRα represents the functionally more relevant isoform for Aβ phagocytosis in primary microglia.
Figure 4.
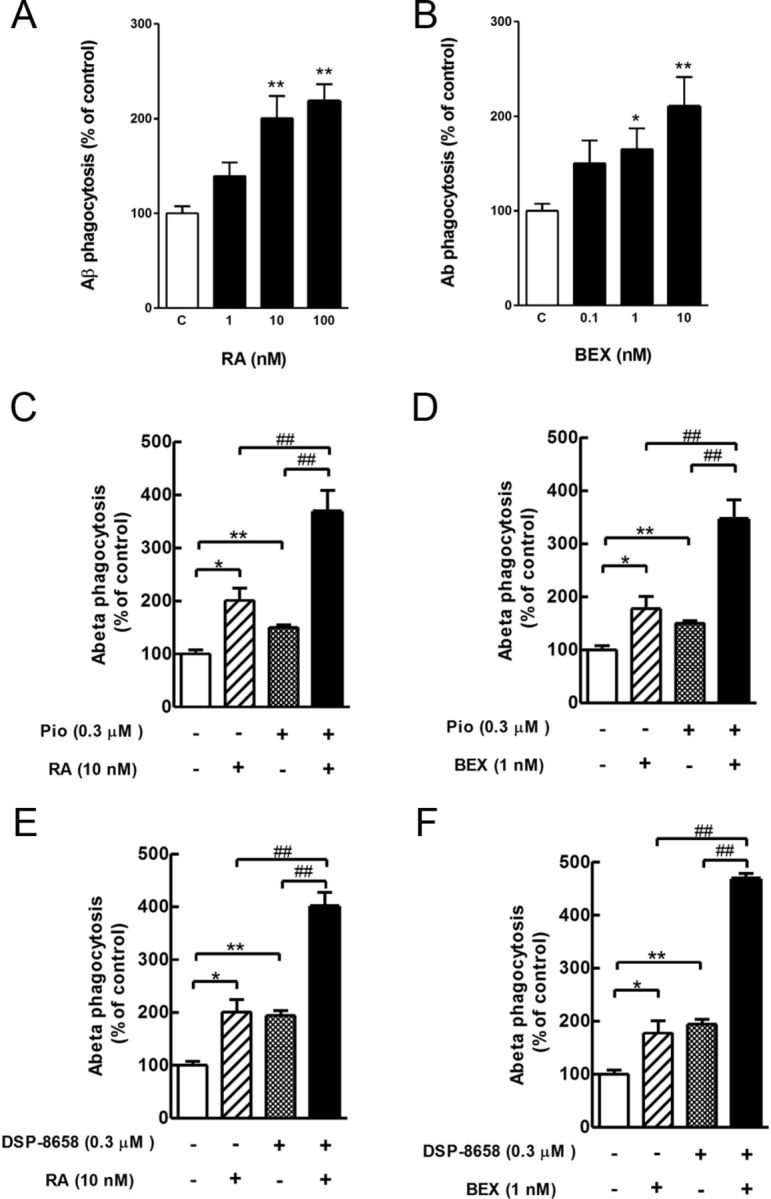
Additive effects dual activation of PPARγ and RXR in primary microglia. Primary microglia were incubated with indicated concentrations of retinoic acid (A; RA) or bexarotene (B; BEX) in the presence of FAM-Aβ (0.5 μm). After 4 h expose to Aβ, intracellular levels of Aβ were determined. RXR agonists enhanced the uptake of Aβ in rat primary microglia in a concentration-dependent manner. The data show the intracellular Aβ uptake as percentage of control (mean ± SEM of n = 3, *p < 0.05, **p < 0.01, one-way ANOVA, Dunnett's multiple comparison test). C–F, Primary microglia was incubated with the combination of pioglitazone (0.3 μm) (C, D; Pio) or DSP-8658 (0.3 μm) (E, F) with the RXR agonists retinoic acid (10 nm) and bexarotene (1 nm) in the presence of FAM-Aβ (0.5 μm). After 4 h expose to Aβ, intracellular levels of Aβ were determined. Coincubation of PPARγ agonists with RXR agonists showed an additive enhancement on Aβ uptake. The data show the intracellular Aβ uptake as percentage of control (means ± SEM of n = 3, *p < 0.05, **p < 0.01 vs control, ##p < 0.01 vs DSP-8658, pioglitazone, retinoic acid, or bexarotene alone, one-way ANOVA, Tukey's post hoc test).
Figure 5.

Effects of RXRα or RXRβ suppression on Aβ phagocytosis in rat primary microglia. A, RXRα and RXRβ detection in rat primary microglia transfected with siRNA for RXRα, RXRβ, or nontarget. B, Primary microglia transfected with siRNA for RXRα or RXRβ were incubated with bexarotene (BEX) in the presence of FAM-Aβ (0.5 μm). After 4 h expose to Aβ, intracellular level of Aβ was determined. Phagocytosis was only suppressed by RXRα siRNA. The data show the intracellular Aβ uptake as percentage of control (mean ± SEM of n = 3, *p < 0.05 vs control, one-way ANOVA, Tukey's post hoc test). C, RXRα and RXRβ detection in murine primary microglia transfected with siRNA for RXRα, RXRβ, or nontarget. D, Primary murine microglia transfected with siRNA for RXRα or RXRβ were incubated with bexarotene (BEX) in the presence of FAM-Aβ (0.5 μm). After 4 h exposure to Aβ, the intracellular level of Aβ was determined. Data show the intracellular Aβ uptake as percentage of control (mean ± SEM of n = 3, *p < 0.05 vs control, one-way ANOVA, Tukey's post hoc test).
DSP-8658 treatment induced in vivo phagocytosis and recruitment of microglia to Aβ plaques
Based on the in vitro data presented above and the superior ability to cross the blood–brain barrier [brain/plasma concentration ratio of 0.13:0.08 for DSP-8658/pioglitazone (Maeshiba et al., 1997)], we chose DSP-8658 for additional in vivo experiments. APP/PS1 transgenic mice received a DSP-8658-enriched diet from 4–6 months of age. Calculated from the monitored daily food intake, each animal received ∼150 mg · kg−1 · d−1 during treatment. So far, evaluation of in vivo Aβ phagocytosis was conducted by immunohistochemical colocalization studies. Recently, our laboratory established a new method using the amyloid dye methoxy-X04 in combination with flow cytometrical measurement of microglial isolated directly from murine brains. For this, mice were injected with methoxy-X04 3 h before they were killed, and microglia were prepared by density gradient fractionation. Methoxy-X04-positive CD11b+/CD45+ cells were detected in vehicle-treated APP/PS1 but almost absent in wild-type mice (Fig. 6A). Interestingly, DSP-8658 treatment increased the percentage of phagocytosing cells in APP/PS1 mice compared with vehicle-treated mice (Fig. 6B). We also analyzed CD36 expression of CD11b+/CD45+/methoxy-X04-negative (non-phagocytosing) cells and CD11b+/CD45+/methoxy-X04+ (phagocytosing) cells. Although non-phagocytosing microglia did not reveal any difference in CD36 expression, DSP-8658 treatment increased CD36 expression in phagocytosing cells (Fig. 6C). To further validate this result, microglial in vivo phagocytosis was analyzed by confocal immunohistochemistry. We therefore analyzed the colocalization of Aβ deposits with the microglial marker CD11b in 6-month-old control and DSP-8658-treated APP/PS1 mice. Aβ deposits were closely surrounded by CD11b+ microglia (Fig. 6D). Although the number of CD11b+ microglia increased with the deposition, confocal microscopy showed that only a minority of microglial cells actually contained Aβ in nontreated APP/PS1 mice (Fig. 6D–F). However, DSP-8658-treated mice revealed a 30% increase in colocalization (Fig. 6F). In addition, short-term DSP-8658 treatment for 3 d also enhanced CD36 expression in vivo (Fig. 6G). Interestingly, the short-term combined treatment of DSP-8658 and bexarotene for 3 d enhanced CD36 expression in vivo as well (Fig. 6H). These results are consistent with our in vitro observations and suggest that PPARγ agonists can be more effective in AD. In addition, the upregulated expression of CD36 may mediate the observed increase of Aβ phagocytosis in vitro and in vivo.
Figure 6.
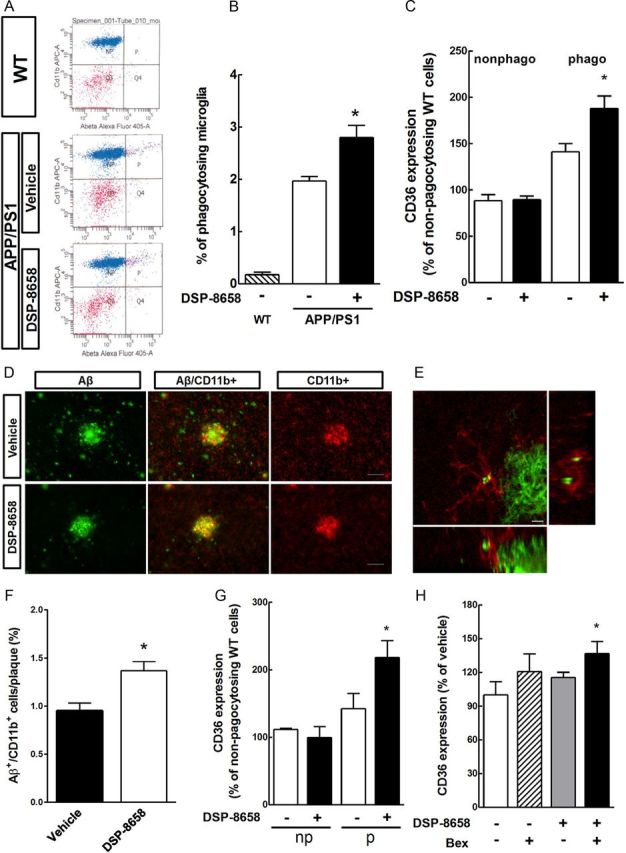
DSP-8658 induces in vivo Aβ phagocytosis and recruitment of microglia to Aβ plaques. APP/PS1 transgenic mice received either DSP-8658 or vehicle containing chow for 3 months. A, Scatter blots of CD11b+/CD45+ isolated microglia after peripheral application of methoxy-X04. Background signal was determined using wild-type (WT) control mice. General microglial background was determined by analysis of unstained, non-injected wild-type mice. B, The percentage of methoxy-X04+/CD11b+/CD45+ cells to CD11b+/CD45+ cells was calculated (mean ± SEM of n = 4, *p < 0.05, Student's t test). C, FACS analysis for CD36 expression was performed in non-phagocytosing (nonphago) and phagocytosing (phago) cells (mean ± SEM of n = 4, *p < 0.05, Student's t test). D, Confocal laser-scanning microscopy detected a higher number of Aβ-plaque-associated and Aβ-containing microglial cells in DSP-8658-treated APP/PS1 transgenic mice compared with vehicle-treated APP/PS1 controls. Scale bar, 50 μm. E, Colocalization of Aβ/Cd11b within microglia visualized by confocal laser-scanning microscopy. Scale bar, 5 μm. F, Ten randomly chosen plaque areas were evaluated for Aβ/Cd11b colocalization per animal (mean ± SEM of n = 4, *p < 0.05, Student's t test). G, APP/PS1 transgenic mice were treated with DSP-8658 for 3 d and injected with methoxy-X04 3 h before analysis. After isolation of microglia, CD36 expression was determined by flow cytometry in non-phagocytosing (np) and phagocytosing (p) cells. Values were normalized to the CD36 expression in untreated microglia from age-matched wild-type mice (mean ± SEM of n = 4, *p < 0.05, Student's t test). H, APP/PS1 transgenic mice were treated with DSP-8658 or bexarotene (Bex) and the combination of these compounds for 3 d. Isolated phagocytosing microglia were analyzed by FACS for CD36 expression per animal (mean ± SEM of n = 4, *p < 0.05, Student's t test).
DSP-8658 reduces brain Aβ levels
To assess whether increased phagocytosis led to an overall reduction or cerebral Aβ levels, cortical and hippocampal levels of Aβ1–40 and Aβ1–42 were determined in APP/PS1 mice after 3 months of DSP-8658 treatment. Determination of cortex Aβ levels revealed 32 and 30% decrease of both SDS-soluble Aβ1–42 and Aβ1–40 levels, respectively, in response to the DSP-8658 treatment (Fig. 7A). DSP-8658 treatment also decreased hippocampal SDS-soluble Aβ1–42 levels (Fig. 7C). RIPA-soluble cortical Aβ1–40 levels were slightly reduced by the same treatment (Fig. 7B,D). In accordance with this, Aβ plaque load determined by thioflavin S staining of sagittal brain sections revealed a decrease in the cortex of DSP-8658-treated APP/PS1 mice (Fig. 7E).
Figure 7.
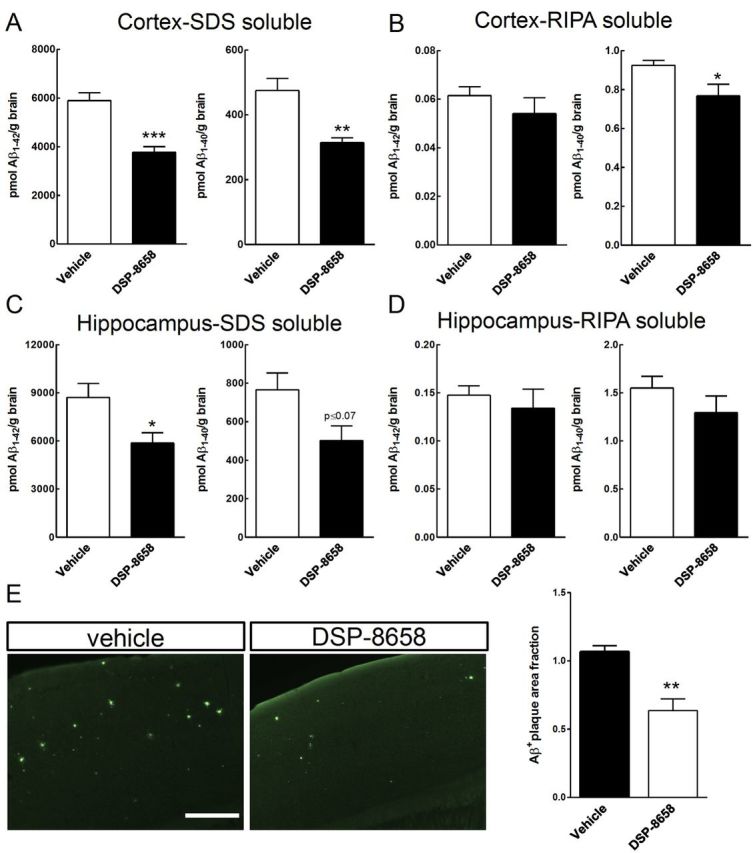
DSP-8658 reduces Aβ burden in the APP/PS1 transgenic mice. APP/PS1 transgenic mice were treated with DSP-8658 in the diet for 3 months. After treatment, cortex and hippocampus were excised and protein was extracted. Concentrations of Aβ1–40 and Aβ1–42 in SDS-soluble (A, C) and RIPA-soluble (B, D) fractions from cortex and hippocampus of APP/PS1 mice were determined by sandwich ELISA. DSP-8658 reduced SDS-soluble Aβ1–40 and Aβ1–42 in cortex and SDS-soluble Aβ1–42 in hippocampus compared with control (mean ± SEM of n = 4, *p < 0.05, **p < 0.01, Student's t test). E, Aβ deposition in the cortex was evaluated by thioflavin S staining and calculated as Aβ-positive area fraction (mean ± SEM of n = 4, *p < 0.05, Student's t test). Scale bar, 250 μm.
Spatial learning of DSP-8658-treated APP/PS1 transgenic mice
Because increased microglial Aβ clearance from the brain may result in improved learning and memory behavior, we assessed spatial memory by the Morris water maze test. Mice were trained to escape to a submerged platform for a maximum of 40 s conducting four trials per day during 8 consecutive days. Data were analyzed with respect to average distance and latency. In DSP-8658-treated APP/PS1 mice, distance traveled and escape latency decreased gradually during training. This decrease in distance and escape latency was more pronounced in DSP-8658-treated APP/PS1 mice than observed in vehicle-treated mice (Fig. 8). A quantitative analysis by calculating the area under the curve (AUC) for distance and latency verified a substantial improvement in response to DSP-8658 treatment. These results indicated that DSP-8658 ameliorated the spatial learning in AD mice.
Figure 8.

DSP-8658 treatment improved spatial memory learning of APP/PS1 transgenic mice. APP/PS1 transgenic mice were treated with DSP-8658 for 3 months. After treatment, all animals were subjected to the Morris water maze test. The time needed to reach the hidden platform (latency, seconds) and distance traveled (distance, centimeters) are depicted over 8 consecutive days. Integrated time and distance traveled (AUC) were determined for the whole observation period. Data represent means ± SEM (n = 9 for vehicle and n = 10 for DSP-8658, **p < 0.01, Student's t test).
Discussion
Impaired cerebral Aβ clearance has been proposed as the leading cause of sporadic AD, the vast majority of cases (Mawuenyega et al., 2010). One of the key functions of microglial cells in the brain is to serve as specialized sensors for brain damage. They represent the first line of defense against invading pathogens and tissue injury (Hanisch and Kettenmann, 2007; Ransohoff, 2009). On pathological stimuli, microglia become activated, migrate to and surround damaged or dead cells, and subsequently clear cellular debris from the area.
In AD, microglia can be found at the site of Aβ deposition, and this assembly is thought to represent an attempt to clear the misfolded and aggregated Aβ peptides from the brain. Aβ itself, however, results in a heterogeneous activation of microglia, including cells that develop a phagocytic phenotype (M1 state or classical activation) and release a variety of pro-inflammatory cytokines and chemokines but also cells that convert to an alternative activation state (M2 state) induced by anti-inflammatory cytokines and chemokines, which is thought to contribute to tissue repair and regeneration (Town et al., 2005; Colton et al., 2006).
However, persisting neuroinflammation and sustained exposure to proinflammatory stimuli compromises microglial clearance functions as AD progresses (Lee and Landreth, 2010). In turn, suppression of inflammation can restore Aβ phagocytosis (Heneka et al., 2010b). There are also indications that inhibition of anti-inflammatory stimuli has beneficial effects on Aβ phagocytosis, β-amyloidosis, and cognitive function (Town et al., 2008).
Activation of PPARγ by endogenous or synthetic ligands modulates insulin sensitivity and exerts profound anti-inflammatory effects in peripheral macrophages and microglia. Less is known, however, about the functional consequences of microglial PPARγ activation under inflammatory stimulation. We hypothesized that the anti-inflammatory action of PPARγ activation would improve microglial clearance function. Currently marketed PPARγ ligands, such as rosiglitazone or pioglitazone, only show a minor blood–brain barrier penetration. They also carry a number of undesirable side effects. Therefore, novel drugs, such as DSP-8658, which acts as a dual PPARα/γ modulator, are currently under clinical development for the treatment of type 2 diabetes and AD. In this study, we investigated the mechanisms and potential of pioglitazone and DSP-8658 to positively modulate microglial Aβ phagocytosis.
Both substances DSP-8658 and pioglitazone enhanced Aβ phagocytosis in primary microglia without affecting its degradation. Because several synthetic PPARγ ligands execute “off-target” actions, we tested the specificity of the observed effects under PPARγ knockdown conditions. Using primary microglia transfected with PPARγ siRNA, we found that neither pioglitazone nor DSP-8658 were able to increase Aβ phagocytosis, thus confirming PPARγ dependency. Of note, this effect was abolished by coincubation with transcriptional or translational inhibitors, suggesting that the observed effect requires de novo protein synthesis.
CD36 is a pivotal target of PPARγ-mediated gene regulation in macrophages and has also been suggested as important receptor for microglial Aβ phagocytosis (Coraci et al., 2002; Zhao et al., 2009). In addition, it mediates the innate immune response caused by fibrillar Aβ, resulting in the proinflammatory secretion of cytokines, chemokines, and reactive oxygen species (El Khoury et al., 2003). Thus, microglia interacts with Aβ through a recently characterized cell-surface receptor complex comprising CD36, α6β1 integrin, and CD47 (Lee and Landreth, 2010). Therefore we tested whether exposure of cells to pioglitazone or DSP-8658 would result in a functionally relevant CD36 expression. DSP-8658 and pioglitazone enhanced CD36 mRNA and protein levels in primary microglia. Importantly, PPARγ agonist-stimulated Aβ phagocytosis was abolished by coincubation of inhibitory CD36 antibody or likewise by transfection of cells with CD36 siRNA. These results indicate clearly that PPARγ agonist-induced upregulation of CD36 promotes microglia-mediated phagocytosis.
PPARγ heterodimerizes with RXRs (Tugwood et al., 1992), and, during ligand activation, the PPAR/RXR heterodimer recruits coactivators and binds to sequence-specific PPAR response elements present in the promoter region of several target genes. Recently, the RXR agonist bexarotene has been reported to facilitate the intracellular degradation of soluble Aβ1–42 in a PPARγ-, liver X receptor-, and apolipoprotein E-dependent manner (Cramer et al., 2012). Here we show that two structurally different RXR agonists, bexarotene and retinoic acid, increase microglial Aβ phagocytosis. Dual activation of both heterodimer partners has been reported previously to show increased efficacy compared with the activation of a single heterodimer partner (Papi et al., 2009). Likewise, the combination of DSP-8658 and pioglitazone with RXR agonists (retinoic acid and bexarotene) showed an additive enhancement of Aβ uptake in the present study. Using siRNA-based knockdown analysis for RXRα or RXRβ, we revealed that RXRα represents the functionally important RXR in primary microglia. Therefore, we suggest that a combination therapy using agonists of PPARγ and RXRα may be more effective in AD.
Several studies analyzed the effect of pioglitazone treatment in animal models of AD. An acute treatment of 10-month-old APPV717I mice with pioglitazone or ibuprofen reduced the number of activated microglia, reactive astrocytes, and overall Aβ deposition in the hippocampus and frontal cortex (Heneka et al., 2005). Yan et al., 2003 found that 6-month treatment of Tg2576 mice with pioglitazone resulted in a modest reduction in soluble Aβ levels with no effect on plaque burden or inflammation. The absence of a substantial effect was postulated to be attributable to poor penetration of pioglitazone into the brain (Maeshiba et al., 1997). DSP-8658 passes the blood–brain barrier more effectively than pioglitazone (data not shown). Therefore, verification of the above described in vitro findings was performed using DSP-8658 in APP/PS1 transgenic mice. Using the amyloid dye methoxy-X04 in combination with flow cytometrical measurement of microglial isolated from brains of adult DSP-8658-treated and untreated mice, we found that DSP-8658 treatment increased the percentage of phagocytosing cells in APP/PS1 mice, confirming our in vitro observations. Moreover, these data were further supported by confocal microscopy showing that only a minority of microglia contained Aβ in nontreated APP/PS1 mice, while DSP-8658-treated mice revealed a 30% increase in colocalization. DSP-8658 finally reduced cortical and hippocampal SDS-soluble Aβ1–42 and Aβ1–40 levels and decreased Aβ plaque load. Increase in phagocytosis and subsequently reduced levels of Aβ were accompanied by a substantial improvement of spatial memory performance.
In summary, PPARγ activation by DSP-8658 or pioglitazone alone or, more effectively, in combination with RXRα activation, enhances microglial Aβ phagocytosis by upregulation of CD36. Moreover, we revealed that DSP-8658 treatment increased microglial Aβ phagocytosis in vivo, resulting in an overall reduction of Aβ levels and improved spatial memory performance. Based on these results, PPARγ/RXR activators that efficiently cross the blood–brain barrier may be considered as future therapeutics for AD.
Footnotes
This research was supported in part by a grant from Dainippon Sumitomo Pharma and German Research Foundation Grant KFO177 (M.T.H.). We are grateful to Drs. C. Pietrzik and S. Weggen for providing antibody IC16.
References
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fleisher-Berkovich S, Filipovich-Rimon T, Ben-Shmuel S, Hülsmann C, Kummer MP, Heneka MT. Distinct modulation of microglial amyloid β phagocytosis and migration by neuropeptides (i) J Neuroinflammation. 2010;7:61. doi: 10.1186/1742-2094-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden AM, Combs CK. β-Amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci. 2006;26:4644–4648. doi: 10.1523/JNEUROSCI.4822-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta. 2007;1771:1031–1045. doi: 10.1016/j.bbalip.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wüllner U, Klockgether T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol. 1999;100:156–168. doi: 10.1016/s0165-5728(99)00192-7. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010a;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm. 2010b;117:919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in t'Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Jäger S, Leuchtenberger S, Martin A, Czirr E, Wesselowski J, Dieckmann M, Waldron E, Korth C, Koo EH, Heneka M, Weggen S, Pietrzik CU. alpha-secretase mediated conversion of the Amyloid Precursor Protein derived membrane stub C99 to C83 limits Abeta generation. J Neurochem. 2009;111:1369–1382. doi: 10.1111/j.1471-4159.2009.06420.x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117:949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshiba Y, Kiyota Y, Yamashita K, Yoshimura Y, Motohashi M, Tanayama S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung. 1997;47:29–35. [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- Papi A, Tatenhorst L, Terwel D, Hermes M, Kummer MP, Orlandi M, Heneka MT. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J Neurochem. 2009;109:1779–1790. doi: 10.1111/j.1471-4159.2009.06111.x. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity. 2009;31:711–721. doi: 10.1016/j.immuni.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer's disease. Neurochem Int. 2001;39:333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JÅ, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992;11:433–439. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009;40:S92–S94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]