

Abstract
Rett syndrome (RTT) is a severe neurological disorder that is associated with mutations in the methyl-CpG binding protein 2 (MECP2) gene. RTT patients suffer from mental retardation and behavioral disorders, including heightened anxiety and state-dependent breathing irregularities, such as hyperventilation and apnea. Many symptoms are recapitulated by the Mecp2-null male mice (Mecp2−/y). To characterize developmental progression of the respiratory phenotype and explore underlying mechanisms, we examined Mecp2−/y and wild-type (WT) mice from presymptomatic periods to end-stage disease. We monitored breathing patterns of unrestrained mice during wake–sleep states and while altering stress levels using movement restraint or threatening odorant (trimethylthiazoline). Respiratory motor patterns generated by in situ working heart–brainstem preparations (WHBPs) were measured to assess function of brainstem respiratory networks isolated from suprapontine structures. Data revealed two general stages of respiratory dysfunction in Mecp2−/y mice. At the early stage, respiratory abnormalities were limited to wakefulness, correlated with markers of stress (increased fecal deposition and blood corticosterone levels), and alleviated by antalarmin (corticotropin releasing hormone receptor 1 antagonist). Furthermore, the respiratory rhythm generated by WHBPs was similar in WT and Mecp2−/y mice. During the later stage, respiratory abnormalities were evident during wakefulness and sleep. Also, WHBPs from Mecp2−/y showed central apneas. We conclude that, at early disease stages, stress-related modulation from suprapontine structures is a significant factor in the Mecp2−/y respiratory phenotype and that anxiolytics may be effective. At later stages, abnormalities of brainstem respiratory networks are a significant cause of irregular breathing patterns and central apneas.
Introduction
Rett syndrome (RTT) is a severe neurological disorder that is frequently (80–90%) associated with mutations in the methyl-CpG binding protein 2 (MeCP2) gene (for review, see Neul and Zoghbi, 2004). The conventional view that loss of MeCP2 during a critical developmental window causes irreversible changes in brain structure and cellular function has been significantly modified with a striking reversal of phenotype when Mecp2 expression is reactivated in an inducible transgenic mouse model (Guy et al., 2007; Cobb et al., 2010; Guy et al., 2011; Lioy et al., 2011). Patients with RTT appear normal at birth but progressively suffer from a number of behavioral disorders, including severe breathing irregularities that can be highly variable, both among affected individuals and within individuals, depending, for example, on the level of behavioral arousal. The fact that patients experience extended periods of normal breathing (e.g., during sleep) suggests that the basis for the respiratory dysfunction is not necessarily within respiratory rhythm generating centers per se. Rather, abnormal respiratory rhythm could be attributable to state-dependent, descending drive to brainstem respiratory networks. Here we test this hypothesis using male mice (Mecp2−/y) that lack the Mecp2 gene product and have clear, progressive respiratory dysfunction as part of their phenotype (for review, see Katz et al., 2009). We focused on the relationship between anxiety levels and respiratory abnormalities because RTT patients have frequent episodes of heightened anxiety and an abnormal physiological response to stress (Sansom et al., 1993; Hagberg, 2002; Mount et al., 2002; Axelrod et al., 2006). Mouse models of RTT similarly show increased anxiety-like behavior (Moretti and Zoghbi, 2006; Fyffe et al., 2008) and an altered stress hormone system (Nuber et al., 2005; McGill et al., 2006; Braun et al., 2012).
We assessed central respiratory control mechanisms by examining individual mice and/or cohorts of mice over a wide development window that covered presymptomatic periods through to end-stage disease. Using whole-body plethysmography, we monitored breathing patterns as well as hypoxic and hypercapnic ventilatory responses in Mecp2−/y and wild-type (WT) mice at different sleep–wake states while systematically altering the levels of stress-related signaling. Furthermore, we tested whether blockade of stress-related signaling could reverse respiratory instabilities and stabilize rhythm. In addition, we compared at different ages rhythmic respiratory activity generated by in situ preparations from WT and Mecp2−/y mice to assess the function of brainstem respiratory networks when they are isolated from the influence of descending suprapontine inputs. Collectively, the data demonstrate that Mecp2−/y mice experience two general stages of respiratory dysfunction through their lifespan. Stress-related modulation of respiratory pattern from suprapontine structures accounts for a significant component of the respiratory phenotype during the early stage of the disease. In the later stage, the respiratory phenotype is dominated by the development of functional abnormalities within brainstem respiratory networks.
Materials and Methods
Animals.
All experimental procedures were approved by the University of Alberta, Faculty of Medicine and Dentistry Animal Welfare Committee. Mecp2 heterozygous female (Mecp2tm1–1Jae) (Chen et al., 2001) and WT male mice were obtained from the Mouse Regional Resource Center (University of California Davis, Davis, CA), and a colony was established at the University of Alberta. B6.129 Mecp2−/y and WT (Mecp2+/y) mice from within the same litter were processed together for comparisons.
Plethysmographic measurements.
Measurements from unrestrained mice were performed in whole-body, Plexiglas plethysmographs that had inflow and outflow ports for the continuous delivery of a steady flow (300 ml/min) of fresh air and removal of expired carbon dioxide. Custom made Plexiglas chambers of various sizes were used to accommodate animals. The large chamber had a volume of 260 ml, a diameter of 5 cm, and a length of 13 cm. The small chamber had a volume of 50 ml, a diameter of 3.5 cm, and a length of 6 cm. Recordings were made in the large chamber (in which mice could move freely) except when indicated. Plethysmographic recordings commenced ∼20 min after placing the mice in the chamber. Pressure changes were detected with a pressure transducer (model DP 103; Validyne), signal conditioner (CD-15; Validyne), an analog-to-digital board (Digidata 1322A), and data acquisition (Axoscope) and analysis (Clampfit) software (Molecular Devices). It should be noted that our plethysmographic recording setup is effective for studying respiratory frequency (fR) and detection of apneas. However, it is not designed for precise quantification of tidal volume (VT; milliliters per grams). The physical principle underlying whole-body plethysmography is the detection of pressure changes in the chamber resulting from the heating and humidification of inspired gas. However, tidal volume measurements may also be influenced by gas compression effects related to the airway resistance. Because of these limitations, our whole-body plethysmographic system only provided semiquantitative measurements of VT and detection of changes relative to control state. As a result, our measurements of minute ventilation (VE; milliliters per minutes per grams), which equates to fR × VT, are also semiquantitative and only reported relative to control. An apnea is defined as the absence of airflow for a period equivalent or greater than two complete respiratory cycles. The chemoreceptive responses (1 min after gas exchange) to changed O2 or CO2 were assessed by switching from room air to hypoxia (8% O2) or hypercapnia (5% CO2) with a flow rate of 3 L/min. Note that the flow of room air was first elevated to 3 L/min, animals were allowed to settle, and then the gas composition was changed without any change in airflow or any other noticeable event that was coincident with the gas switching. It took ∼12 s for the new gas to equilibrate within the large chamber, as confirmed by measurements of oxygen content with Oxychek instrumentation (Critikon). Rectal temperature was measured before and after plethysmographic recordings (model 8402-10 thermistor; Cole-Parmer Instrument Company). Body temperature was maintained in both WT and Mecp2−/y mice during the 1–2 h recording sessions without a significant change (<0.2°C, n = 3 each).
Radio telemetry for monitoring sleep–wake states.
Cerebral cortical electroencephalogram (EEG) and neck electromyogram (EMG) signals were measured by telemetry and used to identify and confirm the reliability of visual inspection of sleep–wake states in Mecp2−/y and WT mice. Combinations of high-amplitude EMG activity and low-amplitude EEG indicated wakefulness. Quiet wakefulness was associated with irregular EEG, and active wakefulness (animals actively walking and turning within the chamber) was associated with regular theta rhythm EEG. Lower levels of EMG activity with regular theta (5–9 Hz) rhythm corresponded to rapid eye movement (REM) sleep. Lower levels of EMG activity with high-amplitude delta (1–4 Hz) waves were classified as non-rapid eye movement (NREM) sleep (Steriade et al., 1993; Brankack et al., 2010). With visual inspection only, one could not reliably differentiate REM from NREM sleep. Thus, we only visually separate the sleep from wake state. Mice were anesthetized with isoflurane (0.5–1% in 5% CO2–95% O2) and instrumented with F20 radio telemetry probes (Data Sciences International) as described previously (Stephenson et al., 2001). Briefly, midline incisions were made in the scalp and abdomen to expose the skull and peritoneal cavity, respectively. Body temperature was maintained at 37°C using a heating pad. The radio transmitter was inserted into the peritoneal cavity and loosely sutured to the rectus abdominus muscle. The electrode leads from the implant were tunneled from the peritoneal cavity and led subcutaneously to the head. The muscle and skin of the abdomen were closed. An EMG electrode was sutured to the neck muscle. A hole was drilled into the skull, and an EEG electrode was placed ∼2 mm to the right and 2 mm anterior to bregma. Dental acrylic was used to anchor the electrodes to the skull. The mice were allowed to recover for at least 1 week before recordings were made. Animals were placed in the plethysmograph from where transmitted neck EMG and EEG signals were recorded continuously while monitoring respiration. Mice slept ∼30% of the 1–2 h period per day in the plethysmograph (n = 3 each for WT and Mecp2−/y).
Measurement of stress-related behavior and serum corticosterone.
Stress-related behavior was monitored in conjunction with recordings of respiratory patterns. Indication of stress was monitored by the accepted criteria of counting fecal boli deposited in the first 10 min after a mouse is put into the plethysmographic chamber (Calvo-Torrent et al., 1999). In addition, serum corticosterone measurements (collected between 12:00 P.M. and 2:00 P.M.) were obtained from WT and Mecp2−/y after exposure to a stress-inducing protocol for 1 h. Specifically, corticosterone levels were obtained from four experimental groups: (1) in the home cage or large chamber (260 ml volume); (2) in the more restraining confines of the small recording chamber (50 ml volume); (3) exposure to 0.5 μl trimethylthiazoline (TMT) (Phero Tech) (TMT is extracted from fox feces and is an experimental model proven to cause acute anxiety and increased corticosterone levels (Blanchard et al., 2003; Vendruscolo et al., 2006) in the large chamber; and (4) after the administration of the corticotropin releasing hormone receptor 1 (CRHR1) antagonist antalarmin (10 mg/kg, i.p.), animals were put back to the home cage for 20 min and then put into the large chamber or small chamber for 1 h. Antalarmin was dissolved in 100% DMSO at a concentration of 60 mg/ml and then diluted with saline to a 3 mg/ml solution for injection. Mice were killed by rapid decapitation, and the trunk blood was collected in prechilled tubes containing EDTA. The blood was centrifuged for 10 min, and serum was collected and frozen at −80°C until analyzed. Total serum corticosterone concentration was measured using ELISA kits as per the instructions of the manufacturer (Immunodiagnostics Systems). In each assay, duplicate wells were assigned for each sample. A microplate reader was used to measure signal intensity from the wells. A standard curve was generated using the KC Junor software (Bio-Tek).
In situ working heart–brainstem preparation.
The in situ decerebrated working heart–brainstem preparation (WHBP) generates a robust respiratory discharge (Paton, 1996) and is well suited for analyzing basic respiratory rhythmogenesis in reduced adult rodents without modulatory state-dependent inputs from suprapontine structures. We followed our previously described protocol (Ren et al., 2006). Briefly, mice were anesthetized with isoflurane, submerged in ice-cold oxygenated perfusate, decerebrated, and transected caudal to the diaphragm. The torso and brainstem were then transferred to a recording chamber, in which the descending aorta was cannulated with a double lumen cannula (one line to deliver perfusate, the second to monitor blood pressure) and perfused with saline [125 mm NaCl, 4 mm KCl, 1.25 mm MgSO4, 24 mm NaHCO3, 1.25 mm KH2PO4, 2.5 mm CaCl2, 10 mm d-glucose, and 1.3% Ficoll (Sigma) bubbled with 95% O2–5% CO2 at a flow rate of 15–22 ml/min] sufficient to generate and maintain arterial pressure at 60 mmHg (<8 min from initial submersion). Once perfusion was initiated and arterial pressure was stabilized, the preparation was gradually warmed to 30°C by heating the perfusate. The preparation was then allowed to stabilize for 1 h, during which time the left phrenic nerve was dissected to monitor inspiratory activity (frequency and burst amplitude). After the 1 h stabilization period, baseline respiratory output was recorded. Respiratory rhythm in WHBP was monitored from the phrenic nerve, which was placed over two platinum hook electrodes. Signals were amplified, rectified, low-pass filtered, and recorded to a computer, using an analog-to-digital converter (Molecular Devices Digidata 1200) and data acquisition (Axoscope) and analysis (Clampfit) software.
Data analyses.
Data are expressed as mean ± SEM. The coefficient of variation of breathing frequency was calculated by dividing the SD by arithmetic mean of breathing frequency. The respiratory parameters were measured over a period of at least 5 min for both in vivo and WHBP except for the chemoreceptive experiments in which we analyzed the reflex response to hypoxia and hypercapnia for 1 min. For minute ventilation measurements, the values were reported as mean relative to WT control during quiet wakefulness. SigmaStat 3.5 (Systat Software) was used to perform statistical analyses. In intact animals and WHBP experiments, the significance of changes in the respiratory parameters, stress hormone corticosterone, fecal boli, and body weight were evaluated by a one-way ANOVA with Tukey's post hoc test (multiple groups) or t test (two groups). Statistical comparisons of data were performed only after verification of normality. The analysis of survival rates was made with the Kaplan–Meier log-rank test. Correlation of respiratory phenotype against measures of corticosterone was examined by Spearman's rank order test, with a correlation coefficient close to 1, and p < 0.001 indicating a strong positive correlation. p < 0.05 was taken to indicate a significant difference between means.
Results
Mecp2−/y mice show progressive respiratory dysfunction in vivo
We initiated the study by performing a longitudinal analysis of the respiratory pattern of individual mice. We observed that Mecp2−/y mice express two stages of respiratory dysfunction. Figure 1A shows representative plethysmographic traces of WT mice during quiet wakefulness and sleep. The recording in Figure 1A is from a postnatal day 32 (P32) WT mouse. Respiratory patterns were similar among WT animals of similar ages (P32–P115). Recordings from Mecp2−/y mice under similar conditions are shown in Figure 1B (all recordings in a large 260 ml chamber). In initial experiments, mice were instrumented for EEG recordings from the cerebral cortex and EMG from neck muscle in combination with whole-body plethysmographic recordings (Fig. 1C; n = 3 each for WT and Mecp2−/y mice) to verify that the subsequently used observational methods of demarcating sleep from wake state were reliable. All of the Mecp2−/y mice studied showed a markedly abnormal breathing pattern by ∼1–2 months of age. At the early stage, Mecp2−/y mice had an unstable breathing pattern characterized by fast breathing interposed by decreased frequency of breathing and marked apneas during quiet wakefulness but not during sleep. Thus, in this study, we defined early stage of respiratory dysfunction as marked apneas during wakefulness but not during sleep. We did not analyze breathing patterns during gross body movements (i.e., walking or turning around within the chamber). Over time, Mecp2−/y mice began to display apneas during sleep as well as wakefulness. The onset and progress of respiratory dysfunction varied among the cohort of Mecp2−/y mice examined. In the majority (9 of 13) of mice, the onset of apneas during wakefulness appeared at approximately P32. Sleep apneas started appearing 1–3 weeks later and became more frequent and longer lasting with age (Fig. 1D shows apneas before death). Two Mecp2−/y mice (of 13 animals tested) had clear wakefulness apnea by P26, sleep apnea from day P34, and died at P43 and P45. The remaining two Mecp2−/y studied had marked wakefulness apnea from P46 to P50. All 13 Mecp2−/y mice died at 6–12 weeks of age. The population data showing age-dependent differences between Mecp2−/y and WT mice in apneas per minute, respiratory frequency, coefficient of variation (f), and relative minute ventilation are shown in Figure 1E. Because of the fact that the onset of the respiratory phenotype varied between individual mice, the population data for age-dependent changes in respiratory parameters are normalized, with day 0 defined as the day immediately before the onset of marked apnea during wakefulness.
Figure 1.

Age- and state-dependent changes in the respiratory patterns of WT and Mecp2−/y mice. Representative whole-body plethysmographic measurements of the respiratory activity of unrestrained WT (A, P32) and Mecp2−/y (B, from one animal of different developmental ages P32, P40, and P48) mice during quiet wakefulness (left) and sleep (right). C, Sleep–wake stage recordings showing plethysmographic traces (Resp), EEG, and neck EMG from Mecp2−/y mice at early and late stages of disease progression. D, Plethysmographic recordings from an Mecp2−/y mouse made during the final few minutes before terminal apnea. E, Population data showing the number of apneas per minute, respiratory frequency, coefficient of variation of respiratory frequency, and minute ventilation (relative to WT control during quiet wakefulness) at different ages and wake–sleep states. n = 7–13 for each data point; +p < 0.05, *p < 0.01, and #p < 0.001 compared with day 0 and between WT and Mecp2−/y mice. Note that days plotted are normalized to start just before the onset of clear wakefulness apneas (the onset varied with age among the population of Mecp2−/y mice studied; for details, see Results).
Altered chemoreceptive responses in Mecp2−/y mice
Chemoreceptive responses to altered levels of ambient O2 and CO2 were also examined via plethysmography. Note that our system allowed for rapid gas exchange (12 s), and we focused our measurements on the primary response (1 min duration after allowing 12 s for equilibration of gases) to hypoxia and hypercapnia. Representative traces from WT and Mecp2−/y mice (during the early and late stages of respiratory dysfunction) in room air and after exposure to hypoxia (8% O2) and hypercapnia (5% CO2) are shown in Figure 2A (during quiet wakefulness). Population data showing frequency (relative to room air) and coefficient of frequency variation are shown in Figure 2B. There was a trend for an increased breathing frequency and coefficient of variation in response to hypoxia and hypercapnia in Mecp2−/y (n = 5) relative to WT (n = 6) mice at early stages, but the differences were not statistically significant. However, by late stages of respiratory dysfunction, Mecp2−/y mice had a significantly altered response to hypoxia (8% O2, n = 5), as evident by significantly increased relative frequency (2.1 ± 0.12, F(2,13) = 12, p = 0.001) and coefficient of variation of respiratory frequency (0.33 ± 0.04, F(2,13) = 4.7, p = 0.02) compared with WT (relative frequency, 1.42 ± 0.07; coefficient of variation of respiratory frequency, 0.18 ± 0.02). Similarly, Mecp2−/y mice had a significantly altered response to hypercapnia (5% CO2, n = 5) as evident by significantly increased relative frequency (1.84 ± 0.15, F(2,13) = 6.8, p = 0.012) and coefficient of variation of respiratory frequency (0.31 ± 0.04, F(2,13) = 9.3, p = 0.003) compared with WT (relative frequency, 1.35 ± 0.08; coefficient of variation of respiratory frequency, 0.14 ± 0.02).
Figure 2.

Ventilatory response to hypoxia and hypercapnia. A, Representative whole-body plethysmographic measurements of breathing from a P52 WT, a P40 Mecp2−/y (with an early-stage of respiratory dysfunction), and a P52 Mecp2−/y (with a late-stage of respiratory dysfunction) mouse in room air, hypoxia (8% O2), or hypercapnia (5% CO2). B, Population data showing the responses of WT (n = 6), early-stage (n = 5), and late-stage (n = 5) Mecp2−/y mice to hypoxia and hypercapnia; frequency (relative to room air) and the coefficient of variation of respiratory frequency are plotted. Relative frequency (frequency in room air delineated as 1) was compared because the baseline respiratory frequency was slow in room air in the late-stage Mecp2−/y mice. NSp ≥ 0.05, +p < 0.05, *p < 0.01 and between two groups.
CRHR1-mediated, heightened anxiety and stress in Mecp2−/y mice
Typically, the size of plethysmographic chambers is kept to a minimum when recording rodent respiratory patterns to maximize the signal-to-noise ratio. However, we noted that, compared with other mouse strains, many of the Mecp2−/y mice showed excessive hyperventilation, followed by apneas when placed in the confining 50 ml chamber, suggesting heightened anxiety. This anxious, hyperventilatory–apneic behavior was markedly decreased when animals were placed in the larger 260 ml plethysmographic chamber. These observations in conjunction with past reports of heightened anxiety in Mecp2−/y and Mecp2308/y mice (Gemelli et al., 2006; Fyffe et al., 2008; Adachi et al., 2009) led us to investigate further the stress response and how it relates to respiratory dysfunction in the mouse model. First, we measured stress-related behavior (fecal bolus) in WT and Mecp2−/y mice (early-stage respiratory phenotype) under the following conditions: basal conditions (in a large plethysmographic chamber); restraint stress (in a smaller plethysmographic chamber); stress induced by exposure to TMT in large chambers; and placement in large or small chambers 20 min after administration of the CRHR1 antagonist antalarmin (10 mg/kg, i.p.). Figure 3A shows the number of fecal boli counted in the first 10 min after a mouse was put in the chamber for plethysmographic recordings. Mecp2−/y mice (4.4 ± 0.53/10 min, n = 7, t(12) = 3.4, p = 0.006) deposited significantly more fecal boli than WT (2.3 ± 0.36/10 min, n = 7) in the large plethysmographic chamber. Within the smaller chamber, both WT (4.2 ± 0.6/10 min, n = 6, F(3,21) = 6.5, p = 0.02) and Mecp2−/y (7.2 ± 0.6/10 min, n = 6, F(3,21) = 15, p = 0.005) mice significantly increased fecal boli deposition relative to the larger chamber. More fecal boli were observed in Mecp2−/y relative to WT (t(10) = 3.5, p = 0.005) mice in the small chamber. Exposure to the TMT odor induced a similar significant increase in fecal bolus deposition in WT (4.0 ± 0.37/10 min, n = 6, F(3,21) = 6.5, p = 0.037) and Mecp2−/y (6.2 ± 0.48/10 min, n = 6, t(11) = 2.4, p = 0.035) mice as the smaller chamber, with the effect on Mecp2−/y mice being significantly greater than in WT mice (t(10) = 3.6, p = 0.005). In the final paradigm, WT and Mecp2−/y were administered antalarmin (10 mg/kg, i.p.) before being placed in the large plethysmographic chamber (n = 3) or small chamber (n = 3). This had no effect on fecal boli deposition in WT mice in large chamber but decreased the fecal boli in small chamber (t(7) = 2.4, p = 0.049). However, antalarmin pretreatment decreased the fecal boli deposition by Mecp2−/y mice in both large and small chambers to a level (2.5 ± 0.43/10 min, n = 6) comparable with WT mice in large chambers. Antalarmin data from large and small chambers were pooled together in counts of fecal boli because they were not significantly different.
Figure 3.

Stress as measured by fecal boli deposition and corticosterone levels in WT and Mecp2−/y mice (early stage). A, Number of fecal boli deposited within the first 10 min after mice were placed in large (n = 7 each for WT and Mecp2−/y groups) or small (n = 6 each for WT and Mecp2−/y groups) plethysmographic chambers, exposed to TMT (0.5 μl; n = 6 each for WT and Mecp2−/y groups) or administered antalarmin before placement in large or small chambers (10 mg/kg, i.p., n = 6 each for WT and Mecp2−/y groups; among them, n = 3 each for large and small chamber). B, Blood corticosterone levels after mice were taken from their home cages (n = 3 each for WT and Mecp2−/y groups), after 1 h in large (n = 3 each for WT and Mecp2−/y groups) or small (n = 6 each for WT and Mecp2−/y groups) plethysmographic chambers, exposed to TMT (0.5 μl, n = 5 each for WT and Mecp2−/y groups) or administered antalarmin before placement in a chamber (10 mg/kg, i.p., n = 6 each for WT and Mecp2−/y groups; among them, n = 3 each for large and small chamber). NSp ≥ 0.05, +p < 0.05, and *p < 0.01 between two groups.
The experimental protocols were repeated and the stress-related hormone corticosterone in blood samples isolated from Mecp2−/y and WT mice was measured 1 h after removing the animal from the plethysmograph chamber (population data shown in Fig. 3B). Note that corticosterone data from animals placed in their home cage were pooled with data from animals placed in the large chamber (Fig. 3B, large) because there was no significant difference between them. These data (home cage and large chamber) pooled for the WT animals were used as the reference point (78 ± 10 ng/ml, n = 6) for all other experimental groups. The corticosterone levels of Mecp2−/y mice taken from their home cages (n = 3) or after spending 1 h in the large plethysmographic chamber (n = 3) were significantly elevated (122 ± 12 ng/ml, t(10) = 2.9, p = 0.017) relative to similarly treated WT mice. Corticosterone levels increased significantly in response to restraint stress in the small plethysmographic chamber (119 ± 11 ng/ml, n = 6, F(3,19) = 6.4, p = 0.046) or to TMT exposure (131 ± 11 ng/ml, n = 5, F(3,19) = 6.4, p = 0.012) in WT. Similarly, corticosterone levels increased significantly in response to restraint stress in the small plethysmographic chamber (176 ± 19 ng/ml, n = 6, t(10) = 2.5, p = 0.032) or to TMT exposure (208 ± 16 ng/ml, n = 5, F(3,19) = 14, p = 0.004) in Mecp2−/y mice. The increase in the mutant mice was significantly greater than in WT mice in both the small (t(10) = 2.7, p = 0.023) or large (t(8) = 4, p = 0.004) chambers when exposed to TMT. Twenty minutes after administration of antalarmin, WT and Mecp2−/y mice were placed in the large (n = 3) or small (n = 3) chamber for 1 h. This had no effect on blood corticosterone levels in WT mice in the large chamber but decreased the corticosterone concentration when placed in the small chamber (t(7) = 2.4, p = 0.049). However, antalarmin pretreatment decreased the corticosterone level in Mecp2−/y mice in both large and small chambers to a level (84 ± 12 ng/ml, n = 6) comparable with WT mice in large chambers. Corticosterone data from animals placed in large and small chambers after antalarmin were pooled because they were not significantly different.
Stress-related respiratory dysfunction in Mecp2−/y mice in vivo
To correlate the respiratory phenotype with the analyses of stress-related responses, we performed plethysmographic recordings from Mecp2−/y and WT mice under basal conditions (in the large chamber), with restraint stress (in the small chamber), after exposure to TMT (0.5 μl), and when animals were placed in large or small chambers after administration of the CRHR1 antagonist antalarmin (10 mg/kg, i.p.). As shown in representative plethysmographic recordings in Figure 4A, Mecp2−/y mice with the early-stage respiratory phenotype had an unstable breathing pattern during wakefulness in the large plethysmographic chamber (n = 6; apneas, 4.2 ± 0.4/min, t(10) = 5.1, p < 0.001; coefficient of frequency variation, 0.44 ± 0.04, t(10) = 3.4, p = 0.007) compared with WT (n = 6, apnea, 1.8 ± 0.26/min; coefficient of frequency variation, 0.27 ± 0.03). Even WT had irregular breathing pattern when placed in the small plethysmographic chamber (n = 6; apnea, 3.9 ± 0.38/min, F(3,19) = 7.2, p = 0.004; coefficient of frequency variation, 0.41 ± 0.04, t(10) = 3.1, p = 0.011) or when exposed to TMT (n = 5; apnea, 3.5 ± 0.47/min, F(3,19) = 7.2, p = 0.027; coefficient of frequency variation, 0.43 ± 0.05, F(3,19) = 5.3, p = 0.04). Similarly, these breathing pattern irregularities were accentuated when placed in the small plethysmographic chamber (n = 6; apnea, 7.8 ± 0.87/min, F(3,19) = 12.5, p = 0.018; coefficient of frequency variation, 0.64 ± 0.05, F(3,19) = 11, p = 0.048) or when exposed to TMT (n = 5; apnea, 7.7 ± 1.15/min, F(3,19) = 12.5, p = 0.019; coefficient of frequency variation, 0.68 ± 0.06, F(3,19) = 11, p = 0.016) in Mecp2−/y mice. The increase in these breathing pattern irregularities in mutant mice was significantly greater than in WT mice in both small (t(10) = 3.9, p = 0.003) and large (t(8) = 3.4, p = 0.009) chamber after exposure to TMT. Note that these data have implications for interpreting the degree of respiratory abnormalities in plethysmographic studies of RTT mouse models because the severity of the respiratory pattern disturbance will relate to amount of stress associated with the measurement apparatus. Administration of antalarmin to WT increased the regularity of respiratory pattern in the small chamber (apnea, 2.2 ± 0.42/min, F(3,19) = 7.2, p = 0.021; coefficient of frequency variation, 0.27 ± 0.04, t(10) = 2.8, p = 0.019), without a significant difference in the large chamber. Administration of antalarmin to Mecp2−/y increased the regularity of respiratory pattern (n = 6; apnea, 2.4 ± 0.45/min, F(3,19) = 12.5, p < 0.001 compared with the small chamber; t(10) = 3, p = 0.014 compared with big chamber; coefficient of frequency variation, 0.34 ± 0.04, F(3,19) = 11, p = 0.002 compared with small chamber). After antalarmin, there is no significant difference in the regularity of respiratory pattern (apnea, t(10) = 0.4, p = 0.69; coefficient of frequency variation, t(10) = 1.2, p = 0.25) between WT and Mecp2−/y. Population data are shown in Figure 4B for the number of apneas and coefficient of variation of frequency of Mecp2−/y mice during early-stage disease under the various experimental conditions. We then plot the severity of the respiratory phenotype (i.e., apneas per minute and coefficient of variation of frequency) against the measure of stress (blood corticosterone level) in Mecp2−/y mice during early-stage disease as measured under the various stress-inducing experimental conditions (n = 20). The correlation coefficients were 0.87 and 0.83 for apneas per minute and the coefficient of variation of respiratory frequency, respectively (Fig. 4C; p < 0.001). All data demonstrate that respiratory abnormalities correlate with the level of stress (as indicated by blood corticosterone levels).
Figure 4.

Respiratory patterns of WT and early-stage Mecp2−/y mice in response to stress. A, Representative whole-body plethysmographic recordings of Mecp2−/y mice during wakefulness at early stage in the large chamber, the small chamber, during exposure to TMT (0.5 μl, in the large chamber), or treatment with antalarmin (Anta; 10 mg/kg, i.p., in the small chamber at 40 min after drug administration). B, Population data showing apneas per minute and coefficient of variation of frequency for WT and early-stage Mecp−/y mice during quiet wakefulness in large (n = 6 each) and small (n = 6 each) plethysmographic chambers, exposure to TMT (in the large chamber, n = 5 each) or administration of antalarmin before placement in the large (n = 3 each) or small chamber (n = 3 each). NSp ≥ 0.05, +p < 0.05, and *p < 0.01 between two groups. C, Correlation between the severity of respiratory dysfunction (apneas minute, coefficient of variation of frequency) and measures of stress (blood corticosterone level) in Mecp2−/y mice. Data from large chamber (n = 3), small chamber (n = 6), TMT exposure (in large chamber, n = 5), and antalarmin treatment (in large and small chamber, n = 3 each) pooled together for analysis. Correlation coefficient was 0.87 and 0.83 for apneas per minute and coefficient of variation of respiratory frequency against blood corticosterone level, respectively, with p < 0.001.
Figure 5A shows the irregular breathing pattern of a Mecp2−/y mouse at later stages when there were clear apneas during sleep. Figure 5B shows population data for the number of apneas and coefficient of variability during quiet wakefulness and sleep in Mecp2−/y mice with late-stage respiratory phenotype. Administration of antalarmin (10 mg/kg, i.p., n = 5) partially decreased the number of apneas (F(2,16) = 22, p = 0.007) and the irregularity of breathing (F(2,16) = 18, p = 0.024) during wakefulness in Mecp2−/y mice. In the presence of antalarmin, Mecp2−/y mice (n = 5) still had an irregular respiratory pattern (apnea, 3.9 ± 0.88/min, F(2,16) = 22, p = 0.048; coefficient of frequency variation, 0.45 ± 0.07, F(2,16) = 18, p = 0.047) compared with WT (n = 9; apnea, 1.7 ± 0.21/min; coefficient of frequency variation, 0.27 ± 0.023) mice during wakefulness. Furthermore, administration of antalarmin did not significantly decrease the number of apneas (F(2,16) = 16, p = 0.86) and the irregularity of breathing (F(2,16) = 13, p = 0.44) during sleep in Mecp2−/y mice (n = 5).
Figure 5.

Respiratory patterns of late-stage Mecp2−/y mice in response to treatment of CRHR1 blocker antalarmin. A, Representative whole-body plethysmographic recordings of Mecp2−/y mice during sleep at late stage in a large chamber under control conditions and 40 min after administration of antalarmin (Anta; 10 mg/kg i.p.). B, Population data showing apneas per minute and coefficient of variation for Mecp2−/y mice during quiet wakefulness and sleep at late stage in large plethysmographic chambers with or without antalarmin pretreatment. n = 9 for WT, n = 5 for Mecp2−/y; NSp ≥ 0.05, +p < 0.05, *p < 0.01, and #p < 0.001 between two groups.
We next tested whether chronic antalarmin treatment (single daily intraperitoneal injection of 10 mg/kg) for 20 d (starting at P32) could prolong the life of Mecp2−/y mice. As shown in Figure 6A, there was a modest but significant increase (p = 0.018, Kaplan–Meier log-rank test) in the lifespan of the antalarmin-treated Mecp2−/y group to P75 ± P4 (n = 13) from P63 ± P2 (n = 25) in the untreated/vehicle-treated group. Antalarmin treatment had no significant effect on the weight loss that occurs with age in Mecp2−/y mice (Fig. 6B).
Figure 6.

Effects of antalarmin treatment on lifespan and weight gain in WT and Mecp2−/y mice. A, Percentage of surviving WT and Mecp2−/y mice with age after treatment with vehicle (5% DMSO in saline) and antalarmin (10 mg/kg, i.p., daily starting at P32). B, Body weight of WT (n = 7), Mecp2−/y mice at P39 and P52 after treatment with vehicle (n = 5) and after treatment of antalarmin (n = 6). NSp ≥ 0.05, +p < 0.05, *p < 0.01, and #p < 0.001 between two groups.
Mecp2−/y mice show progressive respiratory dysfunction in situ
Based on our observations that respiration was normal during sleep and that there was a clear correlation between the degree of respiratory instability and behavioral state (e.g., stress levels) in the early-stage Mecp2−/y mice, we hypothesized that the respiratory dysfunction of Mecp2−/y mice that occurs during the early stage of disease progression was not caused by an inherent problem with respiratory rhythmogenesis. To test this hypothesis, we examined the respiratory pattern generated by the reduced WHBP. The preparation generates a robust respiratory pattern without the influence of behavioral and state-dependent inputs from suprapontine brain structures. As shown in Figure 7, A and B, WHBPs from WT (n = 5) and early-stage Mecp2−/y mice (n = 4) generated regular respiratory patterns as recorded from the phrenic nerve. There was no difference (F(3,13) = 7.3, p = 0.89) in the number of apneas in Mecp2−/y (0.96 ± 0.22/min) and WT (0.67 ± 0.16/min) mice. In contrast, there were more central apneas (F(3,13) = 7.3, p = 0.007 compared with WT, p = 0.039 compared with early stage of Mecp2−/y) in WHBPs isolated from later-stage Mecp2−/y mice (2.2 ± 0.38/min, n = 5), in which apneas occurred during both wakefulness and sleep in intact animals (Fig. 7C). The defects in central rhythmic activity in the WHBP were not affected by administration of antalarmin (1 mg/100 ml, n = 3; apnea, 2.0 ± 0.29/min, F(3,13) = 7.3, p = 0.99). (Fig. 7D). Population data are shown in Figure 7E.
Figure 7.
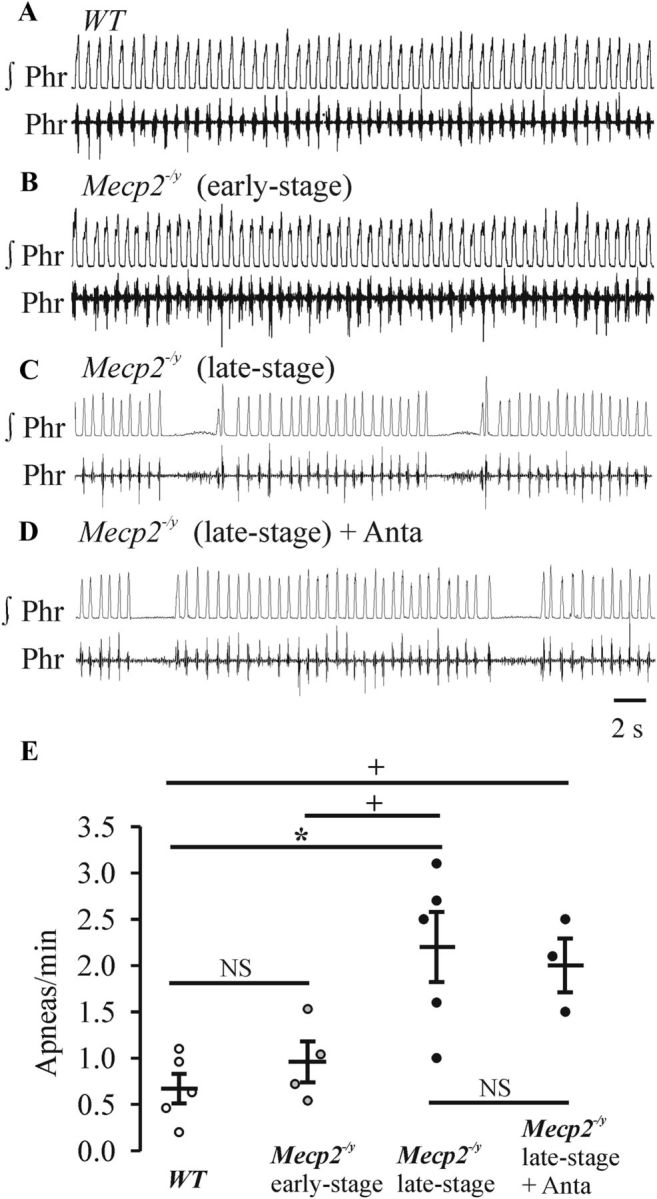
Respiratory rhythm generated by perfused in situ WHBPs of WT and Mecp2−/y mice at early and late stages. Integrated (∫) and raw phrenic (Phr) nerve recordings from a P59 WT male (Mecp2+/y) (A), P36 Mecp2−/y (early-stage) (B), P44 Mecp2−/y (late stage) (C), and same WHBP preparation (D) from after application of CRHR1 antagonist antalarmin (1 mg/100 ml) (C). E, Population data showing the numbers of apneas per minute. n = 5, 4, 5, and 3 for WT, early-stage Mecp2−/y, later-stage Mecp2−/y before antalarmin, and later-stage Mecp2−/y in the presence of antalarmin, respectively. NSp ≥ 0.05, +p < 0.05, and *p < 0.01 between the two groups.
Discussion
This longitudinal study of Mecp2−/y mice characterized disease progression in relation to respiratory behavior and demonstrated two general stages. In the early stage, abnormalities in respiratory pattern were limited to wakefulness, exacerbated by stressful stimuli, and alleviated by the CRHR1 antagonist antalarmin, as well as the removal of descending suprapontine inputs to brainstem respiratory networks. In the later stage, respiratory abnormalities were more severe, no longer state dependent, and manifest at the level of brainstem networks.
Early stage
Consistent with previous studies, the respiratory phenotype of Mecp2−/y mice features a marked breathing dysfunction by 1–2 months of age (Viemari et al., 2005; Ogier et al., 2007; Stettner et al., 2007). Central apneas occur initially only during wakefulness. Data from the WHBP were consistent with normal output from brainstem respiratory networks in the absence of suprapontine structures. There was a clear correlation between respiratory phenotype and the level of anxiety. Even under baseline conditions, Mecp2−/y mice had elevated corticosterone levels and stress-related behavior relative to WT mice. The differences were accentuated with the additional stressors of confinement or exposure to a threatening predator-related odor. Involvement in CRHR1 signaling was evident by the reduction of corticosterone release, stress-related behavior, and respiratory phenotype with administration of antalarmin. These data are consistent with previous reports of heightened anxiety in RTT mouse models (Gemelli et al., 2006; Fyffe et al., 2008; Adachi et al., 2009). Inputs from the amygdala, an origin of major CRH-containing pathways to the brainstem and hypothalamus, and the bed nucleus of the stria terminalis are candidates for providing stress-related modulation of respiration (Swanson et al., 1983; Sakanaka et al., 1986; Gray and Bingaman, 1996; Pitkänen et al., 1997; Chen et al., 2000). This is supported by the markedly enhanced stress levels in mice with targeted Mecp2 deletions within those regions (Fyffe et al., 2008; Adachi et al., 2009).
Similar to antalarmin, several pharmacological interventions reduce respiratory phenotype of RTT mouse models. These include the norepinephrine transporter inhibitor desipramine (Roux et al., 2007; Zanella et al., 2008), benzodiazepines midazolam and diazepam (Voituron and Hilaire, 2011; Abdala et al., 2010), 5-HT receptor antagonist 8-OH-DPAT [8-hydroxy-2(di-n-propylamino)tetralin], GABA uptake inhibitor NO-711 (1-[2([(diphenylmethylene)imino]oxy)ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid hydrochloride), and the positive modulator of GABA receptor function, L-838,417 [3-(2,5-difluorophenyl)-7-(1,1-dimethylethyl)-6-[(1-methyl-1H-1,2,4,-triazol-5-yl)methoxy]-1,2,4-triazolo[4,3-b]pyridazine] (Abdala et al., 2010). It is noteworthy that these agents also have sedative, hypnotic, and/or anxiolytic properties (Brady et al., 1991; De Vry et al., 1993; Dalvi and Rodgers, 1996; Centeno and Volosin, 1997; Rudolph et al., 1999; Santibañez et al., 2006). Thus, it is likely a combination of anxiolytic effects and specific targeting of abnormal neurotransmitter signaling at respiratory nuclei that account for the beneficial effects.
Late stage
As the disease progressed, apneas occurred during sleep and wakefulness. There was also a significant increase in inspiratory frequency and instability of breathing pattern in the presence of hypoxia and hypercapnia. Administration of antalarmin did not alleviate the apneas occurring during sleep. Recordings from the WHBP demonstrated that the apneas originated at the brainstem level and did not depend on disruptive modulatory input from suprapontine structures. These data are consistent with observations by Ward et al., (2011) who selectively deleted and restored Mecp2 expression in mouse brainstem neurons. The baseline hyperventilation of their RTT model was dependent on input from supramedullary inputs, but the abnormal response to hypoxia and profound disturbances of autonomic control at late-stage disease required brainstem Mecp2 deletion.
Stettner et al. (2007) reported central apneas in conjunction with abnormalities of the post-inspiratory vagal nerve discharge in WHBPs isolated from 40-d-old Mecp2−/y mice (Guy et al., 2001). This stage corresponds with the late stage of respiratory phenotype in the Mecp2−/y RTT model and is consistent with the late emergence of a central defect in rhythmogenesis. However, there is a report claiming a central defect in juvenile Mecp2−/y mice well before any signs of apneas or irregular breathing (Viemari et al., 2005). This interpretation is based on recordings of thick medullary slice preparations with undefined rostrocaudal boundaries isolated from P14–P21 Mecp2−/y mice that generate rhythmic bursting in vitro. In vivo, none of the mice from which the medullary slices were isolated demonstrated a respiratory phenotype. Also, there were no statistically significant differences in the number of apneas, frequency, amplitude, or duration of rhythmic motor bursting between Mecp2−/y and WT in vitro preparations. The only significant difference reported was a greater variability of inspiratory frequency in Mecp2−/y slices, which was interpreted as suggesting an early onset respiratory instability.
We propose that a number of factors working in concert with the abnormal stress-related network contribute to the late-stage respiratory phenotype in the mouse model. There are progressive perturbations of multiple neurotransmitter systems in RTT patients and mouse models, including acetylcholine, dopamine, serotonin, glutamate, GABA, and substance P, all of which regulate respiratory rhythmogenesis (for review, see Katz et al., 2009; Roux and Villard, 2010). Surprisingly, deletion of Mecp2 from the majority of a specific class of aminergic neurons does not produce a profound respiratory phenotype or early lethality (Samaco et al., 2008), as proposed from previous studies (Viemari et al., 2005). More likely, the respiratory phenotype results from a combination of multiple neuromodulatory deficits and a wide range of structural and functional changes at multiple levels of brain organization (for review, see Neul and Zoghbi, 2004; Chahrour and Zoghbi, 2007; Weng et al., 2011). This includes reduced brain size and weight and changes in neuronal density and morphology, including reduced dendritic branching, axonal fasciculation, and changes in spine density and morphology. Functionally, there are alterations in neuronal firing properties, synaptic signaling, frequency, and amplitude of synaptic events and basal inhibitory and excitatory synaptic transmission. Deficits in the expression of brain-derived neurotrophic factor in RTT is likely one of the key factors associated with abnormalities in neuronal survival and function, including in key brainstem respiratory nuclei (Katz, 2005; Mironov et al., 2009; Kline et al., 2010; Schmid et al., 2012). Furthermore, loss of Mecp2 expression in astrocytes and microglia appears to play a significant role in the progression of RTT and, of particular relevance, the development of the respiratory and anxiety phenotypes (Maezawa and Jin, 2010; Lioy et al., 2011; Derecki et al., 2012).
Correlation with RTT patient data
Females with an MeCP2 mutation are typically mosaic, with approximately half of the cells randomly expressing the mutant allele. The heterogeneity of the deletion likely explains the spectrum of breathing abnormalities in RTT patients, which includes periods of slow-deep breathing, hyperventilation, central apnea, breath-holding, and Valsalva and Cheynes-Stokes breathing (Julu et al., 2001; Weese-Mayer et al., 2006). The male mouse model with the uniform deletion expresses only a subset of these patterns. With regards to the occurrence of apnea during sleep versus wakefulness, many RTT clinical studies (Lugaresi et al., 1985; Marcus et al., 1994; Julu et al., 2001) report regular breathing during sleep states, similar to what we observe during early-stage disease in the mouse model. However, there are reports of abnormal respiratory rhythms during sleep in a subset of patients (Rohdin et al., 2007; d'Orsi et al., 2009) that correlate with the later stages of Mecp2−/y mouse respiratory phenotype. There are also data showing that breathing irregularities emerge during sleep as patient's age (Schlüter et al., 1995).
Heightened anxiety is reported to occur significantly more frequently in RTT than in other forms of mental retardation (Sansom et al., 1993; Mount et al., 2002). We show a clear correlation between the level of anxiety and the breathing instability in the early stage of the mouse respiratory phenotype. Furthermore, anxiolytic agents normalize respiratory pattern in Mecp2 mouse models. This suggests a potential pharmacological intervention in those patients who have clear elevated anxiety and a respiratory phenotype primarily during wakefulness. A recent human study demonstrated that administration of a well-tolerated CRHR1 antagonist diminished anxiety evoked by 7.5% CO2 exposure (Bailey et al., 2011). The administration of anxiolytics that also act on neuromodulatory systems that are affected in RTT could be advantageous. The challenge is that, because of the mosaic nature of the disorder and potential for age-dependent changes within an individual patient, the specific neuromodulatory imbalances will vary. Longitudinal studies of respiratory patterns during wake–sleep states and reflex responses to hypoxia would differentiate between a stable respiratory phenotype and a progressive one attributable to the development of neuronal–glial abnormalities within medullary respiratory nuclei. In addition, there may be a cohort of RTT patients with a particular mosaic of neuronal defects that leads to a central medullary respiratory defect at an early age that cannot be treated effectively with anxiolytics. It will be of interest to incorporate these concepts into future clinical studies of the respiratory phenotype and the strategic development of effective pharmacological therapies.
Footnotes
This work was supported by the International Rett Syndrome Foundation (IRSF), the Ontario Rett Syndrome Foundation, the Canadian Institutes of Health Research, and the Women and Children's Health Research Institute. J.R. received a fellowship from IRSF. J.J.G. and G.D.F. are Alberta Innovates funded scientists. We thank Sandra Kelly for technical help.
References
- Abdala AP, Dutschmann M, Bissonnette JM, Paton JF. Correction of respiratory disorders in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18208–18213. doi: 10.1073/pnas.1012104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod FB, Chelimsky GG, Weese-Mayer DE. Pediatric autonomic disorders. Pediatrics. 2006;118:309–321. doi: 10.1542/peds.2005-3032. [DOI] [PubMed] [Google Scholar]
- Bailey JE, Papadopoulos A, Diaper A, Phillips S, Schmidt M, van der Ark P, Dourish CT, Dawson GR, Nutt DJ. Preliminary evidence of anxiolytic effects of the CRF(1) receptor antagonist R317573 in the 7.5% CO(2) proof-of-concept experimental model of human anxiety. J Psychopharmacol. 2011;25:1199–1206. doi: 10.1177/0269881111400650. [DOI] [PubMed] [Google Scholar]
- Blanchard DC, Griebel G, Blanchard RJ. Conditioning and residual emotionality effects of predator stimuli: some reflections on stress and emotion. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1177–1185. doi: 10.1016/j.pnpbp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Brady KT, Johnston AL, Cunningham M, Malcolm R. Profiles of hospitalized benzodiazepine abusers. J Psychoactive Drugs. 1991;23:71–72. doi: 10.1080/02791072.1991.10472574. [DOI] [PubMed] [Google Scholar]
- Brankack J, Kukushka VI, Vyssotski AL, Draguhn A. EEG gamma frequency and sleep-wake scoring in mice: comparing two types of supervised classifiers. Brain Res. 2010;1322:59–71. doi: 10.1016/j.brainres.2010.01.069. [DOI] [PubMed] [Google Scholar]
- Braun S, Kottwitz D, Nuber UA. Pharmacological interference with the glucocorticoid system influences symptoms and lifespan in a mouse model of Rett syndrome. Hum Mol Genet. 2012;21:1673–1680. doi: 10.1093/hmg/ddr602. [DOI] [PubMed] [Google Scholar]
- Calvo-Torrent A, Brain PF, Martinez M. Fecal bolus as indicator of stress: effect of predatory stress on sucrose intake and behavior on the plus-maze in male mice. Physiol Behav. 1999;67:189–196. doi: 10.1016/s0031-9384(99)00051-7. [DOI] [PubMed] [Google Scholar]
- Centeno VA, Volosin M. Chronic treatment with desipramine: effect on endocrine and behavioral responses induced by inescapable stress. Physiol Behav. 1997;62:939–944. doi: 10.1016/s0031-9384(97)00255-2. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Müller MB, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotrophin-releasing hormone receptor type-1 (CRF1)-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J Comp Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb S, Guy J, Bird A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans. 2010;38:498–506. doi: 10.1042/BST0380498. [DOI] [PubMed] [Google Scholar]
- Dalvi A, Rodgers RJ. GABAergic influences on plus-maze behaviour in mice. Psychopharmacology (Berl) 1996;128:380–397. doi: 10.1007/s002130050148. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vry J, Benz U, Schreiber R, Traber J. Shock-induced ultrasonic vocalization in young adult rats: a model for testing putative anti-anxiety drugs. Eur J Pharmacol. 1993;249:331–339. doi: 10.1016/0014-2999(93)90530-u. [DOI] [PubMed] [Google Scholar]
- d'Orsi G, Demaio V, Scarpelli F, Calvario T, Minervini MG. Central sleep apnoea in Rett syndrome. Neurol Sci. 2009;30:389–391. doi: 10.1007/s10072-009-0108-9. [DOI] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Gray TS, Bingaman EW. The amygdala: corticotropin-releasing factor, steroids, and stress. Crit Rev Neurobiol. 1996;10:155–168. doi: 10.1615/critrevneurobiol.v10.i2.10. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Julu PO, Kerr AM, Apartopoulos F, Al-Rawas S, Engerström IW, Engerström L, Jamal GA, Hansen S. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85:29–37. doi: 10.1136/adc.85.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM. Regulation of respiratory neuron development by neurotrophic and transcriptional signaling mechanisms. Respir Physiol Neurobiol. 2005;149:99–109. doi: 10.1016/j.resp.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168:101–108. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugaresi E, Cirignotta F, Montagna P. Abnormal breathing in the Rett syndrome. Brain Dev. 1985;7:329–333. doi: 10.1016/s0387-7604(85)80039-5. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus CL, Carroll JL, McColley SA, Loughlin GM, Curtis S, Pyzik P, Naidu S. Polysomnographic characteristics of patients with Rett syndrome. J Pediatr. 1994;125:218–224. doi: 10.1016/s0022-3476(94)70196-2. [DOI] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova E, Hartelt N, Mironova LA, Hasan MT, Kügler S. Remodelling of the respiratory network in a mouse model of Rett syndrome depends on brain-derived neurotrophic factor regulated slow calcium buffering. J Physiol. 2009;587:2473–2485. doi: 10.1113/jphysiol.2009.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Mount RH, Charman T, Hastings RP, Reilly S, Cass H. The Rett Syndrome Behaviour Questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry. 2002;43:1099–1110. doi: 10.1111/1469-7610.00236. [DOI] [PubMed] [Google Scholar]
- Neul JL, Zoghbi HY. Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist. 2004;10:118–128. doi: 10.1177/1073858403260995. [DOI] [PubMed] [Google Scholar]
- Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, Bird A. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:2247–2256. doi: 10.1093/hmg/ddi229. [DOI] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF. A working heart-brainstem preparation of the mouse. J Neurosci Methods. 1996;65:63–68. doi: 10.1016/0165-0270(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE. Organization of intraamygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Ren J, Poon BY, Tang Y, Funk GD, Greer JJ. Ampakines alleviate respiratory depression in rats. Am J Respir Crit Care Med. 2006;174:1384–1391. doi: 10.1164/rccm.200606-778OC. [DOI] [PubMed] [Google Scholar]
- Rohdin M, Fernell E, Eriksson M, Albåge M, Lagercrantz H, Katz-Salamon M. Disturbances in cardiorespiratory function during day and night in Rett syndrome. Pediatr Neurol. 2007;37:338–344. doi: 10.1016/j.pediatrneurol.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Roux JC, Villard L. Biogenic amines in Rett syndrome: the usual suspects. Behav Genet. 2010;40:59–75. doi: 10.1007/s10519-009-9303-y. [DOI] [PubMed] [Google Scholar]
- Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25:1915–1922. doi: 10.1111/j.1460-9568.2007.05466.x. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Möhler H. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K. Distribution and efferent projections of corticotropin-releasing factor-like immunoreactivity in the rat amygdaloid complex. Brain Res. 1986;382:213–238. doi: 10.1016/0006-8993(86)91332-6. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom D, Krishnan VH, Corbett J, Kerr A. Emotional and behavioural aspects of Rett syndrome. Dev Med Child Neurol. 1993;35:340–345. doi: 10.1111/j.1469-8749.1993.tb11646.x. [DOI] [PubMed] [Google Scholar]
- Santibañez M, Gysling K, Forray MI. Desipramine prevents the sustained increase in corticotropin-releasing hormone-like immunoreactivity induced by repeated immobilization stress in the rat central extended amygdala. J Neurosci Res. 2006;84:1270–1281. doi: 10.1002/jnr.21023. [DOI] [PubMed] [Google Scholar]
- Schlüter B, Aguigah G, Buschatz D, Trowitzsch E, Aksu F. Polysomnographic recordings of respiratory disturbances in Rett syndrome. J Sleep Res. 1995;4:203–207. doi: 10.1111/j.1365-2869.1995.tb00216.x. [DOI] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. 2012;32:1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson R, Liao KS, Hamrahi H, Horner RL. Circadian rhythms and sleep have additive effects on respiration in the rat. J Physiol. 2001;536:225–235. doi: 10.1111/j.1469-7793.2001.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Stettner GM, Huppke P, Brendel C, Richter DW, Gärtner J, Dutschmann M. Breathing dysfunctions associated with impaired control of postinspiratory activity in Mecp2-/y knockout mice. J Physiol. 2007;579:863–876. doi: 10.1113/jphysiol.2006.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Vendruscolo LF, Vendruscolo JC, Terenina-Rigaldie E, Raba F, Ramos A, Takahashi RN, Mormède P. Genetic influences on behavioral and neuroendocrine responses to predator-odor stress in rats. Neurosci Lett. 2006;409:89–94. doi: 10.1016/j.neulet.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voituron N, Hilaire G. The benzodiazepine Midazolam mitigates the breathing defects of Mecp2-deficient mice. Respir Physiol Neurobiol. 2011;177:56–60. doi: 10.1016/j.resp.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 is critical within HoxB1-derived tissues of mice for normal lifespan. J Neurosci. 2011;31:10359–10370. doi: 10.1523/JNEUROSCI.0057-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, Ramirez JM. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–449. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]
- Weng SM, Bailey ME, Cobb SR. Rett syndrome: from bed to bench. Pediatr Neonatol. 2011;52:309–316. doi: 10.1016/j.pedneo.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008;160:116–121. doi: 10.1016/j.resp.2007.08.009. [DOI] [PubMed] [Google Scholar]