

Abstract
Pathological examination of dementia with Lewy bodies patients identified the presence of abnormal α-synuclein (αSyn) aggregates in the presynaptic terminals. αSyn is involved in the regulation of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Importantly, αSyn-transgenic mouse and postmortem examination of patients with Parkinson's disease have demonstrated the abnormal distribution of SNARE protein in presynaptic terminals. In this study, we investigated the effects of SNARE dysfunction on endogenous αSyn using Snap25S187A/S187A mutant mice. These mice have homozygous knock-in gene encoding unphosphorylatable S187A-substituted synaptosomal-associated protein of 25 kDa (SNAP-25). The mice displayed a significant age-dependent change in the distribution of αSyn and its Ser129-phosphorylated form in abnormally hypertrophied glutamatergic nerve terminals in the striatum. Electron-microscopic analysis revealed the abnormally condensed synaptic vesicles with concomitant mislocalization of αSyn protein to the periactive zone in the glutamatergic nerve terminals. However, the Snap25S187A/S187A mutant mouse harbored no abnormalities in the nigrostriatal dopaminergic neurons. Our present results suggest that SNARE dysfunction is the initial trigger of mislocalization and accumulation of αSyn, and probably is an important pathomechanism of α-synucleinopathies.
Introduction
α-Synucleinopathies are a subgroup of neurodegenerative diseases including dementia with Lewy bodies (DLB), multiple system atrophy (MSA), and Parkinson's disease (PD). The pathological hallmark of the disorders is the formation of intracellular aggregates composed mainly of α-synuclein (αSyn), which are called Lewy bodies and Lewy neurites (Spillantini and Goedert, 2000; Galvin et al., 2001; Yasuda et al., 2012). It was reported recently that >90% of αSyn aggregates are present in presynaptic terminals in the form of very small deposits of the affected neurons in DLB (Neumann et al., 2002; Kramer and Schulz-Schaeffer, 2007; Schulz-Schaeffer, 2010). However, the mechanisms responsible for presynaptic accumulation of abnormal αSyn remain elusive.
Generally, αSyn is abundantly localized in the presynaptic nerve terminals (Maroteaux et al., 1988; Iwai et al., 1995). While the physiological functions of αSyn have yet to be defined, several lines of evidence implicated this protein in the modulation of neurotransmitter release through the regulation of formation of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex (Chandra et al., 2005; Burré et al., 2010; Darios et al., 2010) and the size of synaptic vesicle pool (Murphy et al., 2000; Cabin et al., 2002; Larsen et al., 2006; Nemani et al., 2010). Vesicle-associated membrane protein-2 (VAMP-2) present in the synaptic vesicles, and syntaxin and synaptosomal-associated protein of 25 kDa (SNAP-25) in the presynaptic plasma membrane form the core SNARE complex, which regulate the docking and fusion of synaptic vesicles to the presynaptic membrane (Südhof, 2004). A recent study showed the physical interaction of αSyn with VAMP-2 to promote SNARE assembly (Burré et al., 2010). Cysteine-string protein-α (CSPα) also participates in SNARE assembly, and mutant mice lacking CSPα displayed impaired SNARE formation and premature death, but both of these phenotypes are counteracted by transgenic expression of αSyn (Chandra et al., 2005; Sharma et al., 2011). However, overexpression of αSyn with no overt toxicity inhibits neurotransmitter release, due to a defective reclustering of synaptic vesicles after endocytosis (Nemani et al., 2010). Additionally, overexpressed αSyn indirectly inhibited SNARE-mediated exocytosis by sequestering arachidonic acid, which upregulates syntaxin and enhances its engagement with SNARE complex (Darios et al., 2010). Importantly, abnormal redistribution of SNARE proteins has been observed in human PD patients and mice overexpressing a truncated form of human αSyn, which showed decreased release of dopamine (DA) in the striatum (Garcia-Reitböck et al., 2010). Therefore, presynaptic SNARE dysfunction is considered an initial pathogenic event in α-synucleinopathies.
Based on the above background, the present study was designed to investigate the effects of impaired SNARE assembly on the distribution of naive αSyn. We exploited Snap25S187A/S187A mutant mice. This strain is resistant to the protein kinase C-mediated phosphorylation of SNAP-25 at Ser187 and represents a concomitant reduction of neurotransmitter release, including serotonin and DA, from the amygdala, and develops convulsive seizures and anxiety-related behavior in general activity and light-and-dark preference tests (Kataoka et al., 2011). The results indicated that dysfunction of SNARE proteins could trigger abnormal localization and accumulation of presynaptic αSyn, possibly representing one of the pathogenic events in DLB or MSA.
Materials and Methods
Mice.
All experimental protocols described in this study were approved by the Animal Experimentation and Ethics Committee of the Kitasato University School of Medicine. Homozygous SNAP-25 knock-in mutant mice bearing S187A mutation and wild-type littermate control mice were generated based on the procedures described in detail by Kataoka et al. (2011). Briefly, embryonic stem cells with the heterozygous Snap25S187A mutant allele were generated and microinjected into C57BL/6N blastocysts to obtain heterozygous mutant mice. Male chimeras were bred with C57BL/6N female mice. After the line was backcrossed 13 times, homozygous mice (designated as Snap25S187A/S187A mice) were obtained by in vitro fertilization. The genotype was checked by PCR. In this experiment, wild-type and Snap25S187A/S187A mice were used at age 11 (females), 54 (females), 60 (males), and 61 (females) weeks.
Tissue processing.
Deeply anesthetized mice (with 250 mg/kg, i.p., sodium pentobarbital) were perfused transcardially with PBS or with 2% paraformaldehyde/2% glutaraldehyde in 0.1 m phosphate buffer (PB). The brains were removed en bloc from the skull and cut sagittally into two brain hemispheres for confocal microscopy, cell counting, electron microscopy, Western blotting, and catecholamine analysis. Tissue processing for conventional electron microscopy is described below (see Conventional electron microscopy). For confocal microscopy and cell counting, hemisphere brain blocks of mice perfused transcardially with PBS or with 2% paraformaldehyde/2% glutaraldehyde in 0.1 m PB were fixed overnight in 4% paraformaldehyde in PBS and immersed in PBS containing 30% sucrose until sinking. Coronal sections of the striatum and the entire rostrocaudal extent of the substantia nigra (SN) were cut serially at 20 μm thickness using a cryostat (CM1850; Leica Microsystems). For Western blotting and catecholamine measurement by HPLC, brain blocks of mice perfused transcardially with PBS were dissected using the method described previously by our group, to yield the striatal tissues and ventral parts of midbrain tissues including the SN (Yasuda et al., 2011). After completing the dissection, the sections were immediately frozen on dry ice and stored at −80°C until analysis.
Antibodies.
The primary antibodies used for confocal microscopy were mouse anti-αSyn (clone 42; diluted at 1:100; BD Biosciences) (Tapia-González et al., 2011), rabbit anti-αSyn (1:500; Millipore Bioscience Research Reagents) (Chung et al., 2003; Herzig et al., 2011), rabbit anti-tyrosine hydroxylase (TH) (1:5000; Calbiochem) (Yasuda et al., 2011), mouse anti-synaptophysin (clone SY38; 1:500; Millipore) (Masliah et al., 2001; Tabuchi et al., 2007; Gimbel et al., 2010), rat anti-dopamine transporter (DAT) (clone MAB369; 1:500; Millipore) (Herzig et al., 2011; Kaushal et al., 2011), rabbit or goat anti-vesicular glutamate transporter-1 (VGLUT1) (1:300; Frontier Institute) (Miyazaki et al., 2003; Kawamura et al., 2006), goat anti-VGLUT2 (1:300; Frontier Institute) (Miyazaki et al., 2003; Kawamura et al., 2006), and rabbit anti-Ser129-phosphorylated αSyn (ab59264; 1:100; Abcam) (Pan-Montojo et al., 2010; Yasuda et al., 2011). For Western blotting, primary antibodies used were mouse anti-αSyn (clone 42; 1:150; BD Biosciences), rabbit anti-Ser129-phosphorylated αSyn (ab59264; 1:500; Abcam), rabbit anti-SNAP25ct (1:1000) (Yamamori et al., 2011), mouse anti-syntaxin 1 (10H5) (1.4 μg/ml) (Yamamori et al., 2011), rabbit anti-VAMP-2 (1.4 μg/ml) (Kataoka et al., 2011), mouse anti-synaptophysin (clone SY38; 1:500; Millipore), rabbit anti-TH (1:500; Calbiochem), and rabbit anti-β-tubulin (1:500; Abcam). For immunoelectron microscopy, the primary antibodies used were mouse anti-αSyn (clone 42; 1 μg/ml; BD Biosciences), guinea pig anti-DAT (1 μg/ml; Frontier Institute), and rabbit anti-VGLUT1 (1 μg/ml; Frontier Institute).
Confocal microscopy.
Free-floating sections were washed in PBS medium containing 0.05% Triton X-100 (PBS-T). As to the sections prepared from mice perfused transcardially with 2% paraformaldehyde/2% glutaraldehyde in 0.1 m PB, they were incubated with Liberate Antibody Binding (L.A.B.) solution (Polysciences) followed by washing in PBS medium and then processed in the same way as other sections. The sections were soaked with blocking agents and then incubated with the primary antibody dissolved in dilution reagent at 4°C for 48 h. Vector M.O.M. Immunodetection Kit (Vector Laboratories) was used for blocking and antibody dilution according to the instructions provided by the manufacturer. Subsequently, for fluorescent visualization of the antigens, the sections were incubated for 2 h in fresh medium containing fluorescein isothiocyanate-conjugated anti-mouse or rabbit IgG, and Cy3-conjugated anti-mouse, rabbit, goat, or rat IgG secondary antibodies (1:200–500; Jackson ImmunoResearch Laboratories). The sections were mounted on slide glass and coverslipped with Vectashield Mounting Medium with or without DAPI (Vector Laboratories). Images were captured using a confocal laser-scanning microscope (model LSM510; Zeiss) equipped with ZEN 2009 software (Zeiss) with identical settings. The software was used for image processing (brightness and contrast adjustment) applied equally to the images of wild-type and mutant mice.
Conventional electron microscopy.
For conventional electron microscopy, the hemisphere brains prepared from mice perfused transcardially with 2% paraformaldehyde/2% glutaraldehyde in 0.1 m PB were postfixed in 2% OsO4 for 1 h. After washing in water, the samples were incubated in 2% uranyl acetate for 30 min. After dehydration using 50, 70, 90, and 100% ethanol solutions and 100% propylene oxide, the samples were embedded in Epon 812 resin (Nissin EM). Ultrathin sections were obtained using a Leica Ultracut UCT and stained with 2% uranyl acetate and lead citrate. Electron micrographs were taken with an H-7650 electron microscope (Hitachi).
Immunoelectron microscopy.
For preembedding immunoelectron microscopy, parasagittal brain sections (60 μm in thickness) were incubated successively with 5% normal goat serum, primary antibodies, and peroxidase- or coloidal gold (1.4 nm)-conjugated secondary antibodies (Invitrogen). Immunoreaction was visualized with 3,3′-diaminobenzidine or silver enhancement kit (HQ silver; Nanoprobes). When combining the two methods for double immunoelectron microscopy, the brain tissue was subjected first to immunoperoxidase and then to silver-enhanced immunogold. Sections labeled by immunoperoxidase and silver-enhanced immunogold were subjected to electron microscopy as described above. The size of the nerve terminal was quantitated by using the ImageJ 1.43 software (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij).
Western blotting.
Frozen striatal and ventral midbrain tissues were sonicated in chilled CelLytic-MT mammalian tissue lysis/extraction reagent (Sigma-Aldrich) mixed with protease inhibitor mixture set I (Calbiochem) and phosphatase inhibitor mixture set V (Calbiochem). The samples were centrifuged (20,000 × g for 10 min at 4°C), and the resulting supernatants were collected and used for Western blotting. The protein concentration in the lysate of all samples was determined using BCA protein assay kit (Pierce). Each protein sample (5–15 μg) was resolved by SDS-PAGE by means of Compact-PAGE-twin (ATTO) and then electrotransferred to Clear Blot Membrane-P (ATTO) using powered BLOTmini (ATTO). The membrane was washed in PBS-T, incubated for 1 h in PBS-T containing 50% ChemiBLOCKER (Millipore), and then incubated for 24 h with the primary antibody in the same fresh medium. Subsequently, the membrane was incubated for 2 h in fresh medium containing horseradish peroxidase-linked anti-mouse or rabbit IgG secondary antibody (1:10,000–20,000; GE Healthcare), followed by development of chemiluminescence using GE Healthcare ECL Plus Western Blotting Detection System (GE Healthcare). The image was captured using LAS-4000 (Fujifilm), and the signal intensity was quantitated using the ImageJ 1.43 software.
Assay for SNARE complex assembly.
SNARE complex assembly was assessed by measuring the levels of the high-molecular-weight SDS-resistant complex in Western blotting performed without boiling of the samples before gel electrophoresis (Hu et al., 2002; Asuni et al., 2008; Sakisaka et al., 2008). Striatal lysate samples (15 μg of protein/sample) of wild-type and Snap25S187A/S187A mice were resolved in the SDS sample buffer. Samples were incubated for 20 min at room temperature and were further incubated at room temperature or boiled at 100°C for 3 min. Subsequently, they were analyzed by SDS-PAGE followed by the immunoblotting using the rabbit anti-SNAP25ct primary antibody (1:1000) (Yamamori et al., 2011) and horseradish peroxidase-linked anti-rabbit IgG secondary antibody (1:10,000; GE Healthcare). Except for the sample preparation before gel electrophoresis, all procedures were the same as described above (see Western blotting).
Determination of striatal levels of DA and its metabolites by HPLC.
Frozen striatal tissues were sonicated in 50 mm sodium acetate. The samples were centrifuged (20,000 × g for 10 min at 4°C), and the resulting supernatants were mixed with equal volumes of 0.2N perchloric acid. The samples were centrifuged (20,000 × g for 10 min at 4°C), and the resulting supernatants were subjected to measurement of DA, homovanillic acid (HVA), and 2-(3,4-dihydroxyphenyl)-acetic acid (DOPAC) concentrations by HPLC, using an HPLC system equipped with electrochemical detection (ECD-100; EICOM; applied voltage, 500 mV) and reverse-phase column (TSK gel ODS-80TM; Tosoh). The HPLC system consisted of a pump (PU-980; Jasco; flow rate, 1.0 ml/min), autosampler (AS-1550; Jasco), column oven (860-CO; Jasco), and degasser (DG-2080–53 Jasco). The mobile phase consisted of a solution, pH 3.72, containing the following: 0.085 m NaH2PO4 · 2H2O, 2.5 mm 1-octanesulfonic acid sodium salt, 20 μm EDTA-2Na, and 15% methanol. The concentrations of DA, HVA, and DOPAC were determined in nanomoles per gram of tissue weight, and the results were expressed relative to the values of wild-type mice.
Cell count.
For DA cell counting in the SN pars compacta (SNpc), every fourth 20-μm-thick serial section of the brain was immunostained for TH and counterstained with cresyl violet (Nissl staining) (Furuya et al., 2004; Yasuda et al., 2011). The numbers of TH- and Nissl-double-positive cells in the SNpc were counted both in wild-type and Snap25S187A/S187A mice in a blind manner. The SNpc cells that have nuclei optimally visible by TH immunostaining, and nuclei, cytoplasm, and nucleoli prominently stained by Nissl staining were counted. To avoid double counting of neurons with unusual shapes, TH- and Nissl-double-positive cells were counted only when their nuclei and nucleoli were optimally visualized. The rostral end of the SNpc was determined to the level where TH- and Nissl-double-positive cells began to appear (∼2.60 mm caudal to the level of the bregma) (Franklin and Paxinos, 2008), and the caudal end of the SNpc was defined to the level where TH- and Nissl-double-positive cells and oculomotor nerves can be observed (∼3.80 mm caudal to the level of the bregma) (Yasuda et al., 2011). In the rostral half region of the SN (∼2.60–3.16 mm caudal to the level of the bregma), the SNpc was distinguished from medial ventral tegmental area (VTA) by the vertical line extended from the medial end of the cerebral peduncle. In the caudal region of the SN where the medial lemniscus can be observed (∼3.16–3.80 caudal to the level of the bregma), the SNpc and medial VTA were divided along the medial lemniscus.
Statistical analysis.
All data are expressed as mean ± SEM, excluding those of VGLUT1-positive nerve area, which are expressed as mean ± SD. The two-tailed Student's t test (for two groups) was applied. A value of p < 0.05 denoted the presence of statistically significant difference.
Results
Abnormal distribution of αSyn protein at presynaptic terminals in the striatum of Snap25S187A/S187A mice
Using immunohistochemistry, we investigated the distribution of endogenous αSyn in the striatum of Snap25S187A/S187A mice, which have unphosphorylatable Ala at position Ser187 in the SNAP-25 protein. A significant decrease of neurotransmitter release was noted in serotonergic and dopaminergic systems in the amygdala, which was associated with behavioral abnormalities, including spontaneous convulsive seizures (Kataoka et al., 2011). Confocal microscopic analysis of the striatal sections of the Snap25S187A/S187A mice showed altered distribution of endogenous αSyn, which resembled coarse granular deposits, at the age of 11 weeks, compared with age-matched wild-type control. These changes were prominent in 60-week-old (54-week-old or more was designated as aged, hereafter) Snap25S187A/S187A mice (Fig. 1A,B), indicating an age-dependent progressive manner of redistribution.
Figure 1.
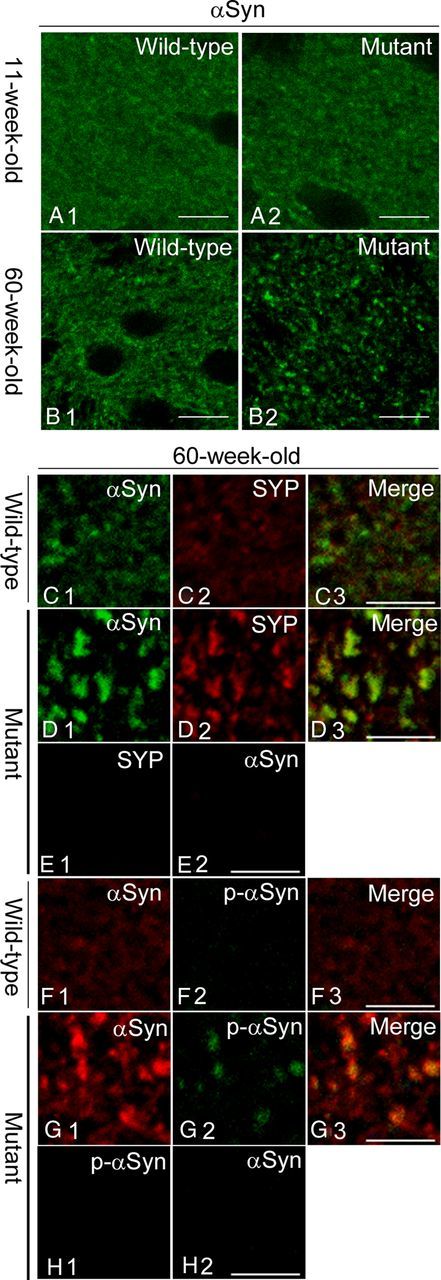
Altered localization of αSyn and its accumulation in presynaptic terminals in the striatum of Snap25S187A/S187A mice. A, B, Striatal sections of wild-type and Snap25S187A/S187A (mutant) mice at postnatal weeks 11 and 60 were immunostained for αSyn (n = 3 for each genotype). Compared with the wild-type mice (A1, B1), αSyn was localized and formed coarse granular deposits in mutant mice at postnatal weeks 11 and 60 (A2, B2). The severity of the change was more prominent at postnatal week 60. Scale bars, 10 μm. C–H, Sections were coimmunostained for αSyn, and synaptophysin (SYP) or p-αSyn. Confocal images showed altered localization of SYP (D2) and p-αSyn (G2) and colocalization with αSyn-positive coarse granular deposits (D1, G1) in mutant mice (D3, G3). No cross-reactivity was observed for the sections of the mutant mice exposed to a single primary antibody followed by the treatment of both secondary antibodies were observed (E1, E2; H1, H2) [i.e., negligible green (E1) and red fluorescence (E2) for the sections treated with anti-SYP and αSyn antibody, respectively]. Scale bars, 5 μm.
Next, we examined the localization of αSyn in the presynaptic terminals by using anti-synaptophysin antibody. Aged Snap25S187A/S187A mice showed similar alternation of the immunoreactivity for synaptophysin as observed for αSyn, compared with the wild-type (Fig. 1C,D). Moreover, αSyn-immunopositive granular structures were largely overlapped with synaptophysin. These results indicate that αSyn accumulates mainly in the presynaptic terminals in the striatum. We also analyzed the immunostaining pattern for Ser129-phosphorylated αSyn (p-αSyn), which was found specifically in human α-synucleinopathies (Fujiwara et al., 2002). Majority of αSyn-positive granular deposits were immunopositive for p-αSyn, which were detected at negligible amounts in wild-type control mice (Fig. 1F,G). These results indicate that the abnormal accumulation of αSyn in the striatum of Snap25S187A/S187A mice represents in part the pathological changes in human DLB brains.
Abnormal accumulation of αSyn and p-αSyn proteins in the striatum of Snap25S187A/S187A mice
Next, we measured αSyn and p-αSyn protein levels in the striatal tissues of aged Snap25S187A/S187A mice. A significant decrease of SNAP-25 protein was found in Snap25S187A/S187A mice (p < 0.0001) compared with age-matched wild-type mice, confirming our previous findings (Kataoka et al., 2011). Importantly, αSyn (∼1.5-fold; p = 0.0047) and p-αSyn (∼1.7-fold; p = 0.0080) protein levels were higher than age-matched wild-type mice (Fig. 2A,B). These results indicate that the abnormal distribution of αSyn and p-αSyn in the presynaptic terminals (Fig. 1F) was reflected by increased amount of these proteins. In addition, high levels of VAMP-2 (∼1.3-fold; p = 0.0011), but neither syntaxin nor synaptophysin, were found in the striatum of aged Snap25S187A/S187A mice (Fig. 2A,B), compared with whole-brain lysate prepared from young Snap25S187A/S187A mice (Kataoka et al., 2011).
Figure 2.

Increase in endogenous αSyn and p-αSyn in the striatum of Snap25S187A/S187A mice. A, B, Western blotting was performed for synaptic marker proteins using the striatal tissues at postnatal week 61. A, Representative Western blotting images of two to five independent experiments, each involving three and five mice for wild-type and Snap25S187A/S187A (mutant), respectively. Mutant mice showed increased band intensity of αSyn, p-αSyn, and VAMP-2 compared with the wild type, while that of SNAP-25 was decreased. B, Relative protein expression levels expressed relative to the loading control (β-tubulin). The band intensity was quantitated using the ImageJ 1.43 software and expressed as the relative protein expression level. Significant increases in α-Syn, p-αSyn, and VAMP-2, and significant decrease in SNAP-25 were observed in mutant mice. No significant differences were noted in other synaptic protein markers. Data are mean ± SEM of two to five independent experiments, each involving three and five mice for wild-type and mutant, respectively. ***p < 0.01 (two-tailed t test).
Ultrastructural analysis of αSyn distribution in presynaptic terminals
To investigate in detail the morphological changes in the presynaptic terminals, we used electron microscopy to analyze the striatum of the aged mice. In Snap25S187A/S187A mice, condensed synaptic vesicles were abundantly present in large excitatory nerve terminals, compared with the normal uniform distribution of the vesicles in wild-type mice (Fig. 3A,B). These results suggest possible decrease of presynaptic neurotransmitter release in Snap25S187A/S187A mice. Subsequent immunoelectron-microscopic examination of αSyn in the synapses showed predominant localization of αSyn proteins in the periactive zone of enlarged excitatory presynaptic nerve terminals in the aged Snap25S187A/S187A mice, compared with the wild type (Fig. 3C,D).
Figure 3.
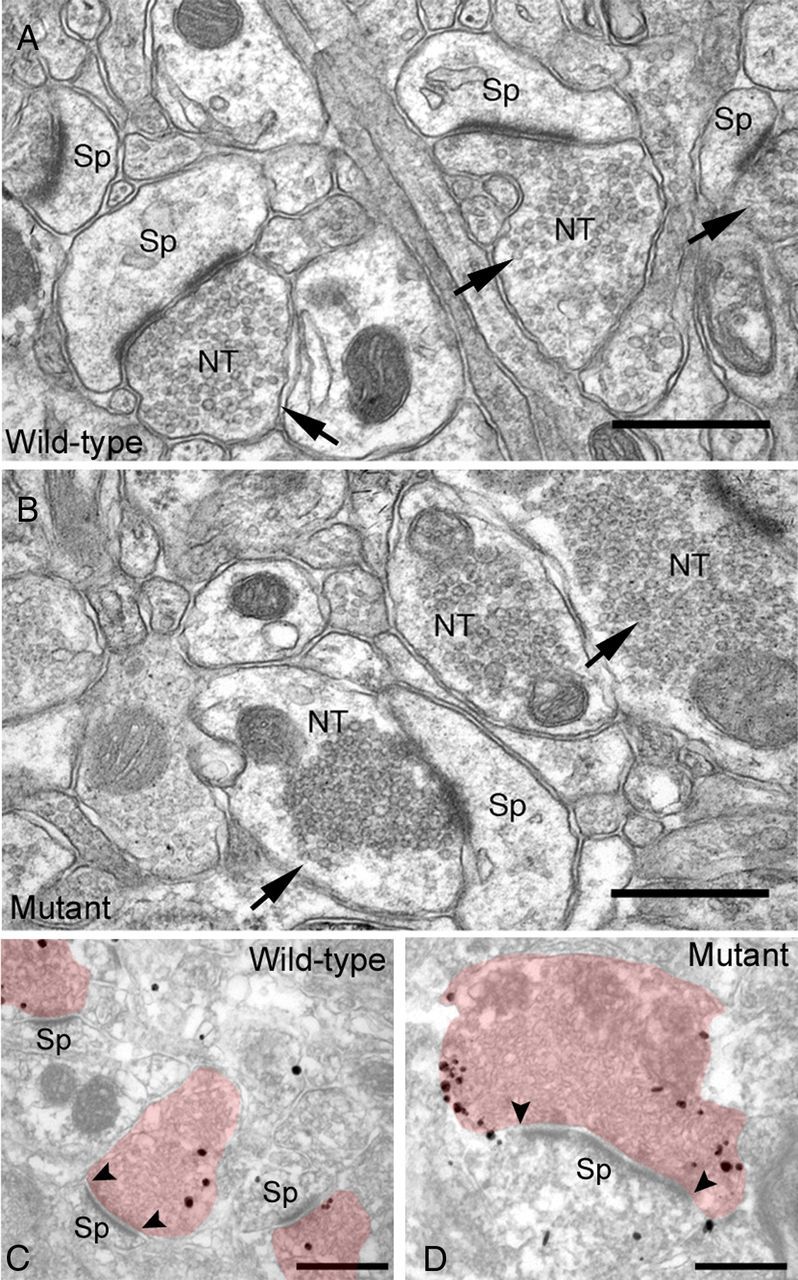
Abnormal excitatory nerve terminals in the striatum of Snap25S187A/S187A (mutant) mice. Representative electron micrographs of wild-type and mutant mice at postnatal week 60 (n = 3 for each genotype). Synaptic vesicles were homogeneously distributed in the cytoplasm of the excitatory nerve terminals in wild-type mice (A), while they were tightly assembled in mutant mice (B). Note the alteration of subcellular localization of α-Syn (immunogold particles) and increased number of αSyn aggregates deposited in association with the cytoplasmic face of the plasma membrane at the periactive zone of the enlarged nerve terminals in mutant mice (D), but not in wild-type mice (C). Arrows, excitatory nerve terminals; arrowheads, edges of active zone. NT, Nerve terminal; Sp, dendritic spine. Scale bars, 500 nm.
Decreased ability of SNARE complex assembly in Snap25S187A/S187A mice
Given that Snap25S187A/S187A mice clearly displayed the significant decrease in the SNAP-25 protein expression and abnormal distribution of the synaptic vesicle in the presynaptic terminals (Figs. 2, 3B), we evaluated the functional ability to produce the SNARE complex in Snap25S187A/S187A mice. SNARE complex assembly was assessed by measuring the levels of the high-molecular-weight heat-sensitive SDS-resistant complex in Western blotting. As shown in Figure 4A, compared with boiled sample of the wild type, SNAP-25-immunoreactive bands were shifted to the higher molecular masses (∼75 to ∼250 kDa) in unboiled samples of wild-type and Snap25S187A/S187A mice. However, the relative signal intensity of higher molecular masses normalized by β-tubulin (loading control) was significantly reduced in Snap25S187A/S187A mice, compared with the wild-type mice (p = 0.00463) (Fig. 4B). These observations indicated the decreased ability of the SNARE complex assembly in Snap25S187A/S187A mice compared with the wild-type mice. This is considered to represent the functional deficit possibly attributable to the decreased level of the SNAP-25 and the insensitivity of protein kinase C-mediated phosphorylation at Ser187 in Snap25S187A/S187A mice.
Figure 4.

Decreased ability of the SNARE complex assembly in Snap25S187A/S187A (mutant) mice. A, B, Western blotting was performed for unboiled or boiled samples of wild-type and mutant mice (at postnatal week 61) before electrophoresis followed by the immunoblotting using SNAP-25ct antibody. A, Representative Western blotting image of two to three independent experiments was provided. Heat-sensitive SDS-resistant SNAP-25ct-immunoreactive bands were detected at high molecular mass (∼75 to ∼250 kDa) in wild-type and mutant mice, indicating the presence of the SNARE complex. However, band intensity of the mutant mice, especially at ∼75 kDa, was decreased compared with the wild-type mice. B, Relative levels of the SNARE complex assembly relative to the loading control (β-tubulin). The band intensity was quantitated using the ImageJ 1.43 software and expressed as the relative protein expression level. Significant decrease in SNARE complex assembly was observed in mutant mice compared with the wild-type mice. Data are mean ± SEM of two to three independent experiments, each involving three mice for each genotype. ***p < 0.01 (two-tailed t test).
αSyn accumulation and enlargement of corticostriatal nerve terminals
The striatal spiny interneurons receive excitatory outputs from cortical and thalamic tracts, and these projections use glutamate as the neurotransmitter (Gerfen and Surmeier, 2011). VGLUT1 and VGLUT2 are localized to the excitatory nerve terminals projecting from the cortical and the thalamic area, respectively, in the striatum. Then, we investigated the distribution of αSyn-immunopositive deposits with VGLUT1 or VGLUT2 to verify the abnormal accumulation of αSyn in the corticostriatal or the thalamostriatal neuronal terminals. As shown in Figure 5B2, we found similar alternation of the immunoreactivity for VGLUT1 as observed for αSyn in the aged Snap25S187A/S187A mice compared with wild-type normal ones (Fig. 5A2). Importantly, such immunoreactivity for VGLUT1, but not for VGLUT2, was exclusively colocalized with αSyn (Fig. 5B3,E3). We also performed double immunostaining with anti-pSyn and anti-VGLUT1 antibodies and found the VGLUT1-immunopositive structures to be immunopositive for p-αSyn (Fig. 5G3). These observations suggest accumulation of αSyn and p-αSyn mainly in the enlarged VGLUT1-positive nerve terminals.
Figure 5.
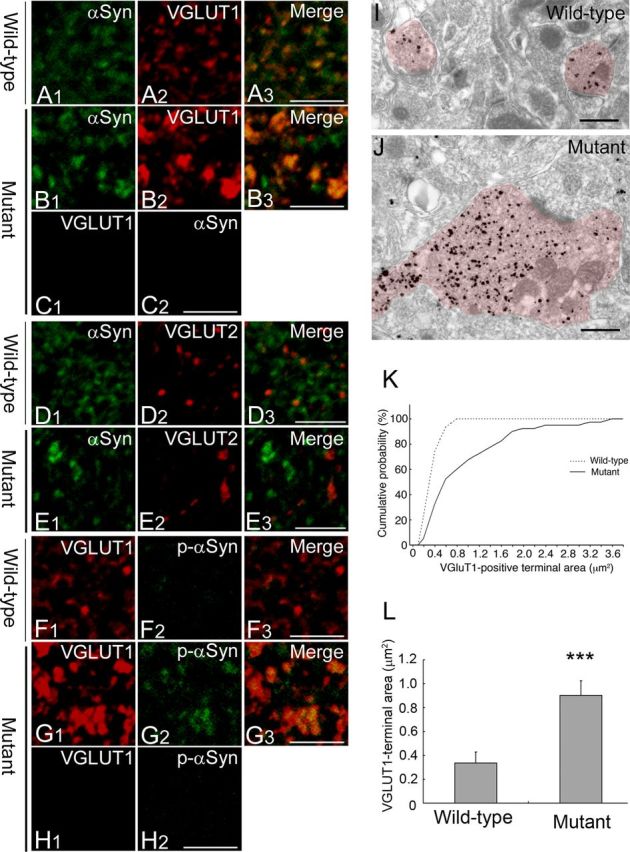
Accumulation of αSyn in the hypertrophied VGLUT1-positive nerve terminals. A–E, Striatal sections of wild-type and Snap25S187A/S187A (mutant) mice at postnatal week 60 were coimmunostained for αSyn (green) and VGLUT1 or VGLUT2 (red) (n = 3 for each genotype). Confocal images showed altered localization of VGLUT1 with more granular appearance in mutant mice (B2) compared with wild-type mice (A2). Immunopositive structures for VGLUT1, but not for VGLUT2, marked colocalization with αSyn-positive granular deposits (B3, E3). No cross-reactivity was observed for the sections of the mutant mice treated with a single antibody followed by both the secondary antibodies, as evidenced by negligible green (C1) and red fluorescence (C2) for the sections treated with anti-VGLUT1 and αSyn antibody, respectively. Scale bars, 5 μm. F–H, Sections were coimmunostained for VGLUT1 (red) and p-αSyn (green) (n = 3 for each genotype). Confocal images showed marked colocalization of VGLUT1 and p-αSyn in mutant mice (G3). Similarly, no cross-reactivity was observed as evidenced by negligible red (H1) and green fluorescence (H2) for the sections treated with anti-VGLUT1 and αSyn antibody, respectively. Scale bars, 5 μm. I, J, Representative electron micrographs showing VGLUT1-positive nerve terminals (labeled with immunogold particles). Note the hypertrophy of VGLUT1-positive nerve terminals in mutant mice (J), but not in wild-type mice (I). Scale bars, 500 nm. K, L, Quantitative analysis of VGLUT1-positive nerve terminal area in three mice per group. The size was measured in 100 nerve terminals per animal. Cumulative probability (K) and group mean value (L) were calculated and showed a larger nerve terminal area in mutant mice than the wild type. Data are mean ± SD. ***p < 0.01 (two-tailed t test).
Immunoelectron-microscopic examination showed massive enlargement of the VGLUT1-positive nerve terminals in the aged Snap25S187A/S187A mice (Fig. 5I,J). Quantitative analysis of the size of the VGLUT1-positive nerve terminals based on the cumulative probability curve confirmed a significantly larger size in Snap25S187A/S187A mice compared with wild-type mice (Fig. 5K). The size of the VGLUT1-positive nerve terminals reached a plateau at 0.8 μm2 in wild-type mice, while that of Snap25S187A/S187A mice reached a plateau at 3.6 μm2. The size of ∼40% of the VGLUT1-positive nerve terminals in the mutant mice was larger than the maximum size measured in wild-type mice. Specifically, the size of the VGLUT1-positive nerve terminals in the mutant mice was approximately fourfold larger than the wild type (p = 0.0030) (Fig. 5L).
No significant changes in nigrostriatal dopaminergic system
Finally, we examined the effect of SNARE dysfunction on the nigrostriatal dopaminergic system, which is affected in parkinsonian brains. The striatal sections of aged Snap25S187A/S187A mice were coimmunostained for αSyn and dopaminergic neuronal markers (TH or DAT). The αSyn-immunopositive deposits appeared adjacent to, but hardly colocalized with both DAT and TH in Snap25S187A/S187A mice. The distribution patterns of DAT- and TH-immunoreactive deposits in the striatum were not significantly different between wild-type and mutant mice (Fig. 6A–D). These results imply that the nigrostriatal dopaminergic neurons are not affected in Snap25S187A/S187A mice.
Figure 6.
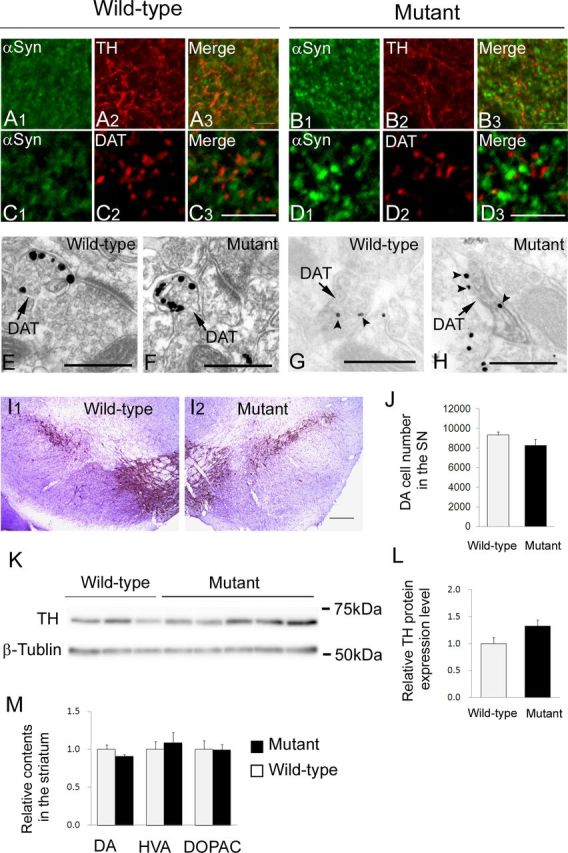
Lack of neurodegenerative changes in nigrostriatal dopaminergic neurons. A–D, Striatal sections of wild-type and Snap25S187A/S187A (mutant) mice at postnatal week 60 were coimmunostained for α-Syn (green) and TH or DAT (red) (n = 3 for each genotype). There were no significant differences in the distribution pattern of TH (A2, B2) and DAT (C2, D2) between wild-type and mutant mice. αSyn-positive granular deposits (B1, D1) were rarely colocalized with TH or DAT (B3, D3). Scale bars, 5 μm. E–H, Representative electron micrographs of dopaminergic nerve terminals at postnatal week 60 (n = 3 for each genotype). They were identified by silver particles (E, F) or DAB labeling (G, H) for DAT (arrows). There were no significant differences in subcellular localization of synaptic vesicles (E, F) or αSyn (arrowheads, silver particles) (G, H) between wild-type and mutant mice. Scale bars, 500 nm. I, J, Representative micrographs of the substantia nigra of wild-type and mutant mice at postnatal week 60 immunostained for TH and counterstained with cresyl violet. Scale bars: I1, I2, 200 μm. Number of DA cell bodies counted in three mice of each genotype. Data are mean ± SEM (J). K, L, Western blotting was performed using substantia nigral tissues at postnatal week 61. Representative images of Western blotting from two independent experiments, each involving three and five mice of wild-type and mutant mice, respectively (K). Relative TH protein expression levels (relative to the expression of β-tubulin) were expressed relative to the value of wild-type mice (L). Data are mean ± SEM of two independent experiments, each involving three and five mice of wild-type and mutant, respectively. M, Quantification of DA, HVA, and DOPAC levels in the striatum of mice at postnatal week 54. Data were expressed relative to those of wild-type mice and represent the mean ± SEM of duplicate measurement of one experiment involving four mice of each genotype. There were no significant differences between wild-type and mutant mice (J, L, M) (two-tailed t test).
To clarify this point further, we conducted immunoelectron-microscopic examination of DAT-positive dopaminergic nerve terminals. There were no significant differences in any structural alternations including condensation of synaptic vesicles (Fig. 6E,F). Furthermore, we found no significant differences in the subcellular localization of αSyn protein between the aged wild-type and Snap25S187A/S187A mice (Fig. 6G,H).
We also investigated the functional preservation of the nigrostriatal dopaminergic neurons in the aged Snap25S187A/S187A mice. No significant difference was found in the number of TH- and Nissl-double-positive cells in the SNpc between wild-type and Snap25S187A/S187A mice (Fig. 6I,J). There were also no significant differences in the levels of TH protein in the midbrain tissue and DA and its metabolites, HVA and DOPAC, in the striatum between the aged wild-type and Snap25S187A/S187A mice (Fig. 6K–M). These results suggest that SNARE dysfunction does not seem to be involved in the initiation of PD-related pathologic changes in nigrostriatal dopaminergic neurons.
Discussion
In this study, we found that SNARE dysfunction leads to presynaptic accumulation of endogenous αSyn, a process that probably represents the initial pathological event in DLB. Previous studies using neural preparations showed that the neurotransmitter release is regulated by protein kinase C, which phosphorylates Ser187 residue in SNAP-25, augmenting exocytosis of synaptic vesicles (Majewski and Iannazzo, 1998; Morgan et al., 2005). Patch-clamp analysis of chromaffin cells that overexpress the S187A form of SNAP-25 inhibited the rate of refilling of presynaptic vesicle pool (Nagy et al., 2002). Recently, we reported that Snap25S187A/S187A mice show reduced DA and serotonin release in amygdala (Kataoka et al., 2011). In human DLB brains, >90% of αSyn aggregates are located in the presynaptic terminals in the form of small deposits (Neumann et al., 2002; Kramer and Schulz-Schaeffer, 2007; Schulz-Schaeffer, 2010). This is consistent with the present findings of abnormal accumulation of αSyn in presynapses, suggesting that this process is the initial pathological event in DLB, eventually leading to the death and degeneration of neuronal cells (Orimo et al., 2008). Another finding that lends support to the role of αSyn aggregates in the presynaptic terminals in DLB was the lack of histopathological changes in the dopaminergic terminals in the present study.
In experiments on glutamate release conducted in hippocampal slices prepared from αSyn knock-out mice (Gureviciene et al., 2007), paired-pulse facilitation was significantly weaker, and high-frequency-induced long-term potentiation and frequency facilitation were not observed. These findings suggest that αSyn contributes to mobilization of glutamate-containing vesicles from the reserve pool (Gureviciene et al., 2007). Thus, αSyn may act as a positive regulator of neurotransmitter release at presynaptic terminals. Therefore, presynaptic accumulation of αSyn observed in Snap25S187A/S187A mice might reflect a compensatory response to a possible SNARE dysfunction-related chronic shortage of neurotransmitter release in the VGLUT1-positive nerve terminals.
In the striatum, the medium spiny neurons, which constitute >90% of all striatal neurons, receive output from glutamatergic axons that contact the spine head and dopaminergic axons that synapse with the dendritic spine neck. DA released from dopaminergic axons regulates the release of glutamate via D2-like receptors on the corticostriatal nerve terminals (Bamford et al., 2004; Wickens and Arbuthnott, 2005). In the present study, we found no significant changes in the striatal tissue levels of DA and its metabolites. These findings confirmed the results reported in our previous study using Snap25S187A/S187A mice, whereas microdialysis analysis study revealed marked reduction of DA release from the amygdala (not measured in other brain regions) (Kataoka et al., 2011). In addition, in another in vitro study using PC12 cells, phosphorylation of SNAP-25 at S187 potentiated calcium-dependent DA release and recruitment of synaptic vesicles containing DA (Shimazaki et al., 1996; Iwasaki et al., 2000; Shoji-Kasai et al., 2002). These observations suggest decreased striatal DA release in Snap25S187A/S187A mice, resulting in increased demand for neurotransmitter release at glutamatergic nerve terminals. Thus, presynaptic accumulation of αSyn might reflect a possible compensatory response to low DA inhibitory control over cortical glutamatergic drive.
Increased expression of VAMP-2 protein accompanied increased αSyn expression in the striatum of Snap25S187A/S187A mice. Binding of the C terminus of αSyn to the N terminus of VAMP-2 primes the subsequent SNARE complex assembly (Burgoyne and Morgan, 2011). Therefore, the increased VAMP-2 level might also reflect a compensatory response to the impaired synaptic vesicle release by enhancing SNARE complex formation in concert with the increased αSyn.
Presynaptic neurotransmitter release is mediated by the synaptic vesicle cycle, consisting of exocytosis followed by endocytosis and recycling. Exocytosis incorporates synaptic vesicles into the presynaptic terminal membranes and increases the surface area, while endocytosis retrieves the excess plasma membrane components followed by recycling to form other synaptic vesicles. Under normal conditions, the dynamics of the balance between exocytosis and endocytosis is well preserved to maintain the correct surface area of the presynaptic terminal (Hayes and Baines, 1996; Haucke et al., 2011). However, excessive accumulation of presynaptic vesicles and enlargement of the VGLT1-positive nerve terminals was observed in Snap25S187A/S187A mice. Taking into consideration the synaptic vesicle cycle, our findings suggest that the balance of the cycle was biased toward decreased exocytosis concomitant with possible decreased endocytosis.
The enlarged VGLUT1-positive nerve terminals of Snap25S187A/S187A mice showed concomitant accumulation of αSyn and p-αSyn. Kramer and Schulz-Schaeffer (2007) have previously reported that 90% or even more of αSyn aggregates in DLB cases were located at the presynapses in the form of very small deposits. In parallel, dendritic spines were retracted, whereas the presynapses were relatively preserved, suggesting that a neurotransmitter deprivation may explain the cognitive impairment in DLB (Kramer and Schulz-Schaeffer, 2007; Schulz-Schaeffer, 2010). While the presynaptic aggregates did not contain much p-αSyn in their examination (Kramer and Schulz-Schaeffer, 2007; Schulz-Schaeffer, 2010), widespread varicosities and dot-like structures containing p-αSyn are commonly observed in mouse model and human DLB brains (Saito et al., 2003; Scott et al., 2010). This may represent axonal transport defects and presynaptic dysfunctions (Saito et al., 2003; Scott et al., 2010). Recent study showed that mutant αSyn (A53T) diminished levels of various motor proteins in neurons (Chung et al., 2009), supporting this scenario. Alternatively, excessive amounts of misfolded αSyn and p-αSyn may aggregate at synapses, physically preventing the targeting of other presynaptic proteins (Kramer and Schulz-Schaeffer, 2007). In experiments using Caenorhabditis elegans overexpressing the human αSyn, four genes related to the endocytosis process were identified as genetic modifiers for αSyn toxicity (Kuwahara et al., 2008). They included the two subunits of the adaptor protein (AP) complex 2, which interacts with clathrin and promotes presynaptic clathrin-mediated vesicle recycling (Morgan et al., 2000). Furthermore, proteomics analysis revealed that p-αSyn also preferentially interacted with the proteins involved in endocytosis, including clathrin heavy chain and subunit of AP-2 and AP-1 complexes, over the nonphosphorylated αSyn (McFarland et al., 2008). Clathrin-mediated recycling of exocytosed synaptic vesicles occurs in the periactive zone, a region adjacent to the active zone where synaptic vesicle is endocytosed (Haucke et al., 2011). Similarly, in Snap25S187A/S187A mice, immunoelectron microscopy showed preferential localization of αSyn at the periactive zone of the excitatory presynaptic nerve terminals (Fig. 3D). This might reflect the interaction of αSyn and p-αSyn with the proteins involved in clathrin-mediated endocytosis. Taking these findings together, presynaptic accumulation of αSyn and p-αSyn could disturb the endocytosis process and consequently contribute to the development of VGLUT1-positive terminal enlargement.
Presynaptic accumulation of αSyn is considered an early event in the pathogenesis of α-synucleinopathies (Neumann et al., 2002; Kramer and Schulz-Schaeffer, 2007; Schulz-Schaeffer, 2010). Mice overexpressing human αSyn showed presynaptic accumulation of αSyn and low DA release in the striatum. These findings were associated with abnormal distribution of SNARE proteins, which colocalized with αSyn aggregates. Similarly, accumulation of SNARE proteins and αSyn were reported in the striatum of PD patients (Garcia-Reitböck et al., 2010). These observations suggest that SNARE dysfunction likely occurs at an early stage of pathogenesis in nigrostriatal dysfunction observed in PD. By considering the findings observed in the VGLUT1-positive nerve terminals, we expected that SNARE dysfunction might have induced presynaptic accumulation of αSyn, which consequently results in the development of neurodegenerative changes in the nigrostriatal system. However, contrary to our expectation, Snap25S187A/S187A mice showed no significant neurodegenerative changes in nigrostriatal dopaminergic neurons, suggesting that SNARE dysfunction alone was insufficient to cause nigrostriatal degeneration as observed in PD, and appeared to be a downstream event associated with abnormal accumulation of αSyn.
In conclusion, the present study demonstrated that SNARE dysfunction leads to accumulation of endogenous αSyn in the corticostriatal nerve terminals. Presynaptic accumulation of αSyn is considered to be a key early event in the pathogenesis of α-synucleinopathies. Although the “prion-like” propagation hypothesis of αSyn, including tau and TAR DNA-binding protein 43 kDa, is currently receiving considerable attention worldwide, our findings provide an insight to our understanding of the possible mechanisms that lead to presynaptic accumulation of endogenous αSyn. Moreover, given that SNAP-25 is reduced in the striatum of MSA brains (Tong et al., 2010), we speculate that a discontinuous pattern of αSyn pathologies usually found in MSA [i.e., glial cytoplasmic inclusions in the putaminal oligodendrocytes, and neuronal cytoplasmic inclusions and neuronal nuclear inclusions in the cortex (Yoshida, 2007; Ubhi et al., 2011)] might be potentially linked through the presynaptic accumulation of αSyn in the corticostriatal neurons. Further investigations on the Snap25 mutant mice with genetic ablation of αSyn would contribute to understanding the essential role of redistributed αSyn and should be a central issue in the following studies.
Footnotes
This work was supported by Grants from the Japan Science and Technology Agency, Core Research for Evolutional Science and Technology (H.M.); Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labour and Welfare of Japan (H.M.); Grant-in-Aid for Research on Applying Health Technology (H23–015) from the Ministry of Health, Labour and Welfare of Japan (H.M.); Ministry of Education, Culture, Sports, Science and Technology of Japan Grant S0801035 (H.M.); and Grant-in-Aid for Scientific Research on Innovative Areas (Brain Environment) from the Ministry of Education, Science, Sports and Culture of Japan (H.M.).
The authors declare no competing financial interests.
References
- Asuni AA, Cunningham C, Vigneswaran P, Perry VH, O'Connor V. Unaltered SNARE complex formation in an in vivo model of prion disease. Brain Res. 2008;1233:1–7. doi: 10.1016/j.brainres.2008.07.083. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Chaperoning the SNAREs: a role in preventing neurodegeneration? Nat Cell Biol. 2011;13:8–9. doi: 10.1038/ncb0111-8. [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YH, Joo KM, Kim MJ, Cha CI. Immunohistochemical study on the distribution of alpha-synuclein in the central nervous system of transgenic mice expressing a human Cu/Zn superoxide dismutase mutation. Neurosci Lett. 2003;342:151–154. doi: 10.1016/s0304-3940(03)00237-4. [DOI] [PubMed] [Google Scholar]
- Darios F, Ruipérez V, López I, Villanueva J, Gutierrez LM, Davletov B. Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010;11:528–533. doi: 10.1038/embor.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. New York: Academic; 2008. [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Furuya T, Hayakawa H, Yamada M, Yoshimi K, Hisahara S, Miura M, Mizuno Y, Mochizuki H. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. J Neurosci. 2004;24:1865–1872. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- Garcia-Reitböck P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, Spano P, Tofaris GK, Goedert M, Spillantini MG. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain. 2010;133:2032–2044. doi: 10.1093/brain/awq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory Impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureviciene I, Gurevicius K, Tanila H. Role of alpha-synuclein in synaptic glutamate release. Neurobiol Dis. 2007;28:83–89. doi: 10.1016/j.nbd.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Haucke V, Neher E, Sigrist SJ. Protein scaffolds in the coupling of synaptic exocytosis and endocytosis. Nat Rev Neurosci. 2011;12:127–138. doi: 10.1038/nrn2948. [DOI] [PubMed] [Google Scholar]
- Hayes NVL, Baines AJ. Small synaptic vesicles. In: Lee AG, editor. Biomembranes: a multi-volume treatise. Greenwich, CT: JAI; 1996. pp. 75–122. [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Carroll J, Rickman C, Davletov B. Action of Complexin on SNARE complex. J Biol Chem. 2002;277:41652–41656. doi: 10.1074/jbc.M205044200. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Kataoka M, Sekiguchi M, Shimazaki Y, Sato K, Takahashi M. Two distinct mechanisms underlie the stimulation of neurotransmitter release by phorbol esters in clonal rat pheochromocytoma PC12 cells. J Biochem. 2000;128:407–414. doi: 10.1093/oxfordjournals.jbchem.a022768. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Yamamori S, Suzuki E, Watanabe S, Sato T, Miyaoka H, Azuma S, Ikegami S, Kuwahara R, Suzuki-Migishima R, Nakahara Y, Nihonmatsu I, Inokuchi K, Katoh-Fukui Y, Yokoyama M, Takahashi M. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS One. 2011;6:e25158. doi: 10.1371/journal.pone.0025158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N, Seminerio MJ, Shaikh J, Medina MA, Mesangeau C, Wilson LL, McCurdy CR, Matsumoto RR. CM156, a high affinity sigma ligand, attenuates the stimulant and neurotoxic effects of methamphetamine in mice. Neuropharmacology. 2011;61:992–1000. doi: 10.1016/j.neuropharm.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Koyama S, Yoshina S, Ren CH, Kato T, Mitani S, Iwatsubo T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet. 2008;17:2997–3009. doi: 10.1093/hmg/ddn198. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski H, Iannazzo L. Protein kinase C: a physiological mediator of enhanced transmitter output. Prog Neurobiol. 1998;55:463–475. doi: 10.1016/s0301-0082(98)00017-3. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland MA, Ellis CE, Markey SP, Nussbaum RL. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics. 2008;7:2123–2137. doi: 10.1074/mcp.M800116-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Morgan A, Burgoyne RD, Barclay JW, Craig TJ, Prescott GR, Ciufo LF, Evans GJ, Graham ME. Regulation of exocytosis by protein kinase C. Biochem Soc Trans. 2005;33:1341–1344. doi: 10.1042/BST0331341. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Hao W, Augustine GJ, Lafer EM. A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J Neurosci. 2000;20:8667–8676. doi: 10.1523/JNEUROSCI.20-23-08667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sørensen JB. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci. 2002;22:9278–9286. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson's disease. Brain. 2008;131:642–650. doi: 10.1093/brain/awm302. [DOI] [PubMed] [Google Scholar]
- Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RH. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 2010;5:e8762. doi: 10.1371/journal.pone.0008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T, Murayama S. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62:644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- Sakisaka T, Yamamoto Y, Mochida S, Nakamura M, Nishikawa K, Ishizaki H, Okamoto-Tanaka M, Miyoshi J, Fujiyoshi Y, Manabe T, Takai Y. Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J Cell Biol. 2008;183:323–337. doi: 10.1083/jcb.200805150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Burré J, Südhof TC. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- Shoji-Kasai Y, Itakura M, Kataoka M, Yamamori S, Takahashi M. Protein kinase C-mediated translocation of secretory vesicles to plasma membrane and enhancement of neurotransmitter release from PC12 cells. Eur J Neurosci. 2002;15:1390–1394. doi: 10.1046/j.1460-9568.2002.01972.x. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A Neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-González S, Giráldez-Pérez RM, Cuartero MI, Casarejos MJ, Mena MÁ, Wang XF, Sánchez-Capelo A. Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol Neurodegener. 2011;6:72. doi: 10.1186/1750-1326-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M, Rajput AH, Muenter MD, Kish SJ, Hornykiewicz O, Furukawa Y. Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson's disease and progressive supranuclear palsy: a comparative investigation. Brain. 2010;133:172–188. doi: 10.1093/brain/awp282. [DOI] [PubMed] [Google Scholar]
- Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011;34:581–590. doi: 10.1016/j.tins.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens JR, Arbuthnott GW. Structural and functional interactions in the striatum at the receptor level. In: Dunnett SB, Bentivoglio M, Björklund A, Hökfelt T, editors. Dopamine, Handbook of Chemical Neuroanatomy. Vol 21. Amsterdam: Elsevier; 2005. pp. 199–236. [Google Scholar]
- Yamamori S, Itakura M, Sugaya D, Katsumata O, Sakagami H, Takahashi M. Differential expression of SNAP-25 family proteins in the mouse brain. J Comp Neurol. 2011;519:916–932. doi: 10.1002/cne.22558. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Hayakawa H, Nihira T, Ren YR, Nakata Y, Nagai M, Hattori N, Miyake K, Takada M, Shimada T, Mizuno Y, Mochizuki H. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2011;70:686–697. doi: 10.1097/NEN.0b013e3182269ecd. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Nakata Y, Mochizuki H. α-Synuclein and neuronal cell death. Mol Neurobiol. 2012 doi: 10.1007/s12035-012-8327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M. Multiple system atrophy: α-synuclein and neuronal degeneration. Neuropathology. 2007;27:484–493. doi: 10.1111/j.1440-1789.2007.00841.x. [DOI] [PubMed] [Google Scholar]