

Abstract
Activated microglia and reactive astrocytes are commonly found in and around the senile plaque, which is the central pathological hallmark of Alzheimer's disease. Astrocytes respond to neuronal activity through the release of gliotransmitters such as glutamate, d-serine, and ATP. However, it is largely unknown whether and how gliotransmitters affect neuronal functions. In this study, we explored the effect of a gliotransmitter, ATP, on neurons damaged by β-amyloid peptide (Aβ). We found that Aβ1-42 (Aβ42) increased the release of ATP in cultures of primary astrocytes and U373 astrocyte cell line. We also found that exogenous ATP protected Aβ42-mediated reduction in synaptic molecules, such as NMDA receptor 2A and PSD-95, through P2 purinergic receptors and prevented Aβ42-induced spine reduction in cultured primary hippocampal neurons. Moreover, ATP prevented Aβ42-induced impairment of long-term potentiation in acute hippocampal slices. Our findings suggest that Aβ-induced release of gliotransmitter ATP plays a protective role against Aβ42-mediated disruption of synaptic plasticity.
Introduction
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder that leads to cognitive dysfunction, memory impairment, and emotional disturbance in elderly persons (McKhann et al., 1984). The extracellular senile plaques, one of pathological hallmarks of AD brain, consist in large part of β-amyloid (Aβ) peptide (Yankner, 1996), surrounded by activated microglia, reactive astrocytes, dystrophic neurites, and degenerating neurons (Wísniewski et al., 1989; Selkoe, 2000).
Astrocytes are the major glial cell subtype and the most numerous cells in the CNS (Raff et al., 1983). Astrocytes are important partners of neighboring cells, including neurons, vascular cells, and other glial cells. They do not play a mere structural or metabolic supportive function but rather are active participants in the regulation of neuronal activity in the brain. One of the central functions of astrocytes is the release and uptake of gliotransmitters/neurotransmitters in the neuronal synaptic cleft (Kimelberg et al., 1997). Gliotransmitters are chemicals released from glial cells (Halassa et al., 2007); astrocytes directly activate neuronal receptors by releasing gliotransmitters such as glutamate (Parpura et al., 1994), d-serine (Mothet et al., 2005), and ATP (Coco et al., 2003). Glutamate released from astrocytes is known to exert numerous effects on neuronal excitability (Angulo et al., 2004; Fellin et al., 2004; Kozlov et al., 2006), whereas astrocyte-derived d-serine contributes to modulation of postsynaptic NMDA receptor-mediated synaptic plasticity (Yang et al., 2003; Panatier et al., 2006). In contrast, astrocyte-derived ATP is a significant extracellular signaling molecule and an endogenous ligand for the purinergic receptor that modulates synaptic transmission, either directly or through ATP metabolic product adenosine (Gordon et al., 2005; Pascual et al., 2005). Astrocyte-derived ATP increases the amplitude of miniature EPSCs through the activation of postsynaptic purinergic receptors (Gordon et al., 2005). In contrast, astrocyte-derived ATP in the hippocampus contributes to GABAergic neuron-mediated heterosynaptic depression of synaptic transmission (Zhang et al., 2003; Bowser and Khakh, 2004). This suggests that astrocytes may differentially modulate neuronal activity by releasing different gliotransmitters and that a given gliotransmitter may have different effects depending on the target neuron or synapse.
Activated astrocytes are found around senile plaques containing Aβ peptide in the AD brain and in mouse models of AD, suggesting potential interactions between astrocytes and senile plaques (Akiyama et al., 2000; Wegiel et al., 2000). However, the role of gliotransmitters, particularly ATP, in the pathogenesis of AD is unclear. We therefore examined whether ATP modulates synaptic activity.
We found that Aβ-related synaptic alteration is prevented by gliotransmitter ATP; Aβ1-42 (Aβ42)-induced disruption of synaptic plasticity, including disruption of synaptic protein levels, dendritic spine density, and long-term potentiation (LTP), was restored by ATP. Furthermore, we found that the protective role of ATP is mediated by the P2 purinergic receptor signaling pathway. Together, these observations suggest that ATP plays a protective role against synaptic dysfunction caused by Aβ42. Thus, neuron–astrocyte crosstalk via gliotransmitter ATP might play an important role in synaptic modulation.
Materials and Methods
Chemicals.
ATP, adenosine 5′-[γ-thio] triphosphate (ATPγS), and PPADS (pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid; a P2X purinergic receptor antagonist) were purchased from Sigma-Aldrich. Aβ1-42 synthetic peptide was purchased from American Peptide.
Electrophysiology.
Field EPSPs (fEPSPs) were recorded from acute hippocampal slices (400 μm thick) obtained from 3–4 month-old male C57BL/6 mice. Acute hippocampal slices were placed in oxygenated (95% O2, 5% CO2) artificial CSF (aCSF) at 37°C for >1 h and maintained in a chamber continuously perfused with oxygenated aCSF. The fEPSPs were recorded in the striatum radiatum of the CA1 subfield by the 3 m NaCl-filled microelectrodes (3–5 MOhm) while the Schaffer collateral afferent fiber was stimulated with a bipolar concentric electrode. Test fEPSPs were evoked by the stimulation intensity that yielded one-third of the maximal fEPSP responses at a frequency of 0.033 Hz, and LTP was induced by five episodes of theta burst stimulation (TBS) that were delivered at 0.1 Hz. In each episode, 10 trains of stimulation consisting of four pulses at 100 Hz were delivered at 5 Hz. The data were acquired through Axopatch 200A amplifier and Digidata 1200 (Axon Instrument).
Cell line and cultures.
The human astroglioma U373 cells (ATCC number: HTB-17) were cultured in DMEM containing 10% fetal bovine serum and 0.1 mg/ml penicillin and streptomycin (P/S; Sigma-Aldrich) at 37°C in humidified 5% CO2 incubator. U373 cells were plated onto 96-multiwell plates (20 × 103 cells in 0.1 ml of culture medium per well) for ATP measurement.
Primary astrocyte cultures.
Astrocytes were prepared from cortices of newborn (1-d-old) ICR mice as previously described (McCarthy and de Vellis, 1980; Benavides et al., 2005). The cells were dissociated gently by several passages and grown in DMEM supplemented with 10% fetal bovine serum and 0.1 mg/ml P/S (Sigma-Aldrich) at 37°C in humidified 5% CO2 incubator for 10–14 d. After trypsinization, the cells were plated onto 96-multiwell plates (50 × 103 cells in 0.1 ml of culture medium per well).
Primary hippocampal neuron cultures.
Primary hippocampal neurons were prepared from newborn (1-d-old) ICR mice and E18 Sprague Dawley rat embryos, as described previously (Brewer et al., 1993), but with some modifications. The neurobasal medium to which B27 supplement (Invitrogen), l-glutamine (0.5 mm), and 0.1 mg/ml P/S (Sigma-Aldrich) were added was changed every 3 d. The experiments were performed in cultures at 21 d in vitro (DIV).
Western blot.
The hippocampal neurons were homogenized in RIPA buffer containing protease inhibitor mixture (Sigma-Aldrich). Homogenated protein samples were separated on 8% SDS-PAGE gradient gels. NMDAR2A was detected with anti-NMDAR2A antibody (1:2000; Millipore). Anti-NMDAR1 antibody (1:2000; Cell Signaling Technology), anti-PSD-95 antibody (1:2000; Abcam), anti-β-actin (1:2000; Sigma-Aldrich), and anti-tubulin antibody (1:2000; Applied Biological Materials) were used for immunoblotting. Immunoreactivity was determined by enhanced chemiluminescence (GE Healthcare). The chemiluminescence signal was quantified with a digital image analyzer (LAS-3000; Fuji).
Measurement of ATP release.
Extracellular ATP in the culture medium of astrocytes was measured with a luciferin-luciferase bioluminescence assay kit (Invitrogen) and luminometer (Infinite M200, Tecan).
Phalloidin staining.
After two washes with prewarmed PBS, cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature (RT), then incubated in 0.1% Triton X-100 in PBS for 3–5 min. After another washing, cells were stained with Alexa Fluor 488-phalloidin fluorescent phallotoxin (Invitrogen) in PBS for 20 min at RT. Stained cells were visualized by laser scanning confocal microscopy (LSCM; Olympus Fluoview 300).
Quantitative spine density analysis.
Images of phalloidin-stained rat primary hippocampal neurons were captured by LSCM (Olympus Fluoview 300), and dendritic spines were identified as small protrusions. The density of the dendritic protrusions (0.4–2.5 μm) was measured from 10 to 30 dendrites of three to six neurons and calculated by quantifying the number of spines per unit length of dendrite and normalized per 10 μm of dendrite length.
Data analysis and statistics.
Data are presented as mean ± SEM. Differences between groups were evaluated by one-way ANOVA with Tukey's multiple-comparisons test using GraphPad Prism 5 software. Significance was accepted at p < 0.05.
Results
Aβ42 increased release of ATP from primary astrocytes and U373 astrocyte cell line
Astrocytes exhibit spontaneous calcium oscillations that can induce the release of glutamate as a gliotransmitter (Halassa et al., 2007). Aβ also disrupts fluctuations in calcium signaling in cultured astrocytes (Stix and Reiser, 1998; Abramov et al., 2003; Chow et al., 2010). To examine whether Aβ42 treatment affects release of ATP from astrocytes, cultures of primary astrocytes and U373 cells were treated with various concentrations of Aβ42 for 3 h, and extracellular ATP concentrations in the culture media were assayed with the luciferin-luciferase enzyme system noted above. With Aβ42 treatment, release of ATP increased significantly in both cultured primary astrocytes and U373 cells (Fig. 1, A and B, respectively), indicating that ATP is released from Aβ42-activated astrocytes.
Figure 1.

Aβ42-induced release of ATP from cultured primary astrocytes and U373 astrocyte cell lines. A, ATP release was measured after treatment of the primary astrocytes with Aβ42 (2 or 4 μm) for 3 h. ATP was significantly released in astrocytes treated with Aβ42 (4 μm). B, ATP was significantly released from U373 cells treated with Aβ42 (2 or 10 μm) for 3 h. Extracellular ATP in the conditioned media was measured by luciferin-luciferase assay. Values are means of three independent experiments ± SD. Statistical significance was tested by one-way ANOVA followed by a Tukey's multiple-comparison test. **p < 0.01 versus vehicle (control) group. Vcl, Vehicle.
ATP prevented Aβ42-mediated dendritic spine loss in primary hippocampal neurons
Because dendritic spine density is associated with synaptic plasticity, learning, and memory (Collin et al., 1997), we investigated the effects of ATP on dendritic spine density in rat primary hippocampal neurons. Cultured hippocampal neurons (21 DIV) were treated with Aβ42 for 48 h or pretreated with ATP for 30 min before Aβ42 treatment. Spines were labeled with green fluorescent Alexa Fluor 488-phalloidin dye. Dendritic spine density was measured at selected segments by means of confocal microscopy (Fig. 2A,B). Dendritic spine density was significantly decreased in the Aβ42-treated primary hippocampal neurons versus control neurons (Fig. 2C). In contrast, pretreatment of the hippocampal neurons with ATP exhibited protective effects on Aβ42-mediated reduction of dendritic spine density.
Figure 2.
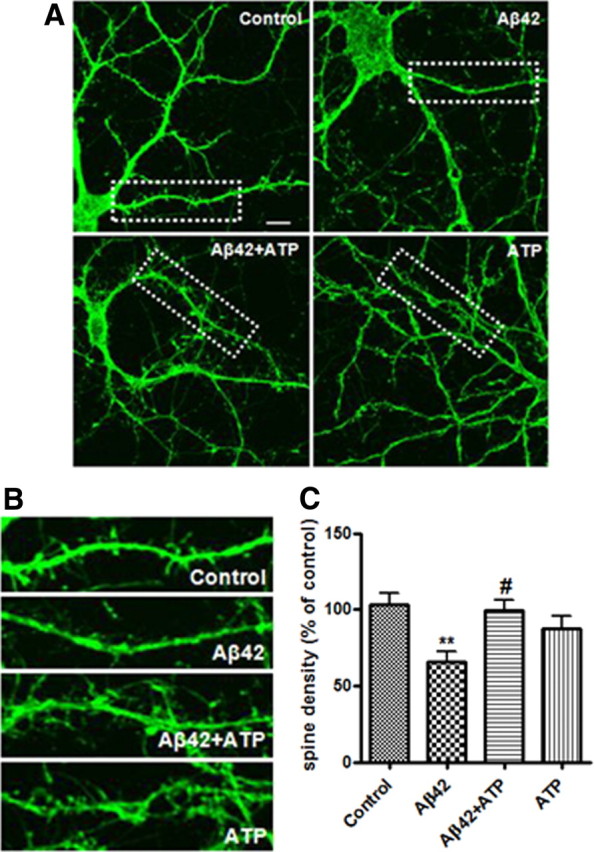
Effect of ATP on Aβ42-mediated dendritic spine loss in cultured primary rat hippocampal neurons. Primary rat hippocampal neurons (21 DIV) pretreated with or without ATP (10 μm) for 30 min were exposed to Aβ42 (2 μm) for 48 h. A, Rat hippocampal neurons were stained with phalloidin Alexa-488 (green) to visualize dendritic spines. B, Magnified images of the areas marked with dotted lines in Figure 2A. C, Quantification of spine density in dendritic segments. Statistical significance was analyzed by one-way ANOVA followed by a Tukey's multiple-comparison test. **p < 0.01 versus vehicle (control) group; #p < 0.05 versus Aβ42-treated group. Scale bar, 10 μm.
ATP restored Aβ42-induced reduction of LTP in hippocampal slices
LTP and dendritic spine density correlate with neuronal synaptic plasticity (Geinisman et al., 1991; Moser et al., 1994; Geinisman, 2000). Previous studies have shown that acute treatment with synthetic Aβ peptides inhibits hippocampal LTP in vitro and in vivo (Cullen et al., 1997; Lambert et al., 1998; Chen et al., 2000). To examine whether ATP protects against Aβ-induced LTP reduction in hippocampal slices, Aβ42 (200 nm) or ATP (5 μm) was perfused into the slice chamber during basal response recording. Basal fEPSP was not affected (data not shown). When TBS was delivered following perfusion with Aβ42, synaptic potentiation was significantly impaired 60 min after TBS (106.31 ± 7.04%, n = 6), as previously reported (Chen et al., 2000). This did not occur in the control group (168.23 ± 8.96%, n = 7; Fig. 3A). However, exposure to ATP alone did not affect basal synaptic transmission or LTP (161.96 ± 8.42%, n = 8; Fig. 3A). Importantly, cotreatment of Aβ42 and ATP prevented the Aβ42-induced reduction in LTP, indicating a protective role of ATP against Aβ42 (Fig. 3A). The protective role of ATP was observed in both the early (10 min post-tetanus) and late (60 min post-tetanus) phases of potentiation (Fig. 3, B and C, respectively). In addition, we investigated the effects of lower concentrations of Aβ42 (200 nm) on dendritic spine density in rat primary hippocampal neurons. As expected, Aβ42 dramatically reduced dendritic spine density at 200 nm concentration in the primary rat hippocampal neurons compared to control (Fig. 3D–F); we also observed reduction of synaptic molecules, NR2A and PSD-95, by treatment of lower Aβ42 concentration (200 nm) in the mouse hippocampal neurons (data not shown). These data indicate that Aβ42 could act to regulate synaptic activity at low dose.
Figure 3.
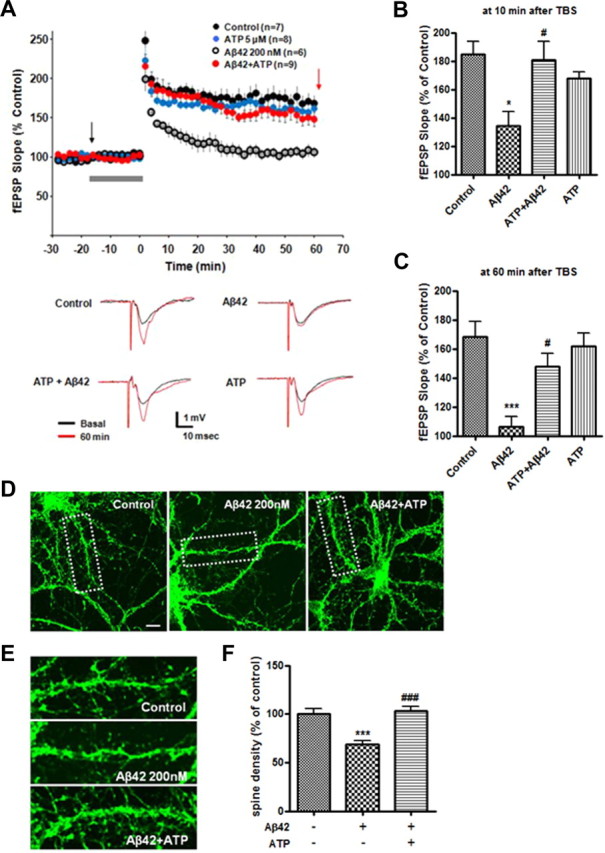
Effects of ATP on Aβ42-mediated alteration of LTP in hippocampal slice. A, fEPSPs were recorded from the CA1 region of the hippocampus. Aβ42 (200 nm) inhibited TBS-induced LTP. In contrast, cotreatment of ATP (5 μm) with Aβ42 prevented the inhibition of LTP. Graph of individual fEPSP slope (top) evoked by stimulation throughout the different drug applications. Traces (below) represent averages of responses taken in different conditions at time points indicated by the black and red arrows of the fEPSP graph (top). B, C, Potentiation of fEPSP at 10 (B) or 60 (C) min after TBS. The gray bar indicates the aCSF (control) or chemicals (Aβ42, Aβ42+ATP, ATP) infusion for 20 min before TBS. p values were calculated using one-way ANOVA followed by a Tukey's multiple-comparison test. *p < 0.01 or ***p < 0.001 versus control group; #p < 0.05 versus Aβ42-treated group. D, Effects of Aβ42 on dendritic spine density at low concentration (200 nm). Primary rat hippocampal neurons (21 DIV) with or without ATP pretreatment (30 min) were exposed to Aβ42 for 48 h. Phalloidin Alexa-488 (green) was used to visualize dendritic spines. E, Magnified images of the areas marked with dotted lines in D. F, Quantification of spine density in dendritic segments. Statistical significance was analyzed by one-way ANOVA followed by a Tukey's multiple-comparison test. ***p < 0.001 versus vehicle (control) group; ###p < 0.001 versus Aβ42-treated group. Scale bar, 10 μm.
ATP prevented Aβ42-induced reduction of synaptic molecule levels in cultured primary hippocampal neurons
Because ATP prevented the Aβ-induced LTP reduction, changes in the levels of synaptic molecules such as the NMDA receptor, which is known to regulate synaptic plasticity (Collingridge et al., 1983; Elgersma and Silva, 1999), and PSD-95, a brain-specific scaffolding protein, were examined. PSD-95 interacts with NMDA receptor subunits NR2A and NR2B and regulates synaptic transmission and plasticity (Kornau et al., 1995; Béïque and Andrade, 2003). NMDA receptor 2A (NR2A) and PSD-95 levels decreased in hippocampal neurons (21 DIV) cultured with Aβ42 (2 μm) for 48 h (Fig. 4A–C), but the NMDA receptor 1 (NR1) level did not change (Fig. 4A,D). To test whether ATP protects neurons from Aβ42-induced decreases in synaptic molecules, we pretreated primary hippocampal neurons for 30 min with ATP (10 μm) before adding Aβ42. ATP prevented the effect of Aβ42 on synaptic molecules. ATP treatment before addition of Aβ42 did not reduce the level of NR2A or PSD-95 (Fig. 4). We performed experiment with the nonhydrolysable ATP analog ATPγS to provide strong evidence that ATP, not its metabolites, play a protective role in the Aβ42-induced synaptic alteration (Fig. 4E,F). Pretreatment of hippocampal neurons with ATPγS (20 μm) before Aβ42 stimulation also had protective effect on the Aβ42-stimulated reduction of NR2A and PSD-95 levels (Fig. 4E,F). In accordance with ATP, we observed that Aβ42-induced reduction of synaptic molecules was restored by ATPγS. Together, these data indicate that ATP can protects against the decrease in neuronal synaptic molecules in response to Aβ42.
Figure 4.

ATP restored Aβ42-mediated reduction of synaptic proteins in primary hippocampal neurons. Primary hippocampal neurons (21 DIV) pretreated with or without ATP for 30 min were exposed to Aβ42 for 48 h. A, Cells were then harvested for immunoblotting with anti-NR2A, anti-PSD-95, anti-NR1, and anti-β-actin antibodies. Equivalent amounts of protein from each sample were analyzed by Western blotting, and β-actin was used as a loading control. The arrowhead indicates a specific band of PSD-95 and the asterisk indicates a nonspecific band. B–D, Quantification of NR2A (B), PSD-95 (C), and NR1 (D). Aβ42-mediated reduction in NR2A and PSD-95 protein levels was prevented by ATP pretreatment. The NR1 protein level was not changed. Statistical significance was analyzed by one-way ANOVA followed by a Tukey's multiple-comparison test. **p < 0.01 and ***p < 0.001 versus vehicle (Veh; control) group (n = 6); #p < 0.05 and ##p < 0.01 versus Aβ42-treated group (n = 6). E, Effect of ATPγS (20 μm) on Aβ42-mediated reduction of synaptic proteins, NR2A and PSD-95. Primary hippocampal neurons (21 DIV) pretreated with or without ATPγS for 30 min were exposed to Aβ42 for 48 h. F, Quantification of the results in E. Values are means of four independent experiments ±SD. Statistical significance was tested by one-way ANOVA followed by a Tukey's multiple-comparison test. **p < 0.01 and ***p < 0.001 versus vehicle (control) group; #p < 0.05 versus Aβ42-treated group.
ATP prevented Aβ42-induced decrease in synaptic molecules via P2 purinergic receptor
Extracellular ATP plays an important role in cellular signaling via P2 type-purinergic receptors (Kanjhan et al., 1996), which are categorized into ionotropic P2X ligand-gated ion channels and metabotropic P2Y G-protein coupled receptors (Bogdanov et al., 1998; Ralevic and Burnstock, 1998). Hippocampal neurons express both P2X and P2Y purinergic receptors. PPADS is a nonselective (but nonuniversal) P2 receptor antagonist and does not block P1 receptor (Lambrecht, 2000; Lambrecht et al., 2002). To determine whether ATP prevents Aβ42-mediated decrease in synaptic molecules by purinergic receptor signaling, we treated primary hippocampal neurons with PPADS before addition of extracellular ATP. Even though ATP prevented the Aβ42-induced decreases in synaptic proteins such as NR2A and PSD-95 (Fig. 4A), treatment of PPADS inhibited the protective effect of ATP on NR2A and PSD-95 (Fig. 5A, lane 4), strongly suggesting that ATP prevents Aβ42-induced reduction of synaptic proteins levels via P2 type-purinergic receptor signaling.
Figure 5.
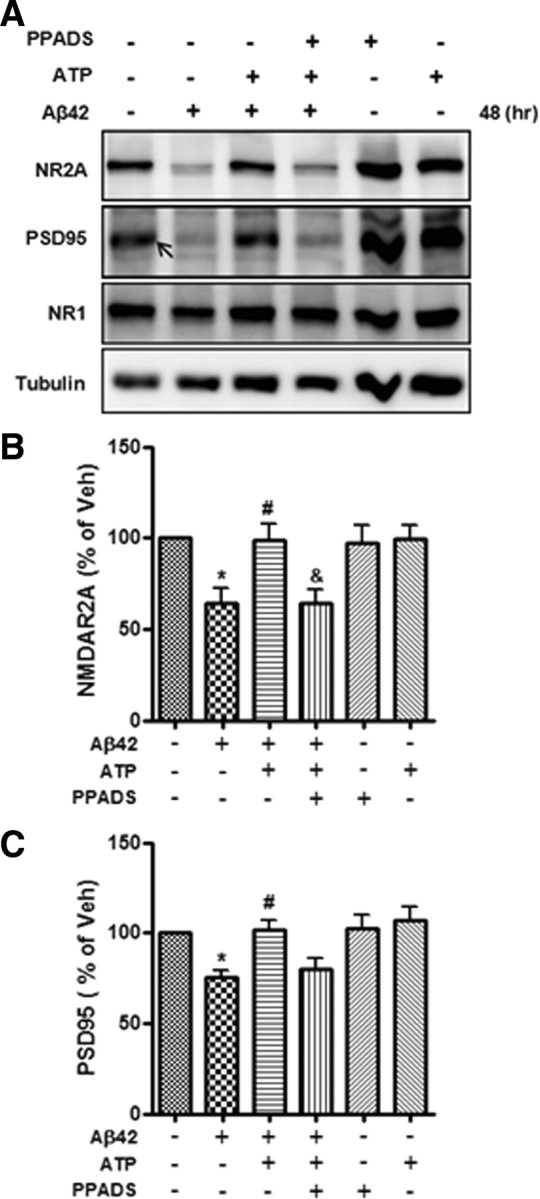
Effects of ATP alone or in combination with PPADS on Aβ42-mediated reduction of synaptic molecules. A, Primary hippocampal neurons treated with Aβ42 (2 μm) and pretreated with or without PPADS (50 μm) for 30 min were exposed to Aβ42 combined with ATP for 48 h. Representative Western blots were detected with antibodies targeting NR2A, PSD-95, NR1, and tubulin. Tubulin was used as a loading control. The arrow indicates a specific band of PSD-95. B, C, Quantification of NR2A (B) and PSD-95 (C). Statistical significance was tested by one-way ANOVA followed by a Tukey's multiple-comparison test. *p < 0.05 versus vehicle (Veh) group; #p < 0.05 versus Aβ42-treated group; &p < 0.05 versus Aβ42 combined with ATP-treated group.
Discussion
Our study provides strong evidence that a gliotransmitter ATP can act against Aβ-induced alteration of synaptic plasticity. First, Aβ42 increased ATP release from cultured primary astrocytes and human astroglioma U373 cell lines. Second, ATP protected Aβ42-mediated dendritic spine loss in primary hippocampal neurons. Third, ATP rescued Aβ42-mediated disruption of LTP in hippocampal slice. Fourth, ATP restored Aβ42-mediated reduction of synaptic proteins through P2 purinergic receptor. These results provide converging evidence for protective roles of ATP against Aβ-induced impairment of synaptic plasticity.
Previous studies on the mechanism underlying Aβ toxicity-associated neuronal death are generally limited to neuronal cells. The most abundant type of glial cell in the CNS is the astrocyte. Astrocytes were originally thought to play a passive role in supporting neuronal cells; however, studies over the last decade have indicated that astrocytes also play active roles in numerous aspects neuronal functions, including synaptic plasticity. Despite the importance of astrocytes in the CNS, the molecular mechanism of interaction between neurons and astrocytes are largely unknown. Calcium signaling in astrocytes is important for astrocyte–neuron communication in the brain (Araque, 2008). Altered neuronal calcium homeostasis is a prominent feature of AD, and Aβ can significantly disrupt intracellular calcium homeostasis in the neurons. Several recent studies showed that Aβ alters astrocytic calcium signaling and gliotransmitter release. Kuchibhotla et al. (2009) reported calcium waves in astrocytes of the APP/PS1 transgenic mouse brain that contains cortical plaques. Aβ25–35-treated microglia and astrocytes exhibit an increase in glutamate and ATP release (Orellana et al., 2011). Therefore, Aβ42-induced ATP release from astrocytes is likely due to altered astrocytic calcium oscillations (Fig. 1). Aβ42-induced LTP reduction has been reported previously (Walsh et al., 2002; Wang et al., 2004; Zhao et al., 2004). Thus, we examined whether ATP can prevent the LTP reduction in the presence of Aβ42. As shown in Figure 3, Aβ42-induced LTP reduction was prevented by ATP at 10 and 60 min after TBS. Because spine formation is strongly associated with neuronal synaptic plasticity and LTP, we explored the effect of the ATP on the density of dendritic spines. However, the effect of gliotransmitter ATP alone on neurons is difficult to explore because astrocyte-conditioned media contain various types of chemicals including ATP, glutamate, proinflammatory cytokines, and chemokines. To examine the pure effect of ATP on neuronal activity and avoid the effects of other astrocyte-derived factors such as glutamate, cytokines, and chemokines, we used primary hippocampal neuron cultures treated with extracelluar ATP. We observed that Aβ42-mediated reduction of dendritic spine density is also significantly restored by pretreatment with ATP (Figs. 2, 3D–F). Together, these results demonstrate a protective role of the ATP against altered synaptic plasticity by Aβ42.
NMDA receptor is a well known key molecule for regulating synaptic plasticity and memory function. It is a heterotetramer complex consisting of 2 NR1 and 2 NR2 subunits (Monyer et al., 1992; Nakanishi, 1992; Ishii et al., 1993) and it plays an important role in the pathophysiology of AD (Hynd et al., 2004). NR2A and NR2B are predominant forms of NR2 NMDA receptor subunits. PSD-95, a postsynaptic scaffolding molecule, is reported to interact with NR2A via the PSD-95/discs large/zona occludens-1 domain (Niethammer et al., 1996), suggesting that changes in NR2A and PSD-95 may follow a similar pattern. As expected, Aβ42-mediated reduction in NR2A and PSD-95 was prevented by ATP (Fig. 4). We also observed that ATP protects against Aβ42-mediated reduction in synaptophysin, a synaptic vesicle glycoprotein, for synaptic transmission in primary hippocampal neurons (data not shown). Extracellular ATP acts as a signal that regulates a variety of cellular processes via binding to P2 purinergic receptors (Burnstock, 1997). The P2 purinergic receptors comprise seven P2X receptor subunits (P2X1–7), which are ligand-gated ion channels (North, 2002), and eight metabotropic P2Y receptors (P2Y1,2,4,6,11,12,13,14), which are G-protein-coupled receptors (Lazarowski et al., 2003). P2X receptor subunits are widely expressed in neurons as well as glial cells (Rubio and Soto, 2001; North, 2002). P2X receptors mediate fast synaptic responses to ATP, whereas P2Y receptors cause slow changes of the membrane potential in neurons (Khakh, 2001; Robertson et al., 2001; Burnstock and Knight, 2004; Illes and Alexandre Ribeiro, 2004). When primary hippocampal neurons were treated with PPADS, ATP did not prevent Aβ42-induced reduction of synaptic molecules (Fig. 5), indicating that P2 receptor mediates protective role of ATP at the neurons. In vitro, PPADS has been shown to block homomeric P2X1, P2X2, P2X3, P2X5, and P2Y1 receptors as well as heteromeric P2X2/3 and P2X1/5 receptors. PPADS, however, does not block homomeric P2X4, P2X6, P2X7, P2Y2, P2Y4, P2Y6, or P2Y11 receptors (Ralevic and Burnstock, 1998). Further studies are needed to clarify the involvement and the exact subtype of P2 receptor. Extracellular ATP is initially converted by ectonucleotidases to ADP or AMP, which are further hydrolyzed to adenosine (Zimmermann, 1994). Adenosine is a signaling molecule that regulates cellular activity in the CNS through P1 purinergic receptor (Kaster et al., 2004; Daré et al., 2007). To exclude the effect of adenosine, we used ATPγS, a nonhydrolyzable ATP analog. In accordance with ATP, Aβ42-induced reduction of synaptic molecules was restored by ATPγS (Fig. 4E,F) and PPADS blocks P2 receptors but not P1 receptors (Lambrecht, 2000).
Our data collectively suggest that ATP from astrocytes regulates synaptic plasticity through activation of P2 purinergic receptors. Furthermore, this effect significantly regulates NR2A and PSD-95 levels, hippocampal dendritic spine density, and LTP. Given the critical involvement of both astrocytes-derived ATP and synaptic plasticity in neurons, the molecules controlling the release of gliotransmitters may provide therapeutic targets for treating AD.
Footnotes
This work was supported by grants from the 21C Frontier Functional Proteomics Project (FPR08K1301-02210), National Research Foundation (2009-0081673), World Class University-Neurocytomics, and the Korean National Institute of Health Research and Development Program Project (2009-0443), and supported in part by the Basic Research Program (2008-05943) and Medical Research Center at Seoul National University (2011-0030738).
References
- Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A. Astrocytes process synaptic information. Neuron Glia Biol. 2008;4:3–10. doi: 10.1017/S1740925X09000064. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia. 2005;52:261–275. doi: 10.1002/glia.20242. [DOI] [PubMed] [Google Scholar]
- Bogdanov Y, Rubino A, Burnstock G. Characterisation of subtypes of the P2X and P2Y families of ATP receptors in the foetal human heart. Life Sci. 1998;62:697–703. doi: 10.1016/s0024-3205(97)01168-5. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Burnstock G. The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology. 1997;36:1127–1139. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res. 2000;60:65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Chow SK, Yu D, Macdonald CL, Buibas M, Silva GA. Amyloid beta-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro. 2010;2:e00026. doi: 10.1042/AN20090035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C. Storage and release of ATP from astrocytes in culture. J Biol Chem. 2003;278:1354–1362. doi: 10.1074/jbc.M209454200. [DOI] [PubMed] [Google Scholar]
- Collin C, Miyaguchi K, Segal M. Dendritic spine density and LTP induction in cultured hippocampal slices. J Neurophysiol. 1997;77:1614–1623. doi: 10.1152/jn.1997.77.3.1614. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Daré E, Schulte G, Karovic O, Hammarberg C, Fredholm BB. Modulation of glial cell functions by adenosine receptors. Physiol Behav. 2007;92:15–20. doi: 10.1016/j.physbeh.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Silva AJ. Molecular mechanisms of synaptic plasticity and memory. Curr Opin Neurobiol. 1999;9:209–213. doi: 10.1016/s0959-4388(99)80029-4. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Geinisman Y. Structural synaptic modifications associated with hippocampal LTP and behavioral learning. Cereb Cortex. 2000;10:952–962. doi: 10.1093/cercor/10.10.952. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, deToledo-Morrell L, Morrell F. Induction of long-term potentiation is associated with an increase in the number of axospinous synapses with segmented postsynaptic densities. Brain Res. 1991;566:77–88. doi: 10.1016/0006-8993(91)91683-r. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Baimoukhametova DV, Hewitt SA, Rajapaksha WR, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Illes P, Alexandre Ribeiro J. Molecular physiology of P2 receptors in the central nervous system. Eur J Pharmacol. 2004;483:5–17. doi: 10.1016/j.ejphar.2003.10.030. [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M. Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- Kanjhan R, Housley GD, Thorne PR, Christie DL, Palmer DJ, Luo L, Ryan AF. Localization of ATP-gated ion channels in cerebellum using P2x2R subunit-specific antisera. Neuroreport. 1996;7:2665–2669. doi: 10.1097/00001756-199611040-00051. [DOI] [PubMed] [Google Scholar]
- Kaster MP, Rosa AO, Rosso MM, Goulart EC, Santos AR, Rodrigues AL. Adenosine administration produces an antidepressant-like effect in mice: evidence for the involvement of A1 and A2A receptors. Neurosci Lett. 2004;355:21–24. doi: 10.1016/j.neulet.2003.10.040. [DOI] [PubMed] [Google Scholar]
- Khakh BS. Molecular physiology of P2X receptors and ATP signalling at synapses. Nat Rev Neurosci. 2001;2:165–174. doi: 10.1038/35058521. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Cai Z, Rastogi P, Charniga CJ, Goderie S, Dave V, Jalonen TO. Transmitter-induced calcium responses differ in astrocytes acutely isolated from rat brain and in culture. J Neurochem. 1997;68:1088–1098. doi: 10.1046/j.1471-4159.1997.68031088.x. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht G. Agonists and antagonists acting at P2X receptors: selectivity profiles and functional implications. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:340–350. doi: 10.1007/s002100000312. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Braun K, Damer M, Ganso M, Hildebrandt C, Ullmann H, Kassack MU, Nickel P. Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr Pharm Des. 2002;8:2371–2399. doi: 10.2174/1381612023392973. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Boucher RC, Harden TK. Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol Pharmacol. 2003;64:785–795. doi: 10.1124/mol.64.4.785. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Moser MB, Trommald M, Andersen P. An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc Natl Acad Sci U S A. 1994;91:12673–12675. doi: 10.1073/pnas.91.26.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, Jiang JX, Naus CC, Sáez JC, Giaume C. Amyloid beta-induced death in neurons involves glial and neuronal hemichannels. J Neurosci. 2011;31:4962–4977. doi: 10.1523/JNEUROSCI.6417-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Raff MC, Abney ER, Cohen J, Lindsay R, Noble M. Two types of astrocytes in cultures of developing rat white matter: differences in morphology, surface gangliosides, and growth characteristics. J Neurosci. 1983;3:1289–1300. doi: 10.1523/JNEUROSCI.03-06-01289.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Robertson SJ, Ennion SJ, Evans RJ, Edwards FA. Synaptic P2X receptors. Curr Opin Neurobiol. 2001;11:378–386. doi: 10.1016/s0959-4388(00)00222-1. [DOI] [PubMed] [Google Scholar]
- Rubio ME, Soto F. Distinct localization of P2X receptors at excitatory postsynaptic specializations. J Neurosci. 2001;21:641–653. doi: 10.1523/JNEUROSCI.21-02-00641.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The origins of Alzheimer disease: a is for amyloid. JAMA. 2000;283:1615–1617. doi: 10.1001/jama.283.12.1615. [DOI] [PubMed] [Google Scholar]
- Stix B, Reiser G. Beta-amyloid peptide 25–35 regulates basal and hormone-stimulated Ca2+ levels in cultured rat astrocytes. Neurosci Lett. 1998;243:121–124. doi: 10.1016/s0304-3940(98)00106-2. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Tarnawski M, Lach B. Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol. 2000;100:356–364. doi: 10.1007/s004010000199. [DOI] [PubMed] [Google Scholar]
- Wísniewski HM, Robe A, Zigman W, Silverman W. Neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol. 1989;48:606–609. doi: 10.1097/00005072-198911000-00001. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U S A. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92:2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Signalling via ATP in the nervous system. Trends Neurosci. 1994;17:420–426. doi: 10.1016/0166-2236(94)90016-7. [DOI] [PubMed] [Google Scholar]