

Abstract
Acetylcholine facilitates long-term potentiation (LTP) and long-term depression (LTD), substrates of learning, memory, and sensory processing, in which acetylcholine also plays a crucial role. Ca2+ ions serve as a canonical regulator of LTP/LTD but little is known about the effect of acetylcholine on intracellular Ca2+ dynamics. Here, we investigated dendritic Ca2+ dynamics evoked by synaptic stimulation and the resulting LTP/LTD in layer 2/3 pyramidal neurons of the rat visual cortex. Under muscarinic stimulation, single-shock electrical stimulation (SES) inducing ∼20 mV EPSP, applied via a glass electrode located ∼10 μm from the basal dendrite, evoked NMDA receptor-dependent fast Ca2+ transients and the subsequent Ca2+ release from the inositol 1,4,5-trisphosphate (IP3)-sensitive stores. These secondary dendritic Ca2+ transients were highly localized within 10 μm from the center (SD = 5.0 μm). The dendritic release of Ca2+ was a prerequisite for input-specific muscarinic LTP (LTPm). Without the secondary Ca2+ release, only muscarinic LTD (LTDm) was induced. D(−)-2-amino-5-phosphopentanoic acid and intracellular heparin blocked LTPm as well as dendritic Ca2+ release. A single burst consisting of 3 EPSPs with weak stimulus intensities instead of the SES also induced secondary Ca2+ release and LTPm. LTPm and LTDm were protein synthesis-dependent. Furthermore, LTPm was confined to specific dendritic compartments and not inducible in distal apical dendrites. Thus, cholinergic activation facilitated selectively compartment-specific induction of late-phase LTP through IP3-dependent Ca2+ release.
Introduction
Interest is growing in the mechanisms that modulate and possibly enhance synaptic plasticity and memory functions. Further stimulating this interest are efforts to address the problem of memory deficit, which occurs in the elderly and in association with degenerative diseases. Acetylcholine (ACh) and muscarinic activation enhance sensory processing, learning, and memory in humans and in experimental animals (Anagnostaras et al., 2003; Gais and Born, 2004). The activation of muscarinic ACh receptors (mAChRs) directly modulates network dynamics by facilitating gamma oscillations (Fisahn et al., 2002; Rodriguez et al., 2004) and reducing the variance of rate responses to sensory stimuli in the visual cortex (Rodriguez et al., 2010). Activation of mAChRs in the hippocampus and the neocortex (Markram and Segal, 1990; Huerta and Lisman, 1995; Auerbach and Segal, 1996; Kirkwood et al., 1999) facilitates long-term potentiation (LTP) and long-term depression (LTD), cellular and molecular substrates of learning and memory (Malenka and Bear, 2004). However, the molecular mechanism that determines the polarity of the long-term synaptic plasticity facilitated by ACh is not clearly understood.
An increase in intracellular Ca2+ via voltage-dependent Ca2+ channels (VDCCs) and Ca2+-permeable NMDA receptors is one of the key determinants of the polarity of long-term synaptic plasticity (Lisman, 1989; Augustine et al., 2003). Released Ca2+ from intracellular stores also contributes to the induction of long-term synaptic plasticity in the cerebellar Purkinje and hippocampal pyramidal neurons (Finch and Augustine, 1998; Raymond and Redman, 2006). In the hippocampal and neocortical pyramidal neurons, activation of mAChR evoked propagating Ca2+ release from inositol 1,4,5-trisphosphate (IP3)-sensitive stores, assisted by an influx of extracellular Ca2+ (Nakamura et al., 2000; Cho et al., 2008). However, it has not been tested whether cholinergically mediated Ca2+ release assisted by the influx of Ca2+ contributes to the induction of long-term synaptic plasticity (Ross et al., 2005). In hippocampal CA1 pyramidal neurons, propagating Ca2+ release from IP3-sensitive stores evoked by puff-applied ACh alone induced long-term enhancement of NMDA and AMPA receptor-mediated transmission (Fernández de Sevilla et al., 2008; Fernández de Sevilla and Buño, 2010), which was a “non-Hebbian” form of LTP, LTPIP3, without correlated presynaptic and postsynaptic activities (Fernández de Sevilla et al., 2008). Because the input specificity of LTP/LTD is crucial in the processing and storage of information and in pathway-specific changes of circuit dynamics (Giocomo and Hasselmo, 2007), it is of prime interest to determine whether muscarinic LTP (LTPm) induced by Ca2+ released from IP3-sensitive stores is input-specific.
Direct dendritic Ca2+ imaging with local synaptic stimulation could resolve these questions. In the present study, we evoked Ca2+ release from IP3-sensitive stores in a localized dendritic area with combined single-shock synaptic stimulation near dendritic trees and cholinergic activation in layer 2/3 pyramidal neurons of the visual cortex. We found that the resulting LTP was input-specific and protein synthesis-dependent. Furthermore, the induction of LTP under mAChR activation was dendritic compartment-specific. Thus, muscarinic cholinergic activation might confer a long-term change in visual cortical circuit dynamics in a pathway-specific manner.
Materials and Methods
Slice preparation.
Animal care and surgical procedures were conducted with the approval of the Ethics Committee of the Catholic University of Korea and were consistent with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Postnatal day 21–27 Sprague Dawley rats of either sex (Orientbio) were anesthetized with chloral hydrate (400 mg/kg, i.p.) before decapitation. Coronal slices (300 μm thick) of primary visual cortex were prepared as described previously (Cho et al., 2008) in ice-cold artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 CaCl2, 2 MgSO4, and 10 d-glucose. This ACSF was aerated with a mixture of 95% O2 and 5% CO2, and allowed to recover at 37°C for 30 min. The slices were then left at room temperature (22–24°C) no longer than 5 h, until recordings were performed. The slices were transferred to the recording chamber and superfused continuously (1.5 ml/min) with the same solution, except for 2 mm CaCl2 and 1 mm MgSO4. All recordings were performed at 31–32°C.
Whole-cell patch clamp.
A standard whole-cell patch-clamp technique with a bridge amplifier (BVC-700A, Dagan) was used to record the membrane potential. The patch electrodes (4–6 MΩ) were pulled from borosilicate glass and filled with a solution containing the following (in mm): 130 K-gluconate, 10 KCl, 3 Mg-ATP, 10 Na2-phosphocreatine, 0.3 Na3-GTP, and 10 HEPES (pH 7.25/KOH), supplemented with 50 μm Oregon Green 488 BAPTA-1 (OGB-1; Kd = 170 nm; Invitrogen) and 25 μm Alexa Fluor 594 (Invitrogen) as a Ca2+-sensitive indicator to monitor Ca2+ concentration changes and a Ca2+-insensitive indicator to visualize thin dendritic structure, respectively. Pyramidal neurons in layer 2/3 of the primary visual cortex were recorded using infrared-differential interference contrast video-microscopy with an upright microscope (BX51-WI fitted with a 40×/0.80 NA water-immersion objective, Olympus). Their regular spiking patterns and the morphology of the bifurcated apical dendrites were confirmed. Access resistance was 12.7 ± 0.4 MΩ (n = 12) as determined from the settings of the bridge balance in experiments. Membrane potentials were not corrected for ∼14 mV junction potential. Command generation, data acquisition, and analyses were performed using the pClamp 9.2 Suite software (Molecular Devices). Data were filtered at 5 kHz, sampled at 20 kHz, and saved to a computer hard drive (Pentium PC).
Cells were used when the membrane potential was sufficiently negative (−76.8 ± 0.7 mV, n = 52). When carbachol (CCh) or muscarine was present in the bath solution, cells were depolarized by 5–7 mV as shown in our previous study (Cho et al., 2010). For local dendritic Ca2+ transients evoked by subthreshold synaptic stimulation, action potential (AP) firing was prevented by offsetting the depolarization with a hyperpolarizing current injection via the recording pipette. For local synaptic stimulation of pyramidal neurons, another patch electrode was filled with ACSF and 50 μm Alexa Fluor 594 and positioned near the dendritic tree of interest (10 μm from dendrite). EPSP was evoked by a brief square current pulse (0.1 ms) with a constant-current isolation unit (A360, World Precision Instruments). In some experiments, EPSP was evoked by stimulation at underlying layer 4 using a concentric bipolar stimulating electrode (100 μm in diameter, SNE-100, David Kopf, Tujunga, CA). Long-term changes in the EPSP amplitude were monitored at 0.05 Hz. After acquisition of stable baseline EPSPs (5–8 mV) for 10 min, muscarine (20 μm) was applied to the bath solution for 5–6 min while synaptic stimulation was continuously delivered. A single electric shock evoking >20 mV EPSP was applied at the end of bath application of muscarine. In some experiments, a burst (3 shocks at 40 or 100 Hz) evoking >20 mV EPSP was applied instead of a single shock. The peak amplitude of the EPSP was measured at 30–40 min after washout of muscarine and compared with that of the baseline EPSP. d(−)-2-amino-5-phosphonopentanoic acid (d-AP5) and anisomycin were applied 10 and 30 min before the baseline recording, respectively, and then washed out with muscarine from the bath. Membrane input resistance was measured by a hyperpolarizing response of ∼5–10 mV from the membrane potential with a negative step current injection into the soma (50 pA, 150 ms).
Ca2+ imaging.
Ca2+ fluorescence imaging was performed by laser-scanning confocal microscopy (FV-300, Olympus). Lights from an argon ion laser (488 nm) and a HeNe laser (543 nm) were used for excitation of OGB-1 and Alexa Fluor 594, respectively, and the emitting fluorescence was filtered using long-pass filters (510 nm and 610 nm, respectively). The glass-stimulating electrode was visualized with the aid of the fluorescence signal of Alexa Fluor 594 in the stimulus electrode. The laser intensity for excitation of the indicator was adjusted to minimize phototoxic damage. Dendritic Ca2+ transients were evoked by synaptic stimulation with a brief square current pulse (0.1 ms) to the stimulus electrode, or by somatic APs generated by current injection (10 ms) into the soma. The fluorescence signal was obtained by using an area-scan mode (every 17–24 ms). Dendritic areas (5–10 μm in length) were averaged for the area-scanned data. The distance of the measured dendritic area from the soma was calculated from the center of the soma. Fluorescence signals were background-corrected and traces were expressed as the relative change in fluorescence [ΔF/F0 = (F − F0)/F0], where F0 was the background-corrected prestimulus fluorescence. The peak amplitude of the Ca2+ transients was determined at the largest ΔF/F0 value of the transients.
Drugs.
d-AP5, 6,7-dinitroquinoxaline-2,3-dione (DNQX) disodium salt, and anisomycin were obtained from Tocris Bioscience. CCh, atropine, muscarine, and all other chemicals were purchased from Sigma-Aldrich. Anisomycin was dissolved in DMSO (0.1%). Inclusion of DMSO at this concentration to ACSF had no effect on the amplitude of EPSP (101.3 ± 2.7% of the baseline, n = 3, p = 0.65).
Statistics.
All data are expressed as the mean ± SEM. A Student's t test was used for statistical comparisons, unless otherwise specified. For multiple comparisons, we used ANOVA (or repeated-measures ANOVA, where appropriate). A p value of <0.05 was considered statistically significant.
Results
Synaptic stimulation evoked secondary Ca2+ transients under carbachol application
To examine the role of synaptically activated Ca2+ release from internal stores on the induction of long-term synaptic plasticity, we initially examined whether a localized synaptic stimulation could evoke the secondary Ca2+ transients under cholinergic stimulation in layer 2/3 pyramidal neurons. A single extracellular electrical shock was applied via a glass electrode located ∼10 μm from the shaft of basal dendrites (65.9 ± 2.4 μm from the soma, n = 45) (Fig. 1A). In normal ACSF, increasing the stimulus intensity gradually increased the amplitude of EPSPs reaching the suprathreshold level, resulting in the generation of a single AP (Fig. 1B). Simultaneous Ca2+ imaging in a nearby dendritic shaft also showed gradual increases in the amplitude of initial fast Ca2+ transients (CaT1). A bath application of CCh (20 μm) reduced the EPSP amplitudes by 40.9 ± 3.3% (n = 13, p < 0.001) without changing peak time (8.0 ± 0.5 ms to 7.8 ± 0.7 ms, p = 0.73) and decay time constant (42.4 ± 3.5 ms to 39.4 ± 3.8 ms, p = 0.23) of EPSP. Membrane resistance (172 ± 14 MΩ to 172 ± 13 MΩ, p = 0.96) and membrane time constant (26.2 ± 1.2 ms to 26.0 ± 1.5 ms, p = 0.89) were not changed by the application of CCh. The reduced synaptic transmission appears to be caused by presynaptic inhibition of glutamate release with mAChR activation (Gil et al., 1997; Qian and Saggau, 1997). The amplitude of Ca2+ transients was reduced similarly to that of EPSP (Fig. 1B–D). The stimulus intensities that evoked EPSPs of 20 mV with the application of CCh (148.7 ± 13.5 μA, n = 13) were ∼1.4 times higher than those in normal ACSF (106.6 ± 10.2 μA) (Fig. 1C). The amplitude of CaT1 was dependent on the amplitude of EPSP, regardless of the application of CCh (Fig. 1D). However, stimulation at intensities evoking EPSPs of >20 mV with the application of CCh evoked slow secondary Ca2+ transients (CaT2) following the CaT1 (Fig. 1B,D). The CaT2 component was isolated by subtracting the CaT1 from the total fluorescence changes (Fig. 1F1,F2), since the decay time constants of CaT1 were changed neither by the application of CCh in each cell (362 ± 32 ms and 392 ± 48, respectively, n = 11, p = 0.30) nor by the amplitude of CaT1 (r2 < 0.001 and r2 = 0.003 in control and with the application of CCh, respectively) (Fig. 1F3). The amplitude of the CaT2 (1.57 ± 0.13 ΔF/F0) correlated with the amplitude of the CaT1 (1.46 ± 0.19 ΔF/F0, r2 = 0.786, n = 13, p < 0.001) (Fig. 1E). The time of peak amplitude of CaT2 was distributed from 46 ms to 2.54 s after the peak of CaT1 (n = 56). These results indicate that, under cholinergic stimulation, subthreshold synaptic stimulation evokes secondary dendritic CaT2 in visual cortical pyramidal neurons.
Figure 1.

Single-shock synaptic stimulation-evoked CaT2 under CCh application in basal dendrites of layer 2/3 pyramidal neurons. A, A reconstructed pyramidal neuron and the stimulating electrode (red). The florescence image was projected from a z-stack of 41 frames at a depth increment of 1.5 μm. The scale is 50 μm. Right, An expanded square region of the left panel, in which the analyzed region is marked with a rectangle. B, Representative EPSPs and dendritic Ca2+ transients evoked by single-shock synaptic stimulation at various current intensities. C, Plot of EPSP amplitude against stimulus intensity. Stimulus intensities were normalized to the stimulus intensity evoking 20 mV EPSP in the ACSF in each cell (open square). D, Peak amplitude of dendritic Ca2+ transients against the amplitude of EPSPs evoked by the single-shock synaptic stimulation. The amplitude of Ca2+ transients were normalized to the maximal amplitude of the CaT1 in each cell. E, Peak amplitude of dendritic CaT2 against the peak amplitude of CaT1 in the presence of CCh. The line represents the yield of a correlation analysis. Data are summaries of response of 5, 12, and 13 cells in C, D, and E, respectively. F, Isolation of CaT2. F1, Overlay of Ca2+ transients evoked by 17 mV EPSP in normal ACSF (control) or in the presence of CCh. F2, Subtraction of the exponential fit of CaT1 decay from Ca2+ transients yielded CaT2. F3, Left, Summary plot of the CaT1 decay time constant (τ) in ACSF (control) and in the presence of CCh for 11 cells along with the mean values. Right, Summary plot of CaT1 decay, τ, against CaT1 amplitude. The lines are fits with a correlation analysis.
NMDA receptors and IP3-sensitive stores are responsible for CaT2
In the following experiments, we investigated which Ca2+ source is responsible for the Ca2+ transients. The application of d-AP5 (50 μm), an NMDA receptor blocker, diminished CaT1 to 4.2 ± 1.0% of the control (n = 10) and abolished CaT2 (Fig. 2A). Coapplication of the AMPA receptor blocker DNQX (20 μm), in addition to d-AP5, abolished the remaining small increase in the initial Ca2+ transients. However, neither CaT1 (p = 0.81, ANOVA) nor CaT2 (p = 0.91, ANOVA) was affected by the inclusion of the T-type and R-type VDCC blocker Ni2+ (50 μm, n = 3), the L-type VDCC blocker nimodipine (10 μm, n = 3), or both (n = 3) in the ACSF (Fig. 2B). Thus, CaT1 evoked by a single-shock synaptic stimulation was due to the influx of extracellular Ca2+, mostly via NMDA receptors. The remaining small proportion of CaT1 might have been elicited by the activation of VDCCs and/or Ca2+-permeable AMPA receptors (Kumar et al., 2002). These results also indicated that EPSP and CaT1 evoked by electrical stimulation near the dendritic tree in the present study were of synaptic origin (Holthoff et al., 2004).
Figure 2.

NMDA receptor-dependent Ca2+ transients. A, Effect of NMDA and AMPA receptor blockers on dendritic Ca2+ transients. d-AP5 (50 μm) and DNQX (20 μm) were applied to the ACSF in the presence of CCh (20 μm) serially in a cell. Right, Summary plot for the amplitude of CaT1 evoked by SES and CCh and in the presence of d-AP5 (n = 10). ***p < 0.001, compared with CCh. B, Effect of voltage-dependent Ca2+ channel blockers on Ca2+ transients. Ni2+ (50 μm) and nimodipine (10 μm) were applied to the bath throughout the experiment. Right, Summary plot for the amplitude of CaT1 and CaT2 evoked by SES and muscarinic stimulation (muscarine, n = 13) and in the presence of Ni2+ (n = 3), nimodipine (n = 3), and Ni2+ plus nimodipine (n = 3).
Since the influx of extracellular Ca2+ via VDCCs evoked a secondary release of Ca2+ from IP3-sensitive stores, which were primed by the activation of mAChR (Nakamura et al., 2000; Larkum et al., 2003; Cho et al., 2008), the CaT2 evoked by synaptic stimulation and muscarinic activation appeared to be a release from IP3-sensitive stores. We examined this mechanism in the next experiment (Fig. 3). CaT2 was abolished by the broad-spectrum mAChR blocker atropine (1 μm) (0.05 ± 0.03 ΔF/F0, n = 5, p < 0.01) or the specific M1 receptor blocker pirenzepine (1 μm; 0.07 ± 0.05 ΔF/F0, n = 3, p < 0.01) in the bath solution. Inclusion of the IP3 receptor blocker heparin (1 mg/ml) in the pipette solution also abolished the CaT2 (0.02 ± 0.01 ΔF/F0, n = 3, p < 0.01). In contrast, the nicotinic AChR blocker mecamylamine (30 μm) or the ryanodine receptor blocker ruthenium red (50 μm) failed to inhibit the CaT2 (1.24 ± 0.06 ΔF/F0, n = 4, p = 0.21; 1.26 ± 0.16 ΔF/F0, n = 4, p = 0.77; respectively). Thus, we confirmed that an influx of extracellular Ca2+ through NMDA receptors evoked by synaptic stimulation induces Ca2+ release from IP3-sensitive stores primed by the activation of M1 mAChR. We did not notice any apparent step-like increase in the amplitude of EPSPs, “NMDA spikes,” or “Ca2+ spikes,” even up to the stimulus intensities generating an AP, which have been reported in other studies (Schiller et al., 2000; Holthoff et al., 2004; Gordon et al., 2006). The difference might be due to pyramidal cells in different layers or the lack of inhibition of inhibitory neurotransmission in our experiment.
Figure 3.
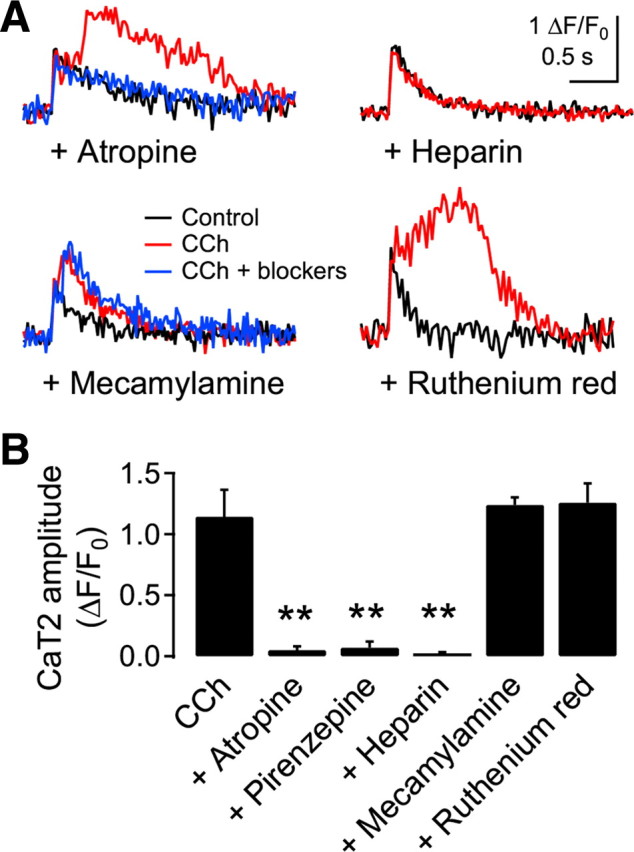
CaT2 released from the IP3-sensitive stores. A, Representative traces of Ca2+ transients in the presence of blockers. Atropine (1 μm, n = 5), pirenzepine (n = 3, data not shown), and mecamylamine (30 μm, n = 4) were applied to the bath solution. Heparin (1 mg/ml, n = 3) and ruthenium red (50 μm, n = 4) were in the pipette solution. EPSP of >∼20 mV amplitude by a single-shock synaptic stimulation was applied to evoke CaT2. B, Summary plot for the effect of blockers. **p < 0.01, compared with CCh.
Spatial profile of secondary Ca2+ transients evoked by activation of multiple synapses
The strong single-shock stimulation appeared to be efficient in recruiting spatially clustered multiple synapses, and thus in summating the intracellular Ca2+ levels to a certain threshold for release from IP3-sensitive stores (Nakamura et al., 2000), in a local dendritic area in a brief time window (Holthoff et al., 2004). The graded Ca2+ influx as the intensity of a single electric shock was increased, as shown in Figure 1, suggested that the focal stimulation activated multiple synapses. We examined Ca2+ signals in multiple spines and dendritic areas in the next experiment. To efficiently detect the fluorescence signal in spines, Ca2+ transients were evoked by two EPSPs (40 Hz) in the presence of a higher concentration of OGB-1 (200 μm) in the pipette solution. Focal electrical stimulation elicited Ca2+ transients in multiple spines near the stimulus pipette and the parent dendrite (Fig. 4A,B). Ca2+ transients in active spines (1.06 ± 0.07 ΔF/F0, n = 3 cells) were abolished by the application of d-AP5 (50 μm) (0.07 ± 0.03 ΔF/F0, p < 0.01), while backpropagating AP-evoked Ca2+ transients remained intact. Thus focal electrical stimulation near the dendritic shaft activated multiple spines in a localized area. Frequent multiple peaks in dendritic CaT2 evoked by strong single-shock stimulation under cholinergic stimulation (Figs. 2, 5) might result from the activation of multiple spines.
Figure 4.

Activation of multiple spines with focal electrical stimulation near dendritic trees. A, Reconstruction of a distal apical dendrite 180 μm from the soma in a layer 2/3 pyramidal neuron. Electrical stimulation was applied to a glass electrode (e) near the dendrite (10 μm from the dendrite, shown with 50 μm fluorescein). Four spines (s1–s4) and two dendritic regions (d1, d2) were analyzed. B, Two shocks at 40 Hz evoked Ca2+ transients in three spines (s1–s3). Ca2+ transients are also shown in the parent dendrite (d1). In contrast, the remote spine (s4) and dendrite (d2) showed no detectable Ca2+ transients. These Ca2+ transients were blocked by the application of d-AP5 (50 μm), while a single somatic AP evoked dendritic Ca2+ transients in all the spine and dendritic areas. C, Summary plot of the amplitude of Ca2+ transients in spines and dendrites (n = 3 cells). OGB-1 (200 μm) was used as a Ca2+ indicator. An Axioskop 2 FS (fitted with a 40×/0.80 NA objective) equipped with a Noran Odyssey XL was used and the fluorescence signal was captured at a video rate of 25 Hz.
Figure 5.

Spatial profile of dendritic Ca2+ transients. A, Reconstructed image of a recorded cell and the location of the stimulus electrode (in red). Inset shows the analyzed dendritic regions. Numbers represent the distance in micrometers from the area showing maximal intensity. Scale bar, 10 μm. B, Representative fluorescence traces evoked by EPSP or pairing of EPSP and somatic AP (+10 ms). The asterisks indicate the peaks analyzed for CaT2. The numbers denote the dendritic areas shown in Figure 1A. C, D, Spatial profiles of CaT1 and CaT2 evoked by EPSP (C) and pairing of EPSP and somatic AP (D). The peak amplitude of each transient was normalized to the maximal amplitude in the respective datasets. Negative and positive values represent the distance toward and away from the soma, respectively, at the maximal fluorescence changes. A Gaussian fitting was applied to all the profiles. Data are a summary of response of five cells.
The extent of Ca2+ release from IP3-sensitive stores evoked by synaptic stimulation was analyzed by examining the spatial profile of CaT2 (Fig. 5). With the application of CCh, EPSP-evoked CaT1 extended up to 20 μm proximally and distally from the site exhibiting maximal amplitude (SD = 8.1 μm, n = 5), while CaT2 extended up to 10 μm in both directions (SD = 5.0 μm), a smaller extension than the CaT1 (p < 0.001, repeated-measures ANOVA) (Fig. 5C). The spatial extent of CaT1 evoked by single EPSP in the present experiment was larger than that evoked by repetitive stimulation of single spine in bitufted interneurons in vitro (Kaiser et al., 2004) or in visual cortical pyramidal neurons in vivo (Jia et al., 2010), which also suggested that strong synaptic stimulation activated multiple spines. To investigate the involvement of Ca2+ influx via VDCCs on the release of Ca2+, we also examined the spatial profile of Ca2+ transients evoked by combined synaptic stimulation and somatic AP. The spatial profile of CaT2 was not changed by pairing EPSP with somatic AP (+10 ms interval, SD = 4.2 μm, p = 0.84), despite widespread and larger elevation in the initial intracellular Ca2+ increases with the contribution of the activation of VDCCs (Fig. 5D). These results indicate that Ca2+ release from IP3-sensitive stores is localized to the activated dendritic areas, which might induce input-specific changes in certain functions dependent on the increase in intracellular Ca2+.
Muscarinic control of the polarity of long-term synaptic plasticity
Ca2+ influx caused by either backpropagating AP or synaptic activation evokes Ca2+ release from IP3-sensitive stores primed by metabotropic receptor activation in pyramidal neurons of the hippocampus and the neocortex (Larkum et al., 2003). However, their functional roles in long-term synaptic plasticity have not yet been demonstrated. We addressed this issue in the next experiment. During the baseline recording, occasional Ca2+ imaging showed no detectable increases in fluorescence in dendritic areas (Fig. 6B1). At the end of the bath application of muscarine (20 μm) for 5 min, a single-shock electrical stimulation (SES) was applied to evoke >20 mV EPSP and CaT2 in basal dendrites. SES at a stimulus intensity that was 1.4 times higher than that evoking a 20 mV EPSP in normal ACSF evoked the same EPSP amplitude in the presence of CCh (Fig. 1C). Thus the intensity of the SES was set 1.4 times higher than that which evoked 20 mV EPSP in a normal ACSF, which was taken before the recording of the test EPSP. The SES evoked CaT2 in basal dendrites in 12 of 19 cells. EPSP amplitude invariably reached a potentiated level (144.5 ± 6.2, n = 12, p < 0.001) in cells showing CaT2 (Fig. 6A,B2,E). However, synaptic responses remained depressed when CaT2 was not evoked in some cells despite SES (60.9 ± 5.4%, n = 7, p < 0.001), or when no SES was applied during the application of muscarine (51.8 ± 3.7%, n = 8, p < 0.001) (Fig. 6A,D). In some cells (n = 3), we applied a single burst (3 shocks at either 40 or 100 Hz) evoking summated EPSP of ∼20 mV, instead of SES, which also evoked CaT2 (1.59 ± 0.20 ΔF/F0) (Fig. 6B3) and LTP in all three cells tested (132.6 ± 8.1%, p < 0.05). Neither CaT2 nor LTP was induced when heparin (1 mg/ml) was included in the pipette solution. Rather, EPSP remained at the depressed level (59.4 ± 7.2%, n = 5, p < 0.01).
Figure 6.

Induction of CaT2 and LTP with single-shock synaptic stimulation under muscarinic activation. A, Representative response of induction of LTP and LTD. The test EPSP was evoked by a glass electrode near basal dendrites as shown in Figure 5A. SES (arrow) was applied at the end of the application of 20 μm muscarine [SES (+), closed circle]. In some cells, only the baseline stimulation was continuously applied during the muscarine application [SES (−), closed triangle]. In another set of cells, a single burst of 3 APs (40 or 100 Hz) was applied instead of the SES (burst, open circle). Insets show the average EPSPs taken at the time indicated. Input resistance (IR) was monitored throughout the experiment, shown with the symbols corresponding to the upper panel for two cells. B1, B2, B3, The dendritic Ca2+ fluorescence and the corresponding voltage recordings evoked by test EPSP (B1), SES (B2), and a burst (B3). A control recording was taken before the EPSP recording. C, Summary plot for LTP induction by SES (n = 12) and in the presence of d-AP5 (n = 6). BAPTA (n = 4) or heparin (n = 5) was included in the pipette solution. LTP induced by the application of a burst (n = 3) instead of the SES is also shown. D, Summary plot for LTD induction. Continuous stimulation at the test intensity was applied during muscarinic stimulation without SES (n = 8) and in the presence of d-AP5 (n = 8), BAPTA (n = 4), or heparin (n = 3). E, Relationship between CaT2 amplitude and the magnitude of LTPm. A fitted line is also shown. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with the baseline recording, respectively.
LTD induced by the application of muscarine without SES was not affected by the inclusion of heparin in the pipette solution (52.3 ± 3.2% of the baseline, n = 3, p < 0.01). However, the application of d-AP5 (50 μm) inhibited LTD induced by the application of muscarine without the SES (92.5 ± 4.8%, n = 8, p = 0.15), as well as LTP induced by muscarine application with SES (96.1 ± 2.6%, n = 6, p = 0.19). Inclusion of the high-affinity Ca2+ chelator BAPTA in the pipette solution (10 mm) blocked both LTP induced by combined SES and muscarine (101.7 ± 0.9%, n = 4, p = 0.15) and LTD induced by muscarine alone (98.1 ± 0.6%, n = 4, p = 0.15).
These results demonstrate that Ca2+ released from IP3-sensitive stores is necessary and specific for the induction of LTPm. However, the magnitude of LTPm was not correlated with the peak amplitude (r2 < 0.001, n = 12, p = 0.99) (Fig. 6E) or with the integrated increase in fluorescence of CaT2 (r2 = 0.032, p = 0.56). These imply that the release of Ca2+ may trigger downstream signaling pathways in an all-or-none fashion and the magnitude of LTP may depend on the proportion of the activated synapses in the local dendritic area to all the activated synapses. On the other hand, the increase in Ca2+ through NMDA receptors efficiently induced muscarinic LTD (LTDm), which was reported previously in the rat visual cortex (Kirkwood et al., 1999).
Because 20 μm ACh or muscarine might not be physiologically relevant (Shinoe et al., 2005), we examined the effect of a lower concentration of muscarine on CaT2 and LTPm. Neither CaT2 nor LTPm/LTDm (100.9 ± 1.7%, n = 4, p = 0.64) was induced by SES in the presence of 0.1 μm muscarine. However, SES evoked CaT2 (1.75 ± 0.18 ΔF/F0, n = 2) and LTPm (135.8 ± 15.5%, p = 0.26) in the presence of 1 μm muscarine in two of five cells. These results suggest that a much lower level of ACh, and thus a more physiologically relevant level of ACh, could evoke CaT2 and LTPm. We further tested whether a synaptically activated Ca2+ release and LTP could be evoked upon the activation of metabotropic glutamate receptors (mGluRs) instead of mAChRs (Ross et al., 2005). SES applied at the end of a 5 min application of the mGluR agonist trans-1-amino-1,3-cyclopentyldicarboxylate (t-ACPD, 15 μm) evoked CaT2 (0.83 ± 0.18 ΔF/F0, n = 3) and LTP (142.9 ± 9.4%, p < 0.05) in three of five cells. This result suggests that activation of mGluR might be synergistic with the activation of mAChR in the induction of CaT2 and LTP (Volk et al., 2007), and thus would reduce the required concentration of ACh for LTPm. However, this conclusion remains to be tested.
Muscarinic activation confers input-specific and protein synthesis-dependent forms of LTP and LTD
Because Ca2+ released from IP3-sensitive stores was confined to highly localized dendritic areas, we next examined input specificity in the induction of LTPm and LTDm. To this end, we positioned another stimulus electrode in underlying layer 4, ∼250 μm away from the recorded cell, through which electrical stimulation was alternately applied (Fig. 7A). In this set of cells, SES applied via the glass electrode near basal dendrites during muscarinic stimulation induced CaT2 (1.07 ± 0.20 ΔF/F0) (Fig. 7B2) and LTPm (155.0 ± 2.3%, n = 4, p < 0.001). Meanwhile, in this same set of cells, EPSP exhibited LTDm after being continuously evoked during the application of muscarine by electrical stimulation of layer 4 at the test intensity with the tungsten electrode (66.1 ± 6.7%, n = 4, p < 0.05) (Fig. 7B3). During stimulation of layer 4, we found no Ca2+ transients in the dendritic area showing Ca2+ transients with SES. These results indicated that LTPm was input-specific, which might result from localized Ca2+ release in dendrites. We further examined the synaptic activity dependence of LTDm. The test stimulation to layer 4 was turned off during the application of muscarine and then turned on 10 min after washout of muscarine (Fig. 7C1), and LTDm was not induced (106.3 ± 6.8%, n = 4, p = 0.21) (Fig. 7C3). SES applied to the basal dendrite-induced CaT2 (0.94 ± 0.08 ΔF/F0) (Fig. 7C2) and LTPm (Fig. 7C1,C3) in these cells. This result indicates that presynaptic activity is required for the induction of LTDm (Kirkwood et al., 1999). The experimental configuration used by Kirkwood et al. (1999) is similar to the stimulation of layer 4 in the present study, showing only LTDm. The stimulation of layer 4 would stimulate distributed synapses in layer 2/3 pyramidal neurons, in which CaT2 might be barely evoked by single shock because a large influx of Ca2+ via NMDA receptors in spatially clustered multiple synapses appeared to be required to trigger the release of Ca2+ from IP3-sensitive stores, as shown in Figure 4.
Figure 7.
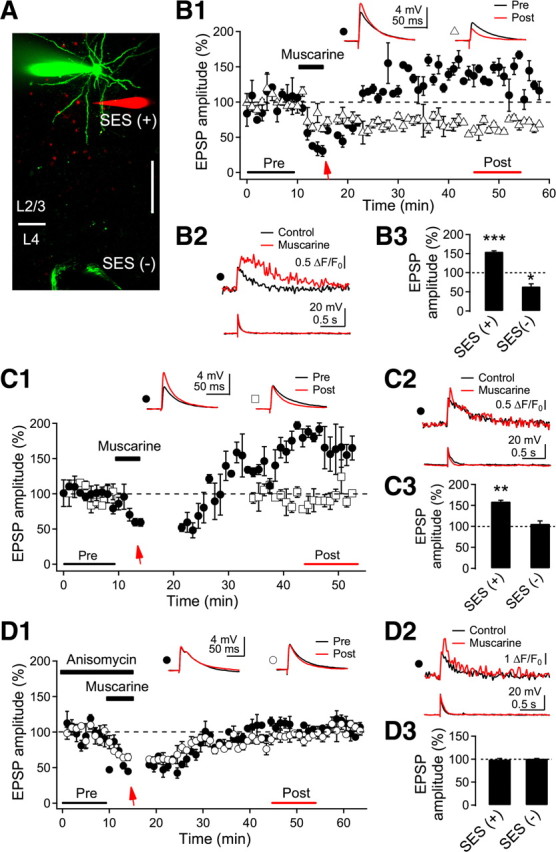
Properties of LTPm and LTDm. A, Fluorescence image of a recorded layer 2/3 pyramidal neuron and electrodes. SES was applied to the glass electrode near basal dendrites (in red). Layer 4 was alternately stimulated in a cell via tungsten electrodes (in green). Layer 4 was stimulated at test intensity without SES. Scale bar, 50 μm. B, Input-specific induction of LTPm and LTDm. B1, Representative EPSP recording by the stimulation with the glass electrode (closed circle) and the tungsten electrode (open triangle) in a cell. The arrow indicates an application of the SES. Insets show average EPSPs taken at the time indicated. B2, Dendritic fluorescence and EPSP evoked by the SES. Control recording was taken before the EPSP recording. B3, Summary plot for LTPm with SES and LTDm without SES (n = 4 cells). *p < 0.05 and ***p < 0.001, compared with the baseline recording, respectively. C, Activity-dependent induction of LTPm and LTDm. C1, Representative EPSP recording by the stimulation with the glass electrode (closed circle) and the tungsten electrode (open square) in a cell. The arrow indicates the application of the SES at the end of muscarine application. Note that the stimulation of layer 4 was discontinued during muscarine application and for 15 min after the washout. Insets show the average EPSPs taken at the time indicated. C2, Dendritic fluorescence and EPSP evoked by the SES. C3, Summary plot for EPSP response with and without SES (n = 4 cells). **p < 0.01, compared with the baseline recording. D, Protein synthesis-dependent induction of LTPm and LTDm. D1, Representative EPSP recordings by the stimulation with the glass electrode (closed circle) and the tungsten electrode (open circle) in a cell. The arrow indicates an application of the SES. Anisomycin (25 μm) was applied to the bath solution 20 min before the experiment. Insets show the average EPSPs taken at the time indicated. D2, Dendritic fluorescence and EPSP evoked by the SES. D3, Summary plot for the effect of anisomycin on LTPm [SES (+)] and LTDm [SES (−)] (n = 4 cells).
Synthesis of new proteins is required for several forms of long-term synaptic plasticity, including muscarinic receptor-dependent LTD in the visual cortex (McCoy and McMahon, 2007), the perirhinal cortex (Massey et al., 2001), and the hippocampus (Volk et al., 2007). Thus, we tested whether LTPm and LTDm were protein synthesis-dependent in our experimental model (Fig. 7D). Application of the protein synthesis inhibitor anisomycin (25 μm) abolished SES-induced LTPm (100.5 ± 1.7%, n = 4, p = 0.81) and LTDm (99.7 ± 1.0%, n = 4, p = 0.79) (Fig. 7D1,D3), while the CaT2 remained unchanged (1.57 ± 0.20 ΔF/F0) (Fig. 7D2). Treatment of anisomycin revealed that protein synthesis-dependent LTPm and LTDm were rapidly induced within 15 min (Fig. 7D1), which might be faster when the time for washout of CCh is taken into consideration. Rapid dendritic protein synthesis and expression of LTD by the activation of mGluR and mAChR have been reported in different brain areas (Huber et al., 2000; Mameli et al., 2007; Volk et al., 2007). In a study by McCoy and McMahon (2007), the expression of protein synthesis-dependent LTDm an hour after the induction might have been a result of the transport of new proteins from the soma, which was suggested in L-type Ca2+ channel-dependent LTP (Raymond, 2007). Thus, Ca2+ released from intracellular stores (CaT2) and Ca2+ entering via NMDA receptors (CaT1) only induced LTPm and LTDm, respectively, if protein synthesis was not impaired. Together, these results indicate that muscarinic activation confers input-specific, activity-dependent, and protein synthesis-dependent (i.e., late-phase) LTP and LTD in layer 2/3 pyramidal neurons of the visual cortex.
Dendritic domain-dependent muscarinic modulation
It is hypothesized that ACh changes circuit dynamics by regulating the relative influence of afferent input and excitatory feedback (Giocomo and Hasselmo, 2007). Thus, we next examined Ca2+ signals in distal apical dendrites, which receive input from nonspecific thalamic nuclei, from ascending collaterals of nearby pyramidal cells, and from feedback projections originating in higher cortical areas (Petreanu et al., 2009). Synaptic activation with SES in the vicinity of distal apical dendrites in layer 1 (169.1 ± 10.7 μm from the soma, n = 14) could evoke >20 mV EPSP at an intensity (112.0 ± 12.9 μA) similar to that in basal dendrites (104.9 ± 6.9 μA, n = 22, p = 0.64). In contrast with the basal dendrites (Fig. 8C), SES never induced CaT2, even at suprathreshold intensities under muscarinic stimulation (n = 14), while the amplitude of CaT1 (1.27 ± 0.20 ΔF/F0) was similar to that found in the basal dendrites (Fig. 8A). Test EPSPs evoked by stimulation of distal apical dendrites in layer 1 (Fig. 8E) were depressed by the application of muscarine (Fig. 8D), similar to the response evoked in basal dendrites, as shown in Figure 1. Consistent with the lack of a CaT2 in distal apical dendrites (Fig. 8A,F), the only observed long-term modification was LTDm (with SES: 60.2 ± 8.6%, n = 5, p < 0.01; without SES: 56.3 ± 4.5%, n = 4, p < 0.001; respectively) (Fig. 8G). This further supports the notion that LTPm induction requires liberation of Ca2+ from intracellular stores and, in addition, suggests an uneven distribution of IP3-sensitive stores in different dendritic domains (Hertle and Yeckel, 2007). When SES was applied in the vicinity of proximal apical dendrites (24.4 ± 3.4 μm from the soma, n = 5), it only evoked CaT2 when it co-occurred with a somatic AP (10 ms after SES) (Fig. 8B). We did not test for LTP/LTD in this dendritic compartment. These results suggest that cholinergic stimulation might modulate long-term synaptic transmission in input-specific and, thus, pathway-specific manners in layer 2/3 pyramidal neurons.
Figure 8.

Dendritic domain-dependent muscarinic induction of CaT2 and LTPm. A–C, A glass stimulating electrode was positioned near dendritic trees in distal apical (A), proximal apical (B), and basal dendrites (C). A somatic AP, synaptic stimulation (EPSP), or EPSP plus AP (10 ms interval) was applied. Left, Reconstructed cells (scale bar, 100 μm) and the stimulating electrodes. Right, representative recordings of the dendritic fluorescence and somatic voltage of nine, five, and nine cells for distal apical, proximal apical, and basal dendrites, respectively. D, Induction of LTDm in distal apical dendrites by muscarinic stimulation. SES (closed circle, indicated by arrow) or stimulation alone at the baseline intensity (open circle) was applied during the application of muscarine. Insets show the average EPSP taken at the time indicated. E, Reconstructed image of a cell (scale bar, 100 μm) and the location of the stimulus electrode (in red). F, Dendritic fluorescence and EPSP evoked by the SES. G, Summary plot of the induction of LTDm with (n = 5) or without (n = 4) the SES in the presence of CCh. **p < 0.01 and ***p < 0.001, respectively.
Discussion
Localized Ca2+ release from IP3-sensitive stores
In previous reports, Ca2+ release from IP3-sensitive stores was widely distributed in the dendrite and propagated into the soma in many brain areas (Nakamura et al., 1999; Larkum et al., 2003; Power and Sah, 2007; Fernández de Sevilla et al., 2008; Hagenston et al., 2008). In the present study, with multiple synaptic activation in a localized dendritic area and with muscarinic activation, we found highly localized release of Ca2+ in the dendrite within 10 μm of the stimulated areas, while the initial Ca2+ influx via NMDA receptors was restricted to within 20 μm. While activation of the muscarinic receptors by itself evoked a propagating Ca2+ wave in basolateral amygdala neurons (Power and Sah, 2007) and in CA1 pyramidal neurons (Fernández de Sevilla et al., 2008), an influx of extracellular Ca2+ appears to be required to evoke release of Ca2+ by bath-application of CCh in neocortical pyramidal cells (Larkum et al., 2003). We noticed no significant increase in the baseline fluorescence intensity upon CCh application alone in the present study and in a previous study (Cho et al., 2008). Immunoreactivity for mAChR is lower in layer 2/3 neocortical pyramidal neurons than in layer 5 or in CA1 pyramidal neurons (van der Zee and Luiten, 1999). The density of IP3 receptors is highest in the soma and the proximal apical dendrites (Fitzpatrick et al., 2009). Thus, the extent of Ca2+ release with cholinergic stimulation appears to be dependent on the local distribution and density of muscarinic and IP3 receptors in many brain areas.
The influx of extracellular Ca2+ via either VDCCs or NMDA receptors caused the release of Ca2+ from the IP3-sensitive stores in cortical pyramidal neurons (Nakamura et al., 2000; Power and Sah, 2002; Larkum et al., 2003; Cho et al., 2008). In the present study, the release evoked by SES under muscarinic stimulation was triggered by the influx of extracellular Ca2+, mostly via NMDA receptors, based on (1) the results with blockers of VDCCs and NMDA receptors (Fig. 2) and (2) similar spatial profiles of CaT2 regardless of combined stimulation with an additional somatic AP (Fig. 5). Ruthenium red failed to block NMDA receptor-dependent dendritic CaT1 (Fig. 2), which indicates that NMDA receptors are the source of the spine Ca2+ signal evoked by synaptic stimulation. These results agree with previous reports concerning NMDA receptor-dependent dendritic CaT1 (Sabatini et al., 2001; Raymond and Redman, 2006), but contrast with results associated with ryanodine receptor-dependent Ca2+ transients in dendritic spines in organotypic hippocampal cultures (Emptage et al., 1999). The delayed appearance of dendritic CaT2 in the present study might have been the result of the diffusion of Ca2+ from NMDA receptors in spines to the IP3 receptors in dendrites, which might explain the difference with nondelayed Ca2+ release in spines (Emptage et al., 1999). Additionally, a single somatic AP in the proximal apical dendrite evoked CaT2 (Fig. 8B). The signaling microdomain for Ca2+ release triggered by the influx of extracellular Ca2+ and its role in long-term synaptic plasticity in different dendritic areas remains to be studied.
Muscarinic control of polarity of long-term synaptic plasticity
The dynamics and magnitude of increase in intracellular Ca2+ are the major determinants of the polarity of long-term synaptic plasticity (Lisman, 1989; Artola and Singer, 1993). As with the influx of extracellular Ca2+, the release of Ca2+ from intracellular stores is also critical for the regulation of cellular responses, such as protein phosphorylation in the spine, local protein synthesis in dendrites, and gene expression in the soma (Augustine et al., 2003; Raymond, 2007). We demonstrated in the present study that Ca2+ release from IP3-sensitive stores was a prerequisite for inducting LTPm and blocking the secondary Ca2+ release with intracellular heparin-converted LTPm to LTDm. IP3-mediated Ca2+ release is involved in LTP in some experimental conditions (Raymond and Redman, 2006; Fernández de Sevilla et al., 2008). However, it is also involved in the induction of mGluR-dependent LTD in different experimental settings (Finch and Augustine, 1998; Bender et al., 2006; Holbro et al., 2009). Furthermore, in layer 2/3 pyramidal neurons of the somatosensory cortex, phospholipase C-dependent synthesis of endocannabinoid and presynaptic NMDA receptors mediates spike-timing-dependent LTD, with (Bender et al., 2006) or without (Nevian and Sakmann, 2006) involvement of IP3-mediated Ca2+ release. Although LTDm depends on protein synthesis, the involvement of endocannabinoid-mediated signaling in LTDm remains to be tested. Thus, enhanced influx of Ca2+ via NMDA receptors is required for the cholinergic-mediated long-term modification of synaptic transmission. Then, an additional large increase in [Ca2+]i via release from the IP3-sensitive stores induces LTPm. Thus, muscarinic activation might function as a detector for synaptic activities to be modified with the differential nature of the increase in [Ca2+]i in neocortical pyramidal neurons.
Induction of late-phase LTPm and LTDm
In the present study, we demonstrated that LTPm was protein synthesis-dependent. Combined activation of muscarinic and NMDA receptors induced protein synthesis in dendrites of hippocampal CA1 pyramidal neurons in vitro (Feig and Lipton, 1993), and cholinergic-dependent late-phase LTP was inhibited by the translation inhibitor anisomycin in vivo (Dringenberg et al., 2004). Release of Ca2+ from endoplasmic reticulum (ER) might be the efficient signal for ER-bound ribosomes for translation of new proteins required specifically for late-phase LTP. However, LTDm induced by the increase in Ca2+ via NMDA receptors was also protein synthesis-dependent in the present study. Thus, we speculate that subcellular localization and calcium sensitivity of each mRNA responsible for LTPm and LTDm might differ.
It is of interest that a burst (3 shocks), as well as SES without prolonged activation of synapses, induced late-phase LTPm under muscarinic activation. A single shock evoked LTD in mouse visual cortical pyramidal neurons (Holthoff et al., 2004). With muscarinic activation, brief synaptic activation of clustered inputs in a localized dendritic area, as shown in Figure 4, might efficiently induce protein synthesis-dependent late-phase LTP, which also supports the “clustered plasticity hypothesis” (Govindarajan et al., 2006, 2011). Stimulation of the medial septum evoked atropine-sensitive LTP in the hippocampal CA1 area (Fernández de Sevilla et al., 2008). CaT2 and LTPm were also induced at a lower concentration of muscarine (1 μm) in the present study. However, it remains to be seen whether synaptic release of endogenous ACh can cause these phenomena in vivo.
Long-term change of cortical circuit dynamics by acetylcholine
Layer 2/3 pyramidal neurons appear to receive afferent and associative inputs largely in the basal and distal apical dendrites, respectively (Cauller and Connors, 1994; Petreanu et al., 2009). In our previous studies, dendritic Ca2+ influx by the backpropagation of somatic APs differs strikingly between basal and distal apical dendrites (Cho et al., 2006) and ACh modulates the Ca2+ dynamics differentially in these domains (Cho et al., 2008). In the present study, we demonstrated that muscarinic activation failed to induce LTPm in inputs to distal apical dendrites, as well as Ca2+ release. High ACh levels in a wakeful state may strengthen forward transmission to set the appropriate dynamics for attention and encoding (Gais and Born, 2004; Giocomo and Hasselmo, 2007). Thus, the study of preferential induction of late-phase LTP in inputs to basal dendrites may be helpful to gain new information.
For the formation of lasting memories, use-dependent strengthening of synaptic connections must be gated in a context-dependent and goal-dependent way. Lasting changes in synaptic gain should not occur in response to accidental local activities, such as might occur during computations that have not yet converged toward a result. A “now print” signal seems to be required, which only permits lasting modifications in response to activity patterns that have been identified as “meaningful” in a more global context. NMDA receptors could contribute to such a gating function as their voltage sensitivity makes them particularly responsive to coherence between feedforward signals and context-dependent horizontal and reentry inputs. The cholinergic projections could complement this gating function by adding more global, state-dependent criteria. The notion that learning is critically dependent on attention and expectancy, and that these functions involve cholinergic modulation, is in agreement with this possibility. Apart from the effect of ACh on the IP3-dependent Ca2+ release described in this study, ACh also directly modulates network dynamics by facilitating and stabilizing gamma oscillations and reducing the variance of rate responses to sensory stimuli (Herculano-Houzel et al., 1999; Rodriguez et al., 2004, 2010). These effects, too, could contribute to memory formation as they enhance signal-to-noise ratios and increase cooperativity and synchronous firing. The latter can be expected to facilitate NMDA receptor activation by generating large compound EPSPs in very much the same way as SES did in the present study. How the various effects of cholinergic modulation interact in sensory processing and memory formation awaits further clarification.
Footnotes
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea, which is funded by the Korea Ministry of Education, Science and Technology (2010-0024775).
References
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. Muscarinic receptors mediating depression and long-term potentiation in rat hippocampus. J Physiol. 1996;492:479–493. doi: 10.1113/jphysiol.1996.sp021323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauller LJ, Connors BW. Synaptic physiology of horizontal afferents to layer I in slices of rat SI neocortex. J Neurosci. 1994;14:751–762. doi: 10.1523/JNEUROSCI.14-02-00751.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KH, Kim MJ, Yoon SH, Hahn SJ, Jo YH, Kim MS, Rhie DJ. Spatial profile of back-propagating action potential-evoked Ca2+ transients in basal dendrites. Neuroreport. 2006;17:131–134. doi: 10.1097/01.wnr.0000198949.85197.ac. [DOI] [PubMed] [Google Scholar]
- Cho KH, Jang HJ, Lee EH, Yoon SH, Hahn SJ, Jo YH, Kim MS, Rhie DJ. Differential cholinergic modulation of Ca2+ transients evoked by backpropagating action potentials in apical and basal dendrites of cortical pyramidal neurons. J Neurophysiol. 2008;99:2833–2843. doi: 10.1152/jn.00063.2008. [DOI] [PubMed] [Google Scholar]
- Cho KH, Jang JH, Jang HJ, Kim MJ, Yoon SH, Fukuda T, Tennigkeit F, Singer W, Rhie DJ. Subtype-specific dendritic Ca2+ dynamics of inhibitory interneurons in the rat visual cortex. J Neurophysiol. 2010;104:840–853. doi: 10.1152/jn.00146.2010. [DOI] [PubMed] [Google Scholar]
- Dringenberg HC, Kuo MC, Tomaszek S. Stabilization of thalamo-cortical long-term potentiation by the amygdala: cholinergic and transcription-dependent mechanisms. Eur J Neurosci. 2004;20:557–565. doi: 10.1111/j.1460-9568.2004.03515.x. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Feig S, Lipton P. Pairing the cholinergic agonist carbachol with patterned Schaffer collateral stimulation initiates protein synthesis in hippocampal CA1 pyramidal cell dendrites via a muscarinic, NMDA-dependent mechanism. J Neurosci. 1993;13:1010–1021. doi: 10.1523/JNEUROSCI.13-03-01010.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Buño W. The muscarinic long-term enhancement of NMDA and AMPA receptor-mediated transmission at Schaffer collateral synapses develop through different intracellular mechanisms. J Neurosci. 2010;30:11032–11042. doi: 10.1523/JNEUROSCI.1848-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Núñez A, Borde M, Malinow R, Buño W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J Neurosci. 2008;28:1469–1478. doi: 10.1523/JNEUROSCI.2723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron. 2002;33:615–624. doi: 10.1016/s0896-6273(02)00587-1. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JS, Hagenston AM, Hertle DN, Gipson KE, Bertetto-D'Angelo L, Yeckel MF. Inositol-1,4,5-trisphosphate receptor-mediated Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots. J Physiol. 2009;587:1439–1459. doi: 10.1113/jphysiol.2009.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gais S, Born J. Low acetylcholine during slow-wave sleep is critical for declarative memory consolidation. Proc Natl Acad Sci U S A. 2004;101:2140–2144. doi: 10.1073/pnas.0305404101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Neuromodulation by glutamate and acetylcholine can change circuit dynamics by regulating the relative influence of afferent input and excitatory feedback. Mol Neurobiol. 2007;36:184–200. doi: 10.1007/s12035-007-0032-z. [DOI] [PubMed] [Google Scholar]
- Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci. 2006;26:12717–12726. doi: 10.1523/JNEUROSCI.3502-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat Rev Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Fitzpatrick JS, Yeckel MF. mGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18:407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S, Munk MH, Neuenschwander S, Singer W. Precisely synchronized oscillatory firing patterns require electroencephalographic activation. J Neurosci. 1999;19:3992–4010. doi: 10.1523/JNEUROSCI.19-10-03992.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro N, Grunditz A, Oertner TG. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:15055–15060. doi: 10.1073/pnas.0905110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff K, Kovalchuk Y, Yuste R, Konnerth A. Single-shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J Physiol. 2004;560:27–36. doi: 10.1113/jphysiol.2004.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Jia H, Rochefort NL, Chen X, Konnerth A. Dendritic organization of sensory input to cortical neurons in vivo. Nature. 2010;464:1307–1312. doi: 10.1038/nature08947. [DOI] [PubMed] [Google Scholar]
- Kaiser KM, Lübke J, Zilberter Y, Sakmann B. Postsynaptic calcium influx at single synaptic contacts between pyramidal neurons and bitufted interneurons in layer 2/3 of rat neocortex is enhanced by backpropagating action potentials. J Neurosci. 2004;24:1319–1329. doi: 10.1523/JNEUROSCI.2852-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Watanabe S, Nakamura T, Lasser-Ross N, Ross WN. Synaptically activated Ca2+ waves in layer 2/3 and layer 5 rat neocortical pyramidal neurons. J Physiol. 2003;549:471–488. doi: 10.1113/jphysiol.2002.037614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R, Lüscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Markram H, Segal M. Long-lasting facilitation of excitatory postsynaptic potentials in the rat hippocampus by acetylcholine. J Physiol. 1990;427:381–393. doi: 10.1113/jphysiol.1990.sp018177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Bhabra G, Cho K, Brown MW, Bashir ZI. Activation of muscarinic receptors induces protein synthesis-dependent long-lasting depression in the perirhinal cortex. Eur J Neurosci. 2001;14:145–152. doi: 10.1046/j.0953-816x.2001.01631.x. [DOI] [PubMed] [Google Scholar]
- McCoy PA, McMahon LL. Muscarinic receptor-dependent long-term depression in rat visual cortex is PKC independent but requires ERK1/2 activation and protein synthesis. J Neurophysiol. 2007;98:1862–1870. doi: 10.1152/jn.00510.2007. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Distribution of IP3-mediated calcium responses and their role in nuclear signalling in rat basolateral amygdala neurons. J Physiol. 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Saggau P. Presynaptic inhibition of synaptic transmission in the rat hippocampus by activation of muscarinic receptors: involvement of presynaptic calcium influx. Br J Pharmacol. 1997;122:511–519. doi: 10.1038/sj.bjp.0701400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR. LTP forms 1, 2 and 3: different mechanisms for the ‘long’ in long-term potentiation. Trends Neurosci. 2007;30:167–175. doi: 10.1016/j.tins.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol. 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Kallenbach U, Singer W, Munk MH. Short- and long-term effects of cholinergic modulation on gamma oscillations and response synchronization in the visual cortex. J Neurosci. 2004;24:10369–10378. doi: 10.1523/JNEUROSCI.1839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Kallenbach U, Singer W, Munk MH. Stabilization of visual responses through cholinergic activation. Neuroscience. 2010;165:944–954. doi: 10.1016/j.neuroscience.2009.10.059. [DOI] [PubMed] [Google Scholar]
- Ross WN, Nakamura T, Watanabe S, Larkum M, Lasser-Ross N. Synaptically activated Ca2+ release from internal stores in CNS neurons. Cell Mol Neurobiol. 2005;25:283–295. doi: 10.1007/s10571-005-3060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Shinoe T, Matsui M, Taketo MM, Manabe T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci. 2005;25:11194–11200. doi: 10.1523/JNEUROSCI.2338-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Zee EA, Luiten PG. Muscarinic acetylcholine receptors in the hippocampus, neocortex and amygdala: a review of immunocytochemical localization in relation to learning and memory. Prog Neurobiol. 1999;58:409–471. doi: 10.1016/s0301-0082(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]