

Abstract
Brain-derived neurotrophic factor (BDNF) plays an important role in neuronal survival through activation of TrkB receptors. The trkB gene encodes a full-length receptor tyrosine kinase (TrkB.FL) and its truncated (T1/T2) isoforms. We investigated the changes in TrkB protein levels and signaling activity under excitotoxic conditions, which are characteristic of brain ischemia, traumatic brain injury, and neurodegenerative disorders. Excitotoxic stimulation of cultured rat hippocampal or striatal neurons downregulated TrkB.FL and upregulated a truncated form of the receptor (TrkB.T). Downregulation of TrkB.FL was mediated by calpains, whereas the increase in TrkB.T protein levels required transcription and translation activities. Downregulation of TrkB.FL receptors in hippocampal neurons correlated with a decrease in BDNF-induced activation of the Ras/ERK and PLCγ pathways. However, calpain inhibition, which prevents TrkB.FL degradation, did not preclude the decrease in signaling activity of these receptors. On the other hand, incubation with anisomycin, to prevent the upregulation of TrkB.T, protected to a large extent the TrkB.FL signaling activity, suggesting that truncated receptors may act as dominant-negatives. The upregulation of TrkB.T under excitotoxic conditions was correlated with an increase in BDNF-induced inhibition of RhoA, a mediator of excitotoxic neuronal death. BDNF fully protected hippocampal neurons transduced with TrkB.T when present during excitotoxic stimulation with glutamate, in contrast with the partial protection observed in cells overexpressing TrkB.FL or expressing GFP. These results indicate that BDNF protects hippocampal neurons by two distinct mechanisms: through the neurotrophic effects of TrkB.FL receptors and by activation of TrkB.T receptors coupled to inhibition of the excitotoxic signaling.
Introduction
Overactivation of glutamate receptors plays an important role in neuronal damage in brain ischemia, epileptic seizures, and in chronic neurodegenerative disorders (Choi, 1994; Lau and Tymianski, 2010). The resulting [Ca2+]i overload stimulates calpains, which target numerous substrates within the cell, including cytoskeletal proteins, kinases and phosphatases, membrane receptors, and transporters (Chan and Mattson, 1999; Xu et al., 2007; Gomes et al., 2011).
Impairment of intracellular signaling induced by trophic factors was suggested to play an important role in neurodegenerative processes (Drukarch and van Muiswinkel, 2001; Gauthier et al., 2004; Tolosa et al., 2009; Counts and Mufson, 2010). However, the underlying mechanisms and the interrelationship with excitotoxic injury remain to be established. Brain-derived neurotrophic factor (BDNF) promotes neuronal survival and provides neuroprotection in various brain regions (Lu et al., 2005; Manadas et al., 2007) through activation of tyrosine kinase TrkB receptors (TrkB.FL). Stimulation of these receptors induces their transphosphorylation on tyrosine residues, setting in motion several signaling cascades. Activation of the Ras/ERK and PI3-K (phosphatidylinositol-3-kinase)/Akt pathways accounted for neuroprotection by BDNF under excitotoxic conditions in studies where cultured hippocampal neurons were preincubated with the neurotrophin before the insult (Almeida et al., 2005). The active TrkB-FL receptor also activates phospholipase Cγ (PLCγ) by tyrosine phosphorylation (Reichardt, 2006). Two additional TrkB splicing isoforms are expressed in the brain, TrkB.T1, and TrkB.T2 (Klein et al., 1990; Huang and Reichardt, 2003; Manadas et al., 2007), which may behave as dominant-negative inhibitors of TrkB-FL in the regulation of cell survival (Dorsey et al., 2006), in addition to their own signaling activity (Takai et al., 2001; Ohira et al., 2006; Manadas et al., 2007). Thus, TrkB.T1 receptors are coupled to inhibition of RhoA in astrocytes (Ohira et al., 2006). This GTPase mediates Ca2+-dependent activation of p38 MAPK, which is coupled to neuronal death under excitotoxic conditions (Semenova et al., 2007).
The TrkB isoforms are differentially regulated under pathological conditions (Manadas et al., 2007). Injection of kainate in the rat hippocampus increased BDNF and TrkB.T protein levels, whereas the full-length receptor levels did not change significantly (Rudge et al., 1998). In a different study kainate injection upregulated BDNF and TrkB.FL protein levels in the surviving neurons in the CA1 and dentate gyrus subregions of the hippocampus. However, neuronal loss in the damaged areas was preceded by a reduction of BDNF and TrkB.FL immunoreactivity (Goutan et al., 1998). The TrkB.FL and BDNF immunoreactivity was also decreased in the infarct core region in a rat model of stroke (Ferrer et al., 2001).
In this work we investigated the mechanisms contributing to the changes in TrkB.FL and TrkB.T under excitotoxic conditions, the resulting alterations in their signaling activity, and the impact in neuroprotection. The results show that excitotoxicity downregulates TrkB.FL protein levels in cultured hippocampal and striatal neurons, and inhibits their signaling activity by a mechanism that correlates with the upregulation of TrkB.T receptors. This shift in expression of TrkB isoforms may partially alter the role of BDNF, from a neurotrophic to a neuroprotective function.
Materials and Methods
Primary neuronal cultures
Cultures of rat hippocampal neurons were prepared from E18 Wistar embryos as previously described (Almeida et al., 2005; Caldeira et al., 2007; Gomes et al., 2011). Striatal neurons were cultured using the same procedures, from E15–16 Wistar rat embryos (Gomes et al., 2011). Hippocampal cultures were maintained in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), glutamate (25 μm), glutamine (0.5 mm), and gentamicin (0.12 mg/ml). Striatal cultures (15 d in vitro; DIV) were maintained in the same medium but in the absence of glutamate. The cells were kept at 37°C in a humidified incubator with 5% CO2/95% air, for 7 d (hippocampal cultures) or 14–15 d (striatal cultures). Cells were cultured at a density of 90,000 cells/cm2 on poly-d-lysine-coated 6-well microplates (MW6) (for Western blot and real-time PCR experiments). Animals used in the preparation of cell cultures were handled according to National and Institutional guidelines. Experiments conducted at the Center for Neuroscience and Cell Biology were performed according to the European Union Directive 86/609/EEC on the protection of animals used for experimental and other scientific purposes. These experiments did not require approval by an Institutional Animal Care and Use Committee.
Western blot
Hippocampal and striatal neuronal cultures were washed twice with ice-cold PBS and once more with PBS buffer supplemented with 1 mm DTT and a mixture of protease inhibitors: 0.1 mm PMSF and CLAP (1 μg/ml chymostatin, 1 μg/ml leupeptin, 1 μg/ml antipain, and 1 μg/ml pepstatin; Sigma). The cells were then lysed with RIPA (150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 5 mm EGTA, 1% Triton, 0.5% DOC, and 0.1% SDS at a final pH 7.5) supplemented with the mixture of protease inhibitors. After centrifugation at 16,100 × g for 10 min, protein in the supernatants was quantified using the BCA method, and the samples were denatured with 2× concentrated denaturing buffer (125 mm Tris, pH 6.8, 100 mm glycine, 4% SDS, 200 mm DTT, 40% glycerol, 3 mm sodium orthovanadate, and 0.01% bromophenol blue) at 95°C for 5 min. Protein samples were separated by SDS-PAGE, in 12% polyacrylamide gels, transferred to PVDF membranes (Millipore), and immunoblotted, as previously described (Caldeira et al., 2007). Blots were incubated with primary antibodies (overnight at 4°C), washed, and exposed to alkaline phosphatase-conjugated secondary antibodies (1:20,000 dilution for anti-rabbit IgG and 1:10,000 dilution for mouse IgG; 1 h at room temperature). Alkaline phosphatase activity was visualized by enhanced chemifluorescence on the Storm 860 gel and blot imaging system, and quantified using the ImageQuant program (GE Healthcare). The following primary antibodies were used: anti-TrkB (1:750, Ab:156–322 aa; BD Biosciences), anti-RhoA (1:1000, Ab: C-terminal; Cell Signaling Technology), anti-spectrin (1:1000, Mab1622; Millipore Bioscience Research Reagents), anti-phospho-Trk (1:1000; Cell Signaling Technology), anti-phospho-Akt (Ser473, 1: 1000; Cell Signaling Technology), anti-Akt (1:1000; Cell Signaling Technology), and anti-PLCγ (1:1000; Cell Signaling Technology). The anti-β-tubulin I (1:500,000; Sigma) antibody was used as loading control.
mRNA semiquantification through real-time PCR
Total RNA was extracted from 7 DIV cultured hippocampal neurons using TRIzol Reagent (Invitrogen), as previously described (Santos and Duarte, 2008; Gomes et al., 2011). RNA quality and integrity was assessed using the Experion automated gel-electrophoresis system (Bio-Rad), as previously described (Santos and Duarte, 2008). Samples showing RNA degradation or contamination by DNA were discarded. RNA concentration was determined using the fluorescent dye RiboGreen (Invitrogen) or NanoDrop 1000 (Thermo Scientific). The samples were aliquoted and stored at −80°C until further use. cDNA synthesis was performed using 2 μg of total RNA and the AMV Reverse Transcriptase (10 U; Roche), as previously described (Santos and Duarte, 2008). Samples were stored at −80°C until further use.
Primers for TrkB isoforms in real-time PCR were as previously described (Silhol et al., 2007). GAPDH was used as the reference gene with the following primers: forward, 5′CTCCCATTCCTCCACCTTTG3′ and reverse, 5′TGTAGCCATATTCATTGTCATACC3′. The annealing temperature was 63.5°C.
For gene expression analysis, 2 μL of 1:100 diluted cDNA was added to 10 μL of 2× SYBR Green Master Mix (Bio-Rad), and the final concentration of each primer was 250 nm in 20 μL total volume. The thermocycling reaction was initiated with activation of the TaqDNA polymerase by heating at 95°C during 30 s, followed by 55 cycles of a 10 s denaturation step at 95°C and 30 s annealing and 30 s elongation steps at 72°C. The fluorescence was measured after the extension step, using the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). After the thermocycling reaction, the melting step was performed with slow heating, starting at 55°C and with a rate of 0.5°C per 10 s, up to 95°C, with continuous measurement of fluorescence. Data analysis was performed using the Pfaffl method for efficiency correction (Pfaffl, 2001). The results were normalized with GAPDH as the internal reference gene, because it showed a stable expression in hippocampal neurons stimulated with glutamate.
DNA constructs/lentivirus production
The rat TrkB isoforms were kindly provided by V. Leßmann (Institut für Physiologie, Otto-von-Guericke-Universität Magdeburg, Magdeburg, Germany) (Hartmann et al., 2004). Briefly, the cDNA sequences from TrkB full-length (TrkB.FL) and TrkB truncated isoform 1 (TrkB.T1) were amplified from the plasmids provided. Fragments were then subcloned into pRRLSIN (Addgene plasmid 12252) using BamHI and SalI restriction sites in 5′ and 3′, respectively. The resulting plasmids were verified by automated sequencing (STABVIDA). Lentivirus were then generated as previously described (Zufferey et al., 1998). Briefly, HEK 293T cells were transfected by the calcium phosphate method and media collected 24 and 48 h after transfection. The virus were then concentrated by centrifugation at ∼80,000 × g for 2 h and the virus pellet resuspended in PBS supplemented with 1% BSA, aliquoted, and stored at −80°C until further use. Viral titers were determined by infection of HEK 293T cells.
Cell death assay
Hippocampal neurons were cultured for 7 d on poly-d-lysine-coated glass coverslips as previously described. After the experiments cells were fixed in 4% sucrose/4% paraformaldehyde (in PBS). The cells were washed twice with PBS and were then incubated with Hoechst 33342 (0.5 μg/ml) to stain nuclei. Analysis of the nuclear morphology was performed on a Zeiss Axiovert 200 florescence microscope, under a 40× objective.
Intrahippocampal injection of kainate
Injection.
Intrahippocampal injection of kainate (Tocris Bioscience) was performed as previously described (Tomioka et al., 2002; Takano et al., 2005), with minor modifications. Briefly, adult male mice (C56BL6) were deeply anesthetized with avertin (2,2,2 tribromoethanol, 2-methyl-2-butanol), placed in a stereotaxic apparatus (Stoelting), and given a unilateral injection of 1 nmol of kainate (in 0.3 μl of PBS) into the hippocampal CA1 region, using a 10 μl motorized syringe (Hamilton), after drilling a small hole with a surgical drill. The coordinates of the injection were anterior–posterior −2.3 mm, mediolateral −1.5 mm, and dorsoventral −1.7 mm from the bregma. Two minutes after the needle insertion, kainate was injected at a constant flow rate of 0.05 μl/min. The needle remained in place for an additional 2 min to prevent reflux of fluid. The body temperature of mice was monitored and maintained at 37°C during surgery and 30 min after the injection, with a homeothermic heating blanket (Harvard Apparatus), with feedback regulation. Experiments were conducted according to the European Union Directive 86/609/EEC on the protection of animals used for experimental and other scientific purposes.
Western blot.
12 h after injection mice were killed and a 2 mm section around the hippocampus was taken with the help of a 1 mm coronal mouse matrix (Harvard Apparatus). The slices were immediately frozen with dry ice and the damaged ipsilateral and contralateral areas of the hippocampal slices were taken using a Harris Unicore 2 mm tip (Pelco International). Samples were then homogenized and processed for Western blot.
Middle cerebral artery occlusion
Focal cerebral ischemia was induced by the transient occlusion of the right middle cerebral artery (MCA), using the intraluminal filament placement technique as described previously (Nygren and Wieloch, 2005; Gomes et al., 2011). Briefly, adult male mice were anesthetized by inhalation of 2.5% isoflurane (IsobaVet; Schering-Plough) in O2:N2O (30:70). Anesthesia was subsequently reduced to 1.5–1.8% isoflurane and sustained throughout the occlusion period. Body temperature was kept at ∼37°C throughout the surgery period. To monitor regional cerebral blood flow (rCBF), an optical fiber probe (Probe 318-I; Perimed) was fixed to the skull at 2 mm posterior and 4 mm lateral to bregma and connected to a laser Doppler flow meter (Periflux System 5000; Perimed). A filament composed of 6–0 polydioxanone suture (PSD II; Ethicon) with a silicone tip (diameter of 225–275 μm) was inserted into the external carotid artery and advanced into the common carotid artery. The filament was retracted, moved into the internal carotid artery, and advanced until the origin of the MCA, given by the sudden drop in rCBF (∼70% of baseline). After 45 min, the filament was withdrawn and reperfusion observed. The animals were placed in a heating box at 37°C for the first 2 h postsurgery and thereafter transferred into a heating box at 35°C to avoid postsurgical hypothermia. Thirty minutes after the onset of reperfusion, 0.5 ml of 5% glucose was administered subcutaneously. Temperature and sensorimotor deficits were assessed at 1 and 2 h and in the morning after the surgery. The Ethics Committee for Animal Research at Lund University approved animal housing conditions, handling, and surgical procedures. C57BL/6J male mice, 9 to 11 weeks old (weight, 21.5–27.9 g; Taconic), were housed under diurnal conditions with free access to water and food before and after surgery.
Western blot.
Mice were killed 6 h after middle cerebral artery occlusion (MCAO) and 1-mm-thick coronal brain sections were generated using a mouse brain matrix. The slices were immediately frozen with dry ice and the damaged ipsilateral and contralateral areas of the slices were taken using a Harris Unicore 2 mm tip (Pelco International). Samples were then homogenized and processed for Western blot as described above.
2,3,5-Triphenyltetrazolium chloride staining
After 24 h of recovery, animals were killed to evaluate the infarct volume. The brain was removed and the forebrain was sliced into 1-mm-thick sections using a mouse brain slicer on ice. The sections were rinsed once in ice-cold 0.9% NaCl for 10 min and subsequently immersed in 50 ml of 0.01% 2,3,5-triphenyltetrazolium chloride (TTC) in 0.9% NaCl at 37°C for 15 min. Slices were fixed in 4% formalin and images were acquired with a MicroPublisher 3.3 RTV CCD camera (QImaging) using standard conditions.
Expression of recombinant GST-Rhotekin-Rho-binding domain
Expression of the pGEX-Rhotekin-Rho-binding domain (amino acids 7–89) fusion protein, kindly provided by Michael Courtney (University of Kuopio, Finland) (Semenova et al., 2007), was induced by treating an Escherichia coli BL21 culture in the exponential phase of growth (37°C, A600 nm = 0.7) with 0.3 mm isopropyl β-d-thiogalactoside, for 4 h at 25°C. The protein was extracted and purified from the bacterial pellet through affinity chromatography on glutathione Sepharose 4B (GE Healthcare), according to the manufacturer's recommendations.
Pull-down assay
Rho activity was measured as previously described (Semenova et al., 2007) with some modifications. Cultured striatal neurons with 15 DIV were treated with or without glutamate (125 μM, 5 or 20 min) and BDNF (100 ng/ml), for the periods of time indicated. Cells were rinsed with ice-cold PBS once and lysed in 900 μl lysis buffer (50 mm Tris, pH 7.2, 500 mm NaCl, 1% (v/v) Triton X-100, 5 mm MgCl2, 1 mm DTT, and protease inhibitors). Three wells, from the 6-well microplates (MW6), were used for each experimental condition. Homogenized lysates were frozen in liquid nitrogen to promote cell disruption and were then centrifuged at 16,000 × g for 10 min. The supernatant was collected and incubated with 30 μg of GST-Rhotekin and glutathione-Sepharose for 1 h at 4°C with constant and gentle rotation. Beads were spun out (5 min, 500 × g) and washed three times with 50 mm Tris, pH 7.2, 150 mm NaCl, 1% (v/v) Triton X-100, 5 mm MgCl2, 1 mm DTT, and protease inhibitors. Finally, beads with GTP-RhoA were denatured with 5× concentrated denaturing buffer and boiled at 95°C for 5 min, followed by a centrifugation in a microtube provided with a filter to separate beads from the supernatant. Active RhoA was detected by immunoblotting with monoclonal anti-RhoA antibody.
Bio-Plex phosphoprotein testing
Bio-Plex phosphoprotein assays and Bio-Plex total target assays are bead-based multiplex assays (Luminex xMAP technology) that detect the phosphorylation of proteins in lysates derived from cell culture. The Bio-Plex total target assay was used to determine the total abundance of Erk in the extracts and the Bio-Plex phosphoprotein assay was used to determine Erk phosphorylation in separate wells. In the phosphoprotein assay antibodies directed against pErk are covalently coupled to internally dyed beads, and the coupled beads then react with the phosphorylated and dephosphorylated forms of Erk. After a series of washes to remove unbound protein, biotinylated detection antibodies for Erk (second antibodies) are added to the reaction forming a sandwich of antibodies around the target proteins (Erk). Streptavidin-PE is finally added to bind to the biotinylated detection antibodies on the bead surface. Data from the reaction are acquired using the Bio-Plex suspension array system (Bio-Rad). The contents of the well are draw up into the reader. The lasers and associated optics detect the internal fluorescence of the individual dyed beads as well as the fluorescent signal on the bead surface. This identifies each assay and reports the level of target proteins (pErk and total protein levels). In the preparation of extracts for the Bio-Plex assays, the cells were washed with cell wash buffer (Bio-Rad) and then lysed with cell lysis buffer (Bio-Plex cell lysis kit, Bio-Rad). After 30 min of agitation, the protein content was determined using the BCA method and the concentration was equalized with assay buffer (Bio-Plex phosphoprotein testing assay buffer). Finally, the samples were analyzed with the Bio-Plex suspension array system.
Statistical analysis
The immunoreactivity obtained in each experimental condition was calculated as a percentage of the control. Data are presented as mean ± SEM of at least three different experiments performed in independent preparations. Statistical analysis of the results was performed using one-way ANOVA analysis followed by either the Dunnett's or Bonferroni's posttest: ***p < 0.001, ** p < 0.01, and * p < 0.05.
Results
Excitotoxicity downregulates TrkB.FL in vitro and in vivo, and upregulates the truncated TrkB isoform
To understand how excitotoxic conditions influence the protein levels of the TrkB receptors, cultured neurons isolated from different brain regions were stimulated with glutamate (125 μm, for 20 min) and further incubated in culture-conditioned medium for different time periods. Stimulation of cultured hippocampal neurons under these conditions induces 40–50% cell death (Almeida et al., 2005), and Western blot experiments using an antibody against an extracellular epitope of TrkB showed a downregulation of full-length TrkB (145 kDa) and upregulation of a truncated form of the receptor with ∼95 kDa (Fig. 1A). TrkB.FL protein levels dropped rapidly by ∼50% within the first hour after the toxic insult, and a secondary slower decrease in the receptor protein levels was observed during the following 7 h. Concomitantly, truncated TrkB protein levels were upregulated with similar kinetics but with a delayed onset. This pattern of change in TrkB protein levels following excitotoxic stimulation was also observed in striatal neuronal cultures, but in this case the upregulation of the truncated TrkB receptors showed a greater magnitude being upregulated to >200% of the control (Fig. 1B).
Figure 1.

TrkB.FL cleavage and upregulation of a truncated TrkB isoform (TrkB.T) in cultured hippocampal and striatal neurons subjected to excitotoxic conditions. Cultured hippocampal (A) and striatal (B) neurons were subjected to excitotoxic stimulation with glutamate (125 μm glutamate, 20 min) and further incubated in culture-conditioned medium. TrkB full-length and truncated TrkB protein levels were determined by Western blot at the indicated time points after excitotoxic stimulation. The results for the hippocampal neurons are the average ± SEM of 3–7 independent experiments, performed in different preparations, whereas 4–15 independent experiments were performed with striatal neurons. Statistical analysis was performed using one-way ANOVA followed by Dunnett's comparison test performed for each condition as compared with the control, not exposed to excitotoxic conditions (***p < 0.001, **p < 0.01, and *p < 0.05). C, Intrahippocampal injection of kainate changes full-length and truncated TrkB immunoreactivity in the ipsilateral (Ipsi) hippocampus of adult mice (C56Bl 6). TrkB and Spectrin breakdown product's (SBDP) protein levels in the injected and contralateral hippocampus were determined by Western blot 12 h after the lesion. The results are representative of two independent experiments, performed in different animals. D, Changes in TrkB isoforms immunoreactivity in the ipsilateral (Ipsi) and contralateral (Contra) brain hemispheres of adult mice (C56Bl6) subjected to a transient 45 min occlusion of the right MCA. TrkB protein levels in the cerebral cortex, hippocampus, and striatum were determined by Western blot 6 h after the lesion. Core and penumbra (Pen) regions were dissected separately in the cerebral cortex and striatum. E, TTC staining of 1-mm-thick sections of the mouse brain (with the bregma stereotaxic coordinates indicated), 24 h after the occlusion, providing an evaluation of the infarct volume and damaged areas in the brain after the 45 min occlusion of the right MCA.
The hippocampus and the striatum are two brain regions particularly sensitive to excitotoxic neuronal death after cerebral ischemia, and the latter region is highly enriched in TrkB receptors (Eide et al., 1996; Dos-Anjos et al., 2009). To address the changes of the TrkB isoform protein levels in an in vivo model of excitotoxicity we tested the effect of kainate injection into the CA1 region of the hippocampus, which leads to excitotoxic neuronal death (Tomioka et al., 2002; Higuchi et al., 2005; Takano et al., 2005). A downregulation of the TrkB.FL protein levels was observed together with an upregulation of the TrkB.T protein levels (Fig. 1C), similar to the results obtained in the neuronal cultures. As expected, kainate injection into the CA1 region of the hippocampus activated calpains, as determined by the formation of 145 kDa spectrin cleavage product; negligible amounts of the 120 kDa cleavage products were detected, indicating a minor contribution of caspase-3 to spectrin cleavage under the experimental conditions used (Wang, 2000).
The pattern of TrkB downregulation in cultured hippocampal and striatal neurons subjected to excitotoxic stimulation was also compared with that observed in the brain of mice subjected to the MCAO model of transient focal ischemia (Traystman, 2003). Adult mice (C56BI6) were subjected to transient 45 min occlusion of the right MCA, and extracts were prepared from the ipsilateral (Ipsi) and contralateral (Contra) brain hemispheres (cortex, striatum, and hippocampus) 6 h after injury. A downregulation of TrkB.FL protein levels was observed in the ipsilateral striatum and cerebral cortex 6 h after the ischemic insult, and this effect was more significant in the core than in the penumbra region (Fig. 1D). Truncated TrkB levels were also downregulated in the two brain regions, in contrast with the upregulation observed in the excitotoxicity studies performed in vitro and in vivo (see above). This may be due to (1) the presence of truncated receptors in astrocytes (Rose et al., 2003; Ohira et al., 2005a,b), which are vestigial in the neuronal cultures used in this work (Brewer et al., 1993), and (2) to a stronger injury induced by kainate in the in vivo experiments when compared with MCAO. MCAO did not change the expression of TrkB isoforms in the hippocampus, a region that is not primarily affected in this model of ischemia, as observed in 1-mm-thick sections of the mouse brain, 24 h after the occlusion, using TTC staining (Fig. 1E).
TrkB.FL is downregulated by a calpain-dependent mechanism
Chemical inhibitors of different classes of proteases were used to determine the mechanism(s) of downregulation of TrkB.FL under excitotoxic conditions. Both calpain inhibitors used, ALLN (50 μm) and MDL28170 (100 μm), prevented TrkB.FL loss as determined 1.5 h after glutamate stimulation (Fig. 2A). Z-VAD-FMK (50 μm), a general inhibitor of caspases, did not affect glutamate-induced downregulation of TrkB.FL (Fig. 2B), indicating that this class of proteases does not contribute to the observed response. Cathepsins, mainly the B and L isoforms, also contribute to protein degradation in ischemic cell death (Yamashima, 2000). The use of specific pharmacological inhibitors of cathepsin B and L (CA-074 Me and Inhibitor III, respectively, at 50 and 10 μm) showed that these proteases are also without effect on the cleavage of TrkB.FL under excitotoxic conditions (Fig. 2C). Similar results were obtained in the presence of the proteasome inhibitor MG132 (1 μm).
Figure 2.

Calpain mediates TrkB.FL downregulation under excitotoxic conditions. A, Cultured hippocampal neurons were preincubated (2 h) with the calpain inhibitors MDL 28170 (100 μm) or ALLN (50 μm), before and during glutamate stimulation (Glu; 125 μm, 20 min). The cells were further incubated in culture-conditioned medium for 1.5 h with calpain inhibitors and extracts were analyzed by Western blot with anti-TrkB antibody. B, The effect of the pan-caspase inhibitor Z-VAD-FMK (50 μm) on glutamate-evoked TrkB.FL downregulation was tested using the experimental conditions described in (A). The effect of cathepsin B (Ca-074, 50 μm) and L (Inhibitor III, 10 μm) inhibitors is shown in (C). D, Experiments where the effect of the proteasome inhibitor MG132 (1 μm) on TrkB.FL degradation were tested. The cells were preincubated for 30 min with the proteasome inhibitor before glutamate (Glu) stimulation. After excitotoxic stimulation the cells were incubated in culture-conditioned medium in the presence or in the absence of the tested inhibitor, for 1.5 h. Extracts were analyzed by Western blot with an antibody against TrkB. The results of (A–D) are the average ± SEM of at least 3 different experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. ***p < 0.001; **p < 0.01; *p < 0.05; n.s., not significant as compared with the control protein levels for TrkB.FL or for the indicated comparisons.
Truncated TrkB is upregulated by a transcription-dependent mechanism
The distinct onset of the TrkB.FL downregulation when compared with the delayed upregulation of a shorter form of the receptor under excitotoxic conditions, both in hippocampal and striatal neurons (Fig. 1A,B), suggests that the latter population of receptors corresponds to a TrkB.T isoform synthesized de novo rather than to a cleavage product. This hypothesis is corroborated by the results showing that inhibition of TrkB.FL downregulation by calpain inhibitors is not associated with a corresponding loss of the low molecular weight form of the receptor (Fig. 2A). In fact, the results rather suggest that the latter population of TrkB receptors corresponds to a TrkB.T isoform that is synthesized de novo upon excitotoxic stimulation. To test this hypothesis, cultured hippocampal neurons were stimulated with glutamate in the presence of the translation inhibitor anisomycin (5 μm), and the protein levels of TrkB.FL and the short form of the receptor were analyzed 8 h later (Fig. 3). Inhibition of protein synthesis abrogated the upregulation of the TrkB truncated form, showing that the effect is dependent on translation activity. Similar results were obtained in the presence of the transcription inhibitor actinomycin D, further indicating that the short form of the TrkB receptor corresponds to a TrkB.T isoform that is synthesized de novo following excitotoxic stimulation (Fig. 3B). Translation inhibition for 8 h, the maximum time period used in this set of experiments, had a minor impact in the total amount of TrkB.FL and TrkB.T isoforms under control conditions, showing their slow turnover rate (Fig. 3A).
Figure 3.

Truncated TrkB is upregulated under excitotoxic conditions by a transcription-dependent mechanism. A, Cultured hippocampal neurons were preincubated (15 min) with the translation inhibitor anisomycin (5 μm) before and during glutamate stimulation (125 μm; 20 min). The cells were then incubated with the inhibitor in culture-conditioned medium for 8 h and extracts were analyzed by Western blot with an anti-TrkB antibody. The results are the average ± SEM of 5–9 different experiments performed in independent preparations. B, Cultured hippocampal neurons were preincubated (30 min) with the transcription inhibitor actinomycin D (1.5 μm), before and during glutamate stimulation (Glu; 125 μm, 20 min). The cells were then incubated with the inhibitor in culture-conditioned medium for 1.5 h and extracts were analyzed by Western blot with anti-TrkB antibody. The results are the average ± SEM of 4–7 different experiments performed in independent preparations. C–E, Cultured hippocampal neurons were subjected to excitotoxic stimulation with glutamate (125 μm, 20 min) and further incubated in culture-conditioned medium for the indicated periods of time. Alternatively, cultures were incubated with the transcription inhibitor actinomycin D (1.5 μm) for 4 h. Total RNA was extracted and Trk.FL (C), TrkB.T1 (D), TrkB.T2 (E), and GAPDH (C–E) mRNA were semiquantified through real-time PCR. The results are the average ± SEM of 3–8 different experiments performed in independent preparations. Statistical analysis from (A–E) was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. ***p < 0.001; **p < 0.01; *p < 0.05; n.s., not significant as compared with the control or for the indicated comparisons.
The inhibition of glutamate-evoked upregulation of TrkB.T upon transcription inhibition suggests that excitotoxicity shifts the splicing of the TrkB pre-mRNA to increase the amount of mature mRNA coding or the truncated form(s) of the receptor. This may not have an impact in the amount of TrkB.FL detected under the experimental conditions used given the low turnover rate of the receptors (see above). This hypothesis was addressed through analysis of the excitotoxicity-induced changes in the mRNA for TrkB.FL and the truncated isoforms, using real-time PCR. TrkB.FL mRNA was downregulated 1.5 and 4 h after glutamate stimulation (125 μm, 20 min) (Fig. 3C). This downregulation was likely due to active degradation of TrkB.FL transcripts since inhibition of transcription with actinomycin D, for the same period of time (4 h), did not significantly change TrkB.FL mRNA (Fig. 3C). In contrast, TrkB.T1 and TrkB.T2 mRNAs were upregulated after the excitotoxic injury, in agreement with the results obtained when TrkB.T protein levels were analyzed (Fig. 3D,E).
BDNF-dependent signaling activity is downregulated under excitotoxic conditions
The TrkB.FL degradation after excitotoxic injury and the upregulation of TrkB.T under the same conditions is likely to have a significant impact in the signaling activity of the full-length receptors. This was addressed at three different levels: (1) TrkB phosphorylation, the first step following binding of BDNF to the receptors and their dimerization; (2) PLCγ phosphorylation, which follows the interaction of the enzyme with the active receptor; and (3) phosphorylation (presumably activation) of ERK and Akt, downstream mediators of two signaling cascades activated by TrkB receptors (Middlemas et al., 1994; Huang and Reichardt, 2003; Manadas et al., 2007). Activation of TrkB.FL by BDNF was evaluated by Western blot, using an antibody against the phosphorylated form of the receptor, and the results were calculated as a ratio to the total amount of TrkB.FL protein levels, under control conditions and in cells subjected to excitotoxic stimulation. Stimulation of hippocampal neurons with BDNF (100 ng/ml) was performed 1.5 h after the excitotoxic insult, a time point when there is a significant degradation of TrkB.FL protein levels and TrkB.T receptors are already upregulated (Fig. 1A). The cells were stimulated with the neurotrophin for 10 min since this time period allows inducing maximal signaling activity (Almeida et al., 2005). Excitotoxic stimulation of hippocampal neurons decreased the total amount of pTrkB detected following stimulation with BDNF (data not shown), due to degradation of a subpopulation of receptors (50%) (Fig. 1A). However, the remaining receptors are still partially responsive to BDNF stimulation, as determined by the increase in the ratio pTrkB /total TrkB.FL (Fig. 4A). When the experiments were performed in the presence of the calpain inhibitor ALLN (50 μm), which prevents the degradation of TrkB.FL (but had no effect on the upregulation of TrkB.T), the ratio pTrkB/total TrkB was still much lower than that obtained under control conditions (Fig. 4A). After excitotoxic injury there was also a decrease in the BDNF-induced PLCγ phosphorylation (Fig. 4B) and ERK activation (Fig. 4C), and the activity of both signaling pathways was still downregulated when the experiments were conducted in the presence of the calpain inhibitor ALLN. In contrast with the results obtained for TrkB, PLCγ, and ERK phosphorylation, excitotoxic stimulation of hippocampal neurons did not significantly change the BDNF-induced accumulation of phosphorylated Akt (Fig. 4D). This may be due to differences in the activity of the Akt phosphatase(s), when compared with the phosphatases responsible for the regulation of TrkB, ERK, and PLCγ phosphorylation state. Together the results clearly show that TrkB.FL signaling activity is downregulated in excitotoxic conditions, but the effect cannot be explained solely based on the degradation of TrkB.FL.
Figure 4.

TrkB.FL-dependent signaling activity is downregulated under excitotoxic conditions. TrkB.FL signaling activity under excitotoxic conditions was assessed by analyzing TrkB.FL phosphorylation (A), PLCγ phosphorylation (B), ERK phosphorylation (C), and Akt phosphorylation (D), following stimulation of hippocampal neurons with BDNF (100 ng/ml) for 10 min, under control condition and after excitotoxic stimulation with glutamate (Glu; 125 μm, 20 min). In the latter experimental conditions the cells were incubated in culture-conditioned medium for 80 min after the toxic insult before stimulation with BDNF. When the effect of the calpain inhibitor ALLN (50 μm) was tested the cells were preincubated with the inhibitor for 2 h before glutamate stimulation, and the inhibitor was also present during all additional experimental manipulation. Erk phosphorylation was analyzed using the Bio-Plex System, and PLCγ, TrkB.FL, and Akt phosphorylation were measured by Western blot. The results are the average ± SEM of 3–9 different experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. ***p < 0.001; *p < 0.05; n.s., not significant as compared with the control or for the indicated comparisons.
Truncated TrkB may act as dominant-negative preventing the activation of TrkB.FL receptors
Truncated TrkB receptors have been described to exert a dominant-negative effect over TrkB.FL, preventing the association of two full-length receptors which is required for their transphosphorylation and activation of signaling pathways (Manadas et al., 2007). The results showing preservation of TrkB.FL receptors but not their signaling activity in hippocampal neurons subjected to excitotoxic stimulation in the presence of ALLN may be partially explained considering that TrkB.T receptors, which are upregulated under the same conditions, may exert such a dominant-negative effect. This may arise from the dimerization of Trk.FL and TrkB.T receptors, which will prevent tyrosine phosphorylation of TrkB.FL and consequently, no signaling activity should be generated (Eide et al., 1996; Li et al., 1998; Haapasalo et al., 2001). This hypothesis was tested by inhibiting translation with anisomycin (5 μm), thereby preventing the upregulation of TrkB.T under excitotoxic conditions (Fig. 3A). Figure 5 shows that translation inhibition prevents the excitotoxicity-induced downregulation of TrkB.FL signaling activity, both at the level of the receptor (pTrkB/TrkB Total; Fig. 5A) and activation of the ERK signaling pathway (Fig. 5B). Together the results strongly suggest that TrkB.FL signaling activity is downregulated under excitotoxic conditions due to the dominant-negative effect exerted by TrkB.T isoforms that are upregulated under the same conditions. The dominant-negative effect of TrkB.T isoforms was further investigated in experiments with cultured hippocampal neurons transduced with lentivirus expressing GFP or TrkB.T1, and the effect of BDNF on TrkB-FL phosphorylation was determined by Western blot. Upregulation of the expression of TrkB.T1 significantly decreased the BDNF-induced TrkB.FL phosphorylation (Fig. 5C) in agreement with the dominant-negative hypothesis.
Figure 5.

Truncated TrkB acts as dominant-negative inhibiting signaling by TrkB.FL. TrkB.FL signaling activity under excitotoxic conditions was measured after blocking the upregulation of truncated TrkB with the translation inhibitor anisomycin (5 μm). Cultured hippocampal neurons were preincubated (15 min) with anisomycin before and during glutamate stimulation (125 μm, 20 min). TrkB.FL phosphorylation (A) and ERK phosphorylation (B) were then measured upon stimulation of hippocampal neurons with BDNF (100 ng/ml) for 10 min under control condition or after excitotoxic stimulation with glutamate (125 μm, 20 min). In the latter experimental conditions, after the excitotoxic insult the cells were incubated in culture-conditioned medium for 80 min and then stimulated with BDNF. When the effect of the translation inhibitor anisomycin (5 μm) was tested the cells were preincubated with the inhibitor for 15 min before glutamate stimulation, and the inhibitor was also present during all additional experimental manipulations. C, TrkB.FL signaling activity under control conditions was also measured in cultured hippocampal neurons infected with lentiviral constructs of GFP or TrkB.T. TrkB.FL phosphorylation was measured upon stimulation of hippocampal neurons with BDNF (100 ng/ml) for 10 min. Erk phosphorylation was analyzed using the Bio-Plex System, and TrkB.FL phosphorylation was measured by Western blot. The results are the average ± SEM of 3–6 different experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. ***p < 0.001, **p < 0.01, *p < 0.05, as compared with the control or for the indicated comparisons.
The dominant-negative effect of TrkB.T receptors may not fully account for the loss of TrkB.FL activity
To evaluate whether the upregulation of TrkB.T receptors may fully account for the loss of TrkB.FL receptor activity in hippocampal neurons subjected to excitotoxic stimulation, a theoretical analysis was performed to compare the observed changes in TrkB.FL phosphorylation with the results predicted based on the changes in the relative abundance of the TrkB.FL and TrkB.T forms.
Addition of BDNF to a system containing TrkB.FL and TrkB.T receptors leads to the formation of TrkB.FL–TrkB.FL, TrkB.T–TrkB.FL, and TrkB.T–TrkB.T dimers. The total number of dimers formed is as follows:
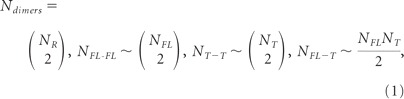 |
where NR is the total number of TrkB receptors, and NFL and NT the total number of TrkB.FL and TrkB.T receptors, respectively. At equilibrium, the total number of receptor homodimers and heterodimers is given by the following equations:
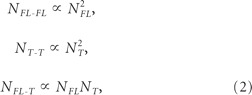 |
The first two terms in Equation 2 state that, from Equation 1, the total number of Trk.FL–TrkB.FL or TrkB.T–TrkB.T homodimers is approximated by proportionality to the square of the total number of receptors belonging to the corresponding class if their expression is large enough.
The last term in Equation 2 states that in the presence of a saturating concentration of BDNF all receptors are in a dimerized form at equilibrium. The model assumes that the formation of the different binary complexes is equally probable and, therefore, the formation of TrkB.FL–TrkB.FL or TrkB.T–TrkB.T homodimers is not preferred to the formation of TrkB.T–TrkB.FL heterodimers.
Under resting conditions (Eqs. 3 and 4) and following stimulation with saturating concentrations of BDNF (Eqs. 5 and 6), the phosphorylation of TrkB.FL receptors under control conditions (Eqs. 3 and 5) and after excitotoxic stimulation (Eqs. 4 and 6) can be described by the following equations:
In these equations krest-e = rate of oligomerization of TrkB.FL–TrkB.T heterodimers, krest-h = rate of oligomerization of TrkB.FL–TrkB.FL homodimers, and kact = rate of activation of TrkB.FL receptors following stimulation with BDNF.
To identify the correct set of parameters we used the constrained least-squares as follows:
and to attain the minimum of the following quantity
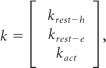 |
where the terms in Equation 7 are defined as:
and
 |
In Mdimers we consider that BDNF-induced TrkB.FL–TrkB.FL homodimerization leads to receptor transphosphorylation with a probability of 1 as follows:
 |
where
and analogously
In the above expressions (Eqs. 8 and 9) we consider that the total number of homodimers and heterodimers is computed as the likelihood of forming a homodimer or heterodimer, respectively (Eq. 2), over the total number of dimers that can be formed. Figure 6 shows the predicted phosphorylation of TrkB.FL receptors considering different expression levels of TrkB.FL and TrkB.T receptors. As expected, maximal TrkB.FL phosphorylation is predicted in the absence of TrkB.T receptors. Based on the quantification of the results of Figure 1A showing the relative expression of TrkB.FL and TrkB.T receptors in cultured hippocampal neurons under control conditions, the phosphorylation levels of TrkB.FL predicted by the model (black circle) were slightly below the immunoreactivity obtained in the Western blot experiments with the p-TrkB.FL receptors (Fig. 5A; Fig. 6, gray bullet). The change in the relative expression of TrkB.FL and TrkB.T receptors following excitotoxic stimulation decreased the predicted p-TrkB.FL response to BDNF stimulation (black circle), in agreement with the dominant-negative hypothesis. In this case, the response to BDNF stimulation was even lower than the predicted results, suggesting that additional factors may contribute to the downregulation of TrkB.FL receptor activity under excitotoxic conditions.
Figure 6.

Prediction of the dominant-negative effect of the TrkB.T receptors over TrkB.FL. The response—measured by TrkB.FL phosphorylation induced by BDNF—is predicted as a function of the number of TrkB.FL and TrkB.T receptors. The surface interpolates the experimental values (gray bullets) in a least-squares sense and predicts the cellular response upon varying the concentration of TrkB.FL and TrkB.T. The gray bullets represent the observed experimental values. The BDNF-induced changes in TrkB.FL phosphorylation 90 min after the excitotoxic insult (3925 a.u. as measured in Western blot experiments; data taken from Fig. 5A) were determined under conditions when the abundance of TrkB.FL = 2050795 (a.u.) and TrkB.T = 7797045 (a.u.), as determined by Western blot (data taken from Fig. 1A). TrkB.FL phosphorylation predicted by the model (51340 a.u.), considering the effects depending on the numbers of TrkB.FL and TrkB.T and their dimerization only, is represented by the black dot (on the surface). A similar analysis was performed for BDNF-induced TrkB.FL phosphorylation under control conditions: 244101 a.u. as measured in Western blot experiments (data taken from Figure 5A) (shown by the gray bullet above the surface). Under these conditions the abundance of TrkB.FL = 4058175 (a.u.) and TrkB.T = 5542307 (a.u.), as determined by Western blot (data taken from Fig. 1A). The predicted increase of TrkB.FL phosphorylation is 233680 (a.u.) (black dot on the surface).
Activation of TrkB.T receptors by BDNF blocks the activation of RhoA GTPase under excitotoxic conditions
TrkB.T1 receptors are coupled to the inhibition of RhoA (Ohira et al., 2006), a small GTPase that mediates the Ca2+-dependent activation of p38 MAPK followed by neuronal death under excitotoxic conditions (Semenova et al., 2007). Therefore, we investigated whether the TrkB.T receptors upregulated under excitotoxic conditions could express their own signaling activity. RhoA activity was assessed by a pull-down assay using a GST fusion protein with the Rhotekin amino acid sequence responsible for binding to the active form of the GTPase. Striatal neurons were subjected to excitotoxic stimulation (125 μm glutamate, 20 min) to induce an upregulation of TrkB.T expression, and 1.5 h later a short stimulation (5 min) with glutamate was performed in the absence or in the presence of BDNF. Alternatively, the effect of BDNF was tested in cells exposed to glutamate for 5 min without previous excitotoxic stimulation. The former (but not the latter) experimental conditions allow the upregulation of TrkB.T before testing for the activity of TrkB.T receptors in response to stimulation with BDNF. In striatal neurons previously exposed to excitotoxic conditions the activation of RhoA induced by 5 min incubation with glutamate was significantly inhibited by BDNF (Fig. 7A). This inhibitory effect was not observed in cells treated with glutamate and BDNF without previous excitotoxic stimulation (Fig. 7A). Although the upregulation of TrkB.T in hippocampal neurons following excitotoxic stimulation is not as robust as in striatal neurons (Fig. 1A,B), the results of Figure 7B show that hippocampal cultures behave similarly to striatal neurons. Interestingly, inhibition of RhoA activity by BDNF in cells previously subjected to excitotoxic stimulation was not observed when the experiments were conducted in the presence of anisomycin to inhibit translation. Since under these conditions there is no upregulation of TrkB.T following the excitotoxic insult (Fig. 3A), the results suggest that the inhibitory effects of BDNF on RhoA GTPase signaling are mediated by activation of the truncated receptors. Accordingly, control experiments showed that the inhibition of RhoA by BDNF is insensitive to the TrkB.FL receptor inhibitor K252A (Fig. 7B). Together these results indicate that the upregulation of TrkB.T following an excitotoxic insult significantly interferes with the activation of RhoA signaling pathways which are known to be coupled to excitotoxic cell death (Semenova et al., 2007).
Figure 7.

Activation of TrkB.T receptors by BDNF inhibits the stimulation of Rho A under excitotoxic conditions. Activated GTP-bound RhoA was measured by pull-down from cell lysates with immobilized recombinant Rhotekin-RBD and detected by immunoblotting with an anti-RhoA antibody. A, RhoA activity was measured in striatal neurons following 5 min stimulation with glutamate, in the presence or in the absence of BDNF (100 ng/ml). Where indicated the cells were subjected to excitotoxic stimulation with glutamate (125 μm, 20 min) before the short incubation with the amino acid. Active RhoA was measured by pull- down and measured as a ratio to the total amount of RhoA. The results are the average ± SEM of 3–5 different experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. **p < 0.01, as compared with the respective control, i.e., cells exposed to excitotoxic stimulation and later exposed to glutamate for 5 min. B, The same experimental procedure was followed in cultured hippocampal neurons. Truncated TrkB upregulation was blocked with anisomycin (5 μm) added to the medium 30 min before glutamate stimulation and present throughout the experiment. When the effect of K252A (200 nm) was tested the cells were preincubated with the inhibitor (30 min) before glutamate stimulation and the inhibitor was present throughout the experiment. The results are representative of two independent experiments performed in distinct preparations.
BDNF protects neurons through TrkB full-length but also through truncated TrkB
To determine how the changes in TrkB protein levels affect the neurotrophic effects of BDNF in hippocampal neurons, we compared the protective activity of the neurotrophin added to hippocampal neurons during the period of glutamate stimulation or after the toxic insult. BDNF protected hippocampal neurons only when incubated during the period of excitotoxic stimulation, and no effect was observed when the neurotrophin was added after the insult (Fig. 8A). The protective effect of BDNF when added during excitotoxic stimulation was insensitive to the TrkB.FL chemical inhibitor SHN722 (1 μm) (Martin et al., 2011), suggesting that it may be mediated by the truncated TrkB receptors. Control experiments showed that 1 μm SHN722 inhibits BDNF-induced TrkB.FL phosphorylation (Fig. 8C). The lack of effect of BDNF when applied after the toxic insult indicates that activation of the truncated receptors expressed after the toxic insult does not provide neuroprotection, possibly because the excitotoxic machinery had been already activated during the period of glutamate stimulation.
Figure 8.

BDNF protects neurons through TrkB.T receptors. A, Cultured hippocampal neurons (DIV 7) were challenged with glutamate (125 μm, 20 min), and BDNF (100 ng/ml) was added during glutamate stimulation or immediately after. Where indicated, 1 μm SHN722 was present during the period of glutamate stimulation, in the presence or in the absence of BDNF, and the cells were preincubated with the inhibitor for 30 min. Cell death was assessed 14 h later by fluorescence microscopy, using the fluorescent dye Hoechst 33342. The results are the average ± SEM of 5–8 different experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test. B, Cultured hippocampal neurons (DIV 7) were transduced with lentiviral constructs of GFP, TrkB.FL, or TrkB.T1 and 48–72 h after infection cultures were challenged with glutamate (125 μm, 20 min) in the presence or absence of BDNF (100 ng/ml), and where indicated the neurotrophin was also present in the postincubation period. Fourteen hours after this stimulation cells were fixed and cell death was assessed as indicated in (A). The results are the average of 3–10 experiments performed in independent preparations. Statistical analysis was performed using one-way ANOVA followed by Bonferroni′s multiple-comparison test. ***p < 0.001; **p < 0.01; and n.s., not significant as compared with glutamate stimulation. ###p < 0.001; ##p < 0.01; #p < 0.05; and n.s., not significant as compared with the control. C, Cultured hippocampal neurons were stimulated or not with BDNF (100 ng/ml) for 10 min, in the presence or in the absence of the inhibitor SHN-711, and TrkB receptor phosphorylation was analyzed by Western blot.
To further investigate putative trophic effects of the TrkB receptor isoforms, cultured hippocampal neurons were transduced with lentivirus expressing GFP, TrkB.FL, or TrkB.T1, and the toxic effects of glutamate were investigated in the presence and in the absence of BDNF. In these experiments the cells were incubated with the neurotrophin only during the period of glutamate stimulation or both during the excitotoxic insult and the postincubation in culture-conditioned medium. Incubation with BDNF during the period of excitotoxic stimulation partially protected hippocampal neurons (Fig. 8B), similarly to the results obtained in nontransduced cells (Fig. 8A). Remarkably, BDNF fully protected hippocampal neurons transduced with TrkB.T when the neurotrophin was added during the period of stimulation with glutamate, and similar results were observed when BDNF was also present during the postincubation period (Fig. 8B). In contrast, overexpression of TrkB.FL did not further increase the neuroprotective effects of BDNF when tested during the period of glutamate stimulation, since the effects were similar to those obtained in hippocampal neurons transduced with GFP (Fig. 8B). Furthermore, when BDNF was also present during the postincubation period no additional neuroprotection was observed.
Discussion
In this work we show that TrkB.FL receptors are degraded under excitotoxic conditions (in vitro and in vivo) and in transient focal cerebral ischemia, and excitotoxicity also upregulated a truncated form of the TrkB receptor. Evidence indicates that the truncated TrkB receptor is not a cleavage product of TrkB.FL: (1) inhibition of calpains prevented glutamate-evoked degradation of the TrkB.FL but had no effect on the upregulation of TrkB.T; (2) transcription and translation inhibitors prevented the upregulation of TrkB.T induced by excitotoxic stimulation; and (3) glutamate stimulation upregulated TrkB.T1 and TrkB.T2 mRNA in cultured hippocampal neurons, with a concomitant decrease in the number of transcripts for TrkB.FL. The latter results suggest that the splicing mechanisms of the TrkB pre-mRNA are changed under excitotoxic conditions, leading to a sustained synthesis of more mature mRNAs coding for TrkB.T rather than TrkB.FL (Barbacid, 1994; Stoilov et al., 2002; Luberg et al., 2010). However, this may not have an impact in the amount of TrkB.FL protein levels detected under the experimental conditions used due to the low turnover rate of the receptors.
Calpains have been shown to play an important role in ischemic neurodegeneration (Bevers and Neumar, 2008), in agreement with the results obtained here showing a role for these proteases in the degradation of TrkB.FL under excitotoxic conditions. Other proteases also involved in excitotoxicity and ischemic cell death do not seem to participate in the downregulation of the receptor as their chemical inhibitors did not affect glutamate-induced TrkB.FL cleavage (Yamashima, 2000). It is not clear at this point how calpains contribute to the degradation of the receptors, since these proteases usually give rise to truncated cleavage products that were not detected in the present work (Haacke et al., 2007; Meary et al., 2007; Xu et al., 2007; Grumelli et al., 2008; Gomes et al., 2011).
In a previous study we showed that TrkB.FL receptors mediate the neuroprotective effects of BDNF when preincubated before excitotoxic stimulation with glutamate, through a mechanism dependent of protein synthesis (Almeida et al., 2005). Activation of the TrkB.FL receptors is followed by receptor dimerization and transphosphorylation on tyrosine residues (Huang and Reichardt, 2003; Manadas et al., 2007). The tyrosine phosphorylated receptors activate the ERK and PI3-K signaling pathways, which account for the neurotrophic effects of BDNF and provide neuroprotection under excitotoxic conditions (Almeida et al., 2005; Manadas et al., 2007). Both signaling pathways were also shown to contribute to the neurotrophic effects of BDNF, promoting neuronal survival (Dudek et al., 1997; Bonni et al., 1999). In the present work we show that BDNF present during the period of excitotoxic stimulation partially protects hippocampal neurons by a mechanism independent of TrkB.FL receptors. Furthermore, hippocampal neurons transduced with TrkB.T receptors were fully protected from excitotoxic stimulation when BDNF was present during the period of incubation with glutamate. This effect was more significant than that observed in neurons transduced with TrkB.FL or with GFP, which gave similar results. Although TrkB.T receptors were upregulated after excitotoxic stimulation, incubation of hippocampal neurons with BDNF after the insult did not prevent cell death, suggesting that the neuroprotective activity of truncated receptors should occur at an initial point, when the excitotoxic signaling cascade is activated.
Activation of TrkB.FL and TrkB.T receptors by BDNF induces independent signaling activity and, therefore, activation of the latter receptors may have distinct modulatory effects on excitotoxic neuronal damage. Activation of TrkB.T1 by BDNF leads to dissociation of Rho guanine nucleotide dissociation inhibitor 1 (Rho GDI 1), which interacts with the C-terminal region of the receptor. Once released from TrkB.T1, Rho GDI 1 acts as a negative regulator of Rho GTPases (Takai et al., 2001), including RhoA. Accordingly, we observed that the upregulation of TrkB.T under excitotoxic conditions was correlated with the induction of a BDNF-induced inhibition of RhoA response in cultured hippocampal and striatal neurons. This response was insensitive to inhibition of TrkB.FL but was blocked by anisomycin, which also prevented the upregulation of the truncated receptor, suggesting that the latter isoform is responsible for effect observed on RhoA signaling. RhoA mediates the Ca2+-dependent activation of p38 MAPK, which is coupled to neuronal death under excitotoxic conditions (Semenova et al., 2007). The GTPase is also coupled to the activation of the stress kinase JNK (Marinissen et al., 2004) and there is evidence that the JNK/c-Jun signaling pathway is important for neuronal death induced by excitotoxicity (Yang et al., 1997). Therefore, the inhibition of RhoA by TrkB.T may account for neuroprotection by BDNF when present during the period of excitotoxic stimulation. This contrasts with the deleterious effects of TrkB.T receptors that have been proposed, resulting from the inhibition of the activity of TrkB.FL receptors through a dominant-negative mechanism (Haapasalo et al., 2001; De Wit et al., 2006).
In contrast with the role of TrkB.T receptors in neuroprotection during excitotoxic stimulation, the TrkB.FL receptors were without effect. Accordingly, neuroprotection by BDNF added during the period of excitotoxic stimulation was insensitive to the TrkB.FL receptor inhibitor SHN722, and no additional protection was observed in hippocampal neurons transduced with the tyrosine kinase receptors. Furthermore, the downregulation of TrkB.FL receptors observed following excitotoxic stimulation and in brain ischemia is expected to decrease their neurotrophic and neuroprotective signaling activity, and this may account, at least in part, for the lack of neuroprotective activity of BDNF when added after excitotoxic stimulation of cultured hippocampal neurons. In vivo, the loss of trophic support provided by TrkB.FL receptors may also contribute to neurodegeneration after the ischemic insult.
The effect of excitotoxicity on the signaling activity of TrkB.FL was investigated by measuring the phosphorylation of the receptor, PLCγ phosphorylation on tyrosine, and activation of the ERK and Akt protein kinases. Analysis of the ratio pTrkB/total TrkB in hippocampal neurons stimulated with BDNF showed a large reduction following excitotoxic stimulation, and similar results were obtained for the activation of PLCγ and ERK (Fig. 4). The reduction in TrkB.FL signaling activity was also observed in experiments conducted in the presence of calpain inhibitors, which prevent the degradation of TrkB.FL receptors, further indicating that under excitotoxic conditions the mechanisms that couple the receptor to the signaling pathways are affected to some extent. This may also partially account for the decrease in the activation of PLCγ and ERK. Since excitotoxic stimulation upregulates TrkB.T, these receptors may have a dominant-negative effect by preventing the dimerization of TrkB.FL thereby modulating the downstream signaling activity (Fig. 9). This hypothesis is supported by (1) the decrease in BDNF-induced TrkB.FL phosphorylation in hippocampal neurons transduced with lentivirus expressing TrkB.T1 and (2) the results showing no difference between the ratio pTrkB/total TrkB in hippocampal neurons stimulated with BDNF under control conditions and after excitotoxic stimulation in the presence of a translation inhibitor (Fig. 5). Similar results were obtained for the activation of the ERK pathway, strongly suggesting that the upregulation of TrkB.T under excitotoxic conditions plays a major role in the inhibition of the TrkB.FL signaling activity. Interestingly, the results showing that inhibition of protein synthesis almost fully restored BDNF-induced ERK activation under excitotoxic conditions suggest that the downregulation of TrkB.FL receptors is a minor player in the deregulation of the receptor signaling activity. The dominant-negative effect of TrkB.T receptors is also supported by the correlation observed between the experimental results and the effects predicted by mathematical modeling of the changes in TrkB.FL and TrkB.T receptors following excitotoxic stimulation of hippocampal neurons. This negative feedback of TrkB.T over TrkB.FL signaling activity in pathological situations has also been described in other studies (Saarelainen et al., 2000; Luikart et al., 2003; Dorsey et al., 2006). However, additional factors may also contribute to the downregulation of BDNF-induced ERK activity since activation of the kinase requires multiple intermediate components (Huang and Reichardt, 2003; Manadas et al., 2007) that may be lost and/or redistributed within the cell upon excitotoxic stimulation.
Figure 9.

Role of TrkB isoforms in cell survival/cell death upon excitotoxic stimulation. Proposed model for effects of TrkB isoforms under excitotoxic conditions, in the absence (B) and in the presence (A,C) of BDNF. A, In normal physiological conditions the signaling activity of TrkB.FL receptors is induced by BDNF contributing to cell survival (trophic support). B, Under excitotoxic conditions TrkB.FL is degraded by a calpain-dependent mechanism, resulting in a loss of the trophic support. At the same time there is an upregulation of TrkB.T which (1) has a dominant-negative effect over the TrkB.FL receptors and (ii) sequesters RhoGDI 1, allowing the activation of RhoA signaling that ultimately may activate p38MAPK and stress signaling pathways, contributing to cell death. C, Under excitotoxic stimulation in the presence of BDNF neuroprotection may result from the activity of the remaining functional TrkB.FL receptors, in addition to the effect of TrkB.T signaling. Binding of BDNF to the latter receptors releases RhoGDI 1 from an intracellular binding domain, allowing the stabilization of GDP-RhoA. This inactivates the RhoA signaling pathway promoting cell survival.
In the absence of BDNF, TrkB.T may also contribute to excitotoxic cell death due to their effect on the sequestration of Rho GDI 1 which interacts with the C-terminal region of the receptor. This sequestration allows the activation of GTP-RhoA, stimulating the stress signaling pathways (Fig. 9B) as shown for pancreatic cancer cells (Li et al., 2009).
Together, the results indicate that calpain mediates TrkB.FL cleavage under excitotoxic conditions, and this is accompanied by an upregulation of the truncated TrkB isoform. Although TrkB.T may have a dominant-negative effect on the signaling activity of the full-length receptors, this may be compensated at an early stage by the neuroprotective signaling activity of the truncated receptors (Fig. 9C). Thus, activation of TrkB.T receptors may constitute an endogenous neuroprotective strategy under conditions characterized by excitotoxic cell death, including brain ischemia, cerebral trauma, epileptic seizures, and in chronic neurodegenerative disorders.
Footnotes
J.R.G, J.T.C, A.R.I., and C.V.M. were supported by FCT, Portugal. This work was supported by FCT and FEDER, Portugal (PTDC/SAU-NEU/65846/2006). We thank Elisabete Carvalho for the assistance in the preparation of cultured hippocampal neurons and Andrea Lobo for the pull-down of active RhoA optimization.
References
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Grãos MM, Carvalho RF, Carvalho AP, Duarte CB. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res. 1999;58:167–190. [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem. 2010;113:649–660. doi: 10.1111/j.1471-4159.2010.06622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit J, Eggers R, Evers R, Castrén E, Verhaagen J. Long-term adeno-associated viral vector-mediated expression of truncated TrkB in the adult rat facial nucleus results in motor neuron degeneration. J Neurosci. 2006;26:1516–1530. doi: 10.1523/JNEUROSCI.4543-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey SG, Renn CL, Carim-Todd L, Barrick CA, Bambrick L, Krueger BK, Ward CW, Tessarollo L. In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron. 2006;51:21–28. doi: 10.1016/j.neuron.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Dos-Anjos S, Martínez-Villayandre B, Montori S, Regueiro-Purriños MM, Gonzalo-Orden JM, Fernández-López A. Transient global ischemia in rat brain promotes different NMDA receptor regulation depending on the brain structure studied. Neurochem Int. 2009;54:180–185. doi: 10.1016/j.neuint.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Drukarch B, van Muiswinkel FL. Neuroprotection for Parkinson's disease: a new approach for a new millennium. Expert Opin Investig Drugs. 2001;10:1855–1868. doi: 10.1517/13543784.10.10.1855. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Martí E, Ambrosio S, Arenas E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001;101:229–238. doi: 10.1007/s004010000268. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntington controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gomes JR, Lobo AC, Melo CV, Inácio AR, Takano J, Iwata N, Saido TC, de Almeida LP, Wieloch T, Duarte CB. Cleavage of the vesicular GABA transporter under excitotoxic conditions is followed by accumulation of the truncated transporter in nonsynaptic sites. J Neurosci. 2011;31:4622–4635. doi: 10.1523/JNEUROSCI.3541-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutan E, Martí E, Ferrer I. BDNF, and full length and truncated TrkB expression in the hippocampus of the rat following kainic acid excitotoxic damage. Evidence of complex time-dependent and cell-specific responses. Brain Res Mol Brain Res. 1998;59:154–164. doi: 10.1016/s0169-328x(98)00156-9. [DOI] [PubMed] [Google Scholar]
- Grumelli C, Berghuis P, Pozzi D, Caleo M, Antonucci F, Bonanno G, Carmignoto G, Dobszay MB, Harkany T, Matteoli M, Verderio C. Calpain activity contributes to the control of SNAP-25 levels in neurons. Mol Cell Neurosci. 2008;39:314–323. doi: 10.1016/j.mcn.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Haacke A, Hartl FU, Breuer P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J Biol Chem. 2007;282:18851–18856. doi: 10.1074/jbc.M611914200. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Koponen E, Hoppe E, Wong G, Castrén E. Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem Biophys Res Commun. 2001;280:1352–1358. doi: 10.1006/bbrc.2001.4296. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Brigadski T, Erdmann KS, Holtmann B, Sendtner M, Narz F, Lessmann V. Truncated TrkB receptor-induced outgrowth of dendritic filopodia involves the p75 neurotrophin receptor. J Cell Sci. 2004;117:5803–5814. doi: 10.1242/jcs.01511. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chang Z, Chiao LJ, Kang Y, Xia Q, Zhu C, Fleming JB, Evans DB, Chiao PJ. TrkBT1 induces liver metastasis of pancreatic cancer cells by sequestering Rho GDP dissociation inhibitor and promoting RhoA activation. Cancer Res. 2009;69:7851–7859. doi: 10.1158/0008-5472.CAN-08-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Luberg K, Wong J, Weickert CS, Timmusk T. Human TrkB gene: novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J Neurochem. 2010;113:952–964. doi: 10.1111/j.1471-4159.2010.06662.x. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Shipman T, Parada LF. In vivo role of truncated trkb receptors during sensory ganglion neurogenesis. Neuroscience. 2003;117:847–858. doi: 10.1016/s0306-4522(02)00719-4. [DOI] [PubMed] [Google Scholar]
- Manadas BJ, Melo CV, Gomes JR, Duarte CB. Neurotrophin signalling and cell survival. In: Rego AC, Cunha RA, Oliveira CR, Malva JO, editors. Interaction between neurons and glia in aging and disease. New York: Springer; 2007. pp. 137–172. [Google Scholar]
- Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell. 2004;14:29–41. doi: 10.1016/s1097-2765(04)00153-4. [DOI] [PubMed] [Google Scholar]
- Martin KJ, Shpiro N, Traynor R, Elliott M, Arthur JS. Comparison of the specificity of Trk inhibitors in recombinant and neuronal assays. Neuropharmacology. 2011;61:148–155. doi: 10.1016/j.neuropharm.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Meary F, Metral S, Ferreira C, Eladari D, Colin Y, Lecomte MC, Nicolas G. A mutant alphaII-spectrin designed to resist calpain and caspase cleavage questions the functional importance of this process in vivo. J Biol Chem. 2007;282:14226–14237. doi: 10.1074/jbc.M700028200. [DOI] [PubMed] [Google Scholar]
- Middlemas DS, Meisenhelder J, Hunter T. Identification of TrkB autophosphorylation sites and evidence that phospholipase C-gamma 1 is a substrate of the TrkB receptor. J Biol Chem. 1994;269:5458–5466. [PubMed] [Google Scholar]
- Nygren J, Wieloch T. Enriched environment enhances recovery of motor function after focal ischemia in mice, and downregulates the transcription factor NGFI-A. J Cereb Blood Flow Metab. 2005;25:1625–1633. doi: 10.1038/sj.jcbfm.9600157. [DOI] [PubMed] [Google Scholar]
- Ohira K, Shimizu K, Yamashita A, Hayashi M. Differential expression of the truncated TrkB receptor, T1, in the primary motor and prefrontal cortices of the adult macaque monkey. Neurosci Lett. 2005a;385:105–109. doi: 10.1016/j.neulet.2005.05.033. [DOI] [PubMed] [Google Scholar]
- Ohira K, Kumanogoh H, Sahara Y, Homma KJ, Hirai H, Nakamura S, Hayashi M. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J Neurosci. 2005b;25:1343–1353. doi: 10.1523/JNEUROSCI.4436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira K, Homma KJ, Hirai H, Nakamura S, Hayashi M. TrkB-T1 regulates the RhoA signaling and actin cytoskeleton in glioma cells. Biochem Biophys Res Commun. 2006;342:867–874. doi: 10.1016/j.bbrc.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature. 2003;426:74–78. doi: 10.1038/nature01983. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- Saarelainen T, Lukkarinen JA, Koponen S, Gröhn OH, Jolkkonen J, Koponen E, Haapasalo A, Alhonen L, Wong G, Koistinaho J, Kauppinen RA, Castrén E. Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons show increased susceptibility to cortical injury after focal cerebral ischemia. Mol Cell Neurosci. 2000;16:87–96. doi: 10.1006/mcne.2000.0863. [DOI] [PubMed] [Google Scholar]
- Santos AR, Duarte CB. Validation of internal control genes for expression studies: effects of the neurotrophin BDNF on hippocampal neurons. J Neurosci Res. 2008;86:3684–3692. doi: 10.1002/jnr.21796. [DOI] [PubMed] [Google Scholar]
- Semenova MM, Mäki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Silhol M, Arancibia S, Maurice T, Tapia-Arancibia L. Spatial memory training modifies the expression of brain-derived neurotrophic factor tyrosine kinase receptors in young and aged rats. Neuroscience. 2007;146:962–973. doi: 10.1016/j.neuroscience.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Stoilov P, Castren E, Stamm S. Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem Biophys Res Commun. 2002;290:1054–1065. doi: 10.1006/bbrc.2001.6301. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, Maki M, Saido TC. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- Tolosa L, Mir M, Olmos G, Lladó J. Vascular endothelial growth factor protects motoneurons from serum deprivation-induced cell death through phosphatidylinositol 3-kinase-mediated p38 mitogen-activated protein kinase inhibition. Neuroscience. 2009;158:1348–1355. doi: 10.1016/j.neuroscience.2008.10.060. [DOI] [PubMed] [Google Scholar]
- Tomioka M, Shirotani K, Iwata N, Lee HJ, Yang F, Cole GM, Seyama Y, Saido TC. In vivo role of caspases in excitotoxic neuronal death: generation and analysis of transgenic mice expressing baculoviral caspase inhibitor, p35, in postnatal neurons. Brain Res Mol Brain Res. 2002;108:18–32. doi: 10.1016/s0169-328x(02)00486-2. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85–95. doi: 10.1093/ilar.44.2.85. [DOI] [PubMed] [Google Scholar]
- Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–295. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincón M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]