

Abstract
Ischemic stroke causes transient increase of neural stem and progenitor cell (NSPC) proliferation in the subventricular zone (SVZ), and migration of newly formed neuroblasts toward the damaged area where they mature to striatal neurons. The molecular mechanisms regulating this plastic response, probably involved in structural reorganization and functional recovery, are poorly understood. The adaptor protein LNK suppresses hematopoietic stem cell self-renewal, but its presence and role in the brain are poorly understood. Here we demonstrate that LNK is expressed in NSPCs in the adult mouse and human SVZ. Lnk−/− mice exhibited increased NSPC proliferation after stroke, but not in intact brain or following status epilepticus. Deletion of Lnk caused increased NSPC proliferation while overexpression decreased mitotic activity of these cells in vitro. We found that Lnk expression after stroke increased in SVZ through the transcription factors STAT1/3. LNK attenuated insulin-like growth factor 1 signaling by inhibition of AKT phosphorylation, resulting in reduced NSPC proliferation. Our findings identify LNK as a stroke-specific, endogenous negative regulator of NSPC proliferation, and suggest that LNK signaling is a novel mechanism influencing plastic responses in postischemic brain.
Introduction
Throughout adult life, neural stem and progenitor cells (NSPCs) in the subventricular zone (SVZ), lining the lateral ventricle, and the subgranular zone (SGZ) of the dentate gyrus proliferate and generate neurons, which integrate into olfactory bulb and granule cell layer, respectively (Zhao et al., 2008). Adult neurogenesis is modulated by pathological conditions, which has raised the possibility that it could contribute to functional recovery. Status epilepticus (SE) leads to increased NSPC proliferation and neurogenesis in SVZ and SGZ (Bengzon et al., 1997; Parent et al., 1997, 2002). Stroke, induced by middle cerebral artery occlusion (MCAO), gives rise to increased proliferation of NSPCs in SVZ and generation of neuroblasts, which migrate toward the damaged striatum where they differentiate to mature striatal neurons (Arvidsson et al., 2002). Also in humans, stroke induces cell proliferation in SVZ and neurogenesis (Jin et al., 2006; Martí-Fàbrega s et al., 2010). Endogenous neurogenesis may be one of the mechanisms underlying the spontaneous recovery after stroke, but its contribution is probably minor. The increased NSPC proliferation in SVZ only lasts up to 2 weeks following the insult, even though the production of new neuroblasts and mature neurons continues for several months (Thored et al., 2006). Little is known about the endogenous mechanisms and molecules negatively regulating NSPC proliferation in the adult brain, which act to suppress the neurogenic response after stroke.
The adaptor protein LNK is an intrinsic negative regulator of hematopoietic stem cell (HSC) proliferation and expansion. Transgenic mice lacking Lnk exhibit increased B-cell lymphopoiesis and number of HSCs (Takaki et al., 2000; Buza-Vidas et al., 2006). LNK also regulates endothelial progenitor proliferation and inhibits neovascularization (Kwon et al., 2009; Kamei et al., 2010). Mutated Lnk was recently detected in leukemic cells in patients with myeloproliferative neoplasms, and loss of Lnk was found to exacerbate myeloproliferative disease (Bersenev et al., 2010; Oh et al., 2010). LNK negatively regulates several cytokine pathways, e.g., C-KIT (Velazquez et al., 2002) and erythropoietin (EPO) (Tong et al., 2005), which are important for hematopoiesis. C-KIT is also a chemoattractant and survival factor for NSPCs in the developing cortex and injured brain, while EPO promotes proliferation and neuronal differentiation in vitro and in SVZ after stroke (Sun et al., 2004; Tsai et al., 2006). Whether LNK is present in the brain and involved in the regulation of NSPCs under normal and pathological conditions has been unknown.
Normal and insult-induced neurogenesis occurs in a multistep process starting with proliferation of NSPCs and continuing with migration and maturation of newly formed neurons and ultimately their integration in established networks. We have previously identified TNF-α signaling through TNF-R1 as a negative regulator of insult-induced NSPC proliferation (Iosif et al., 2006, 2008), establishing TNF-α as an important factor for the neurogenic response to injury. Given the potent action of Lnk in inhibiting cytokine-induced proliferation in the hematopoietic system, and that several known LNK targets including EPO and C-KIT have been shown to regulate NSPCs, we hypothesized that LNK might be a negative regulator also of NSPCs.
Our findings here for the first time identify LNK as a novel endogenous negative regulator of NSPC proliferation, which could have an important role for brain plasticity during recovery after stroke.
Materials and Methods
Animals.
Experimental procedures were approved by the Malmö-Lund Ethical Committee. Generation of Lnk−/− mice has been described (Takaki et al., 2000). Sox2-GFP mice were provided by Dr. L. Pevny, Dcx-GFP mice from Drs L. Aigner and S. Coulliard-Despres, and Iba1-GFP mice from Dr. Kohsaka. Mice were backcrossed into C57BL6 (Charles River), which were used as wild-type (WT) controls. A total of 10 Sox2-GFP, 12 Iba1-GFP, 10 Dcx-GFP mice of either sex, and 55 WT C57BL6 and 35 Lnk−/− male mice were used for this study.
Human tissue.
Ganglionic eminence (GE) was subdissected from brains of dead aborted 6- to 9-week-old human fetuses. Tissue was obtained from Lund University hospital after informed consent in accordance with guidelines set by the Lund/Malmö Ethical Committee. Anonymized SVZ brain samples were obtained, according to national regulations, from autopsies of adult humans in which neuropathological investigations was performed as part of clinical diagnostic routine.
Dissection and cell culture.
Neurospheres were generated from SVZ of intact 8- to 12-week-old mice as described previously (Iosif et al., 2008). For neurosphere formation, cells were grown at clonal density (10 cells/μl). Differentiation of neurospheres was performed by plating on poly-d-lysine (PDL)/laminin coated coverslips and culturing in medium without growth factors but with N2 and 1% fetal bovine serum (FBS). Human fetal GE was mechanically dissociated and cultured in DMEM/F12 with N2, 2 μg/ml heparin, 10 ng/ml leukemia inhibitory factor (Sigma), and 20 ng/ml epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF).
Retroviral infection, transient transfection, and in vivo electroporation.
Expanded neurospheres from WT mice were infected with retroviruses expressing either MSCV-IRES-GFP or Lnk (MIG-Lnk) (Tong and Lodish, 2004) (provided by Drs. J. Lodish and W. Tong,) using a multiplicity of infection of 2. Cells were allowed to grow for an additional 5 days in vitro before cell sorting. Constitutively active (CA)- or dominant-negative (DN)-STAT1 plasmids (provided by Drs. T. Ouchi and J. Darnell, through Addgene, respectively) were transiently expressed in neurospheres using lipofectamine (Invitrogen) and 48 h later expression of Lnk was assessed by Q-PCR. For in vivo transfection of SVZ cells, 6 μg of CA- or DN-AKT (provided by Dr. M. Hung, through Addgene) plasmids were injected into the lateral ventricle of 8- to 12-week-old WT mice. Immediately after injection, mice were electroporated using 5 mm disk electrodes, which were placed laterally on the skin covering the skull parallel to the injection site, after applying conductive gel (Parker Laboratories). A square-wave electroporator CUY21 EDIT (NepaGene) was used to deliver three 50 ms pulses of 200 V with an interval of 950 ms (Barnabé-Heider et al., 2008). After 72 h, mice were injected with bromodeoxyuridine (BrdU) four times with 2 h intervals, and 2 h after last injection mice were perfused.
Fluorescence-activated cell sorting.
Neurospheres were passaged and SVZ tissue dissociated as described above. Cells were washed and resuspended in PBS containing 30 mm glucose and 5% FBS. Suspensions were passed through a 70 μm filter and cells were sorted using a fluorescence-activated cell sorting (FACS) Aria (BD Biosciences). The sorting gate was set around the main GFP+ population, at least 1 log higher than GFP− controls. For BrdU incorporation and cell cycle analysis WT and Lnk−/− neurospheres were pulsed with BrdU and cells harvested at 3, 6, and 10 h and stained for BrdU and labeled with propidium iodide. Data were acquired on an LSR II flow cytometer, with Diva software version 6.0 (Becton Dickinson). Data analysis was done on FlowJo software version 9.4.9 (TreeStar).
Immunocytochemistry in vitro.
Neurospheres or cells attached to slides were fixed with 4% paraformaldehyde (PFA), preincubated in potassium PBS (KPBS) with 0.025% Triton X-100 and 5% serum for 1 h, and incubated with primary antibodies overnight. Primary antibodies included rabbit anti-p-H3 (1:400; Millipore), rabbit anti-SOX2 (1:500; Millipore), and goat anti-LNK (1:100; Santa Cruz Biotechnology). After washing, slides were incubated with goat anti-rabbit Alexa Fluor 488 or donkey anti rabbit Cy3 for p-H3 staining. For LNK/SOX2 double-staining, biotinylated horse anti-goat together with donkey anti-rabbit Cy3 secondary antibodies followed by streptavidin-Alexa Fluor 488 was used.
Proliferation and survival assay.
Neurospheres or sorted cells were plated on PDL/laminin-coated coverslips in proliferative medium. For assessing mitosis, neurospheres or cells were stained for p-H3. The percentage of p-H3+ cells of the total number of Hoechst+ cells was quantified. Apoptosis was quantified with TUNEL staining (Roche) with Hoechst counterstaining. The percentage of TUNEL+ cells with apoptotic Hoechst morphology of the total Hoechst+ cells was estimated.
Induction of stroke.
The MCAO filament model was used (Iosif et al., 2008). Briefly, the left external carotid was ligated and temporary sutures were placed around the common and internal carotid arteries. An 8–0 monofilament (Alcon) coated with silicone was advanced through the internal carotid artery until it blocked the blood flow in the MCA. For reperfusion, mice were re-anesthetized after 40 min of occlusion and the filament was removed. We observed no differences between Lnk−/− and WT mice in initial neurological deficits, or in body weight and motor behavior at 1 week after MCAO (Iosif et al., 2008).
Induction of SE.
Mice were anesthetized and implanted with a stimulating/recording electrode (Plastics One) unilaterally into ventral hippocampal CA1–CA3 region. Ten days later, mice were subjected to electrically induced SE (Iosif et al., 2006). Animals received 1 h of suprathreshold stimulation consisting of 10 s trains of 1 ms biphasic square wave pulses at a frequency of 50 Hz. Stimulation was interrupted for 1 min every 10 min to allow for electroencephalographic (EEG) recording and measurement of afterdischarges (MacLab). After ending the stimulation, all mice exhibited self-sustained, continuous ictal EEG activity, and associated motor behavioral convulsions (Iosif et al., 2006). Epileptic activity was arrested with pentobarbital at 2 h after stimulation offset.
BrdU administration.
BrdU (50 mg/kg, i.p.), dissolved in PBS, was given four times with a 2 h interval to intact mice and at 7 d after SE, and animals were killed 2 h thereafter. Mice subjected to stroke received BrdU once daily for 7 d after the insult and were killed the following day. For BrdU label retention studies, mice received two daily BrdU injections during day 9 and 10 after stroke and were killed 8 weeks later.
Immunohistochemistry in vivo.
Animals were transcardially perfused with ice-cold 4% PFA. Free-floating sections (30 μm) were preincubated in 0.25% Triton X-100 in KPBS containing 5% donkey or goat serum for 1 h. Sections were incubated with rat anti-BrdU (1:100; Sigma), rabbit anti-p-H3, rabbit anti-IBA1 (1:1000; Wako), goat anti-Dcx (1:400; Santa Cruz Technology), rabbit anti-GFAP (1:400; Zymed), mouse anti-Nestin (1:200; Millipore Bioscience Research Reagents), mouse anti-FoxJ1 (1:1000; eBiosciences), rat anti-CD31 (1:200; BD Biosciences), or mouse anti-Sox2 (1:50; R & D Systems) primary antibodies overnight at 4°C. For BrdU staining, DNA was denatured in 1N HCl for 30 min at 65°C or in 2N HCl at room temperature for 2 h. Sections were washed and incubated for 2 h with Cy3-conjugated donkey anti-rat, donkey anti-rabbit (1:200; Jackson ImmunoResearch) or Alexa-Fluor 488-conjugated goat anti-mouse and donkey anti-goat secondary antibodies at room temperature (1:500; Invitrogen). TUNEL staining was performed using the in situ cell death detection kit (Roche) according to manufacturer's suggestions.
Fixed paraffin-embedded 5 μm human brain sections were rehydrated and heat-induced antigen retrieval was performed in 10 mm (pH 6) citrate buffer at 95°C for 10 min. Slides were then blocked with 5% horse and donkey serum for 1 h at room temperature and incubated with goat anti-LNK, and mouse anti-GFAP (1:500; DAKO) antibody overnight. After washing, slides were incubated with biotinylated horse anti-goat and donkey anti-mouse Cy3 secondary antibody for 2 h. Biotinylated antibodies were visualized using streptavidin-Alexa-Fluor 488 and slides were counterstained with Hoechst.
Microscopical analysis.
All assessments were performed by a blinded observer. Immunostainings were examined using fluorescence light microscope. Cells were counted in four evenly spaced sections throughout the SVZ. Results are given as a mean number of cells per section. Assessment of CD31 immunoreactivity was done using cellSens Dimension imaging software (Olympus) in randomly chosen, fixed size areas of SVZ and striatum in WT and Lnk−/− mice subjected to MCAO. In each section, areas of CD31 immunoreactivity were identified using defined representative ranges of threshold for specific signal. Using these defined parameters, the images of each area were analyzed by software, which calculated the total area covered by the specific immunopositive signal.
RNA isolation and RT-PCR.
RNA was isolated from subdissected SVZ primary tissue, expanded neurospheres, or fluorescence-activated sorted cells derived from subdissected SVZ tissue typically pooled from 5 mice. SVZ tissue was subdissected from 1 mm coronal brain sections using microscissors and fine-point forceps. Total RNA was isolated using RNAeasy (Qiagen) with DNase treatment according to manufacturer's instructions. RNA was reverse transcribed using oligoDT primers and superscript-II (Invitrogen). Q-PCR was performed with TaqMan universal master mix and TaqMan Gene expression assays (Applied Biosystems). One hundred nanograms of DNA (tissue) or cDNA from 150 cells (sorted cells) was used for each Q-PCR and all experiments were run in triplicate. cDNA input was normalized to GAPDH. Relative gene expression was calculated using the ΔΔCT method. RT-PCR primer sequences and gene expression assay information are available upon request.
Computational prediction of transcription factor binding sites.
DNA sequences for SH2B3 (LNK) corresponding to conserved regions between the human, mouse, rat, horse, and dog species upstream of the 5′ UTR and including a part of the 5′ UTR were retrieved using the VISTA Browser (http://pipeline.lbl.gov) (Mayor et al., 2000) and the UCSC Genome Browser (http://genome.ucsc.edu/) (Kent et al., 2002). These regions corresponded to the following positions in the human genome: Human SH2B3 March 2006 chr12:110319473–110319590; chr12: 110320884–110320980, chr12:110325532–110325770, and chr12: 110327105–110328342. We applied MotifScanner 3.1.1 (http://med.kuleuven.be/lcb/toucan/help/WebServices/motifscanner.htm) (Aerts et al., 2005) for binding site prediction using matrix files from TRANSFAC and murine and human conserved noncoding sequences as background model.
Chromatin immunoprecipitation PCR.
Neurospheres were fixed with 1% formaldehyde for 10 min at room temperature, and the process stopped by the addition of glycine at a final concentration of 0.0125 m for 5 min. Cells were washed and resuspended in 1 ml lysis buffer with protease inhibitor mixture and 1 mm serine protease inhibitor, phenylmethylsulfonyl fluoride (PMSF; Roche). Sonication was performed with a Branson 450 probe, yielding DNA fragments with a bulk size of 50–300 bp. The debris-cleared lysate was frozen at −80°C. Magnetic Dynabeads (Invitrogen) were incubated (1:2) with blocking buffer (Lonza) containing 1 mg/ml herring sperm DNA (Promega) and 1 mg/ml bovine serum albumin (Sigma-Aldrich) overnight at 4°C on a rotator. For each reaction, beads in 100 μl of the slurry-blocking buffer mix were separated with a DynaMag magnet (Invitrogen) and incubated for 20 min at room with rabbit anti-STAT1, normal rabbit IgG (both from Cell Signaling Technology), or H2O, diluted in 200 μl Binding & Washing Buffer (Invitrogen), and washed with Binding & Washing Buffer. For immunoprecipitation, 100 μl cell lysate was diluted with 900 μl of radioimmunoprecipitation assay buffer containing PMSF and protease inhibitor, and incubated with the prepared beads for 2 h. Beads were washed three times with Washing buffer. Cross-linking was reversed by resuspending beads in 250 μl digestion buffer containing 10 mm Tris, 10 mm EDTA and 1% SDS, addition of proteinase K to a concentration of 50 μg/ml, and incubation for 2 h at 68°C. Ten microliters of raw lysate were diluted in 240 μl digestion buffer and treated equally to the ChIP samples, served as input control. Finally, the material was phenol-chloroform extracted and ethanol precipitated. DNA was resuspended in 50 μl of nuclease-free water and 2 μl was used as a template for PCR. Primers are available upon request.
Luciferase assay.
The regulatory element of mouse Lnk where we detected STAT1 binding, 2000 bp upstream and 300 bp downstream of the predicted Lnk transcription start site, was cloned as WT (PGL4.10LNK) or with mutated (TCCG to GATC) binding sites (PGL4.10LNK-Mut) into PGL 4.10 luciferase reporter plasmid (Promega). PGL4.10, PGL4.10LNK-Mut, or PGL 4.10LNK, alone or in combination with CA-STAT1 or STAT3, was transiently expressed in 3T3 cells or the STAT1-deficient cell line U3A (provided by Dr. G. Stark) using lipofectamine Luciferase activity was assessed 24 h later using the dual luciferase reporter kit and a Glomax 20/20 luminometer (Promega).
ELISA.
Neurosphere cells were grown adherent in 96-well plates in expansion medium until semiconfluent, when growth factors were withdrawn overnight. Cells were stimulated with 10 ng/ml EGF and 10 ng/ml bFGF, 50 ng/ml IGF1, or 5 U/ml EPO for 10 min. Levels of total and phosphorylated ERK1/2 and AKT were analyzed using FACE in-cell Western phospho ELISAs (Active Motif).
Statistical analysis.
Comparisons were performed using Student's unpaired t test, two-way ANOVA followed by Scheffé's post hoc test. Data are presented as means ± SEM, and differences are considered significant at p < 0.05.
Results
LNK is expressed by NSPCs in adult mouse and human SVZ
LNK belongs to an adaptor protein family whose members share NH2-terminal, homologous PH and SH domains, and a conserved C-terminal tyrosine phosphorylation site (Rudd, 2001). Other members of the family, APS and PSM/SH2, have been detected in rodent brain tissue (Welsh et al., 1998; Iseki et al., 2000; Yousaf et al., 2001), and Lnk has been detected in embryonic rat cortex (Wang et al., 2011), but whether LNK is present in the mouse or human brain has been unknown. Using RT-PCR, we found Lnk mRNA in mouse SVZ tissue and neurospheres derived from SVZ (Fig. 1A). In agreement, neurosphere cells expressing the neural stem cell marker SOX2 were immunoreactive for LNK protein (Fig. 1B). To clarify the cellular localization of LNK, we sorted NSPCs, neuroblasts, and microglia from SVZ and hippocampus of Sox2-GFP, Dcx-GFP, and Iba1-GFP reporter mice, respectively. Using RT-PCR and Q-PCR, we detected Lnk mRNA in Sox2-, Dcx-, and Iba1-GFP-positive and -negative cell populations (Fig. 1A), providing evidence that Lnk is expressed not only in NSPCs but in a wide variety of cells in the mouse SVZ and hippocampus.
Figure 1.
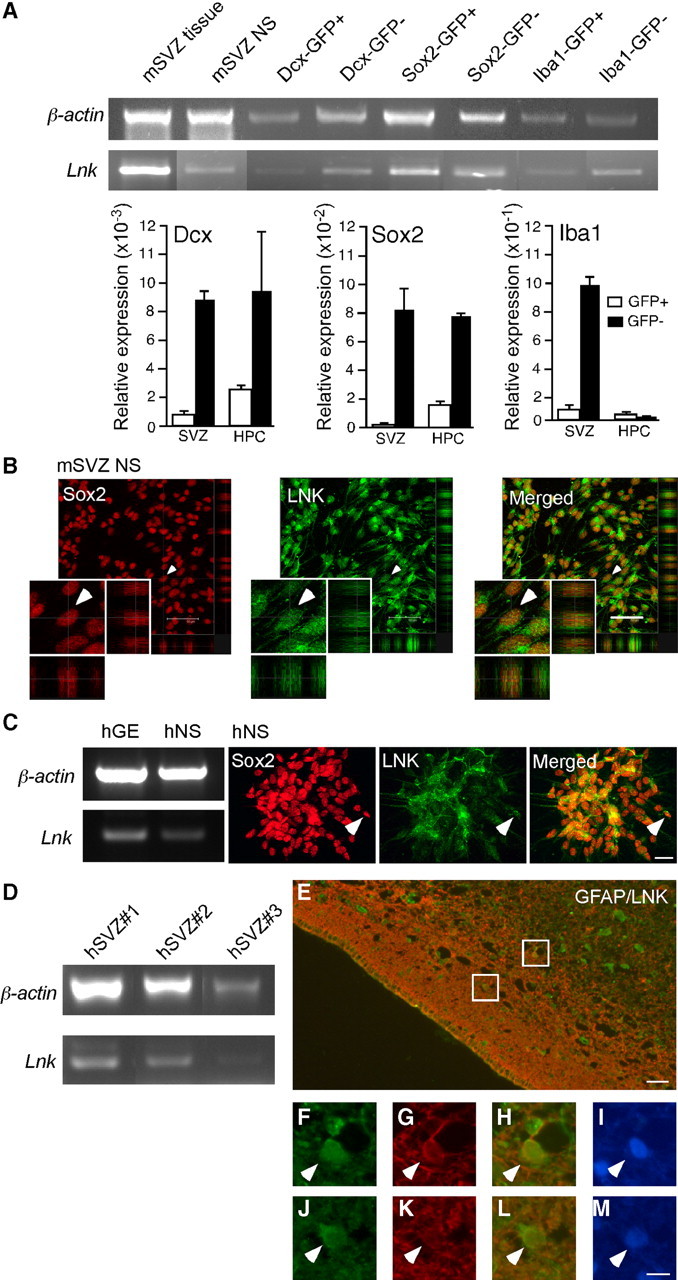
LNK is expressed in SVZ NSPCs in vivo and in vitro. A, RT-PCR and Q-PCR analysis of Lnk expression in tissue and neurospheres derived from mouse SVZ, and in FAC-sorted neuroblasts (Dcx-GFP), NSPCs (Sox2-GFP), and microglia (Iba1-GFP) from SVZ and hippocampus (HPC) of reporter mice. B, Confocal photomicrographs showing LNK immunoreactivity in mouse SVZ neurospheres (NS) costained for SOX2. Double-positive cell depicted by arrowhead on low-magnification images is shown also at higher magnification. C, RT-PCR analysis of LNK expression in GE tissue and NS derived from human fetal GE (hGE). Photomicrographs showing LNK immunoreactivity in human NS (hNS) costained for SOX2. Arrowhead indicates example of double-labeled cell. D, RT-PCR analysis of LNK in three different specimens of adult human SVZ tissue. E, Overview of adult human SVZ stained for LNK (green) and GFAP (red). Boxed areas shown in higher magnification, top box (F–I) and lower box (J–M) depict LNK+ (F, J), GFAP+ (G, K), LNK+/GFAP+ (H, I), and Hoechst+ (I, M) cells. Arrowheads indicate double-labeled cells. Scale bars: B, 50 μm; (in C) C–D, 20 μm; E, 15 μm, and (in M) F–M, 5 μm.
Next we wanted to determine whether LNK is also expressed in the human brain. The human fetal GE gives rise to SVZ in the adult brain and contains NSPCs which generate striatal neurons (Young et al., 2007). LNK mRNA was detected in human fetal GE tissue and neurospheres generated from this structure (Fig. 1C). Moreover, similar to mouse SVZ-derived neurospheres, SOX2 and LNK protein were colocalized in cells of human fetal GE neurospheres (Fig. 1C). We also detected Lnk mRNA and protein in the SVZ of adult human brain using PCR and immunocytochemistry, respectively (Fig. 1D–M). LNK immunoreactivity was observed in GFAP+, putative NSPCs in human SVZ (Fig. 1E–M).Together, our findings clearly demonstrate the presence of LNK in mouse and human brain including the SVZ.
LNK is a negative regulator of NSPC proliferation in vitro
The localization of LNK in the neurogenic SVZ raised the possibility that LNK could influence the properties of NSPCs. We hypothesized that LNK regulates the proliferation of NSPCs similar to what has been described for HSCs (Buza-Vidas et al., 2006; Seita et al., 2007). Lnk−/− mice have previously been reported to exhibit no gross anatomical differences compared with their WT counterparts (Takaki et al., 2000). We observed no obvious changes in the appearance or size of the brain in Lnk−/− mice, nor did we detect any abnormalities in the cytoarchitecture of SVZ, SGZ, hippocampus, and striatum as assessed by Hoechst, NeuN, Sox2, or Dcx staining patterns. Cells from SVZ of Lnk−/− mice were first expanded at clonal density in vitro as neurospheres using epidermal and basic fibroblast growth factor (EGF and bFGF). We did not detect any differences in the number of primary neurospheres between WT and Lnk−/− cells, but we observed a modest 11% increase in the number of secondary neurospheres (Fig. 2A). The size of primary and secondary neurospheres from Lnk−/− and WT mice was also similar (Fig. 2B,C). However, with prolonged time in culture from second passage, cells from Lnk−/− mice exhibited an increased percentage of cells in mitosis as detected by p-H3 staining. The fraction of p-H3+ cells in neurospheres from Lnk−/− mice was 181% higher as compared with that from WT animals (Fig. 2D,E). The increased percentage of p-H3+ cells was not due to improved survival since we did not find any difference in the percentage of apoptotic TUNEL+ cells between WT and Lnk−/− neurospheres (Fig. 2F). To exclude the possibility that the increased number of p-H3+ cells was due to cell cycle arrest in G2/M phase, we pulsed WT and Lnk−/− NSPCs with BrdU and analyzed BrdU incorporation and cell cycle status by FACS at 3, 6, and 10 h. Both WT and Lnk−/− NSPCs increased the percentage of BrdU+ cells over time. Strikingly, in Lnk−/− NSPCs the percentage of BrdU+ cells was higher at all time points increasing to 21.6, 33.4, and 40.6%, respectively (Fig. 2G) as compared with 14.4, 15.2, and 18.1% in WT. The percentage of cells in S-phase increased in WT from 16.6% to 19.2 and 21.2% and in Lnk−/− from 19.9, 30.1, and 35.3% at 3, 6, and 10 h, respectively. The number of WT cells in G0/G1 was similar (74.9, 71.0, and 72.3%) at all time points, but cells in G2/M decreased from 1.5 and 1.4% at 3 and 6 h, respectively, to 0.5% at 10 h. In contrast the number of Lnk−/− cells in G0/G1 decreased from 76.1% to 65.1 and 58.5% at 3, 6, and 10 h, respectively, while the percentage of cells in G2/M remained unchanged (1.5, 1.3, and 1.2%) (Fig. 2H).Together, our data indicate that Lnk−/− cells proliferate faster by shortening the G0/G1 phase. This conclusion is supported by previous reports that decreasing G1 length expands NSPC populations in the developing and adult brain (Lange et al., 2009; Artegiani et al., 2011).
Figure 2.
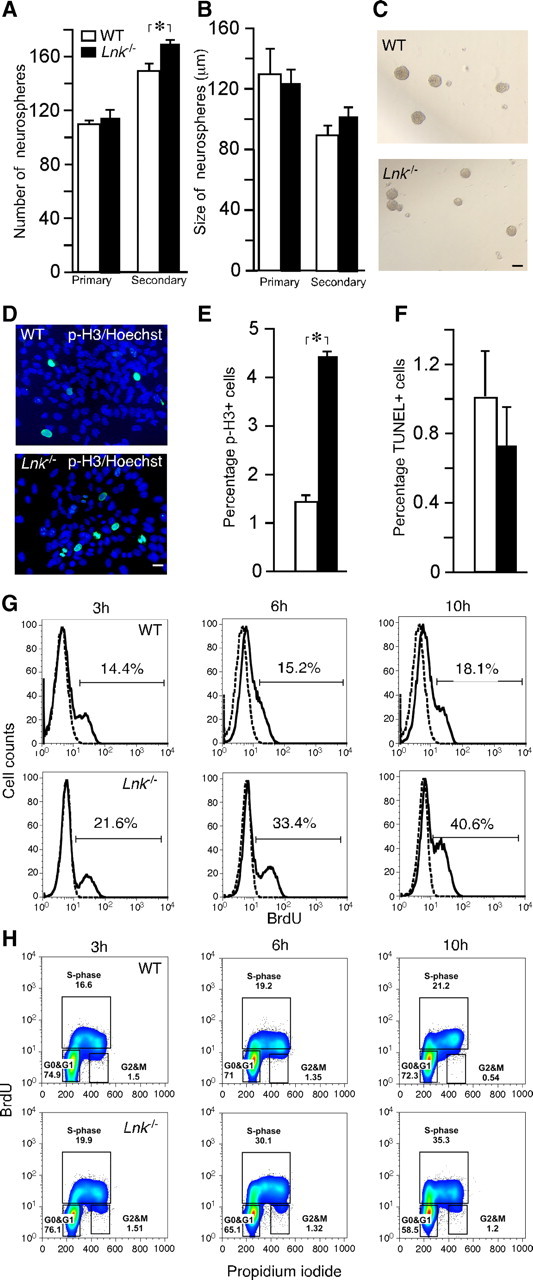
Deletion of Lnk leads to increased in vitro proliferation of SVZ NSPCs by shortening cell cycle duration. Number (A) and size (B) of primary and secondary neurospheres formed from WT and Lnk−/− SVZ. C, Photomicrographs of neurospheres from WT and Lnk−/− mice. Photomicrographs (D) and percentage of p-H3+ (E) and apoptotic TUNEL+ (F) neurosphere cells out of a total number of Hoechst+ cells. (G, H) FACS analysis of BrdU incorporation and cell cycle in WT and Lnk−/− NSPCs. Neurospheres were pulsed with BrdU and analyzed 3, 6, and 10 h thereafter. Histograms showing increased percentage of BrdU+ cells in Lnk−/− (bottom) as compared with WT (top) neurospheres (G). Bivariate (x-axis, propidium iodide; y-axis, BrdU) cell cycle distribution analysis of WT (top) and Lnk−/− neurosphere cells. Density plots showing increased percentage of cells in S- and G2/M-phase in Lnk−/− (bottom) as compared with WT (top) (H). Means ±S EM, n = 5 (A, B) and 4 (E, F). *p < 0.05, Student's unpaired t test. Scale bars: C, 100 μm; D, 10 μm.
To provide further evidence for a suppressant role of LNK in NSPC proliferation, we infected SVZ neurospheres from WT mice with retroviruses to express either GFP alone or GFP together with Lnk. Five days thereafter, GFP+ cells were sorted and Lnk gene expression was assessed by Q-PCR, as well as neurosphere formation, proliferation, and survival were analyzed. In the Lnk-infected cells, we detected a threefold increase in Lnk gene expression (Fig. 3A) and a dramatic, 96 and 61% decrease in the number and size of neurospheres, respectively (Fig. 3B–D). In agreement, the number of cells in mitosis was 63% lower (Fig. 3E,F). Lnk overexpression also influenced survival, the percentage of TUNEL+ apoptotic cells being 107% higher as compared with that in control cells (Fig. 3G). Our findings after deletion and overexpression of Lnk clearly show a role of this adaptor protein as a negative regulator of in vitro proliferation of NPSCs.
Figure 3.

Overexpression of Lnk leads to decreased in vitro proliferation of SVZ NSPCs. A, Relative expression of Lnk in sorted GFP+ cells with or without retrovirally (RV) mediated Lnk-overexpression. Number (B) and size (C) of neurospheres formed from sorted RV-GFP and RV-GFP-Lnk transfected cells. D, Brightfield and fluorescence photomicrographs illustrating size difference between RV-GFP and RV-GFP-Lnk-transfected neurospheres. Percentage and photomicrographs of proliferating p-H3+ cells (E, F), and percentage of apoptotic TUNEL+ cells (G) out of the total number of Hoechst+ cells in sorted GFP+ and GFP+/LNK+ cells. Means ± SEM, n = 5 (B, C) and 4 (E, G). *p < 0.05, Student's unpaired t test. Scale bars: D, 50 μm; F, 10 μm.
Stroke-induced proliferation of NSPCs is negatively regulated by LNK
Next we explored whether LNK also regulates NSPC proliferation in the SVZ in vivo. The proliferative marker BrdU was injected four times 2 h apart in intact Lnk−/− mice and WT controls, and animals were killed 2 h thereafter. To reveal changes in cell cycle duration or cell division of NSPC in SVZ we analyzed BrdU incorporation in replicated DNA and p-H3 expression. No differences in number of p-H3+ or BrdU+ cells were detected between WT and Lnk−/− mice (Fig. 4A,B), indicating that LNK does not regulate proliferation in the healthy, intact SVZ in vivo.
Figure 4.
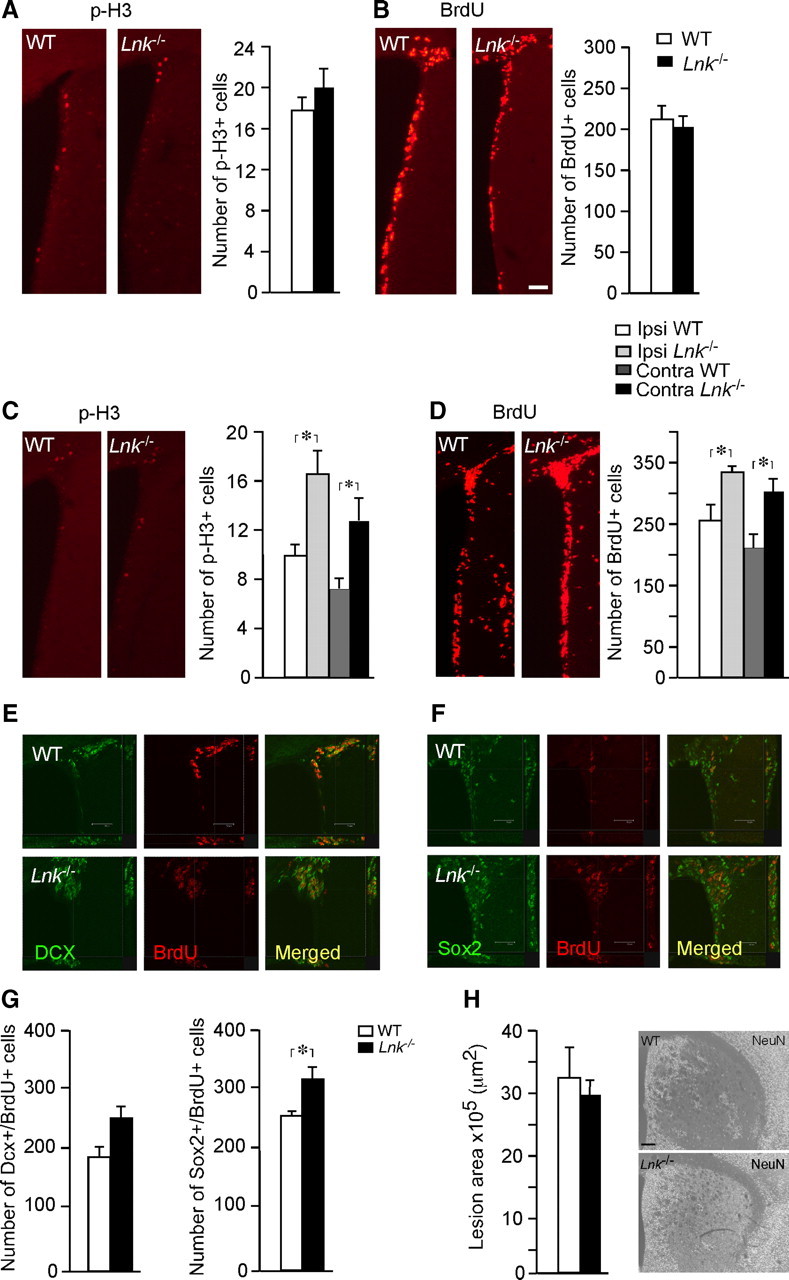
Proliferation of NSPCs in SVZ is enhanced after stroke in Lnk−/− mice. Number and distribution of cells expressing p-H3 (A, C) and BrdU (B, D) in SVZ of intact WT and Lnk−/− mice (A, B) or 7 d after stroke (C, D). Representative confocal images of BrdU+/Dcx+ (E) and BrdU+/Sox2+ (F) cells in the SVZ after stroke. Number of proliferating Dcx+ and Sox2+ cells after stroke (G). Size and distribution of lesioned area after stroke (H). BrdU was injected 4 times 2 h apart in intact animals (A, B) or once daily following MCAO for 7 d (C–F). Ipsilateral denotes side of stroke. Means ± SEM, n = 4 and 5 for WT and Lnk−/− mice, respectively. *p < 0.05, t test (A, B, G, H) or two-way ANOVA with Scheffé's post hoc test (C–D). Scale bar, 100 μm.
We have previously shown that stroke caused by MCAO induces a transient increase of NSPC proliferation in SVZ (Arvidsson et al., 2002; Thored et al., 2006). To determine whether LNK influences NSPC proliferation in SVZ under pathological conditions, we compared NSPC proliferation and early survival of newly formed cells in SVZ of WT and Lnk−/− mice at the peak of the increased proliferation, 7 d after stroke. BrdU was injected once daily following MCAO. In contrast to the lack of change in the intact brain, stroke increased the number of p-H3+ cells by 60 and 63% (Fig. 4C) and BrdU+ cells by 32 and 37% (Fig. 4D) in SVZ of Lnk−/− compared with WT mice, ipsilateral and contralateral to MCAO, respectively.
We then analyzed whether NSPCs and neuroblasts, which were both expressing LNK, contributed to stroke-enhanced cell proliferation in the SVZ of Lnk−/− mice. Sections were double-stained for BrdU and the NSPC marker Sox2 or the neuroblast marker Dcx (Fig. 4E,F). Loss of Lnk increased poststroke proliferation of both NSPCs and neuroblasts in SVZ by 25 and 35%, respectively, although in contrast to the Sox2+ NSPCs, the change in number of Dcx+ cells did not reach statistical significance (Fig. 4G).
To explore the possibility that the higher cell proliferation in SVZ caused by Lnk deletion was not confined to NSPCs but included also other cell types, we analyzed the proliferation of FoxJ1+ ependymal, CD31+ endothelial, and Iba1+ microglial cells using BrdU injections at 1 week after MCAO. However, we did not detect any colabeled cells in the SVZ (data not shown), providing evidence that at this time point after stroke, the vast majority of proliferating cells are NSPCs, and that Lnk deletion enhances the proliferation of this cell type.
Given the importance of the vascular niche in regulating NSPCs, and the previously reported effect of LNK on endothelial cells and vasculature (Kwon et al., 2009; Kamei et al., 2010; Goldman and Chen, 2011), we also assessed the overall staining pattern of CD31 in the SVZ and striatum. We measured the area covered by CD31 immunoreactivity in randomly selected areas of SVZ and striatum of WT and Lnk−/− mice at 1 week after stroke. Arguing against an involvement of an altered vascular environment in the effect on NSPC proliferation, we did not detect any difference in the area of CD31 immunoreactivity in SVZ or striatum between WT and Lnk−/− mice (data not shown).
We found no difference in the area of the ischemic lesion between WT and Lnk−/− mice after stroke (Fig. 4H). This indicated that the effect of Lnk deletion was not mediated indirectly by alteration in the size of the injury, which is known to influence the magnitude of the stroke-induced neurogenic response in the SVZ (Thored et al., 2006).
Our finding that NSPCs from SVZ of intact Lnk−/− mice exhibited no increase in primary and only a modest increase in secondary neurosphere formation indicated that Lnk does not affect NSPC number or self-renewal but negatively regulates proliferative capacity. To further investigate if LNK affects self-renewal or mobilization of NSPCs after stroke, we analyzed the number of BrdU label-retaining cells, which has been used as a marker of stem cells in SVZ (Doetsch et al., 1999; Ferron et al., 2007). Supporting a specific effect of LNK on proliferation, we did not detect any difference in the number of BrdU label-retaining cells in SVZ at 8 weeks after BrdU injections given 1 week after stroke (data not shown). Together, our data suggest that LNK suppresses stroke-induced NSPC proliferation but has minor or no effect on self-renewal.
We then investigated if the enhancement of cell proliferation caused by the loss of LNK function could also occur in other pathological conditions associated with increased proliferation in SVZ. SE gives rise to increased SVZ cell proliferation with low levels of inflammation (Iosif et al., 2008). We induced SE for 2 h by electrical stimulation in the hippocampus. No differences in the percentage of partial and generalized seizures were observed between WT and Lnk−/− mice (Fig. 5A). In agreement with our previous findings (Iosif et al., 2006, 2008), SE increased SVZ cell proliferation in both WT and Lnk−/− mice (55 and 51%, respectively) at 7 d after the insult as assessed by the number of BrdU+ cells. However, we did not detect any differences in SVZ cell proliferation between WT and Lnk−/− mice following SE (Fig. 5B,C).
Figure 5.

Proliferation of NSPCs in SGZ is enhanced after stroke in Lnk−/− mice. Number and distribution of cells expressing p-H3 (A, C) and BrdU (B, D) in SVZ of SE WT and Lnk−/− mice (A, B) or 7 d after stroke (C, D). Means ± SEM, n = 6 and 8 for WT and Lnk−/− mice, respectively for SE (A, B) and n = 7 and 10 for WT and Lnk−/− mice, respectively for stroke (C, D) *p < 0.05, two-way ANOVA with Scheffé's post hoc test. Scale bar, 100 μm.
To determine whether LNK acts as a stroke-specific suppressor of NSPC proliferation also in the hippocampus, we counted the number of p-H3+ and BrdU+ cells in the SGZ of intact and SE- or MCAO-subjected, WT and Lnk−/− mice. We did not detect any differences in SGZ cell proliferation caused by Lnk deletion in intact (data not shown) or SE animals (Fig. 6A,B). Similar to the findings in SVZ, we found higher cell proliferation in SGZ following stroke in Lnk−/− compared with WT mice, as evidenced by the numbers of pH-3+ and BrdU+ cells (Fig. 6C,D, respectively).
Figure 6.

No difference in proliferation between WT and Lnk−/− SVZ NSPCs after SE. WT and Lnk−/− mice display no differences in percentage of partial or generalized seizures during SE (A). SE-induced increase in SVZ proliferation of WT and Lnk−/− mice (B). Number and distribution of cells expressing BrdU (C) and p-H3 (D) in SVZ of WT and Lnk−/− mice 7 d after SE. BrdU was administered 4 times 2 h apart and animals were perfused 2 h after last injection. Ipsilateral denotes side of stimulating electrode. Means ± SEM, n = 7 and 10 for WT and Lnk−/− mice, respectively. p > 0.05, two-way ANOVA with Scheffé's post hoc test..
We have previously shown that microglia activation can influence adult neurogenesis (Ekdahl et al., 2009). Because we found here that also Iba1+ microglia expressed Lnk, we speculated that effects of Lnk deletion on SVZ cell proliferation could potentially be caused by changes in microglia numbers. To test this hypothesis, we quantified the number of Iba+ cells in the SVZ. In agreement with our previous data (Iosif et al., 2008), we found an increased number of Iba1+ microglia in SVZ after stroke but not after SE as compared with intact animals (Fig. 7A,B). However, neither in intact mice nor after SE or stroke did deletion of Lnk cause any changes in the number of Iba1+ microglia the SVZ compared with WT mice (Fig. 7A,B). These findings argue against the possibility that Lnk suppresses NSPC proliferation by influencing microglia activation.
Figure 7.

Deletion of Lnk does not alter number of microglia in SVZ. A, Number of Iba1+ microglia in intact SVZ, and ipsilateral and contralateral to the ischemic lesion or stimulating electrode for stroke and SE, respectively, in WT and Lnk−/− mice. Means ± SEM, n = 8 and 10 (Intact), 9 and 7 (Stroke), and 7 and 9 (SE) for WT and Lnk−/−, respectively. No differences between WT and Lnk−/− mice, p > 0.05 Student's unpaired t test or two-way ANOVA with Scheffé's post hoc test. B, Photomicrograph showing the distribution of IBA+ cells (IBA1 in red, Hoechst in blue) in SVZ of intact WT and Lnk−/− mice. Scale bar, 30 μm. Str, striatum;. LV, lateral ventricle.
Stroke upregulates Lnk expression in NSPCs through STAT1/3 signaling
To determine how stroke regulates Lnk expression, we first measured the level of Lnk mRNA in SVZ tissue from intact mice and at 1 week after stroke or SE using Q-PCR. Consistent with a role of LNK in stroke-induced but not SE-induced SVZ cell proliferation, we found that Lnk expression was upregulated ipsilateral to the damage by 70% after stroke but downregulated by 50% following SE (Fig. 8A). Similar changes were detected in the contralateral SVZ.
Figure 8.
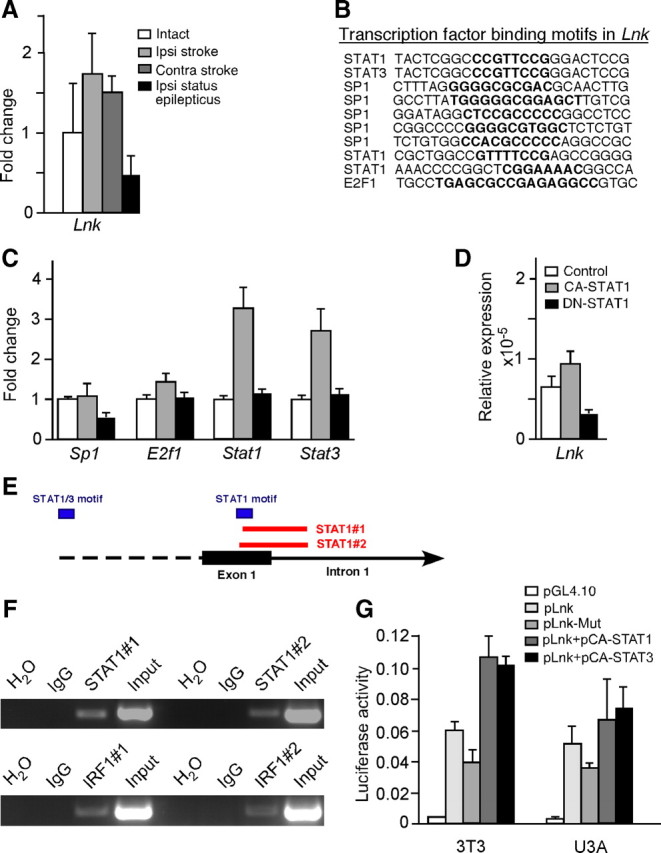
STAT1 regulates expression of Lnk in NSPCs after stroke. A, Relative gene expression of Lnk in SVZ tissue from intact, stroke, and SE mice ipsilateral and contralateral to lesion and stimulating electrode, respectively. B, Occurrence of SP1, E2F1, STAT1, and STAT3 binding motifs (in bold) in mouse Lnk genomic region. C, Relative expression of transcription factor genes in SVZ tissue from intact mice or 7 d after stroke or SE. D, Lnk expression in neurospheres after transfection with CA- and DN-STAT1. E, Schematic representation of the murine Lnk locus with STAT1/3 binding motifs and primers used for ChIP. F, ChIP PCR analysis showing binding of STAT1 to genomic region of the Lnk gene and the positive control gene Irf1. Genomic DNA immunoprecipitated with IgG, as a negative control, or STAT1 antibodies was used for PCR with primers spanning binding motifs of STAT1 in Lnk and Irf1. G, Relative luciferase activity in 3T3 and U3A cell lines transfected with control (pGL4.10), Lnk (pLnk), mutated Lnk (pLnk-Mut), pLnk+pCA-STAT1, and pLnk+pCA-STAT3.
To identify transcription factors that might regulate Lnk expression after stroke we analyzed the promoter regions of Lnk for predicted transcription factor binding motifs. A computational search for motifs revealed the presence of SP1, E2F1, STAT1, and STAT3 transcription factor binding sites in conserved regions of the Lnk gene in five different species (mouse, rat, horse, dog, and human) (Fig. 8B). Using Q-PCR we found that Sp1 was slightly downregulated after SE while E2f1 was slightly upregulated after stroke. The most pronounced changes were observed for Stat1 and 3, which were upregulated 3.1- and 2.7-fold, respectively, after stroke but were virtually unchanged after SE (Fig. 8C), indicating that these transcription factors might regulate Lnk expression in SVZ after stroke. We then transfected SVZ neurospheres with CA-STAT1 or DN-STAT1. Supporting a role of STAT1 in the regulation of Lnk, Q-PCR showed a 51% induction of Lnk expression in CA-STAT1-transfected neurospheres, while DN-STAT1 transfection reduced the expression by 65% (Fig. 8D). To provide further evidence for a regulatory role of STAT family members on Lnk gene expression, we assessed whether STAT1 protein binds to genomic regions of the Lnk gene in SVZ-derived NPSC. Chromatin immunoprecipitation experiments were performed with a STAT1 antibody followed by PCR using primers spanning putative binding motifs in the genomic sequence of Lnk and the known STAT1 target interferon regulatory factor 1 (Irf1). In two independent experiments, using two different primer pairs, we consistently detected binding of STAT1 to one specific motif in the first exon of the Lnk sequence (Fig. 8E,F).
We then cloned the regulatory elements of Lnk where we had detected STAT1 binding into a dual luciferase reporter plasmid. This plasmid was then transiently expressed in murine 3T3 and human STAT1-deficient U3A cell lines. We also cotransfected the luciferase plasmid with CA-STAT1 and CA-STAT3 plasmids and measured luciferase activity 24 h after transfection. These experiments revealed that the regulatory elements of Lnk were active in both mouse and human cells (Fig. 8G). Furthermore, we detected a 1.8- and 1.2-fold induction of luciferase activity in 3T3 and U3A cells, respectively, when the reporter plasmid was cotransfected with CA-STAT1, while CA-STAT3 increased luciferase activity by 1.8- and 1.4-fold compared with reporter plasmid alone (Fig. 8G). In contrast plasmids with mutated STAT1 and STAT3 binding sites decreased the activity by 33 and 29% in 3T3 and U3A cells, respectively, as compared with the WT plasmid. Together, these findings show that Lnk is upregulated in SVZ after stroke and indicate that STAT1 and STAT3 regulate its expression in NSPCs.
LNK suppresses NSPC proliferation by decreasing growth factor signaling through inhibition of AKT phosphorylation
To identify mechanisms through which LNK might suppress stroke-induced NSPC proliferation, we first investigated using Q-PCR the expression of known and potential LNK targets in SVZ tissue from intact mice and at 1 week after stroke or SE. Gene expression of EpoR was marginally decreased whereas levels of Epo were upregulated almost fourfold after stroke but unchanged after SE (Fig. 9A). C-kit and its ligand stem cell factor (Scf) were downregulated after both insults (Fig. 9A). The receptor tyrosine kinases (RTKs) FgfR1, FgfR2, and EgfR were upregulated by 40% after stroke but only slightly changed following SE (Fig. 9B,C). The ligands Egf and transforming growth factor α (Tgf-α) were downregulated after both stroke and SE. Expression of Fgf2 in the intact SVZ was below detection limit and only detected after stroke (Fig. 9B,C). We found a slight upregulation of IGF1 receptor (Igf1R) but a dramatic, 30-fold increase of its ligand (Igf1) after stroke (Fig. 9D). In contrast, SE increased the expression of Igf1 only by 30% and decreased receptor expression by 10%. Based on the specific alterations of gene expression induced by stroke, we hypothesized that LNK may act on the EPO, EGF, FGF, and IGF1, signaling pathways.
Figure 9.
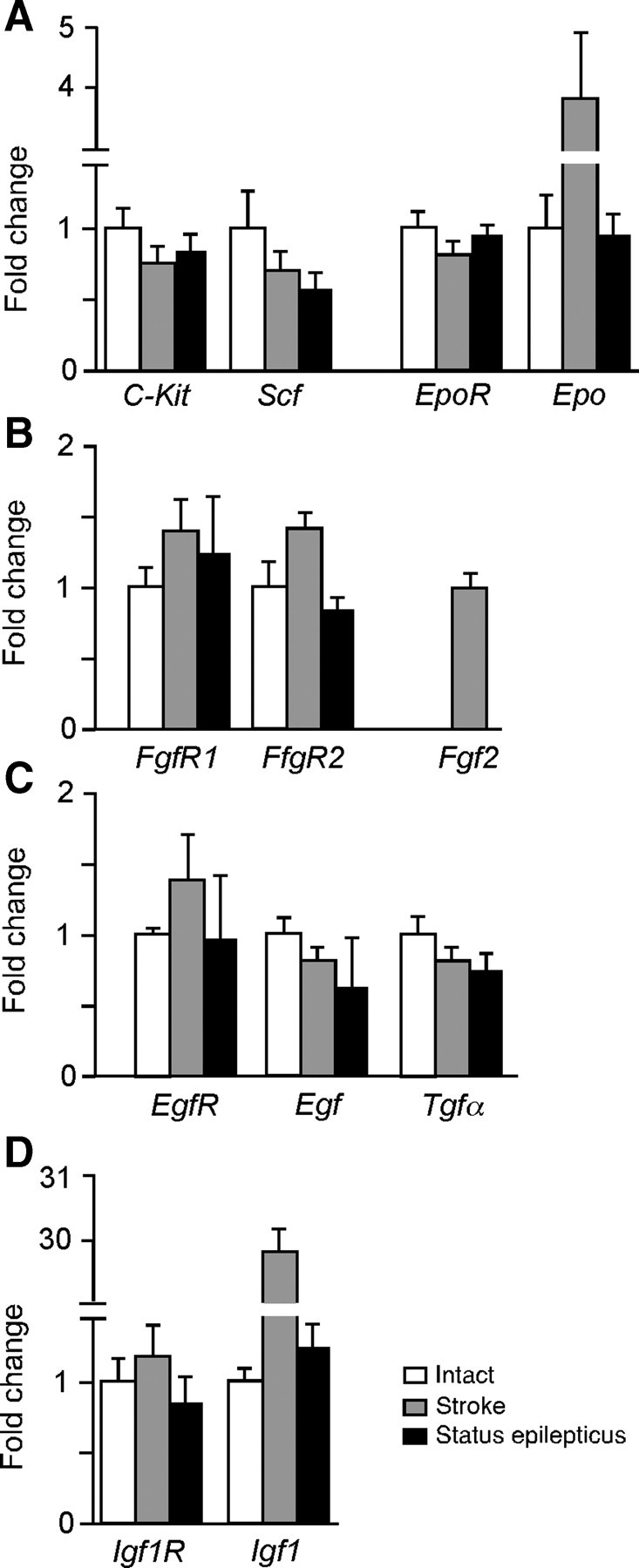
Stroke regulates gene expression of growth factors and their receptors in SVZ. Relative expression of known (A) and potential (B–D) LNK targets. Q-PCR analysis in SVZ tissue from intact mice, and 7 d after stroke or SE. Means ± SEM, n = 3.
LNK is known to inhibit the phosphorylation cascade in RTK signaling by direct interaction with receptors (Takaki et al., 2000; Tong et al., 2005) or with downstream signaling (Bersenev et al., 2008). One common signaling pathway of EGF, FGF, and EPO is the phosphorylation of ERK1/2 through RAS and RAF kinases (Subramaniam and Unsicker, 2006). IGF1 signaling, on the other hand, leads to AKT phosphorylation by PI3K (Ye and D'Ercole, 2006), and AKT is involved in maintenance of neural precursor cells (Oishi et al., 2009). We hypothesized that LNK inhibits phosphorylation of ERK1/2 and AKT downstream of EGF, FGF, EPO, and IGF1 receptors. To test this idea, we first showed expression of these receptors in neurosphere cells (Fig. 10A). Next we analyzed, using ELISA, the degree of phosphorylation of ERK1/2 and AKT in neurosphere cells from SVZ of Lnk−/− and WT mice in response to EGF/FGF, IGF1, and EPO stimulation. We found a modest increase (16%) of ERK1/2 phosphorylation following EGF/FGF stimulation whereas IGF1 gave rise to a more marked increase of AKT phosphorylation (by 32%) in Lnk−/− neurospheres compared with WT controls (Fig. 10B). In contrast, ERK1/2 phosphorylation by EPO stimulation was not altered.
Figure 10.

LNK inhibits phosphorylation of AKT, gene expression in NSPCs, and SVZ cell proliferation. A, RT-PCR analysis of receptors for growth factors and the known LNK target EPOR in mouse SVZ neurospheres. B, Relative level of phosphorylation of ERK1/2 or AKT in WT and Lnk−/− neurospheres after stimulation with EGF + FGF, IGF1, or EPO. Means ± SEM, n = 5, *p < 0.05, Student's unpaired t test. C, Proliferation (expressed as percentage change of p-H3+ and BrdU+ cell numbers) after in vivo transfection of SVZ cells by electroporation of CA- or DN-AKT plasmids. Means ± SEM, n = 3. D, Relative gene expression in RV-GFP or RV-GFP-Lnk transfected neurospheres of factors involved in NSPC maintenance and proliferation. Means ± SEM, n = 4.
To provide proof-of-principle for the role of AKT as a regulator of SVZ proliferation after stroke, we injected CA-AKT or DN-AKT plasmids into the lateral ventricle of WT mice and used in vivo electroporation to transfect the SVZ. Electroporation is a safe and efficient method to target both quiescent ependymal and actively dividing NSPCs without affecting proliferation (Barnabé-Heider et al., 2008, Carlén et al., 2009, Nomura et al., 2010). To control for transfection efficiency, CA-AKT or DN-AKT plasmids were coinjected with red fluorescent protein or GFP plasmids. No differences in efficiency between injections were detected (data not shown). After 3 d, animals were injected with BrdU, and SVZ proliferation was assessed using BrdU and p-H3 immunohistochemistry. Consistent with a regulatory role of AKT on NSPC proliferation, we found 14 and 17% increase in the number of BrdU+ and p-H3+ cells, respectively, in SVZ transfected with CA-AKT, while the number of BrdU+ and p-H3+ cells decreased by 20 and 21%, respectively, in DN-AKT-transfected SVZ (Fig. 10C).
The LNK-induced changes in growth factor signaling could lead to alterations in gene expression and subsequently in cell proliferation. To explore this further, we analyzed the expression of several genes that have been implicated in NSPC proliferation and maintenance in the Lnk-overexpressing neurosphere cells. Interestingly, we found that in parallel to the dramatic reduction of cell proliferation, there was 81 and 68% reduction in Hes5 and Gli1 gene expression, respectively, in Lnk-overexpressing neurospheres (Fig. 10D).
Together, our findings provide evidence that LNK inhibits IGF1 signaling by attenuating phosphorylation of AKT, and possibly also EGF/FGF signaling via ERK1/2, and decreases gene expression of Hes5 and Gli1, resulting in reduction of stroke-induced NSPC proliferation.
Discussion
We show here for the first time that the adaptor protein LNK is expressed and has a functional role in the adult brain. The number of proliferating cells in the neurogenic SVZ was increased after stroke but unchanged in intact brain and following SE in Lnk−/− mice. Thus, LNK suppresses NSPC proliferation specifically after one brain disorder, stroke, but not after another one, SE, despite both pathological conditions giving rise to increased cell proliferation. The previously unknown action of LNK as a suppressant of brain NSPC proliferation is in line with its function in the hematopoietic system, where LNK is a negative regulator of B-cell progenitor (Takaki et al., 2003) and HSC proliferation (Velazquez et al., 2002) and self-renewal (Buza-Vidas et al., 2006; Seita et al., 2007). LNK also inhibits the malignant expansion and transformation of HSC and progenitor cells (Bersenev et al., 2010), and mutations in the Lnk gene are associated with myeloproliferative neoplasms in patients (Oh et al., 2010). Finally, loss of Lnk increases proliferation of endothelial cells and angiogenesis (Kwon et al., 2009; Kamei et al., 2010).
The functional consequences of the LNK-mediated suppression of poststroke NSPC proliferation in SVZ remain to be explored. It should be emphasized, though, that there are several mechanisms which may contribute to recovery in the poststroke brain. These mechanisms include plastic changes in surviving neurons and neurons on the contralateral side, redistribution of brain representation, release of growth factors and anti-inflammatory factors from immune cells, synaptogenesis and changes in synaptic strength, and changes in dendritic arborization and spines, as well as generation of new neurons, glial, and endothelial cells (Moskowitz et al., 2010). Based on its expression in several different cell types, it is conceivable that LNK, in addition to its effect on NSPC proliferation in the SVZ, can influence poststroke functional and structural reorganization through several of these mechanisms. We describe here a very specific effect of LNK in regulating NSPC proliferation after stroke through alteration of IGF1-mediated Akt phosphorylation and gene expression of stem cell regulators. However, this is most likely not the only way that LNK may influence stroke-induced neurogenesis. LNK is also known to affect endothelial cell proliferation and angiogenesis, to inhibit several cytokine signaling pathways in the hematopoietic system, and to exacerbate myeloproliferative disease. Furthermore, LNK is expressed in many cell types other than NSPCs. Therefore, it is conceivable that LNK acts not only on NSPC proliferation but also can influence stroke-induced neurogenesis through other mechanisms. It is possible that LNK acts on a global level by fine-tuning the response to growth factor or cytokine stimulation.
We obtained no evidence for differences between WT and Lnk−/− mice in endothelial cell proliferation or vascularization in SVZ after stroke. However, due to the importance of the vascular niche for NSPCs and the previously reported influence of LNK on endothelial cells, one cannot completely exclude that discrete changes in the vascular niche in Lnk−/− mice might contribute to the NSPC proliferative effect observed here.
The action of LNK as a negative regulator of NSPC proliferation in the SVZ after stroke is most likely exerted in the NSPCs themselves and not indirectly through other cell types. First, we found that the NSPCs express LNK. Second, although our data indicated that also microglia express Lnk, we obtained no in vivo evidence that deletion of Lnk influenced the activation of SVZ microglia. Third, our in vitro data clearly showed that altering the levels of Lnk in NSPCs, in the absence of immune cells and microglia, gave rise to the expected changes of cell proliferation.
We observed that the proliferation of NSPCs in SVZ of intact and SE-subjected animals was not influenced by the deletion of Lnk in the transgenic mice. In contrast, we detected increased proliferation in neurosphere cultures from Lnk−/− mice as well as in the NSPCs in SVZ of animals subjected to stroke. This discrepancy in the effect of LNK deficiency is most probably due to the low levels of growth factors in SVZ of intact WT and SE-subjected animals and the high levels of growth factors in neurosphere culture medium and in the SVZ after stroke. Thus, LNK is known to inhibit RTK signaling by interacting either directly with the growth factor receptors or with downstream kinases, resulting in decreased phosphorylation (Takaki et al., 2000; Tong et al., 2005; Bersenev et al., 2008).
The suppressant effect of LNK on NSPC proliferation was specific for stroke and not observed in intact or SE animals and, accordingly, Lnk was upregulated after stroke but downregulated after SE. This finding is the first evidence that the expression of Lnk is regulated in the adult brain. Our findings indicate that STAT1 and STAT3 induce the elevated Lnk expression in SVZ NSPCs after stroke. We found that Stat1 was upregulated following stroke but not SE in NSPCs, consistent with its previously reported activation in neurons following ischemia (Takagi et al., 2002). Furthermore, STAT3 has been shown to be activated in the peri-infarct areas following ischemia, a process that was inhibited by blocking IL-6 signaling (Yamashita et al., 2005). Stroke induces several cytokines including IL-6 and IFN α and γ (Yamashita et al., 2005; Liesz et al., 2011), which are known to activate STAT family members (Takeda and Akira, 2000). Thus, it is conceivable that the upregulation of LNK seen after stroke is mediated by STAT1/3 activation by cytokines released after the insult.
Stroke but not SE gave rise to increased expression of several growth factor ligands and/or receptors in SVZ which could trigger NSPC proliferation, e.g., EPO, FGF, EGF, and IGF1. For example, Igf1 is upregulated in SVZ both short- and long-term after stroke in rats (Thored et al., 2009) and has been reported to be a mediator of the increased NSPC proliferation after stroke (Yan et al., 2006). The low levels of both Lnk and growth factors in SVZ of intact and SE animals probably explain the lack of change in NSPC proliferation under these conditions in the Lnk−/− mice. In contrast, we detected increased proliferation in the neurosphere cultures from Lnk−/− mice. This in vitro environment is more likely to mimic the SVZ niche after stroke with high levels of growth factor signaling, which could explain why LNK has an effect in vitro but not in vivo under basal conditions. LNK is known to inhibit RTK signaling by interacting either directly with the growth factor receptors or with downstream kinases, resulting in decreased phosphorylation (Takaki et al., 2000; Tong et al., 2005; Bersenev et al., 2008). We found that Lnk deletion in NSPCs gave rise to an increase in the phosphorylation of ERK1/2 in response to EGF receptor and FGF receptor stimulation and a more pronounced increase in the phosphorylation of AKT following IGF1 receptor stimulation. We also showed that changes in AKT activation induced by transfection of CA-AKT or DN-AKT plasmids into SVZ gave rise to increased and decreased NSPC proliferation, respectively. Consistent with our data, LNK inhibits AKT and ERK1/2 signaling in hematopoietic stem/progenitor and endothelial cells, respectively (Tong et al., 2005; Wan et al., 2006; Seita et al., 2007). Thus, LNK can suppress NSPC proliferation following stroke by inhibiting phosphorylation of kinases downstream of growth factor receptors, in particular IGF1R. Hypothetically, the reduction of ERK1/2 and AKT signaling could lead to the decreased gene expression of Hes and Gli1, which are involved in NSPC proliferation (Cayuso et al., 2006; Stecca and Ruiz i Altaba, 2009; Gregory et al., 2010). In support of this hypothesis, ERK and AKT activation positively regulates Gli expression in chicken neural explants and 3T3 cells (Riobó et al., 2006). The most prominent pathway discovered here is IGF1-AKT signaling. However, given the broad spectrum of growth factors and cytokine pathways known to be influenced by LNK, it is possible that LNK also inhibits other pathways in NSPCs following stroke.
Here we have for the first time demonstrated the presence of LNK in the adult brain and identified LNK signaling as a negative regulator of NSPC proliferation. This mechanism has so far been shown to operate only after stroke, i.e., in response to a severe brain injury. Since stroke gives rise to NSPC proliferation in the human brain (Martí-Fàbregas et al., 2010), and we found LNK to be expressed in human NSPCs, it is conceivable that LNK acts to suppress the enhanced NSPC proliferation not only in rodents but also in patients after stroke. Our findings raise the possibility that LNK action is a key player for cellular plasticity and a potential novel therapeutic target in the postischemic brain.
Footnotes
This work was supported by the Swedish Research Council, EU project LSHB-CT-2006-037526 (STEMSTROKE), EU 7th work program through the European Stroke Network (Grant no. 201024), AFA Foundation, and Swedish Government Initiative for Strategic Research Areas (StemTherapy).We thank Drs. W. Tong and H. Lodish for providing the retroviral constructs, Dr. L. Velazquez for providing Lnk antibody, Drs. L. Aigner, S. Couillard-Despres, L. Pevny, and S. Kohsaka for providing transgenic mice, Zhi Ma and Teona Roschupkina for help with cell sorting, Marianne Rissler for virus production, and Camilla Ekenstierna for technical assistance.
The authors declare no conflict of interest.
References
- Aerts S, Van Loo P, Thijs G, Mayer H, de Martin R, Moreau Y, De Moor B. TOUCAN 2: the all-inclusive open source workbench for regulatory sequence analysis. Nucleic Acids Res. 2005;33:W393–W396. doi: 10.1093/nar/gki354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artegiani B, Lindemann D, Calegari F. Overexpression of cdk4 and cyclinD1 triggers greater expansion of neural stem cells in the adult mouse brain. J Exp Med. 2011;208:937–948. doi: 10.1084/jem.20102167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Meletis K, Eriksson M, Bergmann O, Sabelström H, Harvey MA, Mikkers H, Frisen J. Genetic manipulation of adult mouse neurogenic niches by in vivo electroporation. Nat Methods. 2008;5:189–196. doi: 10.1038/nmeth.1174. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmér E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci U S A. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest. 2008;118:2832–2844. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, Chikwava KR, Tong W. Lnk constrains myeloproliferative diseases in mice. J Clin Invest. 2010;120:2058–2069. doi: 10.1172/JCI42032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buza-Vidas N, Antonchuk J, Qian H, Månsson R, Luc S, Zandi S, Anderson K, Takaki S, Nygren JM, Jensen CT, Jacobsen SE. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 2006;20:2018–2023. doi: 10.1101/gad.385606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Göritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabé-Heider F, Yeung MS, Naldini L, Honjo T, Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisén J. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009;12:259–267. doi: 10.1038/nn.2268. [DOI] [PubMed] [Google Scholar]
- Cayuso J, Ulloa F, Cox B, Briscoe J, Martí E. The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development. 2006;133:517–528. doi: 10.1242/dev.02228. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Ferron SR, Andreu-Agullo C, Mira H, Sanchez P, Marques-Torrejon MA, Farinas I. A combined ex/in vivo assay to detect effects of exogenously added factors in neural stem cells. Nat Protoc. 2007;2:849–859. doi: 10.1038/nprot.2007.104. [DOI] [PubMed] [Google Scholar]
- Goldman SA, Chen Z. Perivascular instruction of cell genesis and fate in the adult brain. Nat Neurosci. 2011;14:1382–1389. doi: 10.1038/nn.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory GD, Miccio A, Bersenev A, Wang Y, Hong W, Zhang Z, Poncz M, Tong W, Blobel GA. FOG1 requires NuRD to promote hematopoiesis and maintain lineage fidelity within the megakaryocytic-erythroid compartment. Blood. 2010;115:2156–2166. doi: 10.1182/blood-2009-10-251280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ahlenius H, Ekdahl CT, Darsalia V, Thored P, Jovinge S, Kokaia Z, Lindvall O. Suppression of stroke-induced progenitor proliferation in adult subventricular zone by tumor necrosis factor receptor 1. J Cereb Blood Flow Metab. 2008;28:1574–1587. doi: 10.1038/jcbfm.2008.47. [DOI] [PubMed] [Google Scholar]
- Iseki M, Takaki S, Takatsu K. Molecular cloning of the mouse APS as a member of the Lnk family adaptor proteins. Biochem Biophys Res Commun. 2000;272:45–54. doi: 10.1006/bbrc.2000.2736. [DOI] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Zhu W, Wang Y, Shen J, Mao Y, Banwait S, Greenberg DA. Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A. 2006;103:13198–13202. doi: 10.1073/pnas.0603512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei N, Kwon SM, Alev C, Ishikawa M, Yokoyama A, Nakanishi K, Yamada K, Horii M, Nishimura H, Takaki S, Kawamoto A, Ii M, Akimaru H, Tanaka N, Nishikawa S, Ochi M, Asahara T. Lnk deletion reinforces the function of bone marrow progenitors in promoting neovascularization and astrogliosis following spinal cord injury. Stem Cells. 2010;28:365–375. doi: 10.1002/stem.243. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SM, Suzuki T, Kawamoto A, Ii M, Eguchi M, Akimaru H, Wada M, Matsumoto T, Masuda H, Nakagawa Y, Nishimura H, Kawai K, Takaki S, Asahara T. Pivotal role of lnk adaptor protein in endothelial progenitor cell biology for vascular regeneration. Circ Res. 2009;104:969–977. doi: 10.1161/CIRCRESAHA.108.192856. [DOI] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Martí-Fàbregas J, Romaguera-Ros M, Gómez-Pinedo U, Martínez-Ramírez S, Jiménez-Xarrié E, Marín R, Martí-Vilalta JL, García-Verdugo JM. Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology. 2010;74:357–365. [Google Scholar]
- Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Göritz C, Catchpole T, Henkemeyer M, Frisén J. EphB signaling controls lineage plasticity of adult neural stem cell niche cells. Cell Stem Cell. 2010;7:730–743. doi: 10.1016/j.stem.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD, Jr, Merker JD, Zehnder JL, Nolan GP, Gotlib J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116:988–292. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Watatani K, Itoh Y, Okano H, Guillemot F, Nakajima K, Gotoh Y. Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc Natl Acad Sci U S A. 2009;106:13064–13069. doi: 10.1073/pnas.0808400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Valentin VV, Lowenstein DH. Prolonged seizures increase proliferating neuroblasts in the adult rat subventricular zone-olfactory bulb pathway. J Neurosci. 2002;22:3174–3188. doi: 10.1523/JNEUROSCI.22-08-03174.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobó NA, Lu K, Ai X, Haines GM, Emerson CP., Jr Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2006;103:4505–4510. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd CE. Lnk adaptor: novel negative regulator of B cell lymphopoiesis. Sci STKE. 2001;2001:pe1. doi: 10.1126/stke.2001.85.pe1. [DOI] [PubMed] [Google Scholar]
- Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, Eto K, Takaki S, Takatsu K, Nakauchi H. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc Natl Acad Sci U S A. 2007;104:2349–2354. doi: 10.1073/pnas.0606238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecca B, Ruiz i Altaba A. A GLI1–p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28:663–676. doi: 10.1038/emboj.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Unsicker K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience. 2006;138:1055–1065. doi: 10.1016/j.neuroscience.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113:1364–1374. doi: 10.1172/JCI20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. J Cereb Blood Flow Metab. 2002;22:1311–1318. doi: 10.1097/01.WCB.0000034148.72481.F4. [DOI] [PubMed] [Google Scholar]
- Takaki S, Sauer K, Iritani BM, Chien S, Ebihara Y, Tsuji K, Takatsu K, Perlmutter RM. Control of B cell production by the adaptor protein lnk. Definition of a conserved family of signal-modulating proteins. Immunity. 2000;13:599–609. doi: 10.1016/s1074-7613(00)00060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki S, Tezuka Y, Sauer K, Kubo C, Kwon SM, Armstead E, Nakao K, Katsuki M, Perlmutter RM, Takatsu K. Impaired lymphopoiesis and altered B cell subpopulations in mice overexpressing Lnk adaptor protein. J Immunol. 2003;170:703–710. doi: 10.4049/jimmunol.170.2.703. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Thored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V, Ekdahl CT, Kokaia Z, Lindvall O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells. 2006;24:739–747. doi: 10.1634/stemcells.2005-0281. [DOI] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, Lindvall O. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57:835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200:569–580. doi: 10.1084/jem.20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005;105:4604–4612. doi: 10.1182/blood-2004-10-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, Liu X, Wu H, Carmichael ST. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci. 2006;26:1269–1274. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, Paige CJ, Pawson T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med. 2002;195:1599–1611. doi: 10.1084/jem.20011883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Li Y, Xue H, Li Q, Li J. TNF-alpha induces Lnk expression through PI3K-dependent signaling pathway in human umbilical vein endothelial cells. J Surg Res. 2006;136:53–57. doi: 10.1016/j.jss.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Wang TC, Chiu H, Chang YJ, Hsu TY, Chiu IM, Chen L. The adaptor protein SH2B3 (Lnk) negatively regulates neurite outgrowth of PC12 cells and cortical neurons. PLoS One. 2011;6:e26433. doi: 10.1371/journal.pone.0026433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M, Songyang Z, Frantz JD, Trüb T, Reedquist KA, Karlsson T, Miyazaki M, Cantley LC, Band H, Shoelson SE. Stimulation through the T cell receptor leads to interactions between SHB and several signaling proteins. Oncogene. 1998;16:891–901. doi: 10.1038/sj.onc.1201607. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. 2005;94:459–468. doi: 10.1111/j.1471-4159.2005.03227.x. [DOI] [PubMed] [Google Scholar]
- Yan YP, Sailor KA, Vemuganti R, Dempsey RJ. Insulin-like growth factor-1 is an endogenous mediator of focal ischemia-induced neural progenitor proliferation. Eur J Neurosci. 2006;24:45–54. doi: 10.1111/j.1460-9568.2006.04872.x. [DOI] [PubMed] [Google Scholar]
- Ye P, D'Ercole AJ. Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J Neurosci Res. 2006;83:1–6. doi: 10.1002/jnr.20688. [DOI] [PubMed] [Google Scholar]
- Young KM, Fogarty M, Kessaris N, Richardson WD. Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J Neurosci. 2007;27:8286–8296. doi: 10.1523/JNEUROSCI.0476-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousaf N, Deng Y, Kang Y, Riedel H. Four PSM/SH2-B alternative splice variants and their differential roles in mitogenesis. J Biol Chem. 2001;276:40940–40948. doi: 10.1074/jbc.M104191200. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]