

Abstract
Diabetics are at greater risk of having a stroke and are less likely to recover from it. To understand this clinically relevant problem, we induced an ischemic stroke in the primary forelimb somatosensory (FLS1) cortex of diabetic mice and then examined sensory-evoked changes in cortical membrane potentials and behavioral recovery of forelimb sensory-motor function. Consistent with previous studies, focal stroke in non-diabetic mice was associated with acute deficits in forelimb sensorimotor function and a loss of forelimb evoked cortical depolarizations in peri-infarct cortex that gradually recovered over several weeks time. In addition, we discovered that damage to FLS1 cortex led to an enhancement of forelimb evoked depolarizations in secondary forelimb somatosensory (FLS2) cortex. Enhanced FLS2 cortical responses appeared to play a role in stroke recovery given that silencing this region was sufficient to reinstate forelimb impairments. By contrast, the functional reorganization of FLS1 and FLS2 cortex was largely absent in diabetic mice and could not be explained by more severe cortical infarctions. Diabetic mice also showed persistent behavioral deficits in sensorimotor function of the forepaw, which could not be rescued by chronic insulin therapy after stroke. Collectively these results indicate that diabetes has a profound effect on brain plasticity, especially when challenged, as is often the case, by an ischemic event. Further, our data suggest that secondary cortical regions play an important role in the restoration of sensorimotor function when primary cortical regions are damaged.
Introduction
In North America, ∼8% of the population has diabetes and this number is expected to rise dramatically in the next few decades. Furthermore, many cases of diabetes are completely uncontrolled given that a third of diabetics are unaware of their condition (American Diabetes Association, National Diabetes Fact Sheet, January 26, 2011; www.Diabetes.org/diabetes-basics/diabetes-statistics/). Although diabetes has long been associated with peripheral nerve degeneration and vascular dysfunction in the heart, kidney, and limbs, there is increasing evidence that diabetes directly affects the CNS (Biessels et al., 1999). Studies have shown that diabetic humans and animal models of type 1 and 2 diabetes have impairments in cognitive function, synaptic plasticity, synaptogenesis, and neurogenesis (Manschot et al., 2003; Brands et al., 2005; Francis et al., 2008; Stranahan et al., 2008a; Reijmer et al., 2011).
The effects of diabetes on the brain's vascular system is reflected in the very troubling statistic that diabetics are significantly more likely to suffer a stroke (Iemolo et al., 2002) and are predisposed to poorer recovery (Toni et al., 1994; Kruyt et al., 2010; Wei et al., 2010). At the present time, little is known as to why this is true. In terms of recovery, it is thought that diabetes may exacerbate initial stroke damage by altering the activation of apoptotic or inflammatory signaling pathways (Muranyi et al., 2003; Kumari et al., 2007). However, clinical and experimental studies have not reached a consensus (MacDougall and Muir, 2011), as some reports have shown that diabetes can increase infarct volume (Nedergaard and Diemer, 1987; Duverger and MacKenzie, 1988), decrease it (Ergul et al., 2007; W. Li et al., 2010), or have no effect at all (Mankovsky et al., 1996). Another plausible explanation is that diabetes may impair neural circuit plasticity after stroke. Clinical and animal studies have shown that successful stroke recovery hinges on the brain's ability to remap sensory and motor functions to surviving, functionally homologous brain regions (Nudo and Milliken, 1996; Ward, 2006; Cramer, 2008; Murphy and Corbett, 2009). To our knowledge, whether diabetes affects the reorganization of functional brain maps has never been examined.
To understand why diabetes is associated with poor stroke recovery, we examined sensorimotor function of the forepaw over 10 weeks time after photothrombotic stroke and imaged forepaw evoked sensory responses in diabetic mice that were in the acute (1 week) or chronic (14 weeks) stages of stroke recovery. To our surprise, we found that diabetes did not significantly affect stroke volume, but had a profound effect on the brain's ability to remap forelimb related sensory function onto surviving cortical regions. Further, deficits in forepaw function and cortical map plasticity could not be rescued by normalizing blood glucose levels after stroke with insulin therapy. These results indicate that diabetes limits the brain's innate capacity for repair and rewiring that are critical for successful stroke recovery. Further, functional impairments cannot be reversed simply by reducing blood glucose levels with insulin, which suggests that additional therapies need to be considered if we are to optimize recovery in diabetics.
Materials and Methods
Induction of type I diabetes and monitoring of blood glucose.
Two-month-old male wild-type or GFP-M line mice on a C57BL/6 background (Feng et al., 2000) were used in the present study. All experiments were conducted according to the guidelines laid out by the Canadian Council of Animal Care and the Animal Care Committee. Mice were group housed under a 12 h light/dark cycle and given ad libitum access to water and standard laboratory diet. To induce type 1 diabetes, 2-month-old mice were deprived of food for 4 h, then given a single intraperitoneal injection of streptozotocin (STZ, 140 mg/kg) dissolved in 50 mm citrate buffer, pH = 5.0, or buffer alone. Mice were given 5% sucrose water for 24 h after the injection. Blood glucose levels were measured (Accu-Chek, Aviva, Roche) weekly or every other week by fasting mice for 2–3 h and then withdrawing a drop of blood from the tail vein. To normalize blood sugar levels in diabetic mice, 2–3 slow-release insulin pellets (0.1 U/24 h/implant, LinBit, Linshin Canada) were implanted subcutaneously between the scapulae in the first hour after stroke. Additional insulin pellets were implanted if blood glucose levels exceeded 14 mmol/L.
Targeted photothrombotic stroke.
Focal ischemic stroke of the right forelimb somatosensory cortex was induced in 3-month-old mice using the photothrombotic method (Watson et al., 1985; Brown et al., 2007). Briefly, mice were anesthetized using 1.5–1.8% isoflurane mixed with oxygen. Each mouse was kept on a heating pad during surgery to stabilize body temperature at 37°C, which was measured with a rectal thermoprobe and temperature feedback regulator. The scalp was retracted and the skull overlying the forelimb cortex was carefully thinned to 50% thickness with a high-speed dental drill. Before inducing the stroke, a picture of the surface vasculature of the cortex was taken and then the precise location of the forelimb representation of the somatosensory cortex was identified using intrinsic optical signal (IOS) imaging (Brown et al., 2009). IOS-derived maps of the forelimb representation were superimposed onto the image of the surface vasculature to provide a map for positioning the green laser directly over the primary forelimb somatosensory (FLS1) cortex. Photothrombosis was initiated by exposing the surface vessels over the forelimb cortex to a collimated green laser beam for 15 min after injecting 1% Rose Bengal dye (110 mg/kg, i.p.) dissolved in 0.9% saline. Mice were allowed to recover after surgery under a heating lamp and then returned to their home cages.
Quantification of infarct volume.
Brains were sectioned on a vibratome at 50 μm in the coronal plane. Every sixth section was stained using cresyl violet, and mounted onto glass slides. Serial sections were imaged with a 4× objective under bright-field illumination and quantified using ImageJ software (version 1.44). The area of infarction was measured in each section by a blind observer and an estimate of volume was calculated by summing up the infarct area for each section multiplied by the distance between each section.
Behavioral assessment of forepaw sensory-motor function.
Two behavioral tests were used to assess sensory and motor function of the forepaw. First, removal of adhesive tape from the forepaws is a sensitive test for changes in sensation to the affected paw after stroke (Shanina et al., 2006; Tennant and Jones, 2009). Second, we used the horizontal ladder test as it has previously been used to track recovery of sensory and motor function after stroke (Schallert, 2006; Shanina et al., 2006). These tests were administered at weekly intervals for 2 weeks before stroke and 10 weeks afterward. For the tape removal test, a circular piece of tape (5 mm diameter) was placed on the palm of each forepaw. Mice were then placed in a glass cylinder and filmed for 60 s. This was repeated 3 times per testing session, and the time taken to remove tape from each paw was scored by an observer blind to condition. Sensorimotor function of the forepaw during locomotion was assessed by videotaping mice as they walked across an elevated 70 cm long horizontal ladder that had rungs (1 mm diameter) randomly spaced 1 or 2 cm apart. Forepaw grasping of the rungs was scored on a frame by frame basis using criteria similar to that of previous work (Farr et al., 2006). Briefly, forepaw placements were scored as: (1) “correct” (forepaw placement centered on the rung), (2) “partial” (forepaw partially grasping rung or required a correction of the placement), or (3) “slip/miss.” Due to inherent variability in behavioral measurements, data for each mouse was averaged in 2 week bins.
Voltage-sensitive dye imaging.
Mice were anesthetized using 1–1.5% isoflurane mixed with oxygen. Mice were fitted into a stereotaxic frame, whereupon the eyes were moistened with antibiotic ointment (pentamycetin) and body temperature was maintained at 37°C. For every 2 h of anesthesia, mice were given 0.15 ml of 20 mm glucose dissolved in buffer to maintain proper hydration and glucose levels. To prevent any movement during imaging, the skull was secured to a metal plate using cyanoacrylate glue and dental cement, which was fastened to the surgery stage. A large (∼5 × 5 mm) region of the skull and dura overlying the right cerebral hemisphere was drilled and carefully removed. Gelfoam soaked in HEPES-buffered artificial CSF (ACSF) was used to keep the brain moist throughout the surgical procedure. The exposed brain was bathed in RH1692 dye (Shoham et al., 1999) dissolved in HEPES-buffered ACSF for 90 min (1 mg/ml passed through 0.22 μm syringe filter, Optical Imaging). After the incubation period, the brain was washed thoroughly with brain buffer, covered with 1.3% low-melt agarose dissolved in a HEPES-buffered ACSF and sealed with a glass coverslip. The surgery stage was then mounted underneath an upright Olympus BX51 microscope for imaging.
For voltage-sensitive dye (VSD) imaging, 12-bit image frames (184 × 124 pixels) were captured every 4 ms using a MiCAM02 camera (Brain Vision). The dye was excited with Luxeon K2 red LED (627 nm, ∼20 mW at back aperture) that was passed through a Cy5 filter cube (exciter: 605–650 nm, emitter: 670–720 nm). Red light was focused 200–300 μm below the cortical surface using an Olympus XFluor 2× objective (NA = 0.14). Mechanical stimulation of the forepaw was achieved by gluing a pencil lead to the paw which was connected to a piezoelectric wafer (Q220-AY-203YB, Piezo Systems; ∼300 μm deflection). To eliminate auditory responses, the ears were bilaterally occluded with low-melt agarose and Vaseline. During each trial, images were collected 250 ms before a single 5 ms deflection of the forepaw (or not for null stimulation trials) and then 550 ms afterward. This process was repeated 12–24 times with a 10 s interval between trials. To correct for dye bleaching, stimulation trials were divided by null stimulation trials. VSD images are presented as the percentage change in VSD fluorescence (ΔF/Fo) by dividing frames collected after stimulation by the average of those taken 100 ms before stimulation. Montages of cortical responses were generated by mean filtering ΔF/Fo image stacks (radius = 2) and then binning 2 frames in time (hence 8 ms between each image). Using ImageJ software, forelimb evoked cortical responses were quantified within a circular region of interest (500 μm diameter) centered over the FLS1, hindlimb primary somatosensory cortex (HLS1), primary motor cortex (M1) or secondary forelimb somatosensory (FLS2) cortex (see Fig. 3A, inset). After stroke, the FLS1 was defined as the remaining piece of forelimb cortex that showed the shortest latency to respond, typically immediately anterior or posterior to the center of the original FLS1 region. The peak amplitude, time to peak amplitude and half-width (i.e., duration) of VSD signals in the first 250 ms after stimulation were measured with Clampfit 9.0 software (Molecular Devices).
Figure 3.
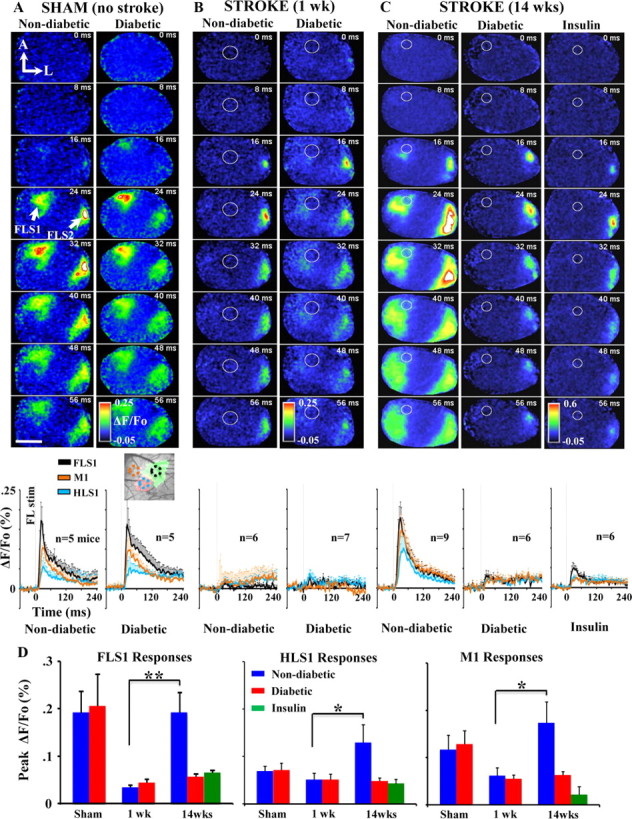
VSD imaging shows that diabetes impairs remapping of the forelimb sensory representation after stroke. A–C, Montages showing the spatiotemporal dynamics of cortical responses to forelimb stimulation. Below each montage are corresponding ΔF/Fo plots showing average group responses to forelimb stimulation in FLS1 (black line), M1 (orange line), and HLS1 (teal line). Inset with dashed circles indicates the location where responses were measured in each region. After stroke, FLS1 responses were measured where short-latency responses first emerged. In the absence of stroke damage (A), forelimb evoked cortical depolarizations were similar between non-diabetic and diabetic mice. One week after stroke (B, white circle denotes infarct), the peri-infarct cortex shows little, if any, response to forelimb stimulation while responses are preserved in FLS2 cortex. Fourteen weeks after stroke (C), forelimb evoked depolarizations reemerge in peri-infarct cortex of non-diabetic mice, but not in diabetic or insulin-treated mice. The FLS2 cortex also appears more responsive to forepaw touch in non-diabetic mice. D, Quantification of forelimb evoked responses in each cortical region in sham operates, or 1 and 14 weeks after stroke. Note that only in non-diabetic mice, peak amplitudes drop in FLS1 cortex 1 week after stroke and then recover or become greater at 14 weeks recovery. *p < 0.05, **p < 0.01. Scale bar, 2 mm.
Reversible inactivation of FLS2 cortex.
To ensure that muscimol injections targeted the FLS2 cortex, we first verified the location of the FLS2 cortex by combining functional imaging with retrograde tracing experiments. Initially, the FLS1 cortex was micro-injected with 0.2 μl of the retrograde tracer cholera toxin β subunit (1% CTB dissolved in 0.1 m PBS; List Biological Laboratories). Five to 7 d later, forepaw evoked responses in putative FLS2 cortex were identified on the brain's surface using IOS imaging and marked with DiI crystals (Invitrogen). Mice were overdosed with sodium pentobarbital and perfused transcardially with 0.1 m PBS followed by 4% paraformaldehyde. Brains were postfixed overnight and cut at 50 μm in the coronal plane on a vibratome. As described previously (Brown and Dyck, 2005), sections were incubated overnight in goat anti-choleragenoid primary antisera (1:10,000; List Biological Laboratories) followed by incubation in biotinylated secondary antibodies for 120 min (1:1000; Vector Labs) and 90 min in streptavidin-HRP (1:1000, Vector Labs). Sections were reacted with chromagen solution (0.2 mg/ml diaminobenzidine, 0.01% H2O2, 0.4 mg/ml nickel chloride) for 10 min, washed in 0.1 m PBS, mounted onto slides, dehydrated in ethanol, and coverslipped with Permount. CTB-labeled neurons in FLS2 were imaged under bright-field illumination and compared with an adjacent brain section where DiI was imaged with epifluorescence.
To inactivate the FLS2 cortex in the right hemisphere, stroke recovered (at 11 weeks recovery) or sham-operated mice were anesthetized with 1.5% isoflurane and a small hole was drilled through the skull ∼1.5 mm posterior and 4 mm lateral of bregma. IOS-derived sensory maps generated initially to target the stroke to the FLS1 cortex were also used to identify the location of FLS2 cortex. A stainless steel Hamilton syringe with a 33 gauge needle was lowered 1.3 mm deep into the brain. Muscimol hydrobromide (5 μg/μl; Sigma G019) dissolved in ACSF with 1% Texas Red dextran (70 kDa, Invitrogen, D-1864) was slowly pressure injected (0.2 μl) over a 5 min period. Inclusion of Texas Red dextran allowed us to verify injection placement. As a control, mice were injected an equivalent volume of ACSF which had no effect on tape removal latency (see Fig. 6D). Four to 5 h after muscimol injection, mice were administered the adhesive tape removal test.
Figure 6.

Inactivating FLS2 cortex after stroke reinstates functional impairments. A, B, Functional imaging and tract tracing were used in combination to delineate the precise location of FLS2 cortex. A, Photomicrograph of a coronal section from a mouse where forelimb evoked responses were imaged and putative FLS2 responses were marked with DiI. B, An adjacent section from the same mouse showing a dense band of CTB-labeled neurons in putative FLS2 cortex (5 d after CTB injection in FLS1) that coregisters with DiI placement. Inset shows higher-magnification image of CTB-labeled neurons in FLS2 cortex. Scale bar: B inset, 200 μm. C, VSD imaging indicated that forelimb evoked responses were intact in FLS1 but were abolished in FLS2 cortex ∼4.5 h after muscimol injection (demarcated by the white circle). Scale bar, 2 mm. D, Histogram shows the average time it took mice to remove tape from the left forepaw in sham-operated controls (black bars) and stroke recovered mice (gray bars, 7 non-diabetic and 2 diabetic) following muscimol (musc) or vehicle (veh) injection in the right FLS2 cortex. Muscimol injection significantly increased tape removal latencies in stroke recovered mice (Stroke + musc) relative to baseline levels of performance (Stroke) or sham-operated mice injected with muscimol (Sham + musc). E, Muscimol injection into the FLS2 cortex was verified in postmortem brain section by imaging the deposition of 1% Texas Red dextran (see red arrow). Note that fluorescent dextran is within the functionally and anatomically defined FLS2 region shown in A and B. Scale bars: A, B, E, 1 mm. **p < 0.01.
Statistics.
Statistical comparisons for the effect of diabetes on stroke recovery were made using an ANOVA with post hoc Student's t tests. Paired t tests were used to examine the effect of muscimol injection on tape removal latency. All p-values ≤0.05 were considered statistically significant. All data are presented as the mean ± SE of the measurement.
Results
Animal model of diabetes and ischemic stroke
Figure 1, A and B, summarizes the two primary experiments described in the present study. To understand why diabetes is associated with poor stroke recovery, we examined changes in the function of the somatosensory cortex using in vivo VSD imaging in non-diabetic and diabetic mice subjected to photothrombotic stroke in the right FLS1 cortex. Mice subjected to sham procedures were exposed to either the laser or the photosensitive dye Rose Bengal (but not both) and showed no signs of ischemic damage. In tandem with functional brain imaging, recovery of forepaw function after stroke was assessed weekly using the adhesive tape removal and horizontal ladder tests (Fig. 1A). Our second aim was to determine whether any diabetes related deficits in cortical plasticity or functional recovery could be reversed by treating diabetic mice with long-term insulin treatment after stroke (Fig. 1B). For this experiment, mice were hyperglycemic for 1 month before stroke and then received insulin treatment for 3 months afterward.
Figure 1.
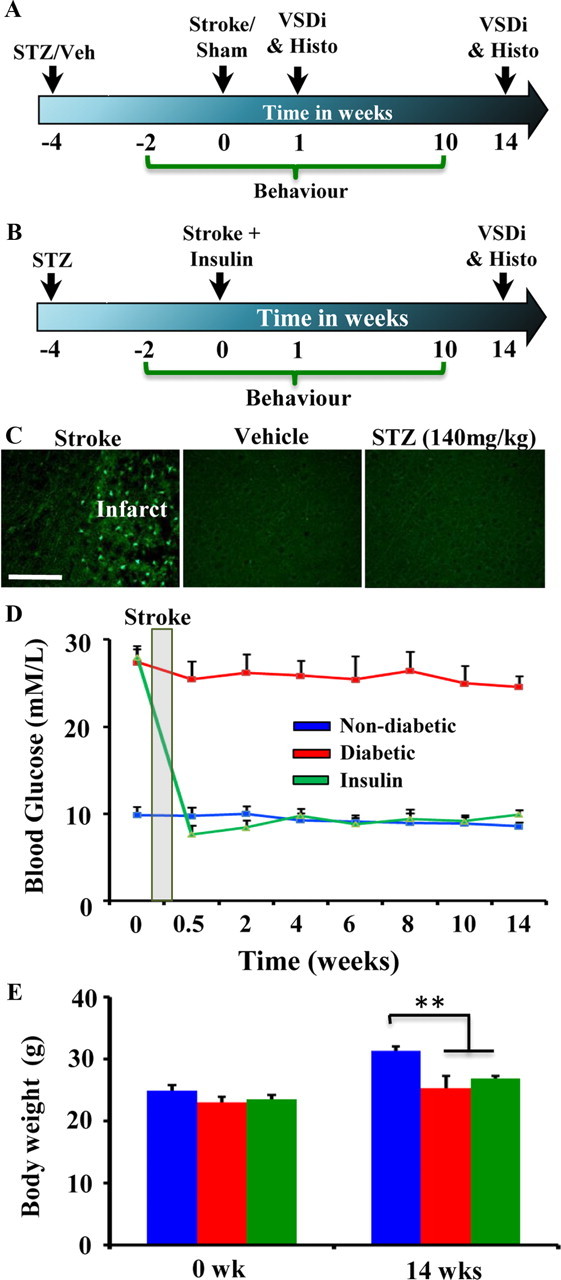
A, Timeline of the experiment investigating the effect of diabetes on stroke recovery, expressed in weeks relative to the induction of stroke. Behavioral tests of sensorimotor function of the forepaw were conducted on a weekly basis before and after photothrombotic stroke (induced at time 0). Mice were imaged and then killed for histological assessment at 1 and 14 weeks. B, To determine whether insulin therapy could normalize stroke recovery, diabetic mice were subjected to stroke and then had slow-release insulin pellets subcutaneously implanted 1 h later. Similar to that described above, somatosensory cortex was imaged at 14 weeks recovery and forepaw function was tested at weekly intervals. C, Photomicrographs show Fluoro-Jade C-labeled degenerating neurons in the infarct core of a mouse killed 24 h after stroke (left image, positive control for Fluoro-Jade C staining). By contrast, no degenerating neurons are found in the cortex 24 h after vehicle (middle image) or STZ injection (right image). These data suggest that STZ does not have a direct toxic effect on the brain. Scale bar, 100 μm. D, Average fasting blood glucose levels (mmol/L) for each group. E, Average body weight at 0 and 14 weeks recovery. **p < 0.01.
To induce diabetes, 2-month-old C57BL/6 male mice were given a single injection of STZ (140 mg/kg, i.p.) or vehicle. STZ administration models type 1 diabetes (Like and Rossini, 1976) as it selectively destroys insulin producing β-cells of the pancreas resulting in chronic hyperglycemia. Importantly, we did not observe any signs of cell death (labeled with Fluoro-Jade C) in the cerebral cortex 24 h after STZ injection, suggesting that it was not having a direct toxic effect on the brain (Fig. 1C). As shown in Figure 1D, blood glucose levels in STZ-injected (“diabetic” group) mice were chronically elevated relative to vehicle-injected controls (“non-diabetic” group) or diabetic mice implanted with slow-release insulin pellets (“insulin” group) 1 h after stroke. We should note that some of the mice in the non-diabetic group consisted of STZ-injected mice that did not develop hyperglycemia (9 of 24 mice; blood glucose levels < 12 mmol/L; none of these mice received insulin). Body weights for all 3 groups were similar at the start of the experiment (Fig. 1E; F(2,19) = 1.41, p = 0.27). However by the end of the 14 week experiment, body weights for non-diabetic mice were significantly greater than diabetic (t(12) = 3.17, p < 0.01) and insulin-treated mice (t(13) = 5.47, p < 0.01). The mortality rate over the 14 week recovery period was 26% and 12.5% for diabetic and insulin-treated mice, respectively.
Diabetes is associated with poorer recovery of forepaw sensation after stroke
The adhesive tape removal test is considered a sensitive method for detecting deficits in forepaw sensation that is commonly found after stroke in the sensorimotor cortex (Schallert, 2006; Tennant and Jones, 2009). In all three groups, stroke increased the time it took for mice to remove tape from the left (impaired) paw in the first 1–2 weeks after stroke (Fig. 2A; F(1,62) = 15.3, p < 0.001; p < 0.01 for all post hoc t tests comparing 0 vs 1–2 weeks recovery). However, there was no significant difference between diabetics and non-diabetics in tape removal latency in the first 6 weeks after stroke (1–2 weeks: t(23) = 0.74, p = 0.23; 3–4 weeks: t(23) = 0.45, p = 0.32; 5–6 weeks: t(23) = 0.96, p = 0.17). From 7–10 weeks after stroke, non-diabetic mice became significantly less impaired on the tape removal test relative to both diabetic and insulin-treated mice (Fig. 2A; Effect of Condition: F(2,182) = 5.53, p < 0.01; 7–8wks Non-diabetic vs Diabetic: t(22) = 1.83, p < 0.05 vs Insulin: t(18) = 2.91, p < 0.05; 9–10wks Non-diabetic vs Diabetic, t(20) = 1.76, p < 0.05 vs Insulin t(20) = 1.77, p < 0.05). For the right forepaw, there was no effect of stroke on tape removal latency (F(1,62) = 0.36, p = 0.54) and there were no significant group differences in latencies during recovery (Fig. 2B; F(2,182) = 1.23, p = 0.29). The fact that only the left forepaw was impaired by stroke suggests that deficits in stroke recovery could not be attributed to a global effect of diabetes on sensory function (i.e., bilateral deterioration of sensation).
Figure 2.

Diabetes impedes the recovery of sensory function after stroke. A, Plot showing the average time it took mice to remove adhesive tape from the left (impaired) forepaw. Time 0 represents data collected before the induction of stroke. Stroke increased tape removal latencies for the left paw, which appeared to slowly recover in non-diabetic mice at 7–8 and 9–10 weeks, but not diabetic or insulin-treated mice. **p ≤ 0.01 for all post hoc t tests comparing 0 vs 1–2 weeks recovery, *p < 0.05 for t tests between non-diabetic and diabetic or insulin-treated mice at 7–8 and 9–10 weeks recovery. B, Latencies for the right forepaw were unaffected by stroke or diabetes. C, Histograms showing summary of left forepaw placements on the horizontal ladder rung test. Before stroke, the majority of forepaw steps on the ladder rung test were considered correct placements, meaning the forepaw was firmly placed and centered over the rung. After stroke, the percentage of partial or incorrect forepaw placements on the rung increased dramatically. There was a slight, albeit nonsignificant trend toward improved performance in non-diabetic mice at later stages of recovery. ***p < 0.001 for comparing percentage correct or partial placements in each group before and 1–2 weeks after stroke.
Sensorimotor function of the left (impaired) forepaw was also examined using the horizontal ladder test. Consistent with previous work (Clarkson et al., 2010), stroke degraded performance on this test reflected by a significant reduction in correct placements and an increase in the incidence of partial (incorrect) forepaw placements (Fig. 2C; p < 0.001 for all t tests comparing percentage correct or partial steps at 0 vs 1–2 weeks after stroke). We should note that even after stroke, more obvious errors such as slips or complete misses were very infrequent and did not differ between groups. However, unlike the tape removal test where group differences became quite obvious, all 3 groups displayed relatively similar deficits in ladder performance after stroke (Fig. 2C; Effect of Condition for correct and partial placements: F(2,134) = 2.63, p = 0.07 and F(2,134) = 2.57, p = 0.08, respectively). For the right forepaw, we found that stroke had no effect on the percentage of partial ladder rung placements (F(1,54) = 1.38, p = 0.24). Collectively, these results suggest that diabetes attenuates the progressive recovery of sensorimotor function on the adhesive tape removal test, but not on the horizontal ladder test.
Diabetes impairs the remapping of sensory function in somatosensory cortex
Recovery of forepaw function is thought to be mediated by adaptive changes in the neural circuitry of peri-infarct and more distant but functionally relevant cortical regions (Dijkhuizen et al., 2001; Dancause et al., 2005; Brown et al., 2009). Accordingly, we investigated whether poor behavioral outcome in diabetic and insulin-treated mice was associated with deficits in cortical plasticity. To do this, we imaged forelimb evoked patterns of brain activity across a large portion of the right cerebral hemisphere in mice that had recovered for either 1 or 14 weeks after right forelimb cortex stroke. This large imaging window allowed us to visualize forelimb responses in both FLS1 and FLS2, which previous studies have not examined. Consistent with previous in vivo VSD imaging data (Ferezou et al., 2007; Brown et al., 2009), brief mechanical stimulation of the forelimb (one 5 ms tap) in sham-operated non-diabetic or diabetic mice produced a strong depolarization in the FLS1 which peaked on average 20–25 ms after stimulation (Fig. 3A; Tables 1 and 2 for peak amplitude and time to peak averages) and then decayed thereafter. As expected, depolarizations that originated in FLS1 cortex spread medially into M1 cortex and posterior-medially into HLS1 cortex. In addition to the FLS1 response, a robust depolarization was observed laterally in FLS2 cortex (Fig. 3A; Benison et al., 2007). We verified that this region was in fact FLS2 cortex, by showing that these lateral forelimb evoked responses (marked with DiI in IOS imaging experiments) were from the same cortical region that shares extensive connections with the FLS1 cortex (labeled with the retrograde tracer CTB; see Fig. 6A,B). The average peak amplitude and latency of FLS1 and FLS2 responses were very similar between sham-operated non-diabetic and diabetic mice (Fig. 3A,D; Tables 1 and 2). These results indicate that diabetes alone (without stroke), does not significantly alter the spatiotemporal dynamics of forelimb evoked sensory responses.
Table 1.
Average peak amplitude of forelimb evoked responses (peak % ΔF/Fo ± SEM) in each cortical region
| Region | Sham Stroke |
Stroke (1 week) |
Stroke (14 weeks) |
||||
|---|---|---|---|---|---|---|---|
| Non-diabetic | Diabetic | Non-diabetic | Diabetic | Non-diabetic | Diabetic | Insulin | |
| FLS1 | 0.19 ± 0.05 | 0.21 ± 0.07 | 0.03 ± 0.01 | 0.04 ± 0.01 | 0.19 ± 0.04 | 0.06 ± 0.01 | 0.07 ± 0.01 |
| HLS1 | 0.07 ± 0.01 | 0.07 ± 0.02 | 0.05 ± 0.01 | 0.05 ± 0.01 | 0.13 ± 0.04 | 0.05 ± 0.01 | 0.04 ± 0.01 |
| M1 | 0.12 ± 0.03 | 0.13 ± 0.03 | 0.06 ± 0.02 | 0.06 ± 0.01 | 0.17 ± 0.04 | 0.06 ± 0.01 | 0.02 ± 0.02 |
| FLS2 | 0.25 ± 0.07 | 0.24 ± 0.08 | 0.33 ± 0.07 | 0.21 ± 0.03 | 0.49 ± 0.05 | 0.36 ± 0.07 | 0.34 ± 0.05 |
Table 2.
Average time to peak forelimb evoked response (ms ± SEM) in each cortical region
| Region | Sham Stroke |
Stroke (1 week) |
Stroke (14 weeks) |
||||
|---|---|---|---|---|---|---|---|
| Non-diabetic | Diabetic | Non-diabetic | Diabetic | Non-diabetic | Diabetic | Insulin | |
| FLS1 | 22.4 ± 0.98 | 24.8 ± 2.0 | 137.3 ± 35.5 | 130.3 ± 37.9 | 28.5 ± 2.4 | 82 ± 28.4 | 42.9 ± 7.9 |
| HLS1 | 74.2 ± 23.2 | 69.3 ± 36.9 | 123.9 ± 35.9 | 86.3 ± 36.2 | 75.1 ± 18.6 | 124.6 ± 26.6 | 113.7 ± 29.7 |
| M1 | 29.9 ± 2.3 | 32.3 ± 2.7 | 127.8 ± 34.0 | 126.3 ± 34.9 | 35.6 ± 2.8 | 142.5 ± 23.9 | 121.7 ± 25.9 |
| FLS2 | 24.5 ± 2.0 | 25.3 ± 2.4 | 24.2 ± 2.2 | 21.1 ± 0.8 | 24.4 ± 0.7 | 23.5 ± 1.2 | 23.4 ± 1.0 |
One week after focal stroke (Fig. 3B, stroke denoted by white circles), stimulation of the forelimb failed to elicit distinct responses in peri-infarct cortex for both non-diabetic and diabetic mice (Fig. 3B). Of note, cortical responses to the forelimb were preserved in FLS2 cortex demonstrating that its responses are not fully dependent on feed forward connections from FLS1 cortex.
Since diabetes did not noticeably alter the course of stroke recovery at 1 week, we then examined cortical responsiveness 14 weeks after stroke when new sensory maps would have already formed (Winship and Murphy, 2008; Brown et al., 2009). In agreement with previous studies examining non-diabetic mice (Brown et al., 2009), forelimb evoked responses reemerged in the remaining fragment of S1FL cortex which then spread to adjacent peri-infarct HLS1 and M1 cortical regions (Fig. 3C). Quantitative analysis of peak cortical depolarizations in non-diabetic mice (Fig. 3D, blue bars) indicated that cortical responses dropped in the first week after stroke and then increased significantly in FLS1 (t(8) = 3.72, p < 0.005), HLS1 (t(10) = 1.94, p < 0.05) and M1 cortex (t(10) = 2.37, p < 0.05) by 14 weeks recovery. In contrast, diabetic mice showed little to no signs of cortical remapping as forelimb evoked depolarizations were barely detectable in FLS1, HLS1 or M1 cortex after stroke (Fig. 3C). Similarly, treating diabetic mice with insulin for 3 months after stroke failed to rescue these defects in cortical remapping (Fig. 3C). The absence of cortical remapping in diabetic and insulin-treated mice can also be noted in Figure 3D, where peak amplitudes drop 1 week after stroke and never recover. Importantly, FLS2 responses were clearly detectable in all groups which suggests that the absence of peri-infarct responses in diabetic and insulin-treated mice was not caused by a global loss of cortical responsiveness due to anesthetic depth or inability to tolerate the surgical/imaging procedure. Furthermore, there were no differences in physiologic parameters such as heart rate (F(2,9) = 0.28, p = 0.75), breath rate (F(2,9) = 2.62, p = 0.12) or O2 saturation (F(2,9) = 2.45, p = 0.14) during VSD imaging.
One obvious explanation for the absence of cortical remapping in diabetics (with and without insulin treatment) is that they may have suffered a much larger stroke than non-diabetics. Accordingly, 1 and 14 weeks after stroke, mice from each group were killed, and their brains were sectioned, stained with cresyl violet, and imaged to assess infarct volume. Our analysis indicated that cortical infarct volumes in non-diabetic and diabetic mice were nearly identical at 1 week recovery (Fig. 4A,B; Non-diabetic = 2.03 ± 0.26 mm3 vs Diabetic = 2.04 ± 0.43 mm3; t(14) = 0.01, p = 0.49). Similarly, there were no group differences in infarct size at 14 weeks recovery (Fig. 4B; Non-diabetic = 1.69 ± 0.22 mm3 vs Diabetic = 1.71 ± 0.28 mm3 vs Insulin = 1.62 ± 0.36 mm3; F(2,20) = 0.03, p = 0.97). It is also worth noting that the position of stroke, based on angle and distance to the FLS2 cortex, was not significantly different between groups (Distance: F(2,18) = 0.35, p = 0.71; Angle: F(2,18) = 0.48, p = 0.62). In light of these data, it would appear that impairments in cortical remapping in diabetics and insulin-treated mice cannot be explained simply by differences in the position or size of the cortical infarction.
Figure 4.

No effect of diabetes on infarct volume at 1 or 14 weeks recovery. A, Representative cresyl violet stained coronal sections of the stroke affected hemisphere in non-diabetic and diabetic mice 1 week after stroke. Scale bar, 2 mm. B, Histogram shows no differences in infarct volume between groups at 1 or 14 weeks recovery (n = 8 mice per group except for insulin group, n = 7).
Local inactivation of FLS2 cortex reinstates functional impairments
An unexpected but intriguing finding from our imaging experiments was that cortical responsiveness in FLS2 cortex increased dramatically in stroke recovered mice (Fig. 3C). Quantitatively, the amplitude of FLS2 responses in non-diabetic mice 14 weeks after stroke was almost double that of sham-operated controls (Fig. 5A,C; Table 1; t(12) = 2.79, p < 0.01). Diabetic mice had slightly elevated FLS2 responses (∼50% increase, Fig. 5B,C; Table 1) but this did not reach statistical significance (t(9) = 1.13, p = 0.14). The timing of peak forelimb evoked responses in FLS2 cortex was not affected by stroke or diabetes (Table 2).
Figure 5.

FLS2 cortex becomes more responsive to forepaw stimulation 14 weeks after stroke. A, B, Plots showing average forelimb evoked responses (% ΔF/Fo) in FLS2 cortex in non-diabetic (black trace in A) and diabetic (gray trace in B) mice after sham stroke (left traces), at 1 week (middle traces) or 14 weeks recovery (right traces). C, The peak amplitude of responses in FLS2 cortex increased significantly in non-diabetic mice 14 weeks after stroke. **p < 0.01.
Currently, there is very little understanding of what role FLS2 cortex might be playing in stroke recovery. We hypothesized that if FLS2 cortex was important in recovery, then silencing this region of cortex should reinstate functional deficits in stroke recovered mice. Therefore we micro-injected 0.2 μl of the GABA agonist muscimol (1 μg total) into the right FLS2 cortex of mice that underwent sham stroke procedure and those that had recovered for 10 weeks after stroke (7 non-diabetic and 2 diabetic mice). Previous studies have shown that muscimol potently inhibits neuronal activity for up to 12 h after injection (Martin and Ghez, 1999; Arikan et al., 2002). We verified the inhibition of FLS2 cortex by imaging forelimb evoked responses 4.5–5 h after muscimol injection (Fig. 6C).
Approximately 4–5 h after muscimol injection, mice were administered the tape removal test. We chose not to examine mice on the horizontal ladder given that spontaneous recovery, at least on this specific test, was relatively limited. As shown in Figure 6D, inactivating FLS2 cortex in sham controls (sham + musc) increased tape removal latencies relative to mice that received no injection (sham) or a vehicle injection (sham + veh), but this did not reach statistical significance (t(10) = 1.54, p = 0.07). By contrast, when FLS2 cortex was silenced in stroke recovered mice, tape removal latencies increased by ∼300% relative to performance at 9–10 weeks recovery (Fig. 6D, gray bars; t(8) = 3.92, p < 0.005) and were significantly greater than that observed for shams injected with muscimol (t(18) = 2.62, p < 0.01). When comparing non-diabetic versus diabetic mice in the stroke recovery group, muscimol increased tape removal latencies by 34 ± 8.6 and 13.2 ± 9.9 s, respectively. The location of muscimol injection into FLS2 cortex was verified in postmortem brain sections by imaging the deposition of Texas red dextran (Fig. 6E). Collectively, these data suggest that FLS2 cortex plays a greater role in forepaw sensorimotor function when FLS1 cortex is damaged by stroke.
Discussion
Here we used in vivo VSD imaging and behavioral tests of sensory-motor function to examine stroke recovery in a mouse model of type 1 diabetes. Although diabetes did not significantly affect cortical responsiveness in the first week after stroke or the volume of cortical infarction, it did however, prevent the reemergence of the forelimb sensory representation onto peri-infarct regions and limited stroke-induced changes in the functional responsiveness of the FLS2 cortex (for summary, see Fig. 7). Furthermore, lowering blood glucose levels with chronic insulin treatment after stroke did not rescue deficits in cortical plasticity or forepaw sensorimotor function. These results provide new insights into the neurobiological mechanisms underlying poor functional outcome after stroke in an animal model of diabetes.
Figure 7.

Summary of functional imaging and behavioral data. Before stroke, forelimb evoked depolarizations in FLS1 and FLS2 cortex were similar between groups. In the acute stage of stroke recovery (1 week), peri-infarct responses were diminished and mice showed severe impairments in forepaw sensorimotor function. After 14 weeks of recovery, forelimb evoked responses remapped onto peri-infarct cortex and became stronger in FLS2 cortex in non-diabetic mice, which also show the greatest improvements in forepaw sensory function. By contrast, cortical plasticity and functional recovery were diminished in both diabetic and insulin-treated mice.
Effect of diabetes on stroke recovery
Recovery from stroke is notoriously variable and difficult to predict. Despite this uncertainty, some common principles have emerged. One is that successful recovery from stroke correlates with the brain's ability to remap sensory and motor functions to unaffected regions (Cramer, 2008; van Meer et al., 2010). Supporting this, numerous studies have shown that spontaneous recovery of hand or paw use after stroke is associated with new patterns of brain activation in cortical regions linked to the damaged zone (Dijkhuizen et al., 2001; Jablonka et al., 2010; van Meer et al., 2010). A second common principle is that certain stroke comorbidities are strongly associated with poor neurological outcome. In particular, clinical studies have shown that diabetes increases the risk of ischemic stroke and is associated with slower or poorer recovery of function/independence (Clavier et al., 1994; Jorgensen et al., 1994; Hankey et al., 2007; Kamouchi et al., 2011), even when adjusting for confounding factors such as age and stroke severity (Wei et al., 2010).
The reasons why diabetics are predisposed to poor stroke recovery is not well understood. One common explanation is that hyperglycemia exacerbates stroke damage, thereby reducing the likelihood of recovering function. As reviewed by MacDougall and Muir (2011), the effects of hyperglycemia on infarct size and stroke recovery in animal models of diabetes are quite variable and depend, in part, on the specific model and duration of diabetes used. In the present study, we attempted to model type 1 diabetes by selectively killing insulin producing cells in the pancreas with STZ. Although this is a widely used model of experimental diabetes, it produces a severe and persistent level of hyperglycemia that in humans could produce life-threatening complications such as hyperglycemic hyperosmolar state (Chaithongdi et al., 2011). This is worth noting since the hyperglycemic hyperosmolar state is associated with serious neurologic problems such as diminished arousal and cognitive function, which could conceivably affect stroke recovery (Chaithongdi et al., 2011). Nevertheless using this model of diabetes, we found virtually identical infarct volumes between our diabetic (treated with and without insulin) and non-diabetic mice at 1 and 14 weeks recovery. This finding is not unprecedented as there are reports stating that diabetes in humans and rat models of type 2 diabetes had either no effect on infarct volume or actually reduced ischemic damage (Mankovsky et al., 1996; Ergul et al., 2007; W. Li et al., 2010). However, our findings contrast with some previous studies showing that diabetes (induced with STZ) aggravates initial stroke damage caused by middle cerebral artery occlusion (MCAO) (Nedergaard and Diemer, 1987; Duverger and MacKenzie, 1988; Li et al., 2004). Similar effects on infarct volume have also been observed in the db/db mouse model of type 2 diabetes after MCAO (Vannucci et al., 2001) and rats chronically fed a high fat diet (Langdon et al., 2011). Given that the present study used photothrombosis to induce ischemic stroke, perhaps some of these differences in present versus past results could be attributed to the model of stroke used. For example, MCAO tends to produce a more slowly evolving infarction (Carmichael, 2005) with a larger “penumbral” region of vulnerable, hypo-perfused tissue around the infarct core (Nedergaard, 1988; Dirnagl et al., 1999), than is found after photothrombotic stroke (Carmichael et al., 2005; Zhang and Murphy, 2007). If diabetes exacerbates the spread of damage into the penumbra then one would likely expect a greater effect of diabetes on infarct volume in MCAO models of stroke.
When examining long-term recovery from stroke, we found that diabetic mice showed poor recovery of sensorimotor function. To date, there have been relatively few studies that have examined the impact of diabetes on recovery of limb use/function in the weeks to months after stroke, and no study to our knowledge has imaged cortical map plasticity. Two studies (Moreira et al., 2007; Langdon et al., 2011) reported greater impairments in forepaw function after cortical injury in rat models of type 2 diabetes. These findings are consistent with our data showing that diabetic mice performed significantly worse on tests of forepaw sensorimotor function in the later stages of stroke recovery. The neural mechanisms underlying the recovery of sensory and motor function are not clear, but our data suggest that functional changes in peri-infarct and FLS2 cortex likely play a role.
Although there is a growing body of data suggesting that intensive insulin therapy reduces the absolute risk of stroke in diabetic humans (Nathan et al., 2005) and possibly acute stroke damage (Auer, 1998), little evidence exists showing that strict glycemic control after stroke can improve stroke recovery (Kruyt et al., 2010; MacDougall and Muir, 2011). Intriguingly, our data show that diabetic mice chronically treated with insulin after stroke did not fare significantly better in terms of functional recovery or cortical plasticity, than their untreated counterparts. We should note that insulin was still beneficial in a general sense as these mice appeared healthier in terms of body weight, appearance and mortality rates which were below that of diabetic mice. The fact that mice in our insulin group were hyperglycemic for at least 4 weeks before the induction of stroke suggest that some pathological processes may have already been in place by the time blood glucose levels were regulated. It is possible that 4 weeks of hyperglycemia could have irreversibly compromised the vascular system's integrity (“leakiness”) or its ability to make adaptive changes after stroke such as redistributing blood flow through collateral vessels or changing vascular tone (Schaffer et al., 2006; Shih et al., 2009; Moskowitz et al., 2010). Our study is not the first to show that normalizing blood glucose levels does not necessarily rescue diabetes related neuropathology. For example, sensory neuron loss in the dorsal root ganglia of type 1 diabetic mice could not be prevented even if mice spontaneously regained islet function and euglycemia (Kennedy and Zochodne, 2005b). Similarly, diabetes-related deficits in spatial learning and hippocampal long-term potentiation could not be reversed by insulin treatment (Biessels et al., 1998).
Mechanisms for impaired cortical plasticity
Since the volume of the cerebral infarction did not dictate the pattern of stroke recovery, it is conceivable that diabetes may have augmented the mechanisms of neuronal and vascular plasticity that support sensory remapping and functional recovery (Kaas, 2002; Murphy and Corbett, 2009). Studies in non-diabetic animals have shown that stroke transiently upregulates the production of growth associated proteins (Carmichael et al., 2005), dendritic spines (Brown et al., 2009; Mostany et al., 2010), axonal sprouting (Carmichael et al., 2001; Dancause et al., 2005; S. Li et al., 2010), microglial activity (Masuda et al., 2011) and angiogenesis (Zhang and Chopp, 2009). Deficits in any one aspect of this coordinate response could explain the impaired cortical plasticity and recovery in our model of diabetes. Indeed, other studies have shown that diabetes is associated with reduced synaptic plasticity (e.g., long-term potentiation), dendritic spine densities in the hippocampus and cerebral cortex (Biessels et al., 1996; Gispen and Biessels, 2000; Malone et al., 2008; Stranahan et al., 2008a,b), as well as impaired peripheral nerve regeneration (Kennedy and Zochodne, 2005a; Ebenezer et al., 2011). Considering that diabetes in both human and animal models is almost invariably linked with vascular disease such as stroke and peripheral arterial disease (Vinik and Flemmer, 2002), deficits in cortical map plasticity could be secondary to widespread vascular dysfunction after stroke.
Role of FLS2 cortex in stroke recovery
It is well established that cortical regions functionally related to the one damaged by stroke can support recovery (Cramer, 2008; Murphy and Corbett, 2009). For example, ablating or disrupting neuronal activity in peri-infarct cortex or homotopic regions in the opposite hemisphere can reinstate functional impairments in the affected hand or forepaw (Castro-Alamancos and Borrel, 1995; Werhahn et al., 2003; Biernaskie et al., 2005). In the present study, we noted that forepaw evoked responses in the FLS2 cortex progressively increased in non-diabetic mice after stroke. Our findings are in agreement with fMRI imaging data showing that improved clinical outcome in stroke patients was paralleled by increased activation volume in S2 cortex (Carey et al., 2002; Nhan et al., 2004). However, our data extend these important clinical observations by showing that inactivation of the FLS2 cortex with GABA-A agonist muscimol is sufficient to unmask profound deficits in sensory function that had appeared to recover. The notion that FLS2 cortex participates in the recovery of sensory functions has some intuitive appeal. Anatomical and imaging studies (including our own), show that S2 cortex is highly interconnected with primary somatosensory cortex (Krubitzer et al., 1997; Chakrabarti and Alloway, 2006; Jablonka et al., 2010) and responds to various aspects of tactile stimuli such as texture, vibration and contour (Romo and Salinas, 2001). Exactly how FLS2 neurons process sensory information from the forepaw after stroke is uncertain and will require further investigation.
Footnotes
This work was supported by operating, salary and equipment grants to C.E.B. from the Canadian Institutes of Health Research, Heart and Stroke Foundation of British Columbia and Yukon, Michael Smith Foundation for Health Research, Natural Sciences and Engineering Research Council of Canada, and Canada Foundation for Innovation. We thank Drs. Stan Bardal, Viera Saly, Gautam Awatramani, and Raad Nashmi for their very insightful comments and suggestions. We thank Jennifer Graham, Maddie Beange, Tribesty Nguyen, Erin Carruthers, and Lisa Fleischauer for aiding with data collection, analysis, and mouse colony management.
References
- Arikan R, Blake NM, Erinjeri JP, Woolsey TA, Giraud L, Highstein SM. A method to measure the effective spread of focally injected muscimol into the central nervous system with electrophysiology and light microscopy. J Neurosci Methods. 2002;118:51–57. doi: 10.1016/s0165-0270(02)00143-7. [DOI] [PubMed] [Google Scholar]
- Auer RN. Insulin, blood glucose levels, and ischemic brain damage. Neurology. 1998;51:S39–S43. doi: 10.1212/wnl.51.3_suppl_3.s39. [DOI] [PubMed] [Google Scholar]
- Benison AM, Rector DM, Barth DS. Hemispheric mapping of secondary somatosensory cortex in the rat. J Neurophysiol. 2007;97:200–207. doi: 10.1152/jn.00673.2006. [DOI] [PubMed] [Google Scholar]
- Biernaskie J, Szymanska A, Windle V, Corbett D. Bi-hemispheric contribution to functional motor recovery of the affected forelimb following focal ischemic brain injury in rats. Eur J Neurosci. 2005;21:989–999. doi: 10.1111/j.1460-9568.2005.03899.x. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes. 1996;45:1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800:125–135. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Cristino NA, Rutten GJ, Hamers FP, Erkelens DW, Gispen WH. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats. Course of development and effects of insulin treatment. Brain. 1999;122:757–768. doi: 10.1093/brain/122.4.757. [DOI] [PubMed] [Google Scholar]
- Brands AM, Biessels GJ, de Haan EH, Kappelle LJ, Kessels RP. The effects of type 1 diabetes on cognitive performance: a meta-analysis. Diabetes Care. 2005;28:726–735. doi: 10.2337/diacare.28.3.726. [DOI] [PubMed] [Google Scholar]
- Brown CE, Dyck RH. Retrograde tracing of the subset of afferent connections in mouse barrel cortex provided by zincergic neurons. J Comp Neurol. 2005;486:48–60. doi: 10.1002/cne.20522. [DOI] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27:4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Aminoltejari K, Erb H, Winship IR, Murphy TH. In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci. 2009;29:1719–1734. doi: 10.1523/JNEUROSCI.4249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LM, Abbott DF, Puce A, Jackson GD, Syngeniotis A, Donnan GA. Reemergence of activation with poststroke somatosensory recovery: a serial fMRI case study. Neurology. 2002;59:749–752. doi: 10.1212/wnl.59.5.749. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis. 2001;8:910–922. doi: 10.1006/nbdi.2001.0425. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193:291–311. doi: 10.1016/j.expneurol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Borrel J. Functional recovery of forelimb response capacity after forelimb primary motor cortex damage in the rat is due to the reorganization of adjacent areas of cortex. Neuroscience. 1995;68:793–805. doi: 10.1016/0306-4522(95)00178-l. [DOI] [PubMed] [Google Scholar]
- Chaithongdi N, Subauste JS, Koch CA, Geraci SA. Diagnosis and management of hyperglycemic emergencies. Hormones (Athens) 2011;10:250–260. doi: 10.14310/horm.2002.1316. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Alloway KD. Differential origin of projections from SI barrel cortex to the whisker representations in SII and MI. J Comp Neurol. 2006;498:624–636. doi: 10.1002/cne.21052. [DOI] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavier I, Hommel M, Besson G, Noèlle B, Perret JE. Long-term prognosis of symptomatic lacunar infarcts. A hospital-based study. Stroke. 1994;25:2005–2009. doi: 10.1161/01.str.25.10.2005. [DOI] [PubMed] [Google Scholar]
- Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272–287. doi: 10.1002/ana.21393. [DOI] [PubMed] [Google Scholar]
- Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Ren J, Mandeville JB, Wu O, Ozdag FM, Moskowitz MA, Rosen BR, Finklestein SP. Functional magnetic resonance imaging of reorganization in rat brain after stroke. Proc Natl Acad Sci U S A. 2001;98:12766–12771. doi: 10.1073/pnas.231235598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8:449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- Ebenezer GJ, O'Donnell R, Hauer P, Cimino NP, McArthur JC, Polydefkis M. Impaired neurovascular repair in subjects with diabetes following experimental intracutaneous axotomy. Brain. 2011;134:1853–1863. doi: 10.1093/brain/awr086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, Elgebaly MM, Middlemore ML, Li W, Elewa H, Switzer JA, Hall C, Kozak A, Fagan SC. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol. 2007;7:33. doi: 10.1186/1471-2377-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr TD, Liu L, Colwell KL, Whishaw IQ, Metz GA. Bilateral alteration in stepping pattern after unilateral motor cortex injury: a new test strategy for analysis of skilled limb movements in neurological mouse models. J Neurosci Methods. 2006;153:104–113. doi: 10.1016/j.jneumeth.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferezou I, Haiss F, Gentet LJ, Aronoff R, Weber B, Petersen CC. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–549. doi: 10.1016/s0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- Hankey GJ, Spiesser J, Hakimi Z, Bego G, Carita P, Gabriel S. Rate, degree, and predictors of recovery from disability following ischemic stroke. Neurology. 2007;68:1583–1587. doi: 10.1212/01.wnl.0000260967.77422.97. [DOI] [PubMed] [Google Scholar]
- Iemolo F, Beghi E, Cavestro C, Micheli A, Giordano A, Caggia E. Incidence, risk factors and short-term mortality of stroke in Vittoria, southern Italy. Neurol Sci. 2002;23:15–21. doi: 10.1007/s100720200018. [DOI] [PubMed] [Google Scholar]
- Jablonka JA, Burnat K, Witte OW, Kossut M. Remapping of the somatosensory cortex after a photothrombotic stroke: dynamics of the compensatory reorganization. Neuroscience. 2010;165:90–100. doi: 10.1016/j.neuroscience.2009.09.074. [DOI] [PubMed] [Google Scholar]
- Jørgensen H, Nakayama H, Raaschou HO, Olsen TS. Stroke in patients with diabetes. The Copenhagen Stroke Study. Stroke. 1994;25:1977–1984. doi: 10.1161/01.str.25.10.1977. [DOI] [PubMed] [Google Scholar]
- Kaas JH. Sensory loss and cortical reorganization in mature primates. Prog Brain Res. 2002;138:167–176. doi: 10.1016/S0079-6123(02)38077-4. [DOI] [PubMed] [Google Scholar]
- Kamouchi M, Matsuki T, Hata J, Kuwashiro T, Ago T, Sambongi Y, Fukushima Y, Sugimori H, Kitazono T. Prestroke glycemic control is associated with the functional outcome in acute ischemic stroke: the Fukuoka Stroke Registry. Stroke. 2011;42:2788–2794. doi: 10.1161/STROKEAHA.111.617415. [DOI] [PubMed] [Google Scholar]
- Kennedy JM, Zochodne DW. Impaired peripheral nerve regeneration in diabetes mellitus. J Peripher Nerv Syst. 2005a;10:144–157. doi: 10.1111/j.1085-9489.2005.0010205.x. [DOI] [PubMed] [Google Scholar]
- Kennedy JM, Zochodne DW. Experimental diabetic neuropathy with spontaneous recovery: is there irreparable damage? Diabetes. 2005b;54:830–837. doi: 10.2337/diabetes.54.3.830. [DOI] [PubMed] [Google Scholar]
- Krubitzer L, Künzle H, Kaas J. Organization of sensory cortex in a Madagascan insectivore, the tenrec (Echinops telfairi) J Comp Neurol. 1997;379:399–414. [PubMed] [Google Scholar]
- Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat Rev Neurol. 2010;6:145–155. doi: 10.1038/nrneurol.2009.231. [DOI] [PubMed] [Google Scholar]
- Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab. 2007;27:710–718. doi: 10.1038/sj.jcbfm.9600382. [DOI] [PubMed] [Google Scholar]
- Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Li S, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, Giger RJ, Coppola G, Geschwind DH, Carmichael ST. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci. 2010;13:1496–1504. doi: 10.1038/nn.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Prakash R, Kelly-Cobbs AI, Ogbi S, Kozak A, El-Remessy AB, Schreihofer DA, Fagan SC, Ergul A. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59:228–235. doi: 10.2337/db09-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZG, Britton M, Sima AA, Dunbar JC. Diabetes enhances apoptosis induced by cerebral ischemia. Life Sci. 2004;76:249–262. doi: 10.1016/j.lfs.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- MacDougall NJ, Muir KW. Hyperglycaemia and infarct size in animal models of middle cerebral artery occlusion: systematic review and meta-analysis. J Cereb Blood Flow Metab. 2011;31:807–818. doi: 10.1038/jcbfm.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JI, Hanna S, Saporta S, Mervis RF, Park CR, Chong L, Diamond DM. Hyperglycemia not hypoglycemia alters neuronal dendrites and impairs spatial memory. Pediatr Diabetes. 2008;9:531–539. doi: 10.1111/j.1399-5448.2008.00431.x. [DOI] [PubMed] [Google Scholar]
- Mankovsky BN, Patrick JT, Metzger BE, Saver JL. The size of subcortical ischemic infarction in patients with and without diabetes mellitus. Clin Neurol Neurosurg. 1996;98:137–141. doi: 10.1016/0303-8467(95)00092-5. [DOI] [PubMed] [Google Scholar]
- Manschot SM, Biessels GJ, Cameron NE, Cotter MA, Kamal A, Kappelle LJ, Gispen WH. Angiotensin converting enzyme inhibition partially prevents deficits in water maze performance, hippocampal synaptic plasticity and cerebral blood flow in streptozotocin-diabetic rats. Brain Res. 2003;966:274–282. doi: 10.1016/s0006-8993(02)04211-7. [DOI] [PubMed] [Google Scholar]
- Martin JH, Ghez C. Pharmacological inactivation in the analysis of the central control of movement. J Neurosci Methods. 1999;86:145–159. doi: 10.1016/s0165-0270(98)00163-0. [DOI] [PubMed] [Google Scholar]
- Masuda T, Croom D, Hida H, Kirov SA. Capillary blood flow around microglial somata determines dynamics of microglial processes in ischemic conditions. Glia. 2011;59:1744–1753. doi: 10.1002/glia.21220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira T, Cebers G, Pickering C, Ostenson CG, Efendic S, Liljequist S. Diabetic Goto-Kakizaki rats display pronounced hyperglycemia and longer-lasting cognitive impairments following ischemia induced by cortical compression. Neuroscience. 2007;144:1169–1185. doi: 10.1016/j.neuroscience.2006.10.054. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci. 2010;30:14116–14126. doi: 10.1523/JNEUROSCI.3908-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi M, Fujioka M, He Q, Han A, Yong G, Csiszar K, Li PA. Diabetes activates cell death pathway after transient focal cerebral ischemia. Diabetes. 2003;52:481–486. doi: 10.2337/diabetes.52.2.481. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861–872. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M. Mechanisms of brain damage in focal cerebral ischemia. Acta Neurol Scand. 1988;77:81–101. doi: 10.1111/j.1600-0404.1988.tb05878.x. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Diemer NH. Focal ischemia of the rat brain, with special reference to the influence of plasma glucose concentration. Acta Neuropathol. 1987;73:131–137. doi: 10.1007/BF00693778. [DOI] [PubMed] [Google Scholar]
- Nhan H, Barquist K, Bell K, Esselman P, Odderson IR, Cramer SC. Brain function early after stroke in relation to subsequent recovery. J Cereb Blood Flow Metab. 2004;24:756–763. doi: 10.1097/01.WCB.0000122744.72175.9C. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Milliken GW. Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J Neurophysiol. 1996;75:2144–2149. doi: 10.1152/jn.1996.75.5.2144. [DOI] [PubMed] [Google Scholar]
- Reijmer YD, van den Berg E, de Bresser J, Kessels RP, Kappelle LJ, Algra A, Biessels GJ. Accelerated cognitive decline in patients with type 2 diabetes: MRI correlates and risk factors. Diabetes Metab Res Rev. 2011;27:195–202. doi: 10.1002/dmrr.1163. [DOI] [PubMed] [Google Scholar]
- Romo R, Salinas E. Touch and go: decision-making mechanisms in somatosensation. Annu Rev Neurosci. 2001;24:107–137. doi: 10.1146/annurev.neuro.24.1.107. [DOI] [PubMed] [Google Scholar]
- Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF, Lyden PD, Kleinfeld D. Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol. 2006;4:e22. doi: 10.1371/journal.pbio.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504. doi: 10.1016/j.nurx.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanina EV, Schallert T, Witte OW, Redecker C. Behavioral recovery from unilateral photothrombotic infarcts of the forelimb sensorimotor cortex in rats: role of the contralateral cortex. Neuroscience. 2006;139:1495–1506. doi: 10.1016/j.neuroscience.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Shih AY, Friedman B, Drew PJ, Tsai PS, Lyden PD, Kleinfeld D. Active dilation of penetrating arterioles restores red blood cell flux to penumbral neocortex after focal stroke. J Cereb Blood Flow Metab. 2009;29:738–751. doi: 10.1038/jcbfm.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham D, Glaser DE, Arieli A, Kenet T, Wijnbergen C, Toledo Y, Hildesheim R, Grinvald A. Imaging cortical dynamics at high spatial and temporal resolution with novel blue voltage-sensitive dyes. Neuron. 1999;24:791–802. doi: 10.1016/s0896-6273(00)81027-2. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008a;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008b;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant KA, Jones TA. Sensorimotor behavioral effects of endothelin-1 induced small cortical infarcts in C57BL/6 mice. J Neurosci Methods. 2009;181:18–26. doi: 10.1016/j.jneumeth.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni D, De Michele M, Fiorelli M, Bastianello S, Camerlingo M, Sacchetti ML, Argentino C, Fieschi C. Influence of hyperglycaemia on infarct size and clinical outcome of acute ischemic stroke patients with intracranial arterial occlusion. J Neurol Sci. 1994;123:129–133. doi: 10.1016/0022-510x(94)90214-3. [DOI] [PubMed] [Google Scholar]
- van Meer MP, van der Marel K, Wang K, Otte WM, El Bouazati S, Roeling TA, Viergever MA, Berkelbach van der Sprenkel JW, Dijkhuizen RM. Recovery of sensorimotor function after experimental stroke correlates with restoration of resting-state interhemispheric functional connectivity. J Neurosci. 2010;30:3964–3972. doi: 10.1523/JNEUROSCI.5709-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Willing LB, Goto S, Alkayed NJ, Brucklacher RM, Wood TL, Towfighi J, Hurn PD, Simpson IA. Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab. 2001;21:52–60. doi: 10.1097/00004647-200101000-00007. [DOI] [PubMed] [Google Scholar]
- Vinik A, Flemmer M. Diabetes and macrovascular disease. J Diabetes Complications. 2002;16:235–245. doi: 10.1016/s1056-8727(01)00212-4. [DOI] [PubMed] [Google Scholar]
- Ward NS. The neural substrates of motor recovery after focal damage to the central nervous system. Arch Phys Med Rehabil. 2006;87:S30–35. doi: 10.1016/j.apmr.2006.08.334. [DOI] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- Wei JW, Heeley EL, Wang JG, Huang Y, Wong LK, Li Z, Heritier S, Arima H, Anderson CS. Comparison of recovery patterns and prognostic indicators for ischemic and hemorrhagic stroke in China: the ChinaQUEST (QUality Evaluation of Stroke Care and Treatment) Registry study. Stroke. 2010;41:1877–1883. doi: 10.1161/STROKEAHA.110.586909. [DOI] [PubMed] [Google Scholar]
- Werhahn KJ, Conforto AB, Kadom N, Hallett M, Cohen LG. Contribution of the ipsilateral motor cortex to recovery after chronic stroke. Ann Neurol. 2003;54:464–472. doi: 10.1002/ana.10686. [DOI] [PubMed] [Google Scholar]
- Winship IR, Murphy TH. In vivo calcium imaging reveals functional rewiring of single somatosensory neurons after stroke. J Neurosci. 2008;28:6592–6606. doi: 10.1523/JNEUROSCI.0622-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Murphy TH. Imaging the impact of cortical microcirculation on synaptic structure and sensory-evoked hemodynamic responses in vivo. PLoS Biol. 2007;5:e119. doi: 10.1371/journal.pbio.0050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]