

Abstract
Seizure-induced release of the neuromodulator adenosine is a potent endogenous anticonvulsant mechanism, which limits the extension of seizures and mediates seizure arrest. For this reason several adenosine-based therapies for epilepsy are currently under development. However, it is not known how adenosine modulates GABAergic transmission in the context of seizure activity. This may be particularly relevant as strong activation of GABAergic inputs during epileptiform activity can switch GABAA receptor (GABAAR) signaling from inhibitory to excitatory, which is a process that plays a significant role in intractable epilepsies. We used gramicidin-perforated patch-clamp recordings to investigate the role of seizure-induced adenosine release in the modulation of postsynaptic GABAAR signaling in pyramidal neurons of rat hippocampus. Consistent with previous reports, GABAAR responses during seizure activity transiently switched from hyperpolarizing to depolarizing and excitatory. We found that adenosine released during the seizure significantly attenuated the depolarizing GABAAR responses and also reduced the extent of the after-discharge phase of the seizure. These effects were mimicked by exogenous adenosine administration and could not be explained by a change in chloride homeostasis mechanisms that set the reversal potential for GABAARs, or by a change in the conductance of GABAARs. Rather, A1R-dependent activation of potassium channels increased the cell's membrane conductance and thus had a shunting effect on GABAAR currents. As depolarizing GABAAR signaling has been implicated in seizure initiation and progression, the adenosine-induced attenuation of depolarizing GABAAR signaling may represent an important mechanism by which adenosine can limit seizure activity.
Introduction
Epilepsy is a severe neurological disorder characterized by recurrent seizures, which reflect a failure of inhibitory systems to contain the generation and spread of neuronal hyperexcitability. Most seizures are terminated either as a result of the depletion of factors necessary for sustaining seizure activity (e.g., energy substrates, ions, neurotransmitters) or as a result of endogenous inhibitory mechanisms that are triggered by elevated network activity. Studying such mechanisms is therefore a promising strategy for developing anti-epileptic therapies (Löscher and Köhling, 2010). One endogenous anticonvulsant mechanism is the seizure-induced release of the neuromodulator adenosine, which regulates synaptic activity via four known guanine nucleotide-binding protein (G-protein)-coupled adenosine receptors (ARs): A1R, A2AR, A2BR, and A3R (Boison and Stewart, 2009). In particular, activation of A1Rs has been shown to inhibit network excitability, limit the extension of seizures, and mediate seizure arrest (Etherington and Frenguelli, 2004; Fedele et al., 2006; Li et al., 2007). The coupling of A1Rs to Gi/Go proteins underlies several mechanisms by which adenosine can modulate synaptic transmission (Dunwiddie and Masino, 2001). In the case of glutamatergic transmission, presynaptic activation of A1Rs causes a reduction in release probability by inhibiting calcium (Ca2+) influx through voltage-gated channels (Yawo and Chuhma, 1993; Wu and Saggau, 1994; Gundlfinger et al., 2007), or by interfering directly with the release process (Scanziani et al., 1992; Thompson et al., 1993). At the same time, adenosine has been shown to function postsynaptically where it activates a series of downstream potassium (K+) channels, including G-protein-coupled, inward-rectifying K+ channels (GIRKs), that can modulate the excitatory effects of postsynaptic glutamate receptors (Takigawa and Alzheimer, 2002).
Relatively less is known about how adenosine modulates inhibitory synaptic transmission. This may be particularly relevant in the context of epilepsy, where a breakdown in effective GABA type A receptor (GABAAR)-mediated synaptic inhibition contributes to the ongoing dynamics of seizures (Wright et al., 2011). In the hippocampus, intense activation of GABAARs during early stages of a seizure results in a large influx of chloride (Cl−) (Isomura et al., 2003b; Fujiwara-Tsukamoto et al., 2006, 2007). The result is a collapse in the trans-membrane Cl− gradient and pronounced depolarizing shift in the reversal potential of the GABAAR (EGABAA) (Thompson and Gähwiler, 1989; Staley et al., 1995; Kaila et al., 1997). This depolarizing shift in EGABAA is thought to establish the conditions for networks of GABAergic neurons to generate synchronized after-discharges that are characteristic of epileptiform activity (Michelson and Wong, 1991; Fujiwara-Tsukamoto et al., 2010).
Recordings from hippocampal neurons have shown that exogenous adenosine does not affect the presynaptic release of GABA (Yoon and Rothman, 1991; Thompson et al., 1992). Whether adenosine contributes to the modulation of GABAAR signaling in the acute setting of a seizure remains unexplored. Here we use gramicidin perforated patch-clamp recordings in an in vitro model of temporal lobe epilepsy to study how adenosine released during seizures influences postsynaptic GABAAR signaling in CA3 pyramidal cells. We show that as seizure activity drives a switch in GABAergic signaling from inhibitory to excitatory, adenosine acts postsynaptically to reduce the resulting GABAAR-mediated depolarization.
Materials and Methods
Slice preparation.
Rat organotypic hippocampal slice cultures were prepared using a method similar to that described by Stoppini et al. (1991). Briefly, 7-d-old male Wistar rats were killed by cervical dislocation in accordance with Schedule 1 methods of UK Animals Scientific Procedures Act 1986. The brains were extracted and placed in cold (4°C) Geys Balanced Salt Solution, supplemented with d-glucose (34.7 mm). All reagents were purchased from Sigma-Aldrich, unless stated. The hemispheres were separated and individual hippocampi were removed and immediately sectioned into 350-μm-thick slices on a McIlwain tissue chopper. Slices were rinsed in cold dissection media, placed onto Millicell-CM membranes, and maintained in culture media containing 25% EBSS, 50% MEM, 25% heat-inactivated horse serum, glucose, and B27 (Invitrogen), and incubated at 36°C in a 5% CO2 humidified incubator. Recordings were performed after 7–14 d in vitro, when the hippocampal slices were comparable to postnatal day 14–21. Previous work in rat hippocampus has shown that EGABAA reaches mature levels within the first two postnatal weeks (Tyzio et al., 2007), and recordings from rat organotypic hippocampal slices have confirmed that GABAergic (Streit et al., 1989) and glutamatergic (De Simoni et al., 2003) synaptic transmission are mature at these stages. Consistent with this, we found that the resting EGABAA was hyperpolarizing (mean resting EGABAA = −82.7 ± 1.2 mV, ranging from −72.5 mV− to −91.2 mV−, n = 17) compared with the resting membrane potential (mean resting membrane potential = −73.5 ± 1.2 mV, ranging from −62.1 mV− to −79.7 mV−, n = 17). These resting EGABAA values are slightly more hyperpolarized than previously reported in the CA3 region (Tyzio et al., 2007), and might reflect differences in the level of endogenous GABAA receptor activity under different recording conditions and/or a bias toward selecting larger, more mature pyramidal neurons in our preparation.
Electrophysiological recordings.
To preserve the intracellular milieu of the cell and particularly the intracellular Cl− concentration ([Cl−]i) we used gramicidin-perforated patch-clamp recordings (Ebihara et al., 1995; Kyrozis and Reichling, 1995; Akerman and Cline, 2006). Patch pipettes of 3–5 MΩ tip resistance were pulled from filamental borosilicate glass capillaries (1.2 mm outer diameter, 0.69 mm inner diameter; Harvard Apparatus), using a horizontal puller (Sutter P-97). The pipettes were filled with a high KCl internal solution containing gramicidin. The composition of internal solution contained (in mm): 135 KCl, 4 Na2ATP, 0.3 Na3GTP, 2 MgCl2, and 10 HEPES. Osmolarity was adjusted to 290 mOsM and the pH was adjusted to 7.35 with KOH. Gramicidin (Calbiochem) was dissolved in dimethylsulfoxide (DMSO) to achieve a stock solution of 10 mg/ml. This was then diluted in internal solution on the day of experimentation to achieve a final concentration of 80 μg/ml. The resulting solution was vortexed for 1 min, sonicated for 30 s, and then filtered with a 0.45 μm pore cellulose acetate membrane filter (Nalgene).
Hippocampal slices were transferred to a recording chamber and continuously superfused with 95% O2/5% CO2 oxygenated artificial cerebrospinal fluid (aCSF), warmed to 30–32°C. The composition of aCSF contained (in mm): 120 NaCl, 3 KCl, 2 MgCl2, 1.5 CaCl2, 1.2 NaH2PO4, 23 NaHCO3, and 11 d-glucose. The pH was adjusted to be between 7.35 and 7.40 using NaOH. Previous studies have shown that hippocampal organotypic slices can be used as a post-traumatic model of epileptogenesis (Dyhrfjeld-Johnsen et al., 2010; Berdichevsky et al., 2012). By 7 d in vitro the tissue has recovered from the acute trauma associated with the slicing process and the neurons exhibit hyperexcitability, probably due to increased connectivity (Berdichevsky et al., 2012). Seizures were induced by perfusion with nominally Mg2+-free aCSF, and the perfusion media was switched back to normal aCSF after the start of a seizure episode. This transient exposure to 0 Mg2+ allowed us to induce reproducible single seizure episodes and prevented repeated seizure episodes that can occur with continuous exposure to 0 Mg2+.
Seizure onset was characterized by a pronounced membrane potential depolarization upon which occurred high-frequency, low-amplitude discharges (see Fig. 1). For analysis purposes the onset of the seizure was defined as the time at which the membrane potential depolarized beyond a “threshold” set at 40 mV above the cell's resting membrane potential (“baseline”). The end of the ictal-like phase and start of the after-discharge phase was defined as the point at which the membrane potential returned to baseline for the first time after the start of the seizure. The after-discharge phase was composed of rhythmic bursts of activity occurring from a relatively hyperpolarized membrane potential. An individual after-discharge event was defined as a burst of activity in which the membrane potential depolarized above the threshold for seizure onset, and then returned to below the baseline value. The end of the after-discharge phase was defined as the end of the last after-discharge event. Postseizure hyperpolarization was measured from the end of the after-discharge phase and expressed relative to the baseline membrane potential. All experiments involving seizure activity (see Figs. 1–3) were performed using uncompensated current-clamp recordings, without any DC injection in the I = 0 mode.
Figure 1.

Endogenous adenosine modulates seizure activity and postseizure recovery. A, Example seizures recorded from a CA3 pyramidal neuron under control conditions (upper trace) and during blockade of ARs with 1 μm CGS 15943 (lower trace). Recordings were performed using uncompensated current-clamp recordings, without DC current injection. Arrows indicate the ictal-like (Ictal) and after-discharge (AD) phases of the seizure. Horizontal dashed lines indicate preseizure resting membrane potential. Note that AR blockade results in an increase in total seizure duration and a faster recovery of the membrane potential to baseline levels following the end of the after-discharge phase. B, Population data (n = 10 cells) for seizure length under control conditions (black symbols) and in the presence of the AR antagonist (open symbols). Square symbols indicate individual experiments; circles indicate population means. C, Population data (n = 10 cells) for postseizure membrane potential recovery under control conditions (black circles) and in the presence of the AR antagonist (open circles). Values indicate the degree of hyperpolarization compared with preseizure baseline, where zero minutes represents the point of maximum hyperpolarization following the end of the after-discharge phase. Error bars indicate SEM, **p < 0.01, paired t test.
Figure 2.

Seizure activity induces depolarizing and excitatory GABAAR signaling. A, Example seizure recorded from a CA3 pyramidal neuron. Postsynaptic GABAAR responses were elicited with a muscimol puff every 15 s (vertical bars) during the seizure episode. Dashed boxes indicate time windows before the seizure (t1) during the after-discharge phase (t2), immediately after the seizure (t3), and during the postseizure recovery period (t4). B, Expanded traces from the time windows represented by the boxes in A. GABAAR responses before the onset of the seizure are hyperpolarizing (t1). During the after-discharge period GABAAR responses become depolarizing and can trigger spikes (t2). Immediately after the seizure, GABAAR responses are still depolarizing but show decreased amplitude and are less likely to trigger spikes (t3). During the postseizure recovery period GABAAR responses return to being hyperpolarizing (t4). C, Recording in which somatic and dendritic EGABAA were measured during seizure activity. Dashed boxes indicate example EGABAA measurements before the seizure (t1), shortly after the ictal phase (t2), and later during the recovery period (t3). Somatic EGABAA values are represented by the circles. To measure EGABAA without contamination from ongoing spiking activity, TTX was applied ∼2 min after seizure onset (horizontal black bar). D, EGABAA was measured by rapidly switching the recording mode from current-clamp (CC) to voltage-clamp and then applying two consecutive voltage ramps, the first under baseline conditions (black line; −GABA) and the second during activation of GABAARs (gray line; + GABA) either on the soma or on the apical dendrites (∼150 μm from the soma). After 2 s in voltage-clamp the cell was returned to current-clamp. EGABAA measurements were made every 10 s. E, I–V plots for somatic (top) and dendritic (bottom) measurements for the three time points represented by the boxes in C. EGABAA was defined as the voltage at which the holding current (generated by the first ramp; black line) and the total current (reflecting the holding current plus the current through the activated GABAARs; gray line) intersected. Note that both somatic and dendritic EGABAA were at hyperpolarized values before the seizure (t1), switched to depolarizing values during the seizure (t2), and recovered to preseizure values (t3).
Figure 3.
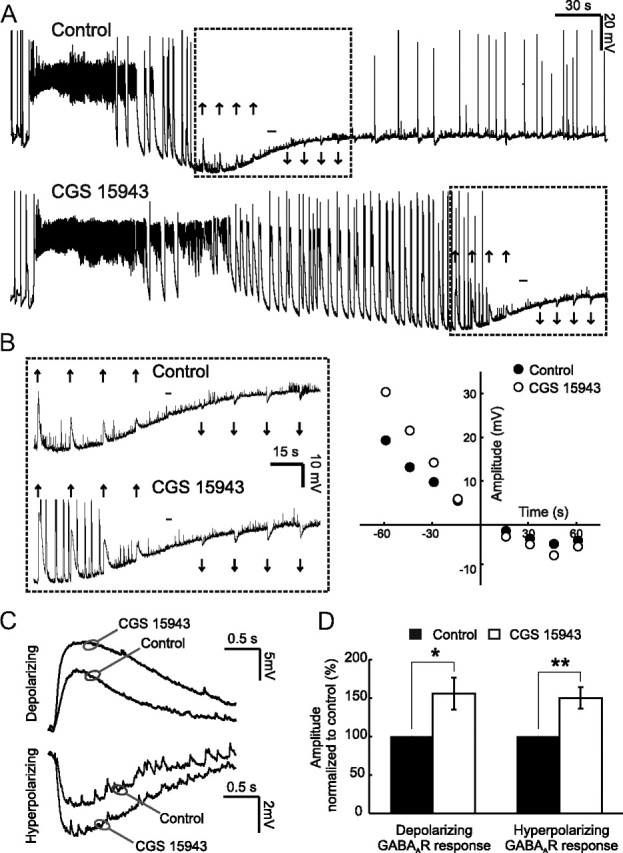
AR activity attenuates postsynaptic GABAergic potentials during seizures. A, Example seizures recorded under control conditions (upper trace) and in the presence of the AR antagonist CGS 15943 (1 μm) (lower trace). Upward arrows indicate depolarizing postsynaptic GABAAR responses, downward arrows indicate hyperpolarizing GABAAR responses, and the hyphen indicates the reversal point, when there is no net response to a muscimol puff. Rectangles indicate a 2 min time window, centered about the reversal for the GABAAR response. B, Traces (left) are expanded versions of the recording in A and illustrate the amplitude and direction of GABAAR responses. The plot (right) shows the amplitude of GABAAR responses in this recording (y-axis), in relation to the time when the GABAAR response switched back from depolarizing to hyperpolarizing (at 0 s on the x-axis); black circles represent GABAAR responses amplitude under control conditions, while open circles represent GABAAR responses amplitude under AR blockade. C, Averaged traces for the cell shown in A confirm that blocking ARs results in an attenuation of both the depolarizing and hyperpolarizing GABAAR responses. D, Population data (n = 10 cells) for the amplitude of depolarizing and hyperpolarizing postsynaptic GABAAR responses. Data are shown for the AR antagonist (open bars), normalized to control values (black bars). Error bars indicate SEM, *p < 0.05, **p < 0.01, paired t test.
For the seizure-free exogenous adenosine experiments, slices were perfused with normal aCSF containing tetrodotoxin (TTX; 1 μm) to prevent spiking activity. To measure hyperpolarizing GABAAR responses under seizure-free conditions, the resting membrane potential was held at −70 mV by somatic DC current injection. Depolarizing GABAAR responses were recorded from a resting membrane potential of −110 mV, established by injecting DC current through the recording pipette in the presence of 200 μm furosemide to block the activity of Cl− transporter proteins. Neurons were visualized under a 60×, water-immersion objective (Olympus BX51WI) and pyramidal neurons from the CA3 region were identified based upon their location within the hippocampus and their triangular somatic morphology. Following formation of a tight seal between the pipette and the underlying plasma membrane (>1 GΩ), the perforation progress was evaluated by continuously monitoring the decrease in the access resistance. Recordings were started once the access resistance had stabilized between 308 and 0 MΩ (mean Ra ∼ 55 MΩ), which usually occurred at ∼20–50 min from gigaseal. During off-line analysis, all membrane potentials were subsequently corrected for the voltage drop across the access resistance. Recordings were made using an Axopatch 1D amplifier (Molecular Devices) and data acquired using WinWCP Strathclyde Whole Cell Analysis software (V.3.9.7; University of Strathclyde) and stored for off-line analysis. GABAARs were activated by delivering short “puffs” of the GABAAR-selective agonist, muscimol (45 μm), through a patch pipette placed in the vicinity of the cell and connected to a picospritzer (5–10 psi for 20–500 ms; General Valve). Puffs were delivered at 15 s intervals to allow recovery of chloride homeostasis (Staley and Proctor, 1999; Jin et al., 2005). At the end of the experiment the integrity of the perforated patch was confirmed by applying suction to rupture the underlying membrane and achieve whole-cell recording mode. This led to a rapid decrease in the access resistance and a shift in EGABAA due to diffusion of Cl− from the pipette into the cell's cytoplasm.
All experiments comparing seizure activity, GABAAR responses, resting EGABAA, GABAAR conductance, and membrane conductance were conducted with a within-cell design, so that each parameter was compared before and after a treatment. To measure the amplitude of individual GABAAR potentials in current-clamp, the membrane potential immediately before the muscimol puff (measured over a 20 ms window) was subtracted from the membrane potential at the peak of the muscimol response (measured over a 5 ms window). During the postseizure period GABAAR potentials were found to be depolarizing and then gradually decreased in amplitude and reversed polarity to become hyperpolarizing. To estimate the kinetics of this recovery of GABAAR responses we fitted an exponential to the amplitudes of depolarizing GABAAR responses during a 1 min time window before their reversal to hyperpolarizing. The recovery rate was calculated as the time constant of the exponential fit.
To measure resting EGABAA, muscimol-evoked currents were recorded in the voltage-clamp configuration at six different holding potentials (between −50 and −100 mV). For each cell, I–V curves were constructed for the “holding current” and the peak of the “total current” (reflecting the holding current plus the current through the activated GABAARs). EGABAA was defined as the intersection of these two I–V curves, which indicated the voltage at which there was no net current through the GABAAR (see Fig. 6A). To estimate peak GABAAR conductance (see Fig. 7A), a 1 s voltage ramp (from −100 to −30 mV) was applied to the membrane to provide a more accurate estimate of the holding current at every membrane potential. “Pure” GABAAR currents were then calculated by subtracting this holding current from the peak of the total current (in response to muscimol) for specific membrane potentials. In this manner I–V plots of the peak GABAAR current were constructed and the peak GABAAR conductance was calculated as the slope of the linear fit. Membrane conductance (see Fig. 5A) was calculated from the membrane current generated by 1 s hyperpolarizing voltage steps (−10 mV), delivered under voltage-clamp conditions.
Figure 6.
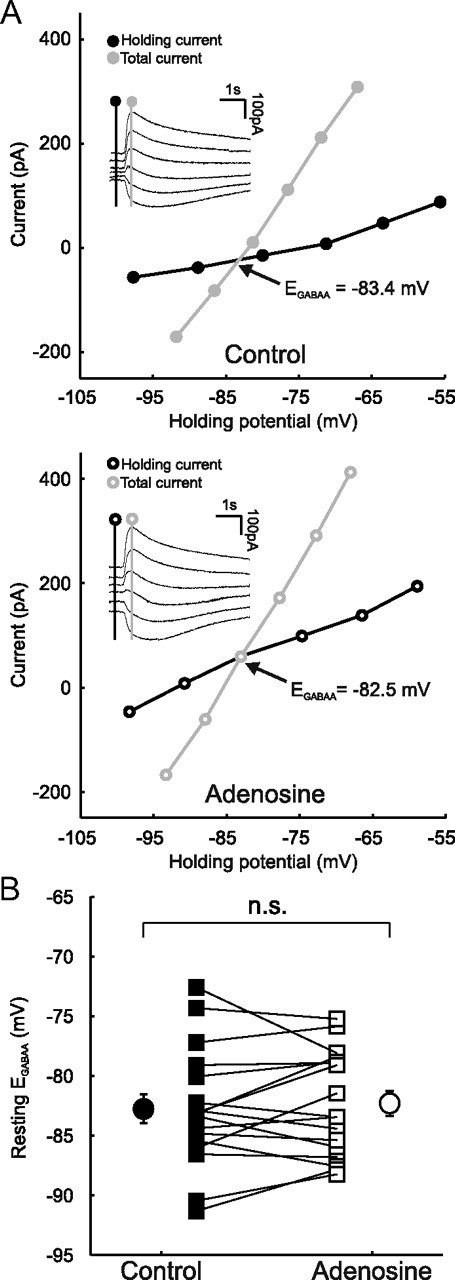
AR activation does not alter Cl− homeostasis. A, Data from a representative CA3 pyramidal neuron in which resting EGABAA was measured under control conditions (top) and in the presence of 100 μm adenosine (bottom). Inserts, Example of GABAAR currents evoked by muscimol puffs at different holding potentials. Holding current (reflecting membrane current) and total current (reflecting membrane current plus the muscimol-evoked current) were measured at the points indicated by the vertical black and gray lines, respectively. I–V plots were constructed and EGABAA was calculated as the potential at which the total current was equal to the holding current. B, Resting EGABAA values for individual cells (n = 17) under control conditions (black squares) and in the presence of adenosine (open squares). Population averages are also shown (black circles, control; open circles, adenosine). Error bars indicate SEM; p > 0.05, paired t test.
Figure 7.
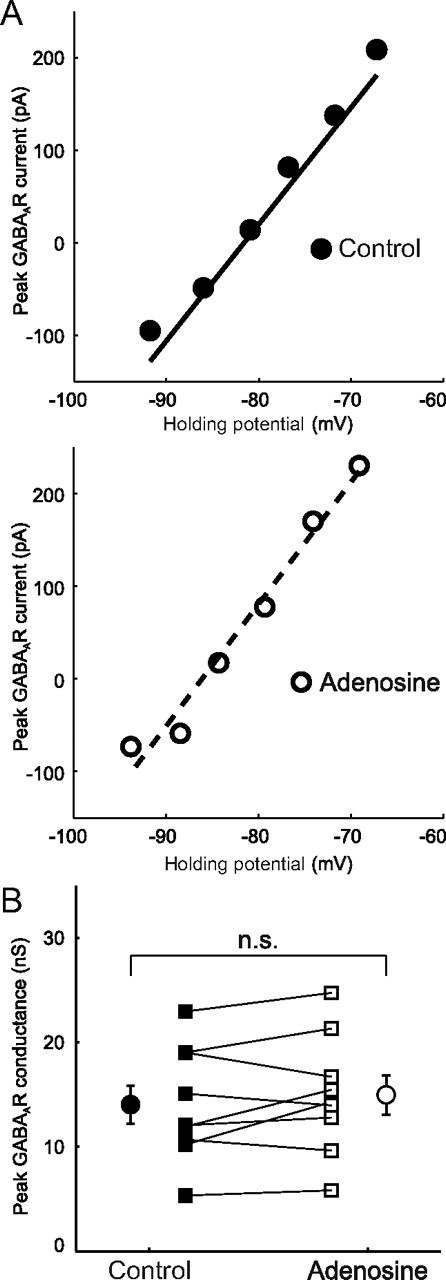
AR activation has no effect on GABAAR conductance. A, Data from a representative CA3 pyramidal neuron in which the GABAAR conductance was measured under control conditions (top) and in the presence of 100 μm adenosine (bottom). The peak GABAAR current was calculated as described in the text and the peak GABAAR conductance was calculated as the slope of the linear fit. B, Peak GABAAR conductance for individual cells (n = 9) under control conditions (black squares) and in the presence of adenosine (open squares). Population averages are also shown (black circles, control; open circles, adenosine). Error bars indicate SEM; p > 0.05, paired t test.
Figure 5.

Adenosine attenuates postsynaptic GABAAR potentials via A1R activation of K+ channels that increase membrane conductance. A, Adenosine (Ado; 100 μm; black bar, n = 23) increased the membrane conductance to ∼220% of control values. Coadministration of adenosine with the selective A1R antagonist DPCPX (5 μm; n = 6) prevented the adenosine-induced increase in membrane conductance. Similar results were obtained when adenosine was coadministered with either a nonspecific blocker of K+ channels (Ba2+ 1.5 mm; n = 6), or with a mixture of K+ channel blockers containing the KATP channel blocker (tolbutamide 1 mm), a GIRK blocker (tertiapin 400 nm), and a small conductance Ca2+-activated K+ channel blocker (apamine 400 nm; n = 10). B, Population data for the amplitude of postsynaptic GABAAR responses for each of the groups in A. When adenosine was delivered alone it reduced the amplitude of GABAAR responses by ∼40%. This effect was blocked when adenosine was applied in the presence of DPCPX, Ba2+, or the K+ channel blocker mixture. All measurements were normalized to control values in the absence of any drugs. Statistical tests reflect comparisons with the adenosine-only group. Error bars indicate SEM, *p < 0.05, **p < 0.01, ANOVA with post hoc Bonferroni's correction. C, D, The effect of 1, 10, and 100 μm adenosine on membrane conductance (C) and GABAAR response amplitude (D). Results from statistical tests indicate within cell comparisons before and after adenosine administration. Error bars indicate SEM, **p < 0.01, ***p < 0.001, paired t test. E, The increase in membrane conductance showed a significant negative correlation with the GABAAR response amplitude (Pearson correlation, r2 = 0.49, p < 0.001, n = 24). Dashed line indicates linear fit of the data; open squares represent individual experiments.
To measure EGABAA during seizure activity, recordings were rapidly alternated between current-clamp and voltage-clamp modes. The current-clamp recordings allowed the monitoring of seizure activity, while the periods in voltage-clamp (2 s duration) were used to measure EGABAA with voltage ramp protocols (see Fig. 2C–E). Each period in voltage-clamp consisted of two consecutive voltage ramps (500 ms duration, ramping from −90 to −40 mV). The first ramp was applied under baseline conditions and the second ramp was applied during activation of GABAARs by local application of GABA (100 μm), in the presence of the GABAB receptor blocker CGP55845 (5 μm). This made it possible to construct I–V plots of the holding current generated by the first ramp and the total current generated by the second ramp (reflecting the holding current plus the current through the activated GABAARs) (Fig. 2D). EGABAA was defined as the voltage at which the holding current and the total current intersected (Fig. 2E).
Drug manipulations.
Pharmacological manipulations were performed by bath application of drugs using a perfusion system. Adenosine was dissolved directly into the aCSF on the day of experiment, whereas all other drugs were prepared in stock solutions, stored at −20°C and added to aCSF on the day of experiment to reach the required concentration. Tolbutamide, CGS 15943, and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) were dissolved in DMSO. All other drugs were dissolved in normal aCSF. Adenosine, BaCl2, furosemide, and tolbutamide were purchased from Sigma-Aldrich. Muscimol, TTX, tertiapin, apamine, CGS 15943, CGP55845, and DPCPX were purchased from Tocris Bioscience.
Data analysis and statistics.
Digital signal processing and presentation were performed using custom-made programs in the MATLAB environment (MathWorks). Numeric results are presented as mean ± SEM. Statistical comparisons were performed using the paired t test or ANOVA, with post hoc Bonferroni's correction.
Results
Endogenous adenosine release modulates seizure duration and the postseizure recovery period
We set out to investigate the postsynaptic effects of adenosine released during epileptic-like seizure activity. A commonly used model of seizure activity is the removal of extracellular Mg2+, which predisposes hippocampal slices to periods of synchronized hyperexcitability (Anderson et al., 1986; Mody et al., 1987; Gutiérrez et al., 1999; Avoli et al., 2002). We monitored seizure activity by performing current-clamp recordings from hippocampal CA3 pyramidal neurons using the gramicidin-perforated patch method. In accordance with previous work, we could distinguish two phases of the seizure episodes: (1) the initial ictal-like phase (also referred to as the “tonic” phase) and (2) the after-discharge phase (also referred to as the “clonic” phase) (Higashima et al., 1996; Trevelyan et al., 2007; Isomura et al., 2008). The ictal phase was characterized by a >40 mV membrane potential depolarization upon which occurred high-frequency, low-amplitude discharges (Fig. 1A). As the seizure progressed, the membrane potential slowly repolarized, reached preseizure resting membrane potential values, and then entered the after-discharge phase. The after-discharge phase was composed of rhythmic bursts of activity that emerged from a relatively hyperpolarized membrane potential background. Beyond the after-discharge phase, the membrane potential remained at relatively hyperpolarized values for several minutes and slowly recovered to preseizure values (Fig. 1A). Comparing two consecutive seizures within the same recordings confirmed that the seizures were highly reproducible. There was no difference between the first and second seizure in terms of seizure length (first seizure mean was 205.2 ± 52.8 s, second seizure mean was 225.1 ± 48.8 s; n = 5, p = 0.58) or the extent of membrane hyperpolarization postseizure (first seizure showed a mean hyperpolarization relative to baseline of −18.3 ± 3.2 mV, second seizure mean was −19.1 ± 1.5 mV; n = 5, p = 0.71). Using this model we were therefore able to explore the contribution of AR signaling during seizure episodes, using pharmacological manipulations in a within-slice paired design.
To test whether endogenous adenosine is released at levels that modulate a seizure episode, we investigated the effect of 1 μm CGS 15943, an antagonist of the four known ARs. Blockade of ARs had a significant effect on the seizures, increasing total seizure duration from 272.8 ± 43.8 s to 666.4 ± 121.8 s (n = 10, p < 0.01, paired t test; Fig. 1B). This effect was primarily due to an increase in the after-discharge period from 185.7 ± 43.8 s to 565.7 ± 129.7 s (n = 10, p < 0.01, paired t test). Meanwhile the duration of the ictal phase was not significantly affected by AR antagonism, as it had a duration of 87.0 ± 19.6 s under control conditions and 100.6 ± 27.3 s following application of CGS 15943 (n = 10, p = 0.61, paired t test). To further investigate the time course of adenosine's release and effects we compared the membrane potential (averaged in 5 s time bins) from the start of each seizure, under control conditions, and during AR antagonism. There was no difference between the two conditions at the start of the seizure. However, over the course of the seizure the membrane potential became more hyperpolarized under control conditions, than during AR antagonism, and this was first evident 25 s from the start of the seizure (absolute membrane potential −42.2 ± 1.6 mV vs −37.9 ± 1.0 mV, n = 10, p < 0.05, paired t test). Blockade of ARs also reduced the degree of postseizure membrane hyperpolarization. For instance, the maximum postseizure hyperpolarization under control conditions was −22.9 ± 1.6 mV relative to baseline, and this was reduced to −15.5 ± 1.9 mV when ARs were blocked (n = 10, p < 0.01, paired t test; Fig. 1C). The adenosine-induced effects persisted for several minutes into the postseizure period, a time course that matches with previous reports of AR antagonism on the recovery of evoked field potentials (Etherington and Frenguelli, 2004; Etherington et al., 2009). More generally, our data demonstrate that ARs are strongly activated during seizures and function to attenuate seizure activity. This is consistent with earlier evidence that adenosine acts mainly as a seizure-suppressing substance (During and Spencer, 1992; Dunwiddie, 1999; Boison, 2006).
AR activation attenuates GABAA receptor-mediated depolarization during epileptiform activity
Previous work has demonstrated strong stimulation of GABAARs during periods of hypersynchronous activity, such as ictal-like episodes. It has been argued that intense activation of GABAARs leads to a pronounced influx and accumulation of intracellular Cl−, which generates a transient-positive shift in EGABAA (Isomura et al., 2008; Wright et al., 2011) that is often followed by a transient increase in extracellular potassium (Kaila et al., 1997; Viitanen et al., 2010). This rapid depolarizing shift in EGABAA can create a situation where GABAAR activity excites pyramidal cells and thus actually promotes their activity further (Köhling et al., 2000; Fujiwara-Tsukamoto et al., 2010). Our gramicidin recordings enabled us to record GABAAR-mediated signals without disturbing intracellular chloride concentration dynamics during seizure episodes. As shown in Figure 2, local somatic application of the selective GABAAR agonist, muscimol (45 μm), hyperpolarized the membrane potential of CA3 pyramidal neurons in the period before the seizure. However, during the after-discharge phase of the seizure and immediately following the seizure, GABAAR activation strongly depolarized neurons (Fig. 2B). Importantly, GABAAR responses during the after-discharge phase were sufficient to trigger action potentials. In the postseizure period, the depolarizing GABAAR responses decreased gradually, were no longer able to trigger action potentials, and eventually reversed polarity and reverted to hyperpolarizing (Fig. 2B). To further investigate the mechanisms underlying the seizure-associated alteration in GABAAR signaling, we performed a separate series of experiments to measure EGABAA during seizure activity. By rapidly switching from current-clamp to voltage-clamp mode during gramicidin recordings we were able to estimate EGABAA using voltage ramp protocols (Fig. 2C–E). EGABAA was measured at two points along the somatodendritic axis by activating GABAARs at both somatic and dendritic sites (apical dendrites, ∼150 μm from the soma) using two separate muscimol puffer pipettes. As shown in Figure 2C and E, ictal-like activity induced a pronounced depolarizing shift in both the somatic and dendritic EGABAA. The fact that the dendritic EGABAA shifted suggests that dialysis of cations via the gramicidin pores was unlikely to contribute significantly to these effects. Furthermore, the time course of the EGABAA changes closely matched the alterations in GABAAR potentials described by Figure 2A. These results are consistent with a seizure-induced increase in intracellular Cl−, which shifts EGABAA to depolarizing values and establishes the conditions for GABAAR activation to trigger action potential activity. Consistent with a recovery of the Cl− gradient in the postseizure period, EGABAA gradually returned to baseline values and GABAAR responses reverted to hyperpolarizing.
To test whether endogenous adenosine modulates these seizure-induced changes in GABAAR transmission, we used a within-cell design to measure GABAAR-mediated potentials during control seizures and during seizures in which ARs were blocked with CGS 15943 (1 μm; Fig. 3). The amplitude of depolarizing GABAAR potentials was measured during the 2 min period before the muscimol response reversed polarity (Fig. 3A,B). AR blockade had a strong and consistent effect on depolarizing GABAAR responses recorded during the seizure episode. Blockade of ARs increased the amplitude of depolarizing GABAAR responses to 155.5 ± 20.9% of control values (n = 10, p < 0.05, paired t test; Fig. 3C,D), indicating that seizure-induced adenosine release could attenuate depolarizing GABAergic potentials during seizures. Consistent with this hypothesis, when we examined the number of after-discharge events per seizure, we found that AR blockade increased their mean number from 45.8 ± 9.22 under control conditions to 87.30 ± 11.31 (n = 10, paired t test, p < 0.05). As depolarizing GABAAR responses can contribute to prolongation of seizures and the generation of after-discharges (Higashima et al., 1996, 2000; Velazquez and Carlen, 1999; Fujiwara-Tsukamoto et al., 2003; Isomura et al., 2003a, 2008), the attenuation of depolarizing GABAAR activity by adenosine may be an important aspect of adenosine's anticonvulsant action. We also examined the modulatory effect of AR activation after GABAAR responses had reverted to hyperpolarizing. Hyperpolarizing GABAAR responses were measured during a 2 min period after the muscimol response reversed polarity (Fig. 3A,B). Similar to the effects on depolarizing GABAAR responses, antagonism of ARs increased the amplitude of the hyperpolarizing GABAAR responses to 150 ± 13.7% of control values (n = 10, p < 0.05, paired t test) (Fig. 3C,D). These results demonstrate that seizure-induced adenosine release attenuates not only the excitatory GABAergic potentials, but can also persist and modulate inhibitory GABAAR-mediated signaling.
Adenosine attenuates postsynaptic GABAA receptor signaling by increasing membrane conductance via activation of K+ channels, downstream of the A1R
The fact that AR activation during a seizure can attenuate both depolarizing and hyperpolarizing GABAAR responses could be explained via an adenosine-induced change in the postsynaptic cell's membrane conductance, which would generate a “shunting” effect (Takigawa and Alzheimer, 2002). First, to establish if AR activation is sufficient to generate the observed effects, we tested whether exogenously applied adenosine (100 μm) is sufficient to attenuate the GABAAR-mediated responses in seizure-free slices (see Materials and Methods; Fig. 4). Bath application of adenosine induced a hyperpolarization of the membrane potential of CA3 pyramidal neurons from −76.5 ± 0.9 mV to −89.7 ± 0.9 mV (n = 6, p < 0.001, paired t test), consistent with the downstream activation of K+ channels (Fredholm et al., 2005; Clark et al., 2009). To maintain the same driving force for the GABAAR, we compensated for the adenosine-induced hyperpolarization by current injection through the patch pipette. The baseline membrane potential was maintained at either −110 mV to compare depolarizing GABAAR responses, or at −70 mV to compare hyperpolarizing GABAAR responses (Fig. 4A,C). In the presence of adenosine, depolarizing GABAAR responses were significantly smaller. The amplitude was reduced to 68.1 ± 4.7% of control values (n = 5, p < 0.01, paired t test), which was reversed following washout of the adenosine (97.7 ± 5.9% of control, p < 0.01, paired t test compared with adenosine; Fig. 4B). Under conditions that generated hyperpolarizing GABAAR responses, we saw a similar reduction in GABAergic signaling. Adenosine caused a reduction in the amplitude of GABAAR responses to 69.5 ± 5.7% of control values (n = 6, p < 0.01, paired t test) and this effect was reversed following washout of the adenosine (104.7 ± 7.8% of control, p < 0.01, paired t test compared with adenosine; Fig. 4D). These results demonstrate that activation of ARs is sufficient to attenuate postsynaptic GABAAR signaling when it is either depolarizing or hyperpolarizing.
Figure 4.

Exogenous adenosine application mimics the inhibitory effect on GABAAR-mediated signaling. Depolarizing and hyperpolarizing GABAAR responses were elected by local application of muscimol under control conditions and in the presence of 100 μm adenosine. A, Depolarizing GABAAR responses recorded from a CA3 pyramidal neuron under control conditions (left) and in the presence of adenosine (right). Cells were maintained at a resting membrane potential of −110 mV by somatic DC current injection (see Materials and Methods). Individual (gray traces) and mean (black traces) GABAAR potentials are shown. B, Population data (n = 5 cells) for the amplitude of depolarizing postsynaptic GABAAR responses in the presence of adenosine (open bars) and after adenosine washout (gray bars), normalized to control values (black bars). Error bars indicate SEM, **p < 0.01, paired t test. C, Hyperpolarizing GABAAR responses recorded from a CA3 pyramidal neuron under control conditions (left) and in the presence of adenosine (right). Cells were maintained at a resting membrane potential of −70 mV by somatic DC current injection. D, Population data (n = 6 cells) for the amplitude of hyperpolarizing postsynaptic GABAAR responses, using the same conventions as above. Error bars indicate SEM, **p < 0.01, paired t test.
To test whether a shunting mechanism can account for the effect of adenosine upon GABAAR responses, we used voltage-clamp and current-clamp recordings to measure the effect of adenosine on membrane conductance and the role of various downstream K+ channels. Adenosine has been reported to activate several K+ channels via A1R signaling (Hosseinzadeh and Stone, 1998; Fredholm et al., 2005; Clark et al., 2009; Kawamura, Jr. et al., 2010) including: GIRKs (Fredholm et al., 2005; Clark et al., 2009), ATP-sensitive K+ channels (KATPs) (Hosseinzadeh and Stone, 1998; Kawamura, Jr. et al., 2010), and small conductance Ca2+-activated K+ channels (SKs) (Clark et al., 2009). As predicted, voltage-clamp recordings showed that the application of adenosine resulted in a significant shift in the membrane conductance of CA3 pyramidal neurons from 4.5 ± 0.4 nS to 9.9 ± 1.0 nS. This represented an increase to 225.6 ± 14.9% of control values (n = 23, p < 0.001, paired t test; Fig. 5A). Furthermore, the effect of adenosine on membrane conductance was substantially reduced by coadministration of the A1R antagonist DPCPX (5 μm; 118.4 ± 8.3% of control, n = 6, p < 0.01), by coadministration of the nonselective K+ channel blocker Ba2+ (1.5 mm; 113.1 ± 5.4% of control, n = 6, p < 0.01), or by coadministration of a mixture of K+ channel blockers designed to block the major downstream targets of A1Rs (GIRKs, 400 nm tertiapin; KATPs, 1 mm tolbutamide; and SKs, 400 nm apamine; 141.2 ± 8.7% of control, n = 10, p < 0.01; Fig. 5A).
Importantly, the same pharmacological manipulations abolished the effect of adenosine on GABAAR responses. The effects of GABAAR activation upon the membrane potential were measured in current-clamp, with the baseline membrane potential maintained at −70 mV. As before, adenosine alone significantly reduced GABAAR responses amplitude to 59.3 ± 3.2% of control (n = 25, p < 0.0001, paired t test compared with control). However, this effect of adenosine was prevented by coadministration of the A1R antagonist, DPCPX (94.2 ± 3.1% of control; n = 5, p < 0.01, ANOVA with post hoc Bonferroni's correction). The effect of adenosine upon GABAAR responses was also abolished by coadministration of the nonselective K+ channel blocker Ba2+ (88.2 ± 11.5% of control; n = 6, p < 0.01, ANOVA with post hoc Bonferroni's correction), or by coadministration of the mixture of K+ channel blockers designed to block the major downstream targets of A1Rs (84.9 ± 4.5% of control; n = 5, p < 0.05, ANOVA with post hoc Bonferroni's correction; Fig. 5B,C). There was no significant difference between the DPCPX, Ba2+, and K+ channel blockers mixture for membrane conductance or GABAAR-mediated responses amplitude (p > 0.05 in all cases, ANOVA with post hoc Bonferroni's correction). These results suggest that adenosine suppresses postsynaptic GABAAR-mediated responses by increasing the cell's membrane conductance via activation of K+ channels that are downstream of A1R signaling.
To better understand how the changes in membrane conductance and GABAAR response relate to extracellular adenosine concentration we tested the effects of 1, 10, and 100 μm adenosine (Fig. 5C,D). Exogenous application of 1 μm adenosine had no statistically significant effect on either the membrane conductance (106.4 ± 6.4% of control, p = 0.37, n = 5, paired t test) or on the amplitude of GABAAR potentials (96.7 ± 3.4% of control, p = 0.44, n = 3, paired t test). In contrast, application of 10 μm adenosine increased the membrane conductance to 157.4 ± 15.0% of control (p < 0.01, n = 8, paired t test) and decreased the amplitude of GABAAR potentials to 78.7 ± 5.2% of control (p < 0.01, n = 7, paired t test). Finally, application of 100 μm adenosine increased the membrane conductance to 228.4 ± 19.7% of control (p < 0.001, n = 14, paired t test, Fig. 5C) and decreased the GABAAR response amplitude to 53.4 ± 3.5% of control (p < 0.001, n = 14, paired t test, Fig. 5D). The importance of extracellular adenosine concentration was also evident from the correlation between the adenosine-induced increase in membrane conductance and the decrease in GABAAR response amplitude (Pearson correlation, r2 = 0.49, p < 0.001, n = 24; Fig. 5E). In terms of kinetics, previous studies using rapid agonist application in dissociated neurons have reported that A1R-induced activation of K+ channels has a latency of ∼0.1 s and is maximal within 5 s (Sodickson and Bean, 1998). In our preparation we found that the peak of the adenosine-induced response was reached after 40.5 ± 6.5 s (n = 10), presumably reflecting the slower diffusion of adenosine into the slice.
GABAAR reversal potential and conductance are not altered by AR activation
Other mechanisms that could contribute to the adenosine-induced effect upon postsynaptic GABAAR-mediated responses include alterations in Cl− homeostasis mechanisms (Kaila et al., 1997; Dzhala et al., 2005, 2010; Jin et al., 2005; Huberfeld et al., 2007) and/or changes in the conductance of the GABAAR (Roseti et al., 2008, 2009). To investigate whether AR activation affects Cl− homeostasis we measured EGABAA under control conditions and following bath application of adenosine. Muscimol-evoked GABAAR currents were recorded at six different holding voltages (from −50 to −100 mV) and EGABAA was calculated from the resulting I–V plots (see Materials and Methods; Fig. 6A). Under control conditions the mean EGABAA was −82.7 ± 1.2 mV and this remained unchanged following activation of ARs, when the mean EGABAA was −82.2 ± 1.0 mV (n = 17, p = 0.48, paired t test; Fig. 6B). These results suggest that adenosine signaling does not modulate Cl−-regulating mechanisms that set the EGABAA. To further explore a potential role of adenosine in modulating Cl− homeostasis mechanisms during seizure activity, we analyzed our seizure data to assess the rate at which GABAAR responses recovered from depolarizing back to hyperpolarizing during the postseizure period (Fig. 3). In agreement with previous studies of chloride homeostasis, we found that the recovery of depolarizing GABAAR responses could be estimated using an exponential fit (Brumback and Staley, 2008; Nardou et al., 2011). Under control conditions we found that the recovery rate had a time constant of 34.7 ± 2.8 s and that this was not affected by AR blockade with CGS 15943, when the mean time constant was 31.6 ± 3.0 (p = 0.22, n = 8, paired t test).Together, these results show that changes in Cl− homeostasis mechanisms are unlikely to contribute to the attenuation of the GABAAR-mediated potentials.
Reducing either the postsynaptic density of GABAAR receptors and/or their permeability would result in a decreased GABAAR conductance, which could also potentially contribute to the adenosine-induced attenuation of GABAAR-mediated responses. To test this possibility we used voltage-clamp recordings and measured the GABAAR conductance across a range of membrane potentials (see Materials and Methods). I–V plots of the GABAAR-mediated currents were constructed and conductance was calculated as the slope of linear fits of the data (Fig. 7A). Under control conditions the peak GABAAR conductance was 14.0 ± 1.8 nS, on average, and remained unchanged following 10 min activation of ARs when the mean was 14.9 ± 1.8 nS (n = 9, p = 0.23, paired t test; Fig. 7B). These results show that changes in the GABAAR conductance are unlikely to contribute to the adenosine-induced attenuation of the GABAAR-mediated potentials.
Discussion
We investigated the role of the endogenous anticonvulsant adenosine in modulating GABAergic signaling during epileptiform activity. Intense activation of the GABAergic system during seizure activity leads to a transient switch in the action of GABAARs from inhibitory to excitatory. Seizure activity also raises the levels of extracellular adenosine. We found that the resulting AR activation reduced the amplitude of excitatory GABAergic potentials during seizures and that the suppression of GABAergic signaling lasted beyond the period in which the EGABAA was shifted positively. These effects of endogenously released adenosine were mimicked by exogenous adenosine administration. In examining the mechanism of adenosine's effects, we established that adenosine does not change chloride homeostasis mechanisms that set EGABAA, or the conductance of GABAARs. Rather, A1R-dependent activation of K+ channels can account for the suppression of depolarizing and hyperpolarizing GABAergic potentials via an increase in postsynaptic conductance and a resulting shunting effect. As depolarizing GABAAR-mediated signaling has been implicated in seizure initiation and progression, the adenosine-induced attenuation of depolarizing GABAAR signaling may represent an important mechanism by which adenosine can limit seizure activity.
Studies using microdialysis probes implanted in the hippocampus of epileptic patients have estimated that seizures lasting 40–200 s trigger substantial (up to 65 μm) increases in extracellular adenosine (During and Spencer, 1992). This adenosine surge correlates with seizure arrest and adenosine levels remain elevated during the postseizure period. In rat hippocampal slices, short seizures are sufficient to elevate extracellular levels of adenosine to ∼5 μm and blocking A1Rs increases seizure duration and intensity (Etherington et al., 2009; Lopatář et al., 2011). Seizures in our study had a mean duration of ∼250 s, which would be predicted to trigger adenosine release. Consistent with this prediction, blockade of ARs doubled seizure duration. These results confirm that adenosine has a net antiepileptic effect and they are consistent with in vivo studies in which focal augmentation of adenosine in the hippocampus of epileptic rats can reduce seizure frequency and duration, and even prevent epileptogenesis (Güttinger et al., 2005a,b; Li et al., 2007, 2009; Boison, 2009; Szybala et al., 2009).
Adenosine had its principal effects during the after-discharge phase of the seizures we recorded. Synchronized after-discharge activity is characteristic of epileptiform activity and it has been shown that a transient change in EGABAA is central to this process. During the ictal phase of seizures, intense activation of GABAARs in combination with membrane depolarization results in a substantial Cl− influx into principal neurons, which overwhelms Cl− homeostasis mechanisms. The resulting positive shift in ECl (and therefore EGABAA) sets the conditions for networks of GABAergic interneurons to generate the synchronized excitation that underlies an after-discharge (Isomura et al., 2003b; Zsiros et al., 2007). The collapse in the Cl− gradient also affects the drive on K+-Cl− cotransporter proteins, which results in transient increases in extracellular K+ that can also influence ongoing activity (Kaila et al., 1997; Viitanen et al., 2010). Consistent with this scenario, we found that ictal activity shifts EGABAA to depolarizing values and that activation of GABAARs during the after-discharge period is able to generate depolarizing excitatory responses and trigger action potentials. The exact time point at which EGABAA becomes depolarizing during a seizure will depend on the balance of ionic conductances and the ability of homeostatic mechanisms to clear Cl−. To address this, future experiments could use imaging techniques to measure temporal and spatial changes in chloride that occur during a seizure.
Our pharmacological experiments showed that blocking ARs during seizures increased the excitatory actions of GABAergic signaling. Consistent with this, we found that when ARs were active under control conditions, seizures had less after-discharge events and a shorter after-discharge period. Other actions of adenosine may affect presynaptic GABAergic transmission during seizure activity. For instance, adenosine may affect the activity of interneurons either directly, or indirectly via reducing their glutamatergic inputs, which would ultimately decrease the amount of GABA released. Nevertheless, it is known that GABAergic interneurons can remain active during seizures and can in fact maintain seizure activity and after-discharge events when glutamatergic transmission is blocked (Fujiwara-Tsukamoto et al., 2006, 2010). Adenosine's effect on the membrane potential could also have complex effects upon excitability of the network, such as by increasing de-inactivation of excitatory cationic voltage-dependent conductances. However, in the context of seizure activity at least, the net effect of adenosine is inhibitory; when ARs were active the postsynaptic membrane potential was more hyperpolarized and the neurons showed less excitability in the form of after-discharges.
The acute effect of adenosine upon GABAAR-mediated signaling could involve changes to Cl− homeostasis mechanisms or changes to the expression and/or properties of GABAARs. Indeed, previous studies have reported seizure-induced alterations in EGABAA that are mediated by changes in the activity of Cl− transporter proteins, including NKCC1 and KCC2 (Rivera et al., 2004; Jin et al., 2005; Dzhala et al., 2010; Khirug et al., 2010). However, we found that adenosine had no direct effect on steady-state EGABAA or the recovery of depolarizing GABAAR responses back to hyperpolarizing. AR activation also did not alter the conductance of the GABAAR, indicating that adenosine release over the timescale relevant to a seizure does not have an acute effect upon the properties of the GABAAR. These observations are consistent with previous recordings following short-term activation of ARs (Yoon and Rothman, 1991; Thompson et al., 1992; Kruglikov and Rudy, 2008) and with the conclusion that adenosine's effects upon GABAAR responses during seizure activity must be via an alternative mechanism.
The parsimonious explanation for adenosine's effects upon GABAAR responses is that they are mediated by a postsynaptic shunting effect that develops under conditions where GABAergic transmission transiently changes from inhibitory to excitatory. In support of such a shunting mechanism, as EGABAA and the postsynaptic membrane potential shifted throughout the seizure period, AR activation consistently reduced the amplitude of GABAAR-mediated potentials. We found that adenosine reduced GABAAR responses via an A1R-dependent activation of K+ channels that hyperpolarize the membrane potential and increase membrane conductance. Adenosine has been reported to activate three types of K+ channels that are downstream of A1R signaling: GIRKs, KATPs, and SK channels, all of which would be expected to increase membrane conductance (Hosseinzadeh and Stone, 1998; Fredholm et al., 2005; Clark et al., 2009; Kawamura, Jr. et al., 2010). Indeed, although we investigated the interaction between adenosine and GABAergic transmission in the context of a model of post-traumatic epilepsy (Berdichevsky et al., 2012), it would seem likely that adenosine's effect upon GABAergic transmission would be a general principle for pathological situations in which GABA becomes transiently depolarizing and adenosine is released at sufficient levels to increase membrane conductance.
The exact mechanisms by which extracellular adenosine increases during seizure activity are still the focus of investigation, and multiple sources appear to contribute in neurons and/or glia, such as the action of equilibrative transporter proteins, local breakdown of ATP, and activity-dependent exocytosis (Dale and Frenguelli, 2009). Direct measurements of the dynamics of extracellular adenosine accumulation are somewhat limited in terms of their spatial and temporal resolution, but previous work indicates that levels of extracellular adenosine are related to seizure duration and intensity (During and Spencer, 1992; Etherington et al., 2009). Consistent with this idea, the effects we observed were evident only while adenosine levels were elevated and could be rapidly reversed following adenosine washout. We also found a relationship between adenosine concentration and response, such that higher concentrations of adenosine resulted in greater increases in membrane conductance and attenuation of GABAAR responses. GABAergic responses were reduced when adenosine was applied at a concentration of 10–100 μm in the aCSF, which is within the range of extracellular concentrations that have been reported during seizures and other pathological states (During and Spencer, 1992; Pearson et al., 2006). Previous studies have also suggested that elevated adenosine levels persist well into the postseizure period, until the adenosine has been cleared from the extracellular space (During and Spencer, 1992; Etherington et al., 2009; Lopatář et al., 2011). Consistent with this, we found that adenosine signaling in the postseizure period continued to attenuate GABAAR signaling once it had returned to hyperpolarizing.
Recordings from hippocampal neurons have revealed that adenosine does not have a direct effect upon the presynaptic release of GABA, although GABAergic synapses in other systems do show evidence of presynaptic modulation (Yoon and Rothman, 1991;Thompson et al., 1992; Kruglikov and Rudy, 2008; Han et al., 2011). Our observations extend this work by demonstrating that adenosine modulates the postsynaptic effects of GABAergic inputs during the dynamic events of a seizure. The increase in extracellular adenosine that results from intense network activity appears to be well placed to attenuate the deleterious effects of depolarizing GABAAR signaling that typically emerge under the same conditions. Adenosine does this without “shutting down” GABAergic transmission, unlike its effect upon the glutamatergic system (Thompson et al., 1993). The net effect is that synaptic inhibition is still functional during the recovery period, as activity throughout the network begins to be reinstated.
Footnotes
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (BB/E0154761) and the Medical Research Council (G0601503). The research leading to these results has received funding from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013), ERC grant agreement number 243273. A.I. was supported by a Wellcome Trust Doctoral Fellowship; J.V.R was supported by a Rhodes Scholarship. We thank Marco Capogna, Karri Lamsa, and members of the Akerman lab for providing insightful comments and critically reading the manuscript.
References
- Akerman CJ, Cline HT. Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci. 2006;26:5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Lewis DV, Swartzwelder HS, Wilson WA. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- Avoli M, D'Antuono M, Louvel J, Köhling R, Biagini G, Pumain R, D'Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Berdichevsky Y, Dzhala V, Mail M, Staley KJ. Interictal spikes, seizures and ictal cell death are not necessary for post-traumatic epileptogenesis in vitro. Neurobiol Dis. 2012;45:774–785. doi: 10.1016/j.nbd.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res. 2009;85:131–141. doi: 10.1016/j.eplepsyres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Stewart KA. Therapeutic epilepsy research: from pharmacological rationale to focal adenosine augmentation. Biochem Pharmacol. 2009;78:1428–1437. doi: 10.1016/j.bcp.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J Neurosci. 2008;28:1301–1312. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BD, Kurth-Nelson ZL, Newman EA. Adenosine-evoked hyperpolarization of retinal ganglion cells is mediated by G-protein-coupled inwardly rectifying K+ and small conductance Ca2+-activated K+ channel activation. J Neurosci. 2009;29:11237–11245. doi: 10.1523/JNEUROSCI.2836-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol. 2009;7:160–179. doi: 10.2174/157015909789152146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV. Adenosine and suppression of seizures. Adv Neurol. 1999;79:1001–1010. [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol. 2010;27:418–424. doi: 10.1097/WNP.0b013e3181fe0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, Kuner T, Augustine GJ, Bacskai BJ, Staley KJ. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010;30:11745–11761. doi: 10.1523/JNEUROSCI.1769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara S, Shirato K, Harata N, Akaike N. Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. J Physiol. 1995;484(Pt 1):77–86. doi: 10.1113/jphysiol.1995.sp020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherington LA, Frenguelli BG. Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner. Eur J Neurosci. 2004;19:2539–2550. doi: 10.1111/j.0953-816X.2004.03355.x. [DOI] [PubMed] [Google Scholar]
- Etherington LA, Patterson GE, Meechan L, Boison D, Irving AJ, Dale N, Frenguelli BG. Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology. 2009;56:429–437. doi: 10.1016/j.neuropharm.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Nambu A, Takada M. Excitatory GABA input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience. 2003;119:265–275. doi: 10.1016/s0306-4522(03)00102-7. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Takada M. Comparable GABAergic mechanisms of hippocampal seizurelike activity in posttetanic and low-Mg2+ conditions. J Neurophysiol. 2006;95:2013–2019. doi: 10.1152/jn.00238.2005. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Fukai T, Takada M. Distinct types of ionic modulation of GABA actions in pyramidal cells and interneurons during electrical induction of hippocampal seizure-like network activity. Eur J Neurosci. 2007;25:2713–2725. doi: 10.1111/j.1460-9568.2007.05543.x. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Ninomiya T, Tsukada M, Yanagawa Y, Fukai T, Takada M. Prototypic seizure activity driven by mature hippocampal fast-spiking interneurons. J Neurosci. 2010;30:13679–13689. doi: 10.1523/JNEUROSCI.1523-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlfinger A, Bischofberger J, Johenning FW, Torvinen M, Schmitz D, Breustedt J. Adenosine modulates transmission at the hippocampal mossy fibre synapse via direct inhibition of presynaptic calcium channels. J Physiol. 2007;582:263–277. doi: 10.1113/jphysiol.2007.132613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R, Armand V, Schuchmann S, Heinemann U. Epileptiform activity induced by low Mg2+ in cultured rat hippocampal slices. Brain Res. 1999;815:294–303. doi: 10.1016/s0006-8993(98)01102-0. [DOI] [PubMed] [Google Scholar]
- Güttinger M, Fedele D, Koch P, Padrun V, Pralong WF, Brüstle O, Boison D. Suppression of kindled seizures by paracrine adenosine release from stem cell-derived brain implants. Epilepsia. 2005a;46:1162–1169. doi: 10.1111/j.1528-1167.2005.61804.x. [DOI] [PubMed] [Google Scholar]
- Güttinger M, Padrun V, Pralong WF, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol. 2005b;193:53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Han TH, Jang SH, Lee SY, Ryu PD. Adenosine reduces GABAergic IPSC frequency via presynaptic A receptors in hypothalamic paraventricular neurons projecting to rostral ventrolateral medulla. Neurosci Lett. 2011;490:63–67. doi: 10.1016/j.neulet.2010.12.026. [DOI] [PubMed] [Google Scholar]
- Higashima M, Kinoshita H, Yamaguchi N, Koshino Y. Activation of GABAergic function necessary for afterdischarge generation in rat hippocampal slices. Neurosci Lett. 1996;207:101–104. doi: 10.1016/0304-3940(96)12496-4. [DOI] [PubMed] [Google Scholar]
- Higashima M, Ohno K, Kinoshita H, Koshino Y. Involvement of GABA(A) and GABA(B) receptors in afterdischarge generation in rat hippocampal slices. Brain Res. 2000;865:186–193. doi: 10.1016/s0006-8993(00)02209-5. [DOI] [PubMed] [Google Scholar]
- Hosseinzadeh H, Stone TW. Tolbutamide blocks postsynaptic but not presynaptic effects of adenosine on hippocampal CA1 neurones. J Neural Transm. 1998;105:161–172. doi: 10.1007/s007020050045. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura Y, Fujiwara-Tsukamoto Y, Takada M. Glutamatergic propagation of GABAergic seizure-like afterdischarge in the hippocampus in vitro. J Neurophysiol. 2003a;90:2746–2751. doi: 10.1152/jn.00057.2003. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sugimoto M, Fujiwara-Tsukamoto Y, Yamamoto-Muraki S, Yamada J, Fukuda A. Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J Neurophysiol. 2003b;90:2752–2756. doi: 10.1152/jn.00142.2003. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Fujiwara-Tsukamoto Y, Takada M. A network mechanism underlying hippocampal seizure-like synchronous oscillations. Neurosci Res. 2008;61:227–233. doi: 10.1016/j.neures.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Jr, Ruskin DN, Masino SA. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci. 2010;30:3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Vreugdenhil M, Bracci E, Jefferys JG. Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci. 2000;20:6820–6829. doi: 10.1523/JNEUROSCI.20-18-06820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglikov I, Rudy B. Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron. 2008;58:911–924. doi: 10.1016/j.neuron.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brüstle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Kaplan DL, Boison D. Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective epileptogenesis. Epilepsy Res. 2009;84:238–241. doi: 10.1016/j.eplepsyres.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatář J, Dale N, Frenguelli BG. Minor contribution of ATP P2 receptors to electrically-evoked electrographic seizure activity in hippocampal slices: Evidence from purine biosensors and P2 receptor agonists and antagonists. Neuropharmacology. 2011;61:25–34. doi: 10.1016/j.neuropharm.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Löscher W, Köhling R. Functional, metabolic, and synaptic changes after seizures as potential targets for antiepileptic therapy. Epilepsy Behav. 2010;19:105–113. doi: 10.1016/j.yebeh.2010.06.035. [DOI] [PubMed] [Google Scholar]
- Michelson HB, Wong RK. Excitatory synaptic responses mediated by GABAA receptors in the hippocampus. Science. 1991;253:1420–1423. doi: 10.1126/science.1654594. [DOI] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- Nardou R, Yamamoto S, Chazal G, Bhar A, Ferrand N, Dulac O, Ben-Ari Y, Khalilov I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134:987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- Pearson T, Damian K, Lynas RE, Frenguelli BG. Sustained elevation of extracellular adenosine and activation of A1 receptors underlie the post-ischaemic inhibition of neuronal function in rat hippocampus in vitro. J Neurochem. 2006;97:1357–1368. doi: 10.1111/j.1471-4159.2006.03823.x. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseti C, Martinello K, Fucile S, Piccari V, Mascia A, Di Gennaro G, Quarato PP, Manfredi M, Esposito V, Cantore G, Arcella A, Simonato M, Fredholm BB, Limatola C, Miledi R, Eusebi F. Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:15118–15123. doi: 10.1073/pnas.0807277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseti C, Palma E, Martinello K, Fucile S, Morace R, Esposito V, Cantore G, Arcella A, Giangaspero F, Aronica E, Mascia A, Di Gennaro G, Quarato PP, Manfredi M, Cristalli G, Lambertucci C, Marucci G, Volpini R, Limatola C, Eusebi F. Blockage of A2A and A3 adenosine receptors decreases the desensitization of human GABA(A) receptors microtransplanted to Xenopus oocytes. Proc Natl Acad Sci U S A. 2009;106:15927–15931. doi: 10.1073/pnas.0907324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. Neurotransmitter activation of inwardly rectifying potassium current in dissociated hippocampal CA3 neurons: interactions among multiple receptors. J Neurosci. 1998;18:8153–8162. doi: 10.1523/JNEUROSCI.18-20-08153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABA(A) receptor function by the kinetics of Cl- and HCO3- transport. J Physiol 519 Pt. 1999;3:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Streit P, Thompson SM, Gähwiler BH. Anatomical and physiological properties of GABAergic neurotransmission in organotypic slice cultures of rat hippocampus. Eur J Neurosci. 1989;1:603–615. doi: 10.1111/j.1460-9568.1989.tb00366.x. [DOI] [PubMed] [Google Scholar]
- Szybala C, Pritchard EM, Lusardi TA, Li T, Wilz A, Kaplan DL, Boison D. Antiepileptic effects of silk-polymer based adenosine release in kindled rats. Exp Neurol. 2009;219:126–135. doi: 10.1016/j.expneurol.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C. Phasic and tonic attenuation of EPSPs by inward rectifier K+ channels in rat hippocampal pyramidal cells. J Physiol. 2002;539(Pt 1):67–75. doi: 10.1113/jphysiol.2001.012883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol. 1989;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Haas HL, Gähwiler BH. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol. 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Baldeweg T, van Drongelen W, Yuste R, Whittington M. The source of afterdischarge activity in neocortical tonic-clonic epilepsy. J Neurosci. 2007;27:13513–13519. doi: 10.1523/JNEUROSCI.3005-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48 [Suppl] 2007;5:96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- Velazquez JL, Carlen PL. Synchronization of GABAergic interneuronal networks during seizure-like activity in the rat horizontal hippocampal slice. Eur J Neurosci. 1999;11:4110–4118. doi: 10.1046/j.1460-9568.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588:1527–1540. doi: 10.1113/jphysiol.2009.181826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R, Raimondo JV, Akerman CJ. Spatial and temporal dynamics in the ionic driving force for GABA(A) receptors. Neural Plast. 2011;2011:728395. doi: 10.1155/2011/728395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsiros V, Aradi I, Maccaferri G. Propagation of postsynaptic currents and potentials via gap junctions in GABAergic networks of the rat hippocampus. J Physiol. 2007;578:527–544. doi: 10.1113/jphysiol.2006.123463. [DOI] [PMC free article] [PubMed] [Google Scholar]