

Abstract
Alterations in the lipid composition of endosomal–lysosomal membranes may constitute an early event in Alzheimer's disease (AD) pathogenesis. In this study, we investigated the possibility that GM2 ganglioside accumulation in a mouse model of Sandhoff disease might be associated with the accumulation of intraneuronal and extracellular proteins commonly observed in AD. Our results show intraneuronal accumulation of amyloid-β peptide (Aβ)-like, α-synuclein-like, and phospho-tau-like immunoreactivity in the brains of β-hexosaminidase knock-out (HEXB KO) mice. Biochemical and immunohistochemical analyses confirmed that at least some of the intraneuronal Aβ-like immunoreactivity (iAβ-LIR) represents amyloid precursor protein C-terminal fragments (APP-CTFs) and/or Aβ. In addition, we observed increased levels of Aβ40 and Aβ42 peptides in the lipid-associated fraction of HEXB KO mouse brains, and intraneuronal accumulation of ganglioside-bound Aβ (GAβ) immunoreactivity in a brain region-specific manner. Furthermore, α-synuclein and APP-CTFs and/or Aβ were found to accumulate in different regions of the substantia nigra, indicating different mechanisms of accumulation or turnover pathways. Based on the localization of the accumulated iAβ-LIR to endosomes, lysosomes, and autophagosomes, we conclude that a significant accumulation of iAβ-LIR may be associated with the lysosomal–autophagic turnover of Aβ and fragments of APP-containing Aβ epitopes. Importantly, intraneuronal GAβ immunoreactivity, a proposed prefibrillar aggregate found in AD, was found to accumulate throughout the frontal cortices of postmortem human GM1 gangliosidosis, Sandhoff disease, and Tay–Sachs disease brains. Together, these results establish an association between the accumulation of gangliosides, autophagic vacuoles, and the intraneuronal accumulation of proteins associated with AD.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease that is characterized neuropathologically by extracellular amyloid-β peptide (Aβ) plaques and intraneuronal neurofibrillary tangles (Selkoe, 2002). Processing of the amyloid precursor protein (APP) is initiated via the proteolytic activities of α- or β-secretase, which cleave the ectodomain of APP to generate soluble N-terminal APP fragments (α- or β-sAPP), and membrane-tethered C-terminal fragments (α- or β-CTFs) of APP. Aβ is then generated by the stepwise proteolytic cleavage of APP by β-secretase [or BACE (β-site APP cleaving enzyme)] and γ-secretase (Selkoe, 2002).
Recent studies highlighted several common neuropathological features shared by lysosomal storage diseases (LSDs) and age-related neurodegenerative diseases such as AD and Parkinson's disease (Boland et al., 2010; Zhang et al., 2010; Mattsson et al., 2011; Shachar et al., 2011; Xu et al., 2011). Glycosphingolipid storage disorders (GSLs) are a subgroup of LSDs caused by mutations in hydrolase or activator protein genes involved in the catabolism of GSLs such as gangliosides (Xu et al., 2010). The GM1 and GM2 gangliosidoses are complex LSDs and, in most cases, present with a progressive and severe neurodegenerative course that includes mental retardation, dementia, motor dysfunction, sensory deficits, and seizures. GM1 gangliosidosis is a neurosomatic disease caused by mutations in the GLB1 (β-galactosidase) gene and is characterized by excessive accumulation of GM1 ganglioside and related sphingolipids (Brunetti-Pierri and Scaglia, 2008). GM2 gangliosidoses are a group of inherited disorders caused by excessive accumulation of GM2 ganglioside and related sphingolipids due to mutations in either the HEXA (β-hexosaminidase A) gene as in Tay–Sachs disease (TS), or the HEXB (β-hexosaminidase B) gene as in Sandhoff disease (SD) (Mahuran, 1999).
Altered ganglioside metabolism in the brain has been observed in association with aging and AD (Ariga et al., 2008; Yamamoto et al., 2008; Chan et al., 2012), and it is proposed that abnormalities in ganglioside metabolism might contribute to AD pathogenesis by promoting Aβ aggregation via the formation of ganglioside-Aβ (GAβ) complexes in membrane microdomains (Yanagisawa, 2011). In addition, ganglioside accumulation in lysosomes has been recently linked to disruption of autophagy (Boland et al., 2008; Tamboli et al., 2011), providing a potential pathway linking altered lipid composition of the lysosomal membranes to aberrant lysosomal–autophagic turnover of proteins in LSDs and AD.
In the current study, we describe the characterization of heterozygous (HEXB HZ) and homozygous (HEXB KO) mouse models of SD by immunohistochemical and biochemical approaches. Immunohistochemical analysis of HEXB KO mice demonstrated extensive accumulation of Aβ-like immunoreactivity (Aβ-LIR) and GAβ-like immunoreactivity (GAβ-LIR) in most brain regions, intraneuronal α-synuclein immunoreactivity in the substantia nigra compacta (SNC), and phosphorylated tau (pTau) immunoreactivity in the medulla and spinal cord. Biochemical analysis of APP metabolism revealed that, although full-length APP levels in the HEXB KO mice are comparable with age-matched wild-type mice, APP-CTFs and Aβ levels are significantly higher in HEXB KO mice. In addition, we show that the frontal cortices of human gangliosidoses brains (GM, SD, TS) accumulate intraneuronal GAβ immunoreactivity, a proposed prefibrillar aggregate found in AD.
Materials and Methods
Mice.
Brains from 4- to 4.5-month-old mice, heterozygous or homozygous for the HEXB gene on a C57BL/6 background, were provided by Dr. Thomas Seyfried (Boston College, Boston, MA). The HEXB mice were made by disruption of the murine HEXB gene and transferring this gene into the mouse genome through homologous recombination and embryonic stem cell technology as previously described (Sango et al., 1995). For Western blotting, six wild-type mice (two males and four females); seven HEXB HZ mice (two males and five females), and seven HEXB KO mice (two males and five females) were used. For immunohistochemistry, three wild-type and three HEXB KO mice of either sex were used.
Immunohistochemistry.
Human and mouse brains were fixed in neutral-buffered formalin (NBF) (Thermo Fisher Scientific), processed in a Tissue-Tek VIP 5 tissue processor (Sakura Finetek), and embedded in paraffin (Tissue-Tek TEC 5; Sakura Finetek). Samples were sectioned at a thickness of 5 μm on an HM 325 microtome (MICROM International). Sections were then dewaxed in xylene (Thermo Fisher Scientific) and rehydrated. The endogenous peroxidase activity was quenched for 30 min in 3% H2O2. Sections were blocked for 30 min with Rodent Block M (Biocare Medical). Epitope retrieval was performed for 20 min in either 70% formic acid for Aβ antibodies or citrate buffer, pH 6.0 (Lab Vision) for pTau antibodies. Sections incubated without primary antibodies were included as negative controls. For peptide competition experiments, 50-fold molar excess rat Aβ1–40 peptide (AnaSpec) or 100-fold molar excess human Aβ1–40 peptide (Covance) was incubated with Aβ antibody 4G8 at room temperature for 30 min before the addition to the mouse or human sections, respectively. Sections were washed in Tris-buffered saline (TBS), incubated with appropriate HRP polymer-conjugated secondary antibodies and developed with Betazoid DAB chromogen kit (Biocare Medical). The nuclei were counterstained in Mayer's hematoxylin (Sigma-Aldrich), and sections were mounted with Cytoseal mounting media (Richard-Allan Scientific). Images were captured on a Nikon Eclipse 90i microscope using NIS Element software (Micron Optics).
For fluorescence staining, sections were permeabilized with 0.25% Triton X-100 for 15 min, blocked with 3% goat serum in TBS for 1 h at room temperature, and incubated with primary antibodies overnight at 4°C. Sections were washed the following day, incubated with the secondary antibodies green fluorescent Alexa Fluor 488 and red fluorescent Alexa Fluor 594 (Invitrogen; 1:500) for 1 h at room temperature, and mounted with Vectasheild (Vector Laboratories). Images were captured on an Olympus BX61 upright microscope with an attached Olympus DP71 camera.
The following primary antibodies were applied overnight at 4°C: mouse anti-PHF Tau-pThr231, clone AT180 (Thermo Fisher Scientific; 1:200); mouse anti-α-Synuclein, clone 4D6 (Abcam; 1:1000); mouse anti-EEA-1, clone 14/EEA-1 (BD Biosciences; 1:500); mouse anti-Lamp-1, clone 25/lamp-1 (BD Biosciences; 1:500); mouse anti-Rab7, clone Rab7-117 (Sigma-Aldrich; 1:500); rat anti-GFAP, clone 2.2B10 (Invitrogen; 1:500); rabbit anti-LC3 (MBL; 1:1000); mouse anti-Aβ, clone 4G8, which recognizes amino acids 17–24 of the human Aβ and detects Aβ, full-length APP, and C-terminal fragments of APP (Covance; 1:500); rabbit anti-Aβ40, clone FCA3340 (1:500), and rabbit anti-Aβ42, clone FCA3542 (1:500) (Barelli et al., 1997); mouse anti-Aβ40, clone 1C8-C8 (1:500), and mouse anti-Aβ42, clone Pan1G6 (1:500), were gifts from Dr. Joseph Buxbaum (Mount Sinai School of Medicine, New York, NY) (Parvathy et al., 2001); mouse anti-GAβ, clone 4396C (1:100), was a gift from Dr. Katsuhiko Yanagisawa (Research Institute of National Center for Geriatrics and Gerontology, Obu, Aichi, Japan) (Hayashi et al., 2004).
Brain tissue homogenization and mouse Aβ extraction.
Brains were homogenized and mouse Aβ was extracted for ELISA analysis as described previously (Schmidt et al., 2005a,b). Brain hemispheres were homogenized by 5 ml glass tissue grinder (20 strokes) at 10% w/v (wet brain weight) in ice-cold tissue homogenization buffer (THB) [250 mm sucrose, 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, 100 mm NaF, 1 μm pepstatin, 1 mm PMSF, and EDTA-free Mini-Complete protease inhibitor mixture tablet (Roche)]. Soluble mouse Aβ was extracted by adding diethylamine (DEA) buffer (0.4% DEA in 100 mm NaCl) to 10% homogenate at ratio of 1:1 (v/v). The samples were then rehomogenized by six strokes using a 5 ml glass grinder and were centrifuged at 100,000 × g for 1 h at 4°C. The resulting supernatant was neutralized with 0.5 m Tris-HCl, pH 6.8, to a final concentration of 0.045 m, vortexed briefly, and divided into three to four aliquots. Lipid-bound Aβ was extracted from the pellet (P1) as described previously (Boland et al., 2010). The pellet was resuspended at 10% w/v of initial wet weight in ice-cold RIPA buffer (10 mm Tris-HCl, pH 8, 150 mm NaCl, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 5 mm EDTA, 1 mm EGTA, 1 mm PMSF, and EDTA-free Mini-Complete protease inhibitor mixture tablet) and sonicated with Branson Sonifier for 10 s on ice. The samples were cleared from debris by centrifugation at 10,000 × g for 10 min at 4°C, and the supernatant was saved for further analysis. The levels of soluble and lipid-bound Aβ40 and 42 were measured using ELISA kits (Wako Chemicals; no. 294-64701 for Aβ40 and 292-64501 for Aβ42), according to the manufacturer's instructions. For Aβ Western blots, 200 μg of protein from each fraction was run on a 10–20% Tricine gel at constant current of 0.04 A for 3 h. The gel was transferred to a polyvinylidene difluoride (PVDF) membrane by wet transfer at 25 V for 1 h. The blot was incubated overnight at 4°C with either rabbit anti-Aβ40, clone FCA3340 (1:1000), or rabbit anti-Aβ42, clone FCA3542 (1:1000).
Protein extraction and Western blotting.
For total protein extraction, RIPA buffer was added to 10% homogenate at a ratio of 1:1 (v/v), vortexed briefly, and kept on ice for 30 min. The samples were then centrifuged at 20,000 × g for 10 min at 4°C, and the supernatant was saved for Western blotting analysis. For quantitative measurements of sAPPα, full-length APP was immunodepleted from 200 μg of whole-tissue extracts using 369 antibody, which recognizes the C terminus of APP. 369 was added to a final dilution of 1:100 and immunoprecipitated using A/G Plus-Agarose beads (Santa Cruz Biotechnology). The supernatant (flow through) was then used to measure the levels of sAPPα using Western blotting after normalization to the levels of GAPDH.
Equal amounts of total protein from each sample were prepared in 5× Laemmli buffer, boiled at 95°C for 5 min, and loaded onto SDS-PAGE gels (4% NuPage–12% Bis-Tris; Bio-Rad) with NuPage MES buffer (Invitrogen) at 200 V for 45 min. The proteins were then blotted onto PVDF membranes (Bio-Rad) for 120 min at 30 V. The blots were blocked overnight in 5% nonfat milk (Santa Cruz Biotechnology) and incubated with the following primary antibodies: mouse anti-GAPDH, clone 6C5 (Santa Cruz Biotechnology; 1:1000); mouse anti-APP, clone 22C11, which recognizes amino acids 66–81 of APP (N terminus) and detects full-length APP and sAPPα (Millipore; 1:1000); rabbit anti-APP (C terminus), clone 369, which recognizes APP (C terminus) and detects full-length APP and C-terminal fragments of APP (Buxbaum et al., 1990) (1:2500); rabbit anti-LC3 (MBL; 1:2000); mouse anti Rab7, clone Rab7-117 (Sigma-Aldrich; 1:1000); guinea pig anti-p62 (American Research Products; 1:1000); goat anti-Lamp-2, clone C-20 (Santa Cruz Biotechnology; 1:1000). The blots were washed the following day with TBS and were then incubated with horseradish peroxidase-conjugated secondary antibodies at a dilution of 1:10,000 (Vector Laboratories) for 1–2 h. Signals were detected by chemiluminescence using the enhanced chemiluminescence system (Pierce) on a Fujifilm LAS-3000 developer.
Thin-layer chromatography analysis of gangliosides.
Human brain tissue was homogenized in 2:1 CHCl3/MeOH using a rotor stator homogenizer at 50% power for 20 s (200 μl of 2:1 CHCl3/MeOH mixture for every 10 mg of brain tissue). The homogenized mixture was vortexed for 30 min at room temperature and centrifuged at 10,000 × g for 5 min at room temperature to separate the solids. The extract was then transferred to a 1.5 ml centrifuge tube, mixed with 20 μl of PBS/100 μl of CHCl3/MeOH, vortexed, and centrifuged at 10,000 × g for 5 min at room temperature. The upper aqueous phase was removed and transferred to a C-18 Sep-Pak cartridge (prewetted with 1 ml of MeOH, preequilibrated with 1 ml of 25% MeOH in deionized H2O). The Sep-Pak cartridge was washed with 1 ml of deionized H2O, and samples were eluted with 0.5 ml of 100% MeOH directly into a 1 ml conical glass vial. The liquid was evaporated using a Speed-Vac for 30 min, and the ganglioside preparations were solubilized in 20 μl of 2:1 CHCl3/MeOH. The ganglioside preparation was spotted on a preheated TLC plate and developed using 200 ml of 60:40:9 CHCl3/MeOH/CaCl2 (0.02% in water), and the ganglioside bands were visualized using resorcinol stain.
Statistical analysis.
The densitometric value of each specific band was obtained using MultiGauge (Fujifilm) software and normalized to GAPDH as a housekeeping gene and presented as mean ± SEM. One-way ANOVA with Bonferroni's post test was performed using GraphPad Prism, version 5.00 for Windows (GraphPad Software).
Results
Aβ-like, α-synuclein-like, and tau-like immunoreactive materials accumulate intraneuronally in the brains of HEXB KO mice
To determine whether a deficiency in lysosomal β-hexosaminidase activity results in the accumulation of pathological proteins associated with AD, we conducted immunohistochemical (IHC) analyses on coronal brain sections of HEXB KO mice and age-matched wild-type controls. We first examined the protein levels and brain distribution of APP and its processing products (APP-CTFs and Aβ) using the monoclonal antibody 4G8, which recognizes residues 17–26 of Aβ. We observed 4G8-positive intraneuronal staining in most brain regions, including the cortex (Fig. 1a), hypothalamus (Fig. 1b), hippocampus (Fig. 1c), olfactory bulb (Fig. 1d), preoptic area (Fig. 1e), superior colliculus (Fig. 1f), midbrain (Fig. 1g), pons (Fig. 1h), medulla (Fig. 1i), and spinal cord (Fig. 1j).
Figure 1.
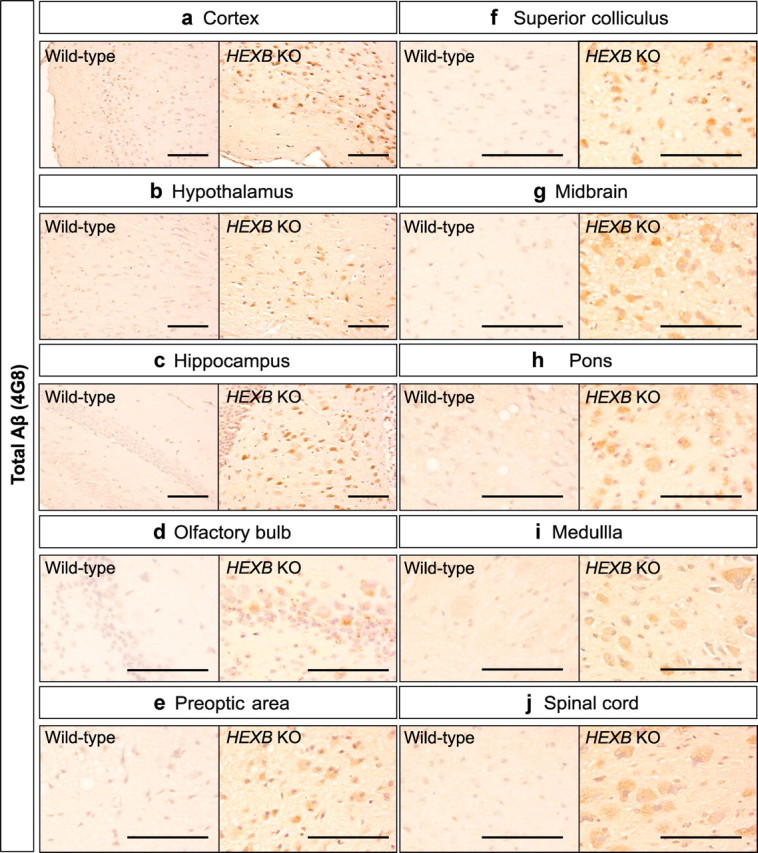
Widespread intraneuronal 4G8 immunoreactivity in the brains of HEXB KO mice. Coronal brain sections of HEXB KO mice were stained with 4G8 antibody, which recognizes amino acids 17–24 of Aβ and detects Aβ, full-length APP and APP-CTFs. Intraneuronal 4G8 signals were observed in most brain regions, including the cortex (a), hypothalamus (b), hippocampus (c), olfactory bulb (d), preoptic area (e), superior colliculus (f), midbrain (g), pons (h), medulla (i), and spinal cord (j). Scale bars: a–j, 200 μm.
Next, we examined the accumulation of α-synuclein-like immunoreactivity (α-syn-LIR) in HEXB KO mice. As shown in Figure 2A, intraneuronal α-syn-LIR was detected in the substantia nigra compacta (SNC) but not in the substantia nigra reticula (SNR). In contrast, 4G8 immunoreactivity was detected in the SNR but not the SNC, indicating that α-synuclein and APP/APP-CTFs/Aβ accumulate in different regions of the SN.
Figure 2.
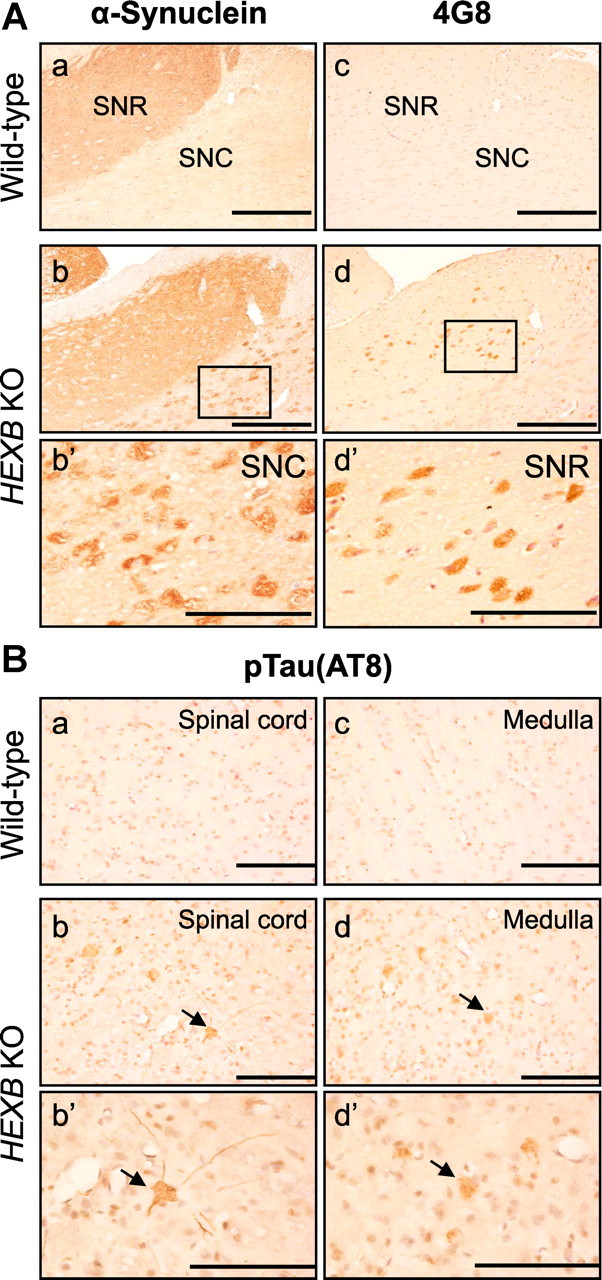
Accumulation of α-synuclein and pTau in HEXB KO mice. A, Brain sections of HEXB KO mice were costained with anti-α-synuclein (a, b) and 4G8 anti-Aβ (c, d) antibodies. α-Synuclein-LIR was detected in the SNC (b, b′), whereas intraneuronal 4G8 signals were detected in the SNR (d, d′). B, Immunohistochemical analysis of wild-type and HEXB KO mice using an antibody (AT8) that detects Tau phosphorylated at serine 202 and threonine 205 shows that pTau accumulates in the gray matter of the spinal cord (b) and the medulla (d) of HEXB KO mice. Arrows point to intraneuronal signals. Scale bars: a–d, 500 μm; b′, d′, 200 μm.
In addition to Aβ and α-synuclein, phosphorylated tau (pTau) is another protein that accumulates in brain neurons in several neurodegenerative diseases. IHC analysis using the antibody AT8, which detects pTau(Ser202) and pTau(Thr205), showed intraneuronal AT8-positive pTau-like immunoreactivity (pTau-LIR) in the medulla and the gray matter of the spinal cord of HEXB KO mice (Fig. 2B).
APP-CTFs and lipid-associated Aβ accumulate in the brains of HEXB KO mice
Since the monoclonal antibody 4G8 detects full-length APP, APP-CTFs, and Aβ, we sought to determine the identity of the intraneuronal 4G8-immunoreactive protein species mentioned above. We measured the levels of APP, sAPPα, APP-CTFs, and Aβ in 4- to 4.5-month-old HEXB HZ and HEXB KO mice. The levels of full-length APP and APP-CTFs were determined in brain homogenates by Western blotting using the APP C terminus-specific antibody 369. To measure sAPPα levels, full-length APP was immunodepleted from cell lysates using the antibody 369, and the depleted lysates were studied by Western blotting using the APP N terminus-specific antibody, 22C11 (as described in Materials and Methods). As shown in Figure 3A, the levels of full-length APP and sAPPα in HEXB KO mice were comparable with those observed in age-matched wild-type mice, whereas the levels of α- and β-CTFs were significantly higher (∼1.5-fold; p < 0.05) in the HEXB KO mice (α-CTFs, 148 ± 9.3%; β-CTFs, 141 ± 11.3%). HEXB HZ mice showed a minor increase in the levels of APP-CTFs (Fig. 3A), indicating that even modest alterations in the activity of β-hexosaminidase may affect the metabolism of APP.
Figure 3.
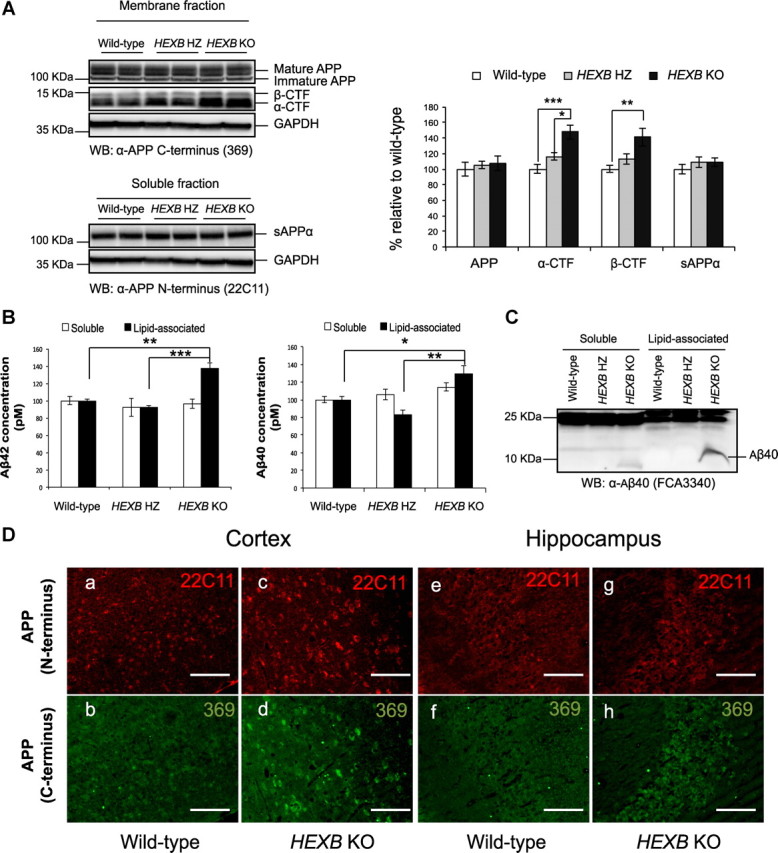
Accumulation of APP-CTFs and lipid-associated Aβ in HEXB KO mice. A, Western blot analysis, using 369 antibody to detect full-length APP and APP-CTFs and 22C11 antibody to detect sAPPα, shows significant increases in the levels of α-CTF and β-CTF in HEXB KO mice relative to wild-type (WT). No significant changes were observed in the levels of full-length APP or sAPPα levels. (For APP, α- and β-CTF analysis, n = 6 for WT, n = 7 for HZ, n = 7 for KO. For sAPPα, n = 3 for WT, n = 4 for HZ, n = 4 for KO). B, ELISA measurement of Aβ40 and Aβ42 levels shows significant increase in the levels of lipid-associated Aβ40 and Aβ42 in the HEXB KO mice (n = 6 for WT; n = 7 for HZ; n = 7 for KO). Shown are mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001). C, Western blot analysis using Aβ40-specific antibody (FCA3340) shows increase in the levels of Aβ40 in the soluble and lipid-associated fractions of HEXB KO mice. The levels of Aβ42 were not detectable by Western blotting. D, Brain sections were costained with antibodies that recognize either the N terminus (22C11) or C terminus (369) of APP. Although the signals from APP-C terminus (369) antibody seem to accumulate in the cortex (d) and hippocampus (h) of HEXB KO mice, no change is observed in the signals from APP-N terminus (22C11) antibody (c, g). Scale bars: a–h, 200 μm.
The levels of Aβ40 and Aβ42 were measured in DEA-soluble (soluble) and RIPA-soluble (lipid-associated) fractions prepared from brain homogenates, as described previously (Boland et al., 2010). Aβ levels in both fractions were measured by ELISA (Fig. 3B) and Western blotting (Fig. 3C). Interestingly, the levels of both Aβ40 and Aβ42 were significantly (p < 0.05) elevated in the lipid-associated fraction of HEXB KO mice (Aβ40, 129 ± 9%; Aβ42, 137 ± 7%). In the soluble fraction, Aβ40 levels were only slightly higher in HEXB KO mice (114 ± 5%), and Aβ42 levels were similar to wild type (Fig. 3B). In addition, immunoblotting using Aβ40-specific antibody (FC3340) shows that the levels of Aβ40 were elevated in the both the soluble and lipid-associated fractions of HEXB KO mice (Fig. 3C).
A recent study demonstrated that intraneuronal APP, rather than intraneuronal (iAβ), accumulates in a mouse model of AD (Winton et al., 2011). However, our results show that the levels of APP-CTFs and Aβ increase significantly in HEXB KO mice, without an increase in the levels of full-length APP. These results indicate that the accumulated intraneuronal 4G8-positive Aβ-LIR signals represent either APP-CTFs, Aβ, or both. To confirm that full-length APP does not accumulate in HEXB KO mice, we costained coronal brain sections with APP N terminus-specific (22C11) and C terminus-specific (369) antibodies (Fig. 3D). Signals from the C terminus (369) antibody, but not the N terminus (22C11), increased in the cortex and hippocampus of HEXB KO mouse brains relative to age-matched wild-type mice, indicating that APP-CTFs (rather than full-length APP) accumulate in the brains of HEXB KO mice.
Intraneuronal Aβ-LIR accumulates in a region-specific manner in HEXB KO mice
Based on the finding that Aβ40 and Aβ42 levels increase in HEXB KO mice, and since 4G8 recognizes residues 17–26 of Aβ, we sought to determine whether some of the intraneuronal 4G8 signals observed in HEXB KO mice represent authentic Aβ. Coronal brain sections from 4-month-old HEXB KO mice and age-matched wild-type mice were stained with Aβ40- and Aβ42-specific antibodies (FC3340 and FC3542, respectively). In addition, serial sections were stained using an antibody for NeuN, a marker for neurons. As shown in Figure 4, NeuN-positive neurons in the cortex (Fig. 4b,b′), subiculum (Fig. 4d,d′), dentate gyrus (Fig. 4f), and CA3 region of the hippocampus (Fig. 4h) appeared swollen with accumulated storage material. Similarly, accumulation of intraneuronal Aβ40- and Aβ42-like immunoreactivity (iAβ40-LIR and iAβ42-LIR, respectively) was observed in the cortex (Fig. 4b1,b1′,b2,b2′), subiculum (Fig. 4d1,d1′,d2,d2′), dentate gyrus (Fig. 4f1,f2), and CA3 region of the hippocampus (Fig. 4h1,h2) of HEXB KO mouse brains.
Figure 4.
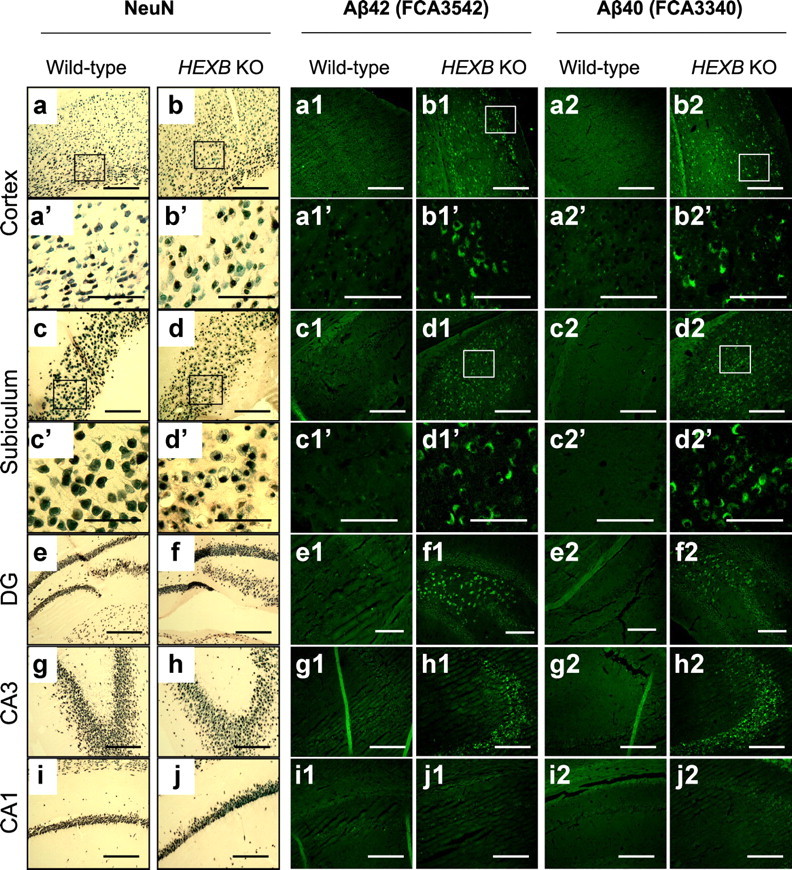
Region-specific accumulation of Aβ-like immunoreactivity in HEXB KO mice. Coronal brain sections of wild-type and HEXB KO mice were stained with NeuN antibody (a–j), Aβ42-specific antibody (FCA3542) (a1–j1), or Aβ40-specific antibody (FCA3340) (a2–j2). NeuN-positive neurons appeared swollen with vacuolar changes in the cortex (b, b′), subiculum (d, d′), dentate gyrus (f), and CA3 region of the hippocampus (h) of HEXB KO mice. Similarly, iAβ-LIR accumulated in the cortex (b1, b1′, b2, b2′), subiculum (d1, d1′, d2, d2′), dentate gyrus (f1, f2), and CA3 region of the hippocampus (h1, h2). The morphology of NeuN-positive neurons appeared normal in the CA1 region of the hippocampus (j), and iAβ-LIR was not observed in this region (j1, j2). DG, Dentate gyrus. Scale bars: a–j, a1–d1, g1–j1, a2–d2, g2–j2, 500 μm; a′–d′, a1′–d1′, a2′–d2′, e1, f1, e2, f2, 200 μm.
Notably, the neurons of the CA1 region of the hippocampus appeared normal (Fig. 4j). Similarly, iAβ-LIR was found to accumulate in the CA3 region (but not the CA1 region) of the hippocampi of HEXB KO mice (Fig. 4, compare h1, h2, with j1, j2). Interestingly, Suzuki et al. (2003) reported similar preferential accumulation of α-synuclein in the CA3, but not the CA1, region of the hippocampi of HEXB KO mice, suggesting regional differences in neuronal susceptibility.
Ganglioside-bound Aβ accumulates in HEXB KO mice
The observed increase in the levels of Aβ40 and Aβ42 in the lipid-associated fraction suggested that the increase in GM2 levels in HEXB KO mice may account for increased murine Aβ association with cellular membranes. Hence, we studied the accumulation of GAβ in the HEXB KO mice by immunohistochemistry. Coronal brain sections of HEXB KO and age-matched wild-type mice were stained with 4396C antibody, an antibody that specifically detects GAβ. As shown in Figure 5A, intraneuronal 4396C immunoreactivity increased in the subiculum and cortex of HEXB KO mice relative to wild-type, indicating that GAβ-LIR accumulates in these mice. In addition, 4396C immunoreactivity was found to colocalize with iAβ42-LIR (Fig. 5A) and with iAβ40-LIR (data not shown), indicating that some of the accumulated iAβ in HEXB KO mice is bound to gangliosides.
Figure 5.
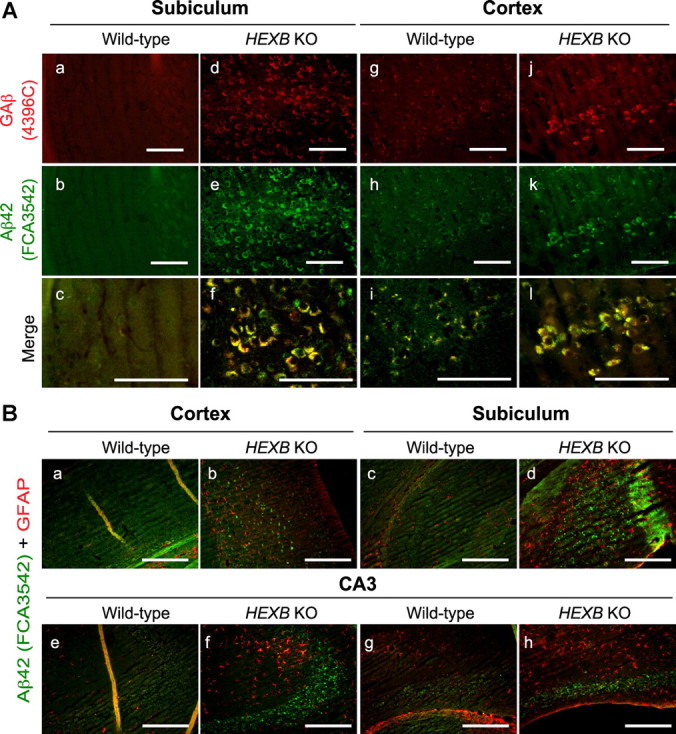
Ganglioside-bound Aβ (GAβ) immunoreactivity colocalizes with iAβ-LIR. A, Coronal brain sections from wild-type (a–c, g–i) and HEXB KO mice (d–f, j–l) were costained with GAβ antibody (4396C) and Aβ42-specific antibody (FCA3542). GAβ immunoreactivity as detected by 4396C antibody colocalized with iAβ-LIR in the subiculum and cortex of HEXB KO mice (f, l). Scale bars: a–l, 200 μm. B, Coronal brain sections from wild-type and HEXB KO mice were costained with GFAP (red)- and Aβ40 and Aβ42 (green)-specific antibodies (FCA3340 and FCA3542, respectively). Increased GFAP immunoreactivity and reactive gliosis were observed in the cortex (b), subiculum (d), and CA3 region of the hippocampus (f, h) of HEXB KO mice. Scale bars: a–h, 500 μm.
Furthermore, we conducted colocalization studies for iAβ-LIR and glial fibrillary acidic protein-like immunoreactivity (GFAP-LIR) to confirm that Aβ-LIR accumulates in neurons rather than in glia (Fig. 5B). GFAP-LIR-positive cells did not accumulate iAβ-LIR. In addition, the morphology of the GFAP-positive cells in the cortex (Fig. 5Bb), subiculum (Fig. 5Bd), and CA3 region of the hippocampus (Fig. 5Bf,Bh) of HEXB KO mice indicated reactive gliosis.
Intraneuronal Aβ-LIR accumulates in endosomal–lysosomal compartments and in undegraded autophagosomes
The accumulation of iAβ-LIR in the HEXB KO mice could be a result of either increased Aβ generation as a result of prolonged presence of β-CTFs in endocytic compartments or reduced Aβ clearance by the lysosomes, or both. To determine whether iAβ-LIR accumulates in the endosomal–lysosomal system, we tested for colocalization of iAβ42-LIR (Fig. 6A) and iAβ40-LIR (data not shown) with markers of early endosomes (EEA1), late endosomes (Rab7), or lysosomes (Lamp-1) by IHC. In the wild-type mice, iAβ-LIR colocalized with the endosomal marker EEA1 and the lysosomal marker Lamp-1. In contrast, in the HEXB KO mice, enlarged EEA1-, Rab7-, and Lamp-1-positive compartments colocalized in part with iAβ-LIR (Fig. 6A). These results indicate that iAβ-LIR accumulates in different compartments of the endosomal–lysosomal system in HEXB KO mice.
Figure 6.
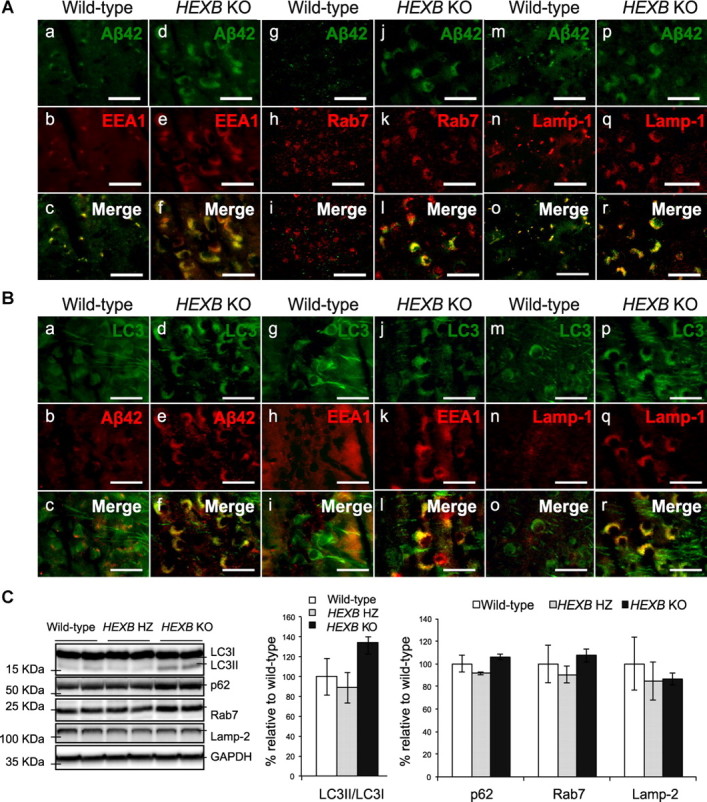
Dysfunction in lysosomal proteolysis leads to the accumulation of iAβ-LIR in the endosomal–lysosomal system and undegraded autophagosomes. A, Coronal brain sections of wild-type and HEXB KO mice were costained with Aβ42-specific antibodies (FCA3542) and markers of early endosomes (EEA1), late endosomes (Rab7), and lysosomes (Lamp-1). iAβ-LIR colocalized partially with EEA1 (a–f), Rab7 (g–l), and Lamp-1 (m–r) in HEXB KO mice. Images were taken from the subiculum. Scale bars, 50 μm. B, Immunohistochemical analysis shows the colocalization of iAβ42 (Pan1G6) with LC3 in undegraded autophagosomes in HEXB KO mice (d–f). The autophagosomal marker (LC3) colocalizes with the early endosomal marker (EEA1) (j–l) and with the lysosomal marker (Lamp-1) (p–r) in HEXB KO mice. Scale bars, 50 μm. C, Western blot analysis shows an increase in the LC3II/LC3I ratio and slight increases in the levels of p62 and Rab7 in HEXB KO mice relative to wild type (n = 3 for WT; n = 3 for HZ; n = 3 for KO). Shown are mean ± SEM.
In addition, we tested the colocalization of iAβ-LIR with the autophagosomal marker (LC3II) to test the possibility that iAβ might accumulate in undegraded autophagosomes (Fig. 6B). The antibody used recognizes the cytoplasmic (LC3I) and the membrane-bound (LC3II) forms of LC3. Upon induction of autophagy, LC3I gets converted to LC3II, which in turn binds to the membrane of forming autophagosomes (Noda et al., 2009). LC3II is normally degraded rapidly upon fusion of the autophagosome with lysosomes. Hence, the accumulation of LC3II-positive vesicular structures is indicative of defects in the fusion of autophagosomes with lysosomes, clearance of autophagosomes, or both. As shown in Figure 6Ba–Bc, LC3-positive material in the wild-type mice was mostly soluble, and this material was distributed throughout the cell bodies and axons, indicating that the majority of LC3 is present as LC3I. iAβ-LIR localized to small vesicular compartments that failed to colocalize with the compartments that stained for LC3I. In contrast, LC3 immunoreactivity in HEXB KO mice was present within small vesicular structures that clustered in the cell body (Fig. 6Bd), indicating that LC3II is accumulating in HEXB KO mice, an observation consistent with the hypothesis that autophagy may be defective in the neurons of these mice. Most importantly, iAβ-LIR colocalized with LC3II immunoreactivity in HEXB KO mice, suggesting that iAβ may be accumulating in undegraded autophagosomes (Fig. 6Bf).
Recently, Boland et al. (2010) suggested that the accumulation of autophagosomes in mouse models of LSDs is due to the impaired lysosomal clearance of the autophagic vacuoles (AVs) rather than enhanced induction of autophagy. The inefficient clearance of the autophagosomes by the lysosomes may occur due to impaired fusion with the lysosomes or the inability of the lysosomes to degrade the vacuoles. To examine fusion between the autophagosome and endosomal/lysosomal compartments, we monitored colocalization of the autophagosomal marker (LC3II) with the early endosomal marker (EEA1) and the lysosomal marker (Lamp-1). As shown in Figure 6B, LC3II colocalized with EEA-1 (Fig. 6Bj–Bl) and Lamp-1 (Fig. 6Bp–Br) in HEXB KO mice, indicating that autophagosomes fuse with endosomes and lysosomes in HEXB KO mice.
To confirm that HEXB KO mice have dysfunctional autophagy and reduced lysosomal clearance, we measured the protein levels of LC3I, LC3II, p62, Rab7, and Lamp-2 by Western blotting (Fig. 6C). Although p62 and Rab7 were only slightly elevated in HEXB KO mice (p62, 106 ± 2.5%; Rab7, 108 ± 6%), the ratio of LC3II/LC3I increased by 1.35-fold relative to age-matched wild-type mice (135 ± 12%), consistent with the presence of dysfunctional autophagosomal–lysosomal turnover in HEXB KO mice.
Human Sandhoff brains accumulate APP-CTFs, intraneuronal Aβ, and ganglioside-bound Aβ
To test whether human gangliosidoses brains (Table 1), including SD, exhibit obvious Aβ histopathology, fixed cortical sections obtained from three SD brains (1, 1, 2 years), two GM1 gangliosidosis brains (1, 19 years), and three TS brains (4, 27, 45 years) were immunostained using the monoclonal antibody 4G8. The total ganglioside levels in the cortices of the different brain samples were analyzed by thin-layer chromatography (Fig. 7A). Purified GM1, GM2, and GM3 gangliosides were loaded as markers to monitor the electrophoretic mobility of the individual ganglioside species (labeled as “standards”).
Table 1.
List of human brain tissue samples analyzed in this study
| ID no. | Disorder | Age (years) | Sex | Cause of death | Brain regions |
|---|---|---|---|---|---|
| C1 | Control | 1 | M | Dehydration | Frontal cortex, hippocampus |
| C2 | Control | 2 | F | Intussusception of the bowel | Frontal cortex, hippocampus |
| C4 | Control | 4 | F | Asthma | Frontal cortex, hippocampus |
| C27 | Control | 27 | F | Respiratory failure | Frontal cortex, hippocampus |
| SD1a | Sandhoff disease | 1 | M | Complication of the disorder | Frontal cortex, hippocampus |
| SD1b | Sandhoff disease | 1 | F | Unknown | Frontal cortex |
| SD2 | Sandhoff disease | 2 | Unknown | Unknown | Frontal cortex, hippocampus |
| GM1 | GM-1 gangliosidosis | 1 | F | Complication of the disorder | Frontal cortex, hippocampus |
| GM19 | GM-1 gangliosidosis | 19 | M | Complication of the disorder | Frontal cortex |
| TS4 | Tay–Sachs disease | 4 | Unknown | Unknown | Frontal cortex |
| TS27 | Tay–Sachs disease | 27 | F | Complication of the disorder | Frontal cortex, hippocampus |
| TS45 | Tay–Sachs disease | 45 | M | Complication of the disorder | Frontal cortex |
| AD75 | Alzheimer's disease | 75 | M | Unknown | Frontal cortex |
Human Sandhoff, GM1 gangliosidosis, Tay–Sachs, AD, and control brains analyzed in this study are designated as SDn, GMn, TSn, ADn, CAAn, and Cn, respectively. In each case, “n” represents the age of the patient at the time of death. Brains from the two 1-year-old SD patients analyzed are designated as SD1a and SD1b. In each sample, genetic deficiency in enzyme activity and the resulting substrate (ganglioside) accumulation was confirmed by enzyme activity and thin-layer chromatography analyses.
Figure 7.
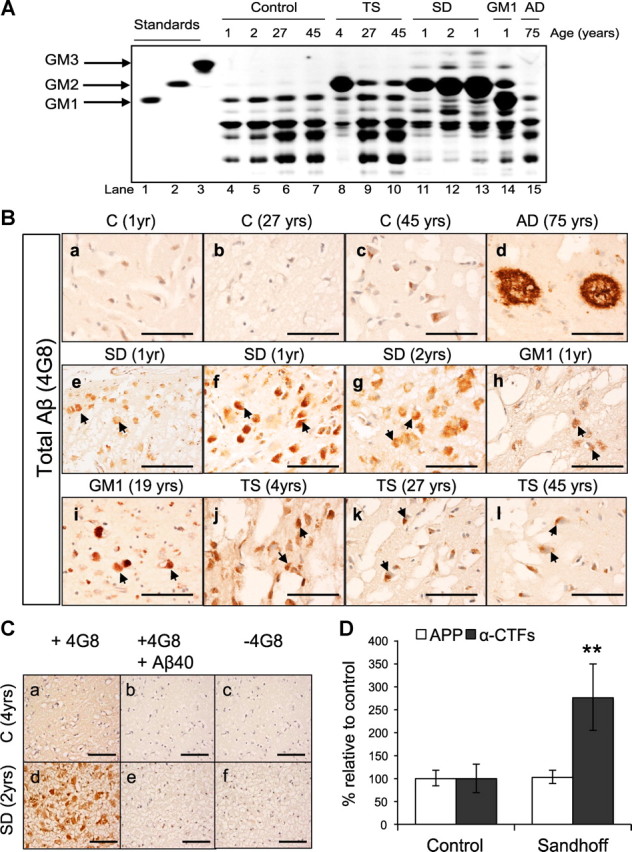
Human Sandhoff brains accumulate intraneuronal 4G8 immunoreactivity and APP-CTFs. A, Thin-layer chromatography analysis of cortices derived from GM1 gangliosidosis (GM1), SD, TS, AD, and age-matched controls, shows the levels of different gangliosides in each brain sample. B, GM1, SD, TS, AD, and age-matched control-derived cortical sections were immunostained using the monoclonal antibody 4G8. The arrows indicate accumulation of intraneuronal APP/Aβ-LIR in 1-year-old (e, f) and 2-year-old (g) SD brains; 1-year-old (h) and 19-year-old (i) GM1 brains; 4-year-old (j), 27-year-old (k), and 45-year-old (l) TS brains. Scale bars: a–l, 200 μm. C, The specificity of 4G8 staining was confirmed by immunostaining cortical sections derived from human SD and control brains with 4G8 antibody preincubated with 100× molar excess of Aβ1–40 peptide (b, e), or with secondary antibody only (c, f). Scale bars: a–f, 200 μm. D, Western blot analysis of seven fibroblast cell lines derived from SD patients or healthy controls show significant accumulation of α-CTFs in the SD fibroblasts (**p = 0.007). Values are the mean of three individual experiments. Error bars indicate SEM.
As shown in Figure 7B, the SD brains (Fig. 7Be–Bg), GM1 gangliosidosis brains (Fig. 7Bh,Bi), and the TS brains (Fig. 7Aj–Al) showed obvious intraneuronal 4G8 immunoreactivity. Typical amyloid plaques were observed in the AD brain (75 years), as expected (Fig. 7Bd). To confirm that 4G8 staining of the human gangliosidoses brains is specific, cortical sections derived from human SD and control brains were immunostained either with 4G8 only, with 4G8 preincubated with 100× molar excess of Aβ1–40 peptide, or with secondary antibody only (Fig. 7C). No staining was observed if the cortical sections were not treated with the primary antibody (Fig. 7Cc,Cf), or if the 4G8 primary antibody was preincubated with Aβ1–40 peptide (100× molar excess) (Fig. 7Cb,Ce).
Since 4G8 also recognizes APP, α-CTFs, and β-CTFs, it is likely that at least some of the 4G8 staining observed in the human gangliosidoses brains could result from APP-CTFs. We measured the levels of APP and α-CTFs in fibroblast cell lines derived from seven different SD patients and seven healthy controls. As shown in Figure 7D, α-CTF levels increase significantly in the SD cell lines relative to the healthy controls (**p < 0.007). Supporting our findings, Tamboli et al. (2011) reported recently strong accumulation of APP-CTFs in primary fibroblasts derived from patients with different sphingolipid storage diseases.
Furthermore, we performed additional experiments using Aβ40- and Aβ42-specific antibodies (FC3340 and FC3542, respectively) to test whether the intraneuronal 4G8-immunoreactive material reflects accumulation of Aβ40, Aβ42, or both. Unlike 4G8, which reacts with an array of APP fragments, FC3340 and FC3542 are specific for Aβ peptides. As shown in Figure 8A, the Aβ42-specific antibody (FC3542) showed significant intraneuronal staining of the 1-year-old SD brain similar to the staining observed with 4G8 (Fig. 8Aa,Ab). Similar results were obtained for the other human gangliosidoses brains stained with FC3542 and FC3340 (data not shown). FC3542 also stained rare extracellular Aβ-like immunoreactivity in the 45-year-old TS brain (Fig. 8Ad,Ae), and numerous plaques in the 75-year-old AD brain (Fig. 8Ag,Ah).
Figure 8.
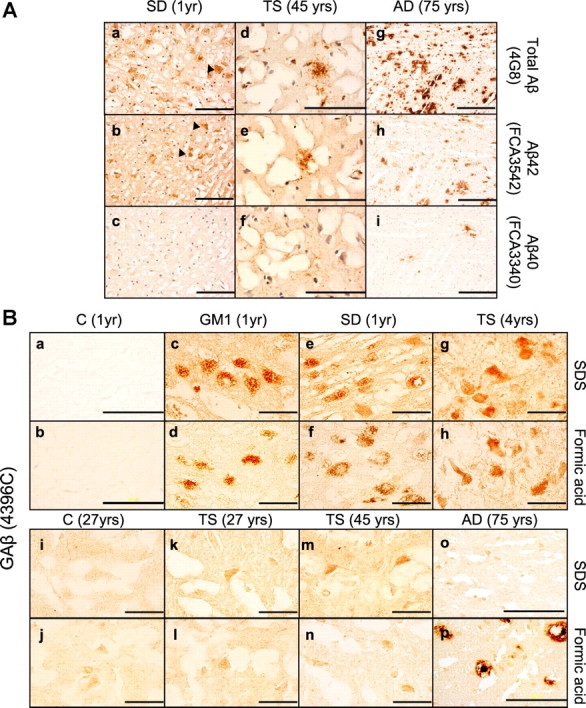
Human gangliosidoses brains accumulate intraneuronal ganglioside-bound Aβ. A, Immunostaining of human brain cortical sections using the antibodies 4G8, FC3542 (Aβ42-specific antibody), and FC3340 (Aβ40-specific antibody) demonstrates that Aβ42 accumulates in SD (b) and TS brains (e). Extracellular Aβ-like immunoreactivity structures were observed in the 45-year-old TS brain (e). The arrows indicate accumulation of intraneuronal Aβ-LIR. Scale bars: a–f, 200 μm; g–i, 500 μm. B, Cortical sections obtained from control (1 year) (a, b), control (27 years) (i, j), GM1 (1 year) (c, d), SD (1 year) (e, f), TS (4 years) (g, h), TS (27 years) (k, l), TS (45 years) (m, n), and AD (75 years) (o, p) were stained with 4396C antibody to detect ganglioside-bound Aβ. Scale bars: a, b, o, p, 200 μm; c–n, 50 μm.
Finally, to test whether the human gangliosidoses brains accumulate GAβ, the species that has been proposed to play a key role in seeding Aβ aggregation, fixed cortical sections were immunostained using the 4396C monoclonal antibody that recognizes GAβ (Fig. 8B). 4396C immunoreactivity was not observed in the control brains (Fig. 8Ba,Bb,Bi,Bj). In contrast, the young human gangliosidoses brains (1–4 years of age) showed prominent 4396C staining (Fig. 8Bc–Bh). Interestingly, in the case of the TS brains, the intensity of 4396C staining (TS4 > TS27 > TS45) correlated with the extent of GM2 ganglioside accumulation (Fig. 7A), consistent with the interpretation that the relative intensity of 4396C staining parallels the relative concentrations of GAβ. It is also worth noting that the intraneuronal 4396C immunoreactivity was observed when the gangliosidoses brain sections were subjected to detergent pretreatment as well as formic acid, albeit the signal was stronger after using the detergent. In contrast, 4396C intraneuronal staining was not observed in the AD brain after the detergent or formic acid pretreatments; only extracellular amyloid plaques were observed after formic acid pretreatment (Fig. 8Bp).
Discussion
Several recent studies have highlighted the neuropathological similarities between AD and LSDs (Ohm et al., 2003; Treiber-Held et al., 2003; Saito et al., 2004; Ohmi et al., 2009; Boland et al., 2010; Zhang et al., 2010; Mattsson et al., 2011). AD and LSDs are both characterized by defects in the endosomal–lysosomal and lysosomal–autophagosomal pathways (Nixon, 2007; Pickford et al., 2008), all of which are major pathways for APP processing and Aβ generation (Caporaso et al., 1992, 1994). These defects may contribute to the pathogenesis and progression of AD and LSDs since they disrupt the sorting and recycling of lipids and proteins and result in the accumulation of mutant and oxidized proteins, protein aggregates, and damaged organelles in affected neurons (Bahr and Bendiske, 2002; Nixon et al., 2008).
Lysosomal lipid accumulation has been linked to autophagic dysfunction (Boland et al., 2008; Tamboli et al., 2011), suggesting that changes in lipid composition of the lysosomal membranes may alter the lysosomal–autophagic turnover of proteins in AD and LSDs. In addition, a growing body of evidence suggests that age-related changes in the metabolism of gangliosides may contribute to the pathogenesis of AD (Ariga et al., 2008; Yamamoto et al., 2008). Gangliosides are sialic acid-containing glycosphingolipids that are predominantly present in the outer leaflet of the plasma membrane. Deficiencies in the activities of lysosomal enzymes that degrade gangliosides lead to ganglioside accumulation in cellular membranes. Neuronal accumulation of gangliosides in LSDs may result in neurodegeneration by multiple mechanisms, including alteration of calcium homeostasis, disruption of endocytic trafficking, perturbation of the sorting and recycling of lipids and proteins, impairment of autophagy, and/or reduction in lysosomal proteolysis of material intended for terminal degradation (Pagano, 2003; Sillence and Platt, 2004; te Vruchte et al., 2004; Boland et al., 2010; Lloyd-Evans et al., 2010).
To investigate whether the accumulation of gangliosides can result in the accumulation of neuropathogenic proteins associated with AD, we examined the accumulation of APP-CTFs, Aβ, α-synuclein, and pTau in a mouse model of SD, which was developed through targeted disruption of the HEXB gene (Sango et al., 1995). The resulting deficiency in the both β-hexoaminidase A and B in SD mice resulted in extensive accumulation of GM2 and asialo-GM2 (GA2) throughout the central and peripheral nervous systems (Kolter and Sandhoff, 1998; Mahuran, 1999). Beginning 3 months after birth, HEXB KO mice exhibited progressive neurologic disturbances that started with defects in balance and coordination and progressed into an almost complete loss of hindlimb movement. HEXB KO mice die at the age of 4–5 months due to their inability to eat, drink, or move (Sango et al., 1995, 1996; Phaneuf et al., 1996). The neurologic features and pathological findings of the SD mice are consistent with human SD (Sango et al., 1995) and are thus considered to be a valuable tool to study disease pathogenesis.
We observed extensive accumulation of 4G8-immunopositive signals in most brain regions and in the spinal cords of HEXB KO mice (Fig. 1). We also observed accumulation of iAβ-LIR in specific brain regions (Fig. 4). In addition, we observed accumulation of pTau in the medulla and spinal cords of HEXB KO mice, and accumulation of α-synuclein in the SNC (Fig. 2). Interestingly, 4G8 immunoreactivity in the SN did not overlap with α-synuclein, suggesting that APP/APP-CTFs/Aβ and α-synuclein accumulate in different regions of the SN. The latter may indicate different mechanisms of accumulation or turnover pathways for APP/APP-CTFs/Aβ and α-synuclein in different subregions of the SN. Regional differences in ganglioside metabolism have been suggested to underlie the distribution of neuronal storage (Sango et al., 1995), indicating that the ganglioside species involved in the accumulation of APP/APP-CTFs/Aβ may be different from those involved in accumulation of α-synuclein.
Because APP and Aβ share many epitopes, the identity of iAβ-LIR has been recently attributed, at least in part, to intracellular APP and/or β-CTFs (Winton et al., 2011). To determine the molecular identity of the 4G8 signals, we performed immunohistochemical and biochemical analyses of homozygous (HEXB KO) and heterozygous (HEXB HZ) mouse models of SD. The biochemical analyses of APP metabolism in HEXB HZ and KO mice demonstrated that the levels of APP-CTFs and Aβ were significantly increased, whereas the levels of full-length APP were similar to those observed in wild-type littermates (Fig. 3). The authenticity of the Aβ-LIR was established by ELISA measurements (Fig. 3), Western blotting analysis (Fig. 3), and immunohistochemistry using Aβ-specific C-terminal neoepitope antibodies (Fig. 4). In addition, immunohistochemical analysis using an APP N terminus antibody (22C11) and an APP C terminus antibody (369) demonstrated that 22C11 immunoreactivity did not increase in the HEXB KO mice (Fig. 3). Based on all of the above, we conclude that the accumulated intraneuronal 4G8 signals represent accumulated APP-CTFs and/or Aβ rather than full-length APP. We cannot exclude the possibility that amyloid-β precursor-like protein 2 (APLP2) might have contributed to part of the signal from antibody 369 (Slunt et al., 1994), but the Aβ-specific antibodies would not be expected to cross-react with APLP2 since APLP2 lacks the Aβ domain.
Worth mentioning, we did not observe iAβ accumulation in mouse models of other LSDs, such as Gaucher disease (L444P/L444P GCase), Fabry disease (GLA−/−), and Pompe disease (GAA−/−) (our unpublished observations).
Under normal conditions, Aβ generated in endosomes and autophagosomes is cleared by lysosomes (Nixon, 2007). Recent studies have shown that the accumulation of GSL in human H4 cells (Tamboli et al., 2011) and in mouse models of LSDs (Boland et al., 2010) leads to impairment in the clearance of AVs. As a result, we investigated the possibility that iAβ might be accumulating in endosomal–lysosomal compartments and in undegraded AVs. As expected, iAβ-LIR was found to colocalize with the autophagosomal marker LC3II, the late endosomal marker Rab7, and the lysosomal marker Lamp-1. In addition, we observed that, although autophagosomes fuse with both the endosomes and lysosomes in HEXB KO mice, iAβ remains undegraded, suggesting that impaired lysosomal clearance of AVs contributes to the accumulation of iAβ.
However, Tamboli et al. (2011) reported that the accumulation of GSL in human H4 and SHSY-5Y cells leads to the accumulation of APP-CTFs in undegraded AVs and stimulate γ-secretase activity, which, in turn, would be predicted to cause increased production of intracellular and secreted Aβ peptides. Together, these results suggest that the accumulation of iAβ in HEXB KO mice is probably due to the following: (1) increased generation in the autophagosomes and endosomes, (2) reduced degradation by the lysosomes, or (3) some combination of (1) and (2).
Moreover, Tamboli et al. (2011) reported the accumulation of APP-CTFs in human fibroblasts derived from different LSD patients. In this study, we found that intraneuronal Aβ-LIR accumulates throughout the frontal cortices of GM1, SD, and TS patients (Fig. 7B), in a pattern similar to that observed in the HEXB KO mice. Similarly, we observed intraneuronal accumulation of GAβ, a proposed prefibrillar aggregate found in AD (Fig. 8B). These results indicate that the inefficient Aβ degradation by the lysosomes in LSDs and its accumulation in compartments that favor Aβ aggregation might be a primary reason for the pathology and neurodegeneration.
The relative contributions of intraneuronal and extracellular Aβ to the overall neurotoxicity of the accumulated Aβ remain unclear. Our data suggest that iAβ plays at least some role in disease pathogenesis. Neurotoxicity by iAβ42 has been attributed to Aβ42 accumulation in various organelles, including autophagosomes (Yu et al., 2005), endosomes/lysosomes (D'Andrea et al., 2001; Takahashi et al., 2002), and mitochondria (Caspersen et al., 2005; Manczak et al., 2006). It has been suggested that iAβ42 exerts its neurotoxic effects by compromising the organelle membrane integrity, causing organelle leakage and eventually leading to neuronal death (Yang et al., 1998; Glabe, 2001; Ling et al., 2009). Furthermore, it has been proposed that the release of iAβ into the extracellular space by exocytosis contributes to extracellular amyloid deposition (Rajendran et al., 2006). In support of this formulation, Yang et al. (2011) recently highlighted the pathogenic importance of the lysosomal system by demonstrating that improving neuronal autophagic–lysosomal proteolytic function in a mouse model of AD reduced levels of intracellular and extracellular Aβ and reversed multiple cognitive deficits.
In summary, our results suggest that the accumulation of gangliosides may increase the accumulation of proteins associated with AD by interfering with the lysosomal–autophagic turnover of proteins (Boland et al., 2008), by enhancing the generation of Aβ by activating γ-secretase (Tamboli et al., 2011), or by a combination of both mechanisms. In addition, the detection of GAβ in HEXB KO mice and human LSD samples indicates the presence of a specific Aβ conformer that is more resistant to degradation and raises the question whether toxic Aβ oligomers form in the cortices of the LSD patients. Together, these results establish an association between the accumulation of gangliosides and the accumulation of aggregated proteins associated with AD. If the linkage of these phenomena is due, at least in part, to the promotion of accumulation of aggregated AD-related proteins, it is possible that modulating ganglioside metabolism may delay or prevent AD.
Footnotes
This work was supported by Alzheimer's Drug Discovery Foundation Grant 291003 VEN and Amicus Therapeutics. F.C. was supported by a grant from the Fondation pour la Recherche Médicale. Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, MD). We thank Dr. Thomas Seyfried (Boston College, Boston, MA) for providing the brain samples from mice that were heterozygous or homozygous for the HEXB gene.
Y.L., A.C.S., H.N.W., E.R.S., R.K., K.J.V., D.J.L., and B.A.W. are full-time employees and stockholders of Amicus Therapeutics. S.G. is an advisor to DiaGenic and to the Pfizer Janssen Alzheimer Immunotherapy Alliance and has grants from Amicus Therapeutics and Baxter.
References
- Ariga T, McDonald MP, Yu RK. Role of ganglioside metabolism in the pathogenesis of Alzheimer's disease—a review. J Lipid Res. 2008;49:1157–1175. doi: 10.1194/jlr.R800007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 2002;83:481–489. doi: 10.1046/j.1471-4159.2002.01192.x. [DOI] [PubMed] [Google Scholar]
- Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Smith DA, Mooney D, Jung SS, Walsh DM, Platt FM. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010;285:37415–37426. doi: 10.1074/jbc.M110.186411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab. 2008;94:391–396. doi: 10.1016/j.ymgme.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Greengard P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci U S A. 1992;89:2252–2256. doi: 10.1073/pnas.89.6.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer β/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–3138. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurons accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci. 2001;17:137–145. doi: 10.1385/JMN:17:2:137. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, Yamamoto N, Michikawa M, Yoshikawa Y, Terao K, Matsuzaki K, Lemere CA, Selkoe DJ, Naiki H, Yanagisawa K. A seed for Alzheimer amyloid in the brain. J Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Glycosphingolipid degradation and animal models of GM2-gangliosidoses. J Inherit Metab Dis. 1998;21:548–563. doi: 10.1023/a:1005419122018. [DOI] [PubMed] [Google Scholar]
- Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Waller-Evans H, Peterneva K, Platt FM. Endolysosomal calcium regulation and disease. Biochem Soc Trans. 2010;38:1458–1464. doi: 10.1042/BST0381458. [DOI] [PubMed] [Google Scholar]
- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Zetterberg H, Bianconi S, Yanjanin NM, Fu R, Månsson JE, Porter FD, Blennow K. Gamma-secretase-dependent amyloid-beta is increased in Niemann-Pick type C: a cross-sectional study. Neurology. 2011;76:366–372. doi: 10.1212/WNL.0b013e318208f4ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4:590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16:984–990. doi: 10.1038/cdd.2009.54. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Treiber-Held S, Distl R, Glöckner F, Schönheit B, Tamanai M, Meske V. Cholesterol and tau protein—findings in Alzheimer's and Niemann Pick C's disease. Pharmacopsychiatry. 2003;36:S120–6. doi: 10.1055/s-2003-43060. [DOI] [PubMed] [Google Scholar]
- Ohmi K, Kudo LC, Ryazantsev S, Zhao HZ, Karsten SL, Neufeld EF. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci U S A. 2009;106:8332–8337. doi: 10.1073/pnas.0903223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano RE. Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philos Trans R Soc Lond B Biol Sci. 2003;358:885–891. doi: 10.1098/rstb.2003.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathy S, Davies P, Haroutunian V, Purohit DP, Davis KL, Mohs RC, Park H, Moran TM, Chan JY, Buxbaum JD. Correlation between Abetax-40-, Abetax-42-, and Abetax-43-containing amyloid plaques and cognitive decline. Arch Neurol. 2001;58:2025–2032. doi: 10.1001/archneur.58.12.2025. [DOI] [PubMed] [Google Scholar]
- Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR, Ritter G, Igdoura SA, Morales CR, Benoit G, Akerman BR, Leclerc D, Hanai N, Marth JD, Trasler JM, Gravel RA. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1996;5:1–14. doi: 10.1093/hmg/5.1.1. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Suzuki K, Hulette CM, Murayama S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J Neuropathol Exp Neurol. 2004;63:323–328. doi: 10.1093/jnen/63.4.323. [DOI] [PubMed] [Google Scholar]
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- Sango K, McDonald MP, Crawley JN, Mack ML, Tifft CJ, Skop E, Starr CM, Hoffmann A, Sandhoff K, Suzuki K, Proia RL. Mice lacking both subunits of lysosomal beta-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat Genet. 1996;14:348–352. doi: 10.1038/ng1196-348. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantitation. Methods Mol Biol. 2005a;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Methods Mol Biol. 2005b;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord. 2011;26:1593–1604. doi: 10.1002/mds.23774. [DOI] [PubMed] [Google Scholar]
- Sillence DJ, Platt FM. Glycosphingolipids in endocytic membrane transport. Semin Cell Dev Biol. 2004;15:409–416. doi: 10.1016/j.semcdb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP) J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- Suzuki K, Iseki E, Katsuse O, Yamaguchi A, Katsuyama K, Aoki I, Yamanaka S, Kosaka K. Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. Neuroreport. 2003;14:551–554. doi: 10.1097/00001756-200303240-00004. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer Abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM, Saftig P, Sandhoff K, Walter J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Aβ generation. J Neurosci. 2011;31:1837–1849. doi: 10.1523/JNEUROSCI.2954-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM, van Blitterswijk WJ, Sillence DJ. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279:26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- Treiber-Held S, Distl R, Meske V, Albert F, Ohm TG. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J Pathol. 2003;200:95–103. doi: 10.1002/path.1345. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu YH, Barnes S, Sun Y, Grabowski GA. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab. 2011;102:436–447. doi: 10.1016/j.ymgme.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Matsubara T, Sato T, Yanagisawa K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochim Biophys Acta. 2008;1778:2717–2726. doi: 10.1016/j.bbamem.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K. Pathological significance of ganglioside clusters in Alzheimer's disease. J Neurochem. 2011;116:806–812. doi: 10.1111/j.1471-4159.2010.07006.x. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1–42 pathogenesis. J Neurosci Res. 1998;52:691–698. doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy—a novel beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang X, Jiang F, Wang W, Vincent I, Bu B. Mitotic epitopes are incorporated into age-dependent neurofibrillary tangles in Niemann-Pick disease type C. Brain Pathol. 2010;20:367–377. doi: 10.1111/j.1750-3639.2009.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]