

Abstract
sn2-15-HpETE-PE generated by mammalian 15-lipoxygenase/phosphatidylethanolamine binding protein-1 (PEBP1) complex is a death signal in a recently identified type of programmed cell demise, ferroptosis. How the enzymatic complex selects sn2-ETE-PE as the substrate among 1 of ~100 total oxidizable membrane PUFA phospholipids is a central yet unresolved question. To unearth the highly selective and specific mechanisms of catalytic competence, we used a combination of redox lipidomics, mutational and computational structural analysis to show they stem from: i) reactivity towards readily accessible hexagonally organized membrane sn2-ETE-PEs, ii) relative preponderance of sn2-ETE-PE species vs. other sn2-ETE-PLs, iii) allosteric modification of the enzyme in the complex with PEBP1. This emphasizes the role of enzymatic vs. random stochastic free radical reactions in ferroptotic death signaling.
Graphical Abstract
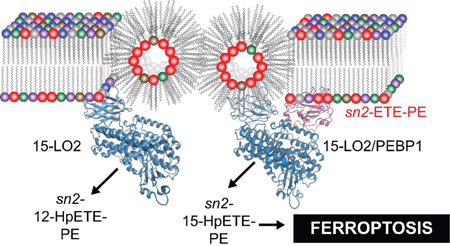
Ferroptosis is a recently identified cell death program1 whose significance relates to a potential role in pathogenesis of several diseases.2–9 Ferroptotic program includes three major metabolic routes dependent on: i)lipid peroxidation, ii)intracellular iron, and iii) glutathione peroxidase-4 insufficiency.10 While details of these three (dys)regulated pathways begin to emerge, there are gaps in understanding of the reactions involved in ferroptosis execution. Recent identification of selective lipid peroxidation products, 15-hydroperoxy-eicasotetraenoyl-phosphatidylethanolamines (sn2-15-HpETE-PE), as death signals suggests involvement of specific enzymatic mechanisms.11 Indeed, two isoforms of 15-lipoxy-genases (15-LO1 and 15-LO2) were identified as catalysts of these reactions.12 The typical substrates of different LOXes are poly-unsaturated fatty acids (PUFA), particularly eicosatetraenoic acid (ETE), whose precise location within the catalytic site determines regio-specificity towards oxidation at the 5, 8, 12, or 15th carbons.13 While 15-LOX can also oxidize membrane phospholipids (PLs), the mechanisms underlying selectivity towards individual molecular species of PE as well as positional specificity of the hydroperoxy-products remain enigmatic. Recently, we discovered that a scaffold protein, PE binding protein-1 (PEBP1), forms a complex with 15-LOX and allosterically regu-lates the enzyme to accommodate sn2-ETE-PE at the catalytic site.5
To unveil the mechanisms of specific peroxidation of sn2-ETE-PE during ferroptosis, we applied redox lipidomics, mutational analysis and computational protocols. We used 15-LO2, which is similar to 15-LO1 and involved in pro-ferroptotic diseases (brain trauma, acute kidney injury (AKI)).5 15-LOX selects sn2-ETE-PE among highly diversified sn2-ETE-PLs and lodges the substrate juxtaposed to the catalytic iron. Pro-ferroptotically stimulated cells and kidney tissue, respond by robust oxidation of a variety of PLs, among which PE dominates in terms of the number of oxygenated species and amounts of oxidized products (Figure 1A–B, S1A–B). MS/MS fragmentation identified these species as sn2-15-HpETE-PE (Figure 1C–D, S1C–D), confirming the involvement of 15-LOX. Oxygenated PE associated with ferroptosis were determined as “surviving” oxidized PE species–increased after pro-ferroptotic stimulation and decreased following treatment with a ferroptosis inhibitor, ferrostatin-1 (Fer-1). Only two Fer-1-sensitive sn2-ETE-PE peroxidation species, sn1-18:0/sn2-HpETE-PE and sn1-18:1/sn2-HpETE-PE were detected in cells and tissues (Figure 1E).
Figure 1.
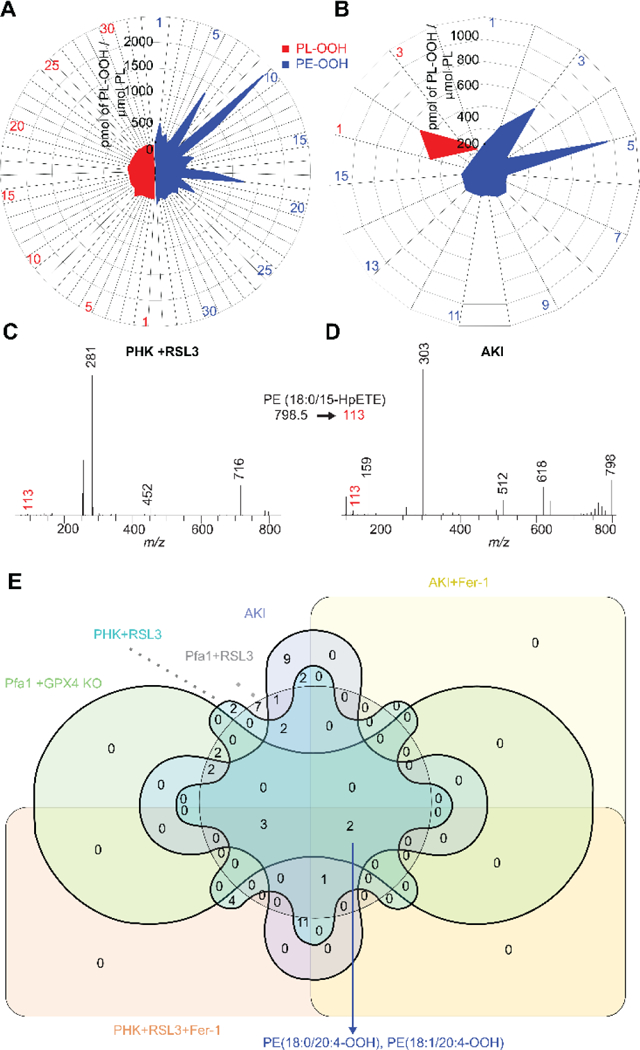
(A, B) Accumulation of HOO-PL products in pro-ferroptotic cells (RSL3 treatment) and tissue (acute kidney injury) compared to normal conditions. (refer Table S1 for the species list). (C, D). Fragmentation of sn1-18:0/sn2-20:4-OOH showing sn2-15-HpETE-PE formation in pro-ferroptotic conditions. (E) Venn diagram of significant signals from pro and anti ferroptotic conditions showing “surviving” PE-OOH products
To characterize selectivity of sn2-ETE-PE oxidation by 15-LOX, we compared two ferroptotic substrates, sn1-18:0/sn2-ETE-PE and sn1-18:1/sn2-ETE-PE, with: i) total number of oxidizable PUFA-PLs in mouse kidney and Pfa1 cells (Figure S2), and ii) total amount of oxidizable PUFA-PLs in Pfa1 cells (Figure S3). The analysis showed selectivity of 15-LOX towards the two ferroptotic substrates among all oxidizable PLs. This prompted us to explore the mechanisms responsible for the specificity/selectivity of 15-LOX catalysis.
Using a simplified LC-MS/MS method (Figure S4), we compared oxidation of major PL classes in liposomes containing sn1-18:0/sn2-ETE-PL by human recombinant 15-LO2 (Figure S5). Oxidation rates varied for each PL, with phosphatidylserine (PS) exhibiting the lowest and phosphatidic acid (PA), the highest rates (Figure 2A). No correlation was observed between oxidation rate and charge of the PL head group. We next performed molecular docking14 to assess whether PL head groups affected binding of PLs to the 15-LO2 active site. Sn2-ETE docked into the active site pocket (Figure S6) with similar binding affinities for different PLs (Figure S7A). No correlation was found between oxidation rates and binding affinities (Figure S7B). Docking of abundant PUFA-containing PLs–phosphatidylcholine (PC), PE, and phosphatidyl-inositol (PI)–revealed no differences in binding affinities with varying PUFAs (Figure S8A–C). However, binding to the active site pocket was limited by increasing acyl chain length and unsaturation (Figure S8D–F). Accordingly, docosahexaenoic acid (DHA)-containing PLs were least likely to dock, while sn2-ETE-PE fell in optimal range for docking. Considering cellular abundance of ETE-PL and experimentally determined PL oxidation rates (Figure 2A), a theoretical PL oxidation rate in cells was calculated demonstrating a greater likelihood of 15-LO2-mediated oxidation of PE vs. other PLs (Figure S9A). Considering PUFA docking efficiencies in the theoretical rate calculation further supported specific oxidation of ETE/docosatetraenoic acid (DTE)-PLs in ferroptosis (Figure S9B).
Figure 2.
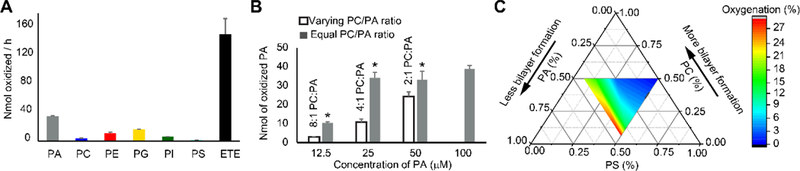
(A) Oxidation rates of various PLs by 15-LO2 in liposomes. (B) PA oxidation in liposomes with variable sn1–18:1/sn2–18:1-PC:sn1–18:0/sn2–20:4-PA ratios compared to liposomes with constant sn1–18:1/sn2–18:1-PC:sn1–18:0/sn2–20:4-PA ratio. *p<0.05 vs. liposome of variable sn1–18:1/sn2–18:1-PC:sn1–18:0/sn2–20:4-PA ratio. (C) Ternary plot showing PS oxidation in liposomes with varying content of non-oxidizable PA and PC.
Given that polar heads did not affect interactions with 15-LOX, we explored whether different structural arrangements of sn2-ETE-PLs in the membrane could play a role in their oxidation by 15-LOX. Conical-shaped lipids (PE and PA) form hexagonal phase, whereas spherical-shaped lipids such as PC form bilayers.15 We previously showed that sn2-ETE-PE oxidation by 15-LO2 is more efficient compared to sn2-ETE-PC and suggested that this is associated with the physical state of the membrane.11 Therefore, we examined correlation of head group surface areas with PL oxidation rates. This confirmed that conical- and inverted conical-shaped lipids which form hexagonal phase-II (HII) and hexagonal phase-I (HI), respectively, showed higher oxidation rates than spherical, bilayer-forming lipids (Figure S10). Then, we experimentally manipulated structural organization of liposomes by altering PC (spherical lipid) to PA or PE (conical lipids) ratios. Oxidation of PA and PE was less effective in the liposomes with the higher fraction of the bilayer organization than in HII phase (Figure 2B, S11). Similarly, cylindrically shaped PS underwent greater oxidation with increasing fraction of non-bilayer arrangements due to greater content of non-oxidizable conically shaped PA (Figure 2C). Knocking out one of the major enzymatic regulators of phospholipid biosynthesis, acyl CoA synthetase long chain family member-4 (ACLS4), makes cells resistant to ferroptosis.16 Phospholipidomics analysis revealed that Acsl4 KO cells had a lower percentage of PUFA containing PLs and a lower percentage of non-bilayer producing lipids (PE and PI) than WT cells (Fig. S12A). We then analyzed how the changes in percentage of non-bilayer lipids and the percentage of PUFA containing lipids affected the oxidation of two major ETE containing phospholipids, sn1-18:0/sn2-20:4-PE and sn1-18:0/sn2-20:4-PC. Both of these hexagonal phase associated properties strongly correlated with the formation of sn1-18:0/sn2-HpETE-PE and sn1-18:0/sn2-HpETE-PC (Fig. S12B, C, R >0.95, p <0.01). This indicated hexagonal phase regulated oxidation in physiological condition. The increase in the formation of HpETE-PE upon changes in percentage of PUFA containing PLs and non-bilayer producing lipids was 3.4 times higher than that of PC (Fig. S12B, C).
Assuming that 15-LOX/PEBP1 complex is essential for ferroptosis, we explored the role of the scaffold protein in sn2-ETE-PL oxidation. PEBP1 increased oxidation of PE ~2-fold, whereas oxidation of PC and PI remained unchanged and PA oxidation was decreased by ~30% (Figure S13). We further assessed 15-LO2±PEBP1 catalyzed oxidation of phospholipid mixtures closely resembling lipid composition of Pfa cells (Table S2). PE was maximally oxidized vs other classes of phospholipids and sn2-ETE-PE oxidation was 5 times more effective than that of the closest oxidized competitor, ETE-PI. The levels of oxidized ETE-PA species were incomparably lower (< 0.01pmol/nmol phospho-lipids) in this model system (Figure 3A). The amounts of oxidized ETE-PL strongly correlated with the predicted value (Figure S9C) with a correlation coefficient of 0.99 (p<0.001).
Figure 3.
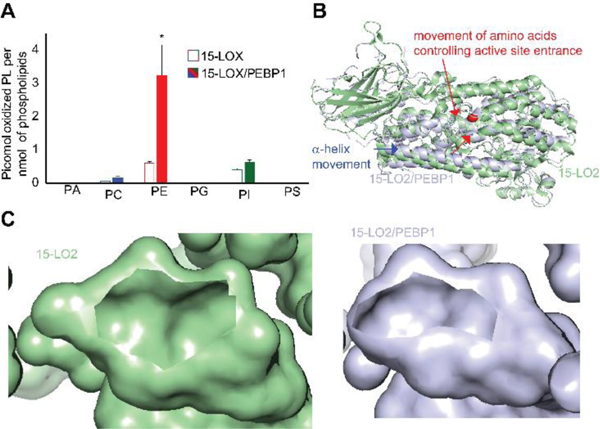
(A) Oxidation rates of phospholipids by 15-LO2 and 15-LO2/PEBP1 complex in liposomes prepared from a phospholipid mixture. *p<0.05 vs. 15-LO2. (B) ANM structure of 15-LO2 showing the movement of lid helix upon binding with PEBP1. (C) Images showing the reduction in the size of the active site entrance in 15-LO2 (left) and 15-LO2/PEBP1 complex (right).
Assuming that PEBP1 is an allosteric modifier of 15LOX, we performed computational analysis using anisotropic network model (ANM).17 This showed that PEBP1 interactions with 15-LO2 altered the binding cavity to facilitate insertion of sn2-ETE-PE (Video S1). While sn2-ETE-PE effectively docked into the active site of 15-LO2/PEBP1 complex, sn2-ETE-PI and sn2-ETE-PC did not dock into the active site pocket (Figure S14). Cooperative binding of PEBP1 caused movements of the flexible helix towards the enzyme interior (Figure 3B) decreasing the active site entrance hence restricting accessibility of PLs with large head groups (PC and PI) (Figure 3C).
To test the effects of 15LOX/PEBP1 complexes in more physiological conditions, we analyzed lipid oxidation in the samples not-treated with RSL3. The latter causes inhibition of GPX4, blocks the reduction of lipid hydroperoxides to the respective alcohols and enhances the production of oxidatively truncated electrophiles readily interacting with nucleophilic (protein) targets. To manipulate the availability of PEBP1 for the interactions with 15LOX, we employed two approaches: genetic knock down and chemical modification of the protein by a small molecule, locostatin, known to covalently interact with His86 and liberate it from its association with Raf1 kinase.5 In cells with active GPX4, PEBP1 KD resulted in the selectively lower levels of eight HETE-PE species (Table S4). Expectedly, locostatin treated cells had elevated levels of HETE-PE in 7 species.
For free-ETE, oxidation by 15-LOX is mostly restricted to carbon 15 (C15).18 We previously demonstrated a predominance of C15 oxidation of ETE-PE under ferroptotic conditions.5, 11 Assuming that changes in the organization of the active site can alter regio-specificity of oxidation, 19–21 we evaluated 15-LO2/PEBP1 regio-specificity towards ETE-PE oxidation. Fragmentation analysis (Figure S15) established that, unlike free-ETE oxidized at C15, sn2-ETE-PE was oxidized by 15-LO2 primarily at C12. In the presence of PEBP1, the regio-specificity of 15-LO2 towards sn2-ETE-PE was re-instated back to C15 (Figure 4A). Thus specificity of sn2-ETE-PE oxidation at C15 in ferroptosis is likely associated with catalysis by 15-LO2/PEBP1 complexes.
Figure 4.
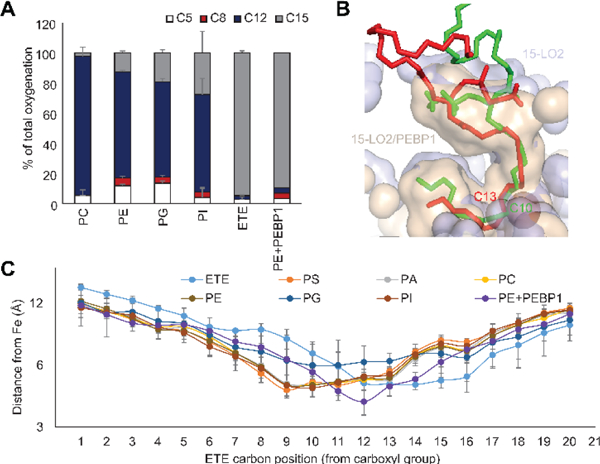
(A) Percentage of oxidation occurring at each oxidizable carbon position of ETE esterified to PE. (B) Distance of all carbon atoms of sn2-ETE present in various PL from 15-LO2 catalytic iron. (C) Docking of sn1-18:0/sn2-20:4-PE on 15-LO2 with (red) and without (green) PEBP1, showing the change in the nearest carbon to the catalytic iron.
Regio-specificity of LOX is typically determined by exact positioning of ETE in the active site which affects the distance between the bis-allylic carbon and catalytic iron.22 Oxidation by 15-LO2 follows “+2 rearrangement”, meaning that the hydro-peroxy-group is inserted two carbons farther from the carboxyl group than the carbon of the initial proton abstraction.23 We there-fore examined the position of free and esterified ETE docking relative to the 15-LO2 catalytic iron. Expectedly C12 and C13 of free ETE were closest to the catalytic iron of 15-LO2, thus indicating oxidation at C15 (Figure 4B). Conversely, ETE-residues of all PLs were positioned so that C9 and C10 were closest to the catalytic iron, suggesting oxidation at C12. 15-LO2 conformational change due to PEBP1 binding restored positioning of sn2-ETE-PE in the active site similar to free ETE (Figure 4B). PEBP1, caused repositioning of sn2-ETE-PE and shifted the carbon nearest to the catalytic iron from C10 to C13 corresponding to a change from production of 12- to 15-sn2-HpETE-PE (Figure 4B, C).
We further generated five PEBP1 mutants with altered amino acids at the interaction site (Figure S17A). Four mutants suppressed while one mutant enhanced 15-LO2 activity towards ETE-PE vs WT PEBP1 (Figure 5A). Fragmentation analysis showed that the mutants changed the regio-specificity to 15-HpETE-PE (Figure S16) thus confirming the role of PEBP1 as a regulator of 15-LO2 specificity. Overexpression of WT (but not mutant (H86E)) PEBP1 increased RSL3-induced ferroptosis in MLE cells (Figure 5B).
Figure 5.
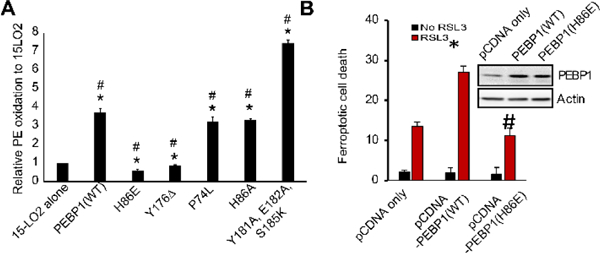
(A) PE oxidation by complexes of 15-LO2 with PEBP1 mutants. N=3, *p<0.05 vs 15-LO2 alone, #p<0.05 vs 15-LO2/PEBP1 (WT). (B) Effect of WT and mutant (H86E) PEBP1 overexpression on RSL3 induced ferroptosis. *p<0.05 vs pCDNA only, #p<0.05 vs pCDNA1-PEBP1(WT).
In conclusion, pro-ferroptotic PE oxidation is controlled by the PEBP1/15LO complex. Three independent but mutually enhancing factors define selective and specific production of sn2-15-HpETE-PE by 15LOX/PEBP1 complexes in ferroptosis: i) greater availability and higher enzyme reactivity towards hexagonally organized ETE-PE species, ii) allosterically induced modification of the substrate preference in the 15-LO2/PEBP1 complex, and iii) relative preponderance of sn2-ETE-PE species vs. other sn2-ETE-PLs.
Supplementary Material
ACKNOWLEDGMENT
Supported by NIH (P01HL114453, U19AI068021, NS076511, NS061817).
Footnotes
ASSOCIATED CONTENT
Supporting Information
1. Supporting_document.pdf
2. Supplementary_video.avi
The authors declare no competing financial interests.
Ferroptosis is initiated by the selective and specific peroxidation of sn2-ETE-PE to sn2-15HpETE-PE by 15-LOX/PEPB1 complex.
REFERENCES
- (1).Dixon SJ; Lemberg KM; Lamprecht MR; Skouta R; Zaitsev EM; Gleason CE; Patel DN; Bauer AJ; Cantley AM; Yang WS; Morrison B 3rd; Stockwell BR Cell 2012, 149, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Angeli JPF; Schneider M; Proneth B; Tyurina YY; Tyurin VA; Hammond VJ; Herbach N; Aichler M; Walch A; Eggenhofer E Nature cell biology 2014, 16, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gao M; Monian P; Quadri N; Ramasamy R; Jiang X Molecular cell 2015, 59, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tuo Q; Lei P; Jackman K; Li X; Xiong H; Liuyang Z; Roisman L; Zhang S; Ayton S; Wang Q Molecular psychiatry 2017, 22, 1520. [DOI] [PubMed] [Google Scholar]
- (5).Wenzel SE; Tyurina YY; Zhao J; St Croix CM; Dar HH; Mao G; Tyurin VA; Anthonymuthu TS; Kapralov AA; Amoscato AA; Mikulska-Ruminska K; Shrivastava IH; Kenny EM; Yang Q; Rosenbaum JC; Sparvero LJ; Emlet DR; Wen X; Minami Y; Qu F; Watkins SC; Holman TR; VanDemark AP; Kellum JA; Bahar I; Bayir H; Kagan VE Cell 2017, 171, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Guiney SJ; Adlard PA; Bush AI; Finkelstein DI; Ayton S Neurochemistry international 2017, 104, 34. [DOI] [PubMed] [Google Scholar]
- (7).Hambright WS; Fonseca RS; Chen L; Na R; Ran Q Redox Biology 2017, 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Li Q; Han X; Lan X; Gao Y; Wan J; Durham F; Cheng T; Yang J; Wang Z; Jiang C JCI insight 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zille M; Karuppagounder SS; Chen Y; Gough PJ; Bertin J; Finger J; Milner TA; Jonas EA; Ratan RR Stroke 2017, 48, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stockwell BR; Friedmann Angeli JP; Bayir H; Bush AI; Conrad M; Dixon SJ; Fulda S; Gascon S; Hatzios SK; Kagan VE; Noel K; Jiang X; Linkermann A; Murphy ME; Overholtzer M; Oyagi A; Pagnussat GC; Park J; Ran Q; Rosenfeld CS; Salnikow K; Tang D; Torti FM; Torti SV; Toyokuni S; Woerpel KA; Zhang DD Cell 2017, 171, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kagan VE; Mao G; Qu F; Angeli JP; Doll S; Croix CS; Dar HH; Liu B; Tyurin VA; Ritov VB; Kapralov AA; Amoscato AA; Jiang J; Anthonymuthu T; Mohammadyani D; Yang Q; Proneth B; Klein-Seetharaman J; Watkins S; Bahar I; Greenberger J; Mallampalli RK; Stockwell BR; Tyurina YY; Conrad M; Bayir H Nature chemical biology 2017, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shintoku R; Takigawa Y; Yamada K; Kubota C; Yoshimoto Y; Takeuchi T; Koshiishi I; Torii S 2017, 108, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Newcomer ME; Brash AR Protein science : a publication of the Protein Society 2015, 24, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Trott O; Olson AJ Journal of computational chemistry 2010, 31, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cullis PR; Hope MJ; Tilcock CP Chemistry and physics of lipids 1986, 40, 127. [DOI] [PubMed] [Google Scholar]
- (16).Doll S; Proneth B; Tyurina YY; Panzilius E; Kobayashi S; Ingold I; Irmler M; Beckers J; Aichler M; Walch A; Prokisch H; Trümbach D; Mao G; Qu F; Bayir H; Füllekrug J; Scheel CH; Wurst W; Schick JA; Kagan VE; Angeli JPF; Conrad M Nature chemical biology 2016, 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Eyal E; Lum G; Bahar I Bioinformatics 2015, 31, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Green AR; Barbour S; Horn T; Carlos J; Raskatov JA; Holman TR Biochemistry 2016, 55, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sloane DL; Leung R; Craik CS; Sigal E Nature 1991, 354, 149. [DOI] [PubMed] [Google Scholar]
- (20).Gan QF; Browner MF; Sloane DL; Sigal E The Journal of biological chemistry 1996, 271, 25412. [DOI] [PubMed] [Google Scholar]
- (21).Borngräber S; Kuban R-J; Anton M; Kühn H Journal of molecular biology 1996, 264, 1145. [DOI] [PubMed] [Google Scholar]
- (22).Gillmor SA; Villaseñor A; Fletterick R; Sigal E; Browner MF Nature Structural Biology 1997, 4, 1003. [DOI] [PubMed] [Google Scholar]
- (23).Kuhn H; Saam J; Eibach S; Holzhütter H-G; Ivanov I; Walther M Biochemical and Biophysical Research Communications 2005, 338, 93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.