

Abstract
IMPORTANCE
Previous phase 1 and 2 trials of PANVAC, a poxviral-based cancer vaccine, suggested clinical efficacy in some patients with breast, ovarian, and colorectal cancer, and evidence of immunologic activity. Preclinical data showed docetaxel can modify tumor phenotype, making tumor cells more amenable to T-cell-mediated killing.
OBJECTIVE
The goal of this study was to determine if the combination of docetaxel and PANVAC could provide evidence of improved clinical outcomes in patients with metastatic breast cancer vs. docetaxel alone.
DESIGN
This open-label randomized phase 2, dual-center trial (5/06 – 2/12) was designed to enroll 48 patients with metastatic breast cancer to receive docetaxel with PANVAC (Arm A) or docetaxel alone (Arm B).
SETTING
National Cancer Institute and the Department of Breast Medical Oncology, MD Anderson Cancer Center.
PARTICIPANTS
Patients with metastatic breast cancer, all subtypes, no limitations on prior lines of therapy.
MAIN OUTCOME MEASURES
The primary endpoint was progression-free survival (PFS), using a phase 2.5 statistical design, with the intent of identifying a trend toward benefit (defined as 1-sided p ≤ 0.10) to guide a larger trial design. Secondary endpoints included safety and immunologic studies.
RESULTS
Forty-eight subjects were enrolled (arm A, n=25; arm B, n=23). No patient remained on study at the time of the final analysis. Patient and tumor characteristics were well matched. Analysis of adverse events on both arms demonstrated very little difference between the 2 groups. Statistically significant differences were increases in the frequency of grade 1 and 2 edema (p = 0.018, likely related to greater median number of docetaxel cycles) and injection site reactions (p <0.0001) in the combination arm. Final data analysis indicates median PFS is 7.9 vs. 3.9 months in A vs. B (one-sided p=0.09, hazard ratio (HR = 0.65 (95% CI: 0.34 – 1.14)).
CONCLUSIONS/RELEVANCE
This pilot study was powered to detect a trend of approximately this magnitude toward improvement in PFS. The results suggest the combination of PANVAC with docetaxel in metastatic breast cancer may provide a clinical benefit. This study was hypothesis generating and provides both rationale and statistical assumptions for a larger definitive randomized study.
TRIAL REGISTRATION
Introduction
Breast cancer is expected to cause approximately 40,000 deaths in 2015.1 Despite improvements in treatment, an overwhelming majority of patients diagnosed with metastatic breast cancer will die as a result of their disease. Standard chemotherapy agents are capable of shrinking tumors, but treatment usually needs to be stopped due to tumor progression or toxicity. At that point the likelihood of response with each subsequent therapy decreases. We and others have previously demonstrated safety, immunoreactivity, and potential clinical activity of a recombinant poxviral-vector therapeutic cancer vaccine, designated PANVAC (CEA-MUC-1-TRICOM), which consists of a priming dose with recombinant vaccinia vector, and subsequent doses with recombinant fowlpox vector.2–4 Each vector encodes the transgenes for CEA and MUC-1 as well as transgenes for 3human co-stimulatory molecules (B7.1, ICAM-1, and LFA3).5
Docetaxel is a commonly used agent in treating patients with metastatic breast cancer. We have previously reported preclinical data indicating a synergistic effect of docetaxel with vaccine.6,7 Docetaxel was shown to be capable of altering human and murine carcinoma cell phenotypes, making them more amenable to T cell-mediated killing. Additionally, a previous clinical trial demonstrated that docetaxel (with glucocorticoid co-administration) in combination with vaccine did not cause decreased immune responses in patients when compared to vaccine alone.8
As single agents, therapeutic cancer vaccines have often failed to impact short-term endpoints, such as median progression-free survival (PFS), despite improvement in long-term outcomes, such as overall survival (OS).9–13 This pattern has also been seen with other immunotherapeutic agents, such as ipilimumab.14 A tumor growth rate kinetics model created with clinical trial data may help to explain the improvement in OS without impact on PFS. Clinical trial data using effective therapeutic cancer vaccines appears to induce slowing of the tumor growth rate over time.15 This process may take months to occur. We have hypothesized that the lag between initial vaccination and eventual slowing of tumor growth rate may explain the lack of improvement in PFS in previous vaccine monotherapy trials despite improvement in OS. If that hypothesis is correct, then combining therapeutic cancer vaccines with standard-of-care agents that can induce an improvement in PFS and do not negatively impact the immune response may provide adequate time for the vaccine effect to occur, resulting in improved PFS when compared to the standard agent alone.16
Subjects and Methods
Patient Eligibility
Subjects were required to have radiographically evaluable metastatic breast cancer that had not been treated with docetaxel in the metastatic setting. There was no limitation on prior lines of systemic therapy or subtype of breast cancer. Subjects were required to be ≥18 years of age, have acceptable hematologic parameters and organ function, have an ECOG performance status of ≤1, have no other malignancies within 12 months, have no clinically active brain metastases, or have other significant medical illnesses or autoimmune diseases. Due to vaccinia vector used in priming, subjects with a history of prior allergy or reaction to vaccinia-based vaccination or an open skin wound were also excluded.
Trial Design and Treatment
The primary endpoint was PFS, and was evaluated using a phase 2.5 trial design.17 This design is intended to provide evidence of a strong trend towards a clinical benefit with the goal of informing a larger randomized clinical trial in the future. This pilot trial had an 80% power to demonstrate a trend toward benefit (defined as 1-sided p-value < 0.10) estimated to be 4.2 months with docetaxel alone, and 8 months with docetaxel and PANVAC, with 24 patients per arm, a total of 48 subjects. The treatment protocol was approved by the National Cancer Institute (NCI) and the University of Texas MD Anderson Cancer Center (MDACC) Institutional Review Boards and subjects were enrolled at both institutions. Subjects were randomized 1:1 to receive either docetaxel alone (Arm B), given 3 out of every 4 weeks at a dose of 35 mg/M2, or in combination with PANVAC (Arm A). PANVAC priming dose (vaccinia) was given 3 weeks prior to the first cycle of docetaxel. All booster doses (fowlpox) were given on day 1 of each docetaxel cycle. Docetaxel was given on day 2, 9, and 16 of each 28-day cycle. Treatment was continued until disease progression or unacceptable toxicity. On Arm A, if docetaxel was discontinued for toxicity, PANVAC could be continued until disease progression. PANVAC was manufactured by Therion Biologics Corporation (Cambridge, MA) as part of a Cooperative Research and Development Agreement between Therion and the Laboratory of Tumor Immunology and Biology, NCI. Vaccines were provided by the Cancer Therapy Evaluation Program, NCI. Subjects were enrolled from May 2005 to February 2012. Subjects enrolled at NCI assigned to Arm A received low dose GM-CSF (100 μg s.c. on the day of each vaccination and for 3 consecutive days thereafter, all near the vaccination site designed as a vaccine adjuvant) with PANVAC, while those at MDACC did not receive adjuvant GM-CSF with PANVAC on Arm A, due to issues of cost and preclinical and preliminary clinical data indicating there was no clear benefit with the addition of GM-CSF in poxviral TRICOM vector vaccines.13 Subjects with Her2+ disease who had previously progressed on trastuzumab were allowed to continue trastuzumab while participating in this study. Progression was defined by RECIST criteria 1.0. Imaging studies (CT chest, abdomen, and pelvis and bone scan) were performed at baseline, after 3 months on study, and then every 2 months thereafter. For subjects with evaluable disease only at baseline, progression was defined by new lesions or measurable growth of existing, previously unmeasurable lesions. At progression, subjects assigned to Arm B (docetaxel alone) had the option to receive vaccine alone if their clinical status met all eligibility criteria.
Immune Assays
Peripheral blood mononuclear cells (PBMC) were separated by Ficoll-Hypaque density gradient separation, washed 3 times, and preserved in 90% heat inactivated human AB serum and 10% DMSO in liquid nitrogen at a concentration of 1×107 cells/mL until assayed. Serum samples were collected in serum separator tubes, spun down, and stored at −80° C. The analysis of the frequency of PBMC immune cell subsets in patients before therapy (baseline) and at the time of restaging (post-cycle 3 of docetaxel) was assessed by multi-parametric flow cytometry. One vial of cryopreserved PBMC per time point was defrosted, counted, and 1×106 cells immediately stained to identify CD8+ T lymphocytes, CD4+ T lymphocytes, and T regulatory (Treg) lymphocytes using the following antibodies: anti-CD8-PE-Cy7 (clone RPA-T8, BD Biosciences, San Jose, CA), anti-CD4-alexa fluor 700 (clone RPA-T4, BioLegend, San Diego, CA), anti-CD25-APC-Cy7 (clone M-A251, BD Biosciences), anti-CD127-FITC (clone A019D5, BioLegend), and anti-FoxP3-PE-Cy7 (clone PCH101 (eBioscience, San Diego, CA).
Analysis of antigen-specific responses was assessed by intracellular cytokine staining (ICS) following a period of in vitro stimulation with overlapping 15-mer peptide pools encoding the tumor-associated antigens (TAAs) CEA, MUC-1, and brachyury. The TAA peptide pools were designed to contain agonist epitopes that have been previously identified;18–20 peptide pools encoding for HLA and CEFT (a mixture of CMV, EBV, Flu, and Tetanus toxin) served as negative and positive controls, respectively. Peptide mixes were purchased from JPT (Berlin, Germany), reconstituted in DMSO, and utilized immediately. Cryopreserved PBMC from patients before therapy and at the time of restaging were thawed and rested overnight at 37°C, 5% CO2 in complete media (IMDM supplemented with 10% Human AB, 2mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin). The next day (Day 0), PBMC were seeded in 12 well plates (2.5 ×106 in 1 mL), and stimulated with peptide mixes (0.1μg/mL per peptide); cultures were supplemented on days 3 and 5 with cytokines (IL7 and IL15, 10 ng/mL, PeproTech, Rocky Hill, NJ) and fresh media, and on day 7 were rested (with removal of cytokines and peptide). On day 11, 1×106 cells were restimulated for 24 hours in 96 well plates with peptide mixes in the presence of anti-CD107a-APC (clone H4A3, BD Biosciences); brefeldin A (1μl/mL) and monensin (0.7μl/mL) (BD Biosciences) were added to cultures 2 hours after the start of the restimulation and incubated for the final 22 hours. PBMC were then stained with anti-CD4-PerCP-Cy5.5 (clone OKT4, Biolegend), anti-CD8-AF700 (clone OKT8, Ebioscience), and anti-TNF-PE (clone MAb11), anti-IFNγ-PE-Cy7 (clone 4SB3), and anti-IL-2-BV521 (clone 5344.111) (BD Biosciences).
For all flow cytometry experiments, at least 3×105 events in the live gate were acquired with a BD LSR-II flow cytometer equipped with a UV, violet, blue, and red laser. FCS files were imported and analyzed with FlowJo V.9.7 for Macintosh (TreeStar, Ashland, OR). Fluorescence minus one (FMO) controls were used for gating, and non-viable cells were excluded. The development of immune responses following therapy to a given TAA were calculated from ICS experiments as follows: We first calculated the absolute number of CD4+ or CD8+ lymphocytes that produced IFNγ, TNF, or IL2 at the end of the IVS following stimulation with CEA, MUC-1, Brachyury or the negative control 15-mer peptides, per 1×106 cells plated at the start of the IVS. Next, background values (i.e. any signal obtained from the IVS with the negative control peptide pool HLA) were subtracted from the signal obtained with CEA, MUC-1 or Brachyury 15-mer peptide pools, and then pre therapy values were subtracted from post therapy values [(Post TAA – Post HLA) – (Pre TAA – Pre HLA)]. An antigen specific immune response to a given TAA was scored as positive if a patient had >250 CD4+ or CD8+ T lymphocytes that produced IFNγ, IL2, or TNF at the end of the in vitro stimulation (IVS) per 1×106 cells plated at the start of the IVS (following subtraction of any background and pre therapy signal). The reproducibility of the IVS assay to measure antigen-specific immune responses has been tested by repeatedly assessing immune responders and non-responders in independent experiments with similar results obtained in each assay. Additionally, when the number of samples was large, requiring samples to be run in batches, internal controls were always included, and importantly, the pre- and post-PBMC from a given patient were always run simultaneously to reduce assay variability.
Assays for the presence of soluble CD27 (sCD27) in sera were as described in detail previously.21
Statistical Methods
Analysis of the primary endpoint was performed with all eligible, randomized subjects included, with the probability of PFS as a function of time estimated by the Kaplan-Meier method and the arms compared using a log-rank test. Secondary objectives included comparison of overall survival, specific T-cell responses to the target antigens of the vaccine and other evidence of immune response, and comparison of the subjects who did and did not receive GM-CSF adjuvant with vaccine.
An analysis of factors associated with time to progression was performed, initially using Kaplan-Meier curves and a log-rank test to determine the impact of clinical and pre-treatment immune response parameters. Continuous parameters were divided into three or four groups initially for this evaluation. When the initial analysis identified that combining patients into two groups would result in a potentially important difference with respect to progression, the p-values were adjusted by multiplying the unadjusted p-value by the number of implicit tests used to arrive at the final grouping. Those factors with unadjusted p<0.05 in this univariate analysis were then included in a Cox proportional hazards model for evaluation of the effect of the joint effect of treatment along with the other potentially important factors.
Time since last chemotherapy was considered as the number of days between the last treatment and the day of enrollment to the trial, and the association between number of days since last chemotherapy (among those with last chemotherapy in the past 3 months on the combination arm) and PFS determined by the Kaplan-Meier method and a log-rank test with the hazard ratio based on a Wald test from a Cox model.
Comparisons between groups with respect to continuous parameters were performed using an exact Wilcoxon rank sum test, and paired differences between time points of parameters were tested for their difference from zero using a Wilcoxon rank sum test. Fisher’s exact test was used to compare dichotomous parameters between groups. Except as noted, all p-values are 2-tailed and reported without any adjustment for multiple comparisons. Analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC).
Randomization Methods
Patients were randomized centrally, using a locally-written SAS software program to generate a random 1:1 sequence of assignments to treatment, using variable block sizes (2 or 4), with parameters for assignment determined by the study statistician (SMS). Patients were originally randomized without stratification, but after later amendments, randomization was stratified according to Herceptin as well as Sargromostim use. The randomization assignment sheets were maintained confidentially in a central registration office, which enrolls the patients; the treatment assignment for a given patient is only disclosed to the study research team by a member of the central registration staff after confirming full eligibility.
Results
Patient Baseline Characteristics
Forty-eight subjects were enrolled (CONSORT diagram, Figure 1) on the study between May 2006 and February 2012: 25 on Arm A (combination of vaccine and docetaxel) and 23 on Arm B (docetaxel alone). Final clinical data was collected September 16, 2013. There was similar heterogeneity with regard to prior cytotoxic chemotherapy for treatment of metastatic disease between arms (Arm A median 2, range 0–7, Arm B median 1, range 0–8). Thirty-two percent and 35% of patients in Arms A and B, respectively, previously received ≥3 prior lines prior to enrollment (Table 1). There was no statistical difference in any baseline characteristic (all p-values for comparisons >0.20, Table 1). The patient population for the study was heterogeneous by current breast cancer clinical trial design standards in terms of biologic characteristics and prior treatments, but there were no statistical differences between groups when comparing all baseline characteristics (all p-values > 0.20, Table 1).
Figure 1.
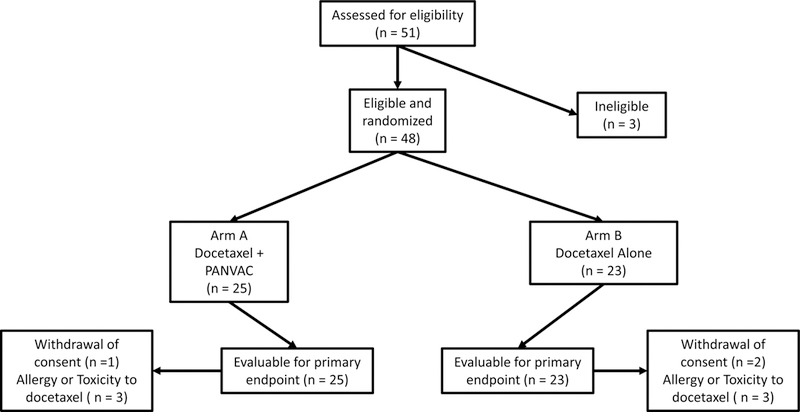
CONSORT diagram. Fifty-one patients screened, 48 were eligible and randomized. Twenty-five randomized to Arm A; 23 to Arm B. All patients on Arm A (25) and Arm B (23) were evaluable for the primary endpoint.
Table 1.
Patient baseline characteristics
| Arm A (n=25) | Arm B (n=23) | |
|---|---|---|
| Median Age | ||
| on study (range) | 55 (33-72) | 52 (34-76) |
| at diagnosis (range) | 48 (31-61) | 45 (26-74) |
| ECOG Performance Status 0 1 |
17 (68%) 8 (32%) |
11 (48%) 12 (52%) |
| Receptor status | ||
| Number of ER-/PR- patients (%) | 7 (28%) | 7 (30%) |
| Number of Her 2 +patients (%) | 3 (12%) | 3 (13%) |
| Number of Female patients (%) | 25 (100%) | 22 (96%) |
| Number of patients enrolled at NCI (%) | 13 (52%) | 12 (52%) |
| Median number of prior chemotherapy regimens for metastatic disease (range) | 2 (0-7) | 1 (0-8) |
| < 3 prior regimens (%) | 18 (68%) | 15 (65%) |
| ≥ 3 prior regimens (%) | 7 (32%) | 8 (35%) |
| Days since prior chemotherapy (range) | 68 (19 – 1,583) | 45 (10 – 2,760) |
| < 30 days since last chemo (%) | 4 (17%) | 6 (29%) |
| > 30 days since last chemo (%) | 20 (83%) | 15 (71%) |
| Days since diagnosis (range) | 1,910 (377 – 6,839) | 1,834 (279 - 6,236) |
| Days from diagnosis to metastasis (range) | 1006 (0 - 6,063) | 1133 (0 - 4,916) |
| Number of patients with metastatic disease at diagnosis (%) | 2 (8%) | 3 (13%) |
All demographic characteristics were compared between Arm A and B and there were no statistical differences. All comparison p-values were >0.20.
Safety
There were no differences in toxicity between Arm A and Arm B, with the exception of injection site reactions and edema in Arm A (Table 2). The incidence of edema corresponded to the number of cycles of docetaxel received, which was higher in Arm A (median number of cycles was 5, range 0–17) than Arm B (median number of cycles was 3, range 1–15) due to prolonged progression-free survival (see Clinical Outcomes). There were no Grade 4 toxicities in either arm. Dose reductions and delays were similar between the two arms. In Arm A, 9 patients had dose reductions, 13 had dose delays, and 14 had dose delay or reduction. In Arm B, 7 patients had dose reductions, 11 had dose delays, and 13 had dose delays or reductions. The likelihood of toxicity causing delay or reduction in dose was more likely in patients who received multiple cycles, related to the cumulative toxicity of docetaxel.
Table 2.
Adverse Events
| Arm A (n=25) | Arm B (n=23) | |||||||
|---|---|---|---|---|---|---|---|---|
| Gr 1 (%) | Gr 2 (%) | Gr 3 (%) | Gr 1 (%) | Gr 2 (%) | Gr 3 (%) | P value | ||
| Hematologic | ||||||||
| Anemia | 8 (32) | 6 (24) | 1 (4) | 6 (26.1) | 6 (26.1) | 1 (4.3) | NS | |
| Elevated PTT | 1 (4) | 0 | 0 | 0 | 1 (4.3) | 1 (4.3) | NS | |
| Neutropenia | 2 (8) | 6 (24) | 3 (12) | 2 (8.7) | 1 (4.3) | 4 (17.4) | NS | |
| Thrombocytopenia | 4 (16) | 0 | 0 | 2 (8.7) | 0 | 0 | NS | |
| Leukocytopenia | 3 (12) | 3 (12) | 3 (12) | 5 (21.7) | 6 (26) | 0 | NS | |
| Lymphocytopenia | 5 (20) | 3 (12) | 3 (12) | 5 (21.7) | 4 (17.3) | 4 (17.4) | NS | |
| Infections | ||||||||
| URI/Sinusitis | 0 | 1 (4) | 0 | 1 (4.3) | 5 (21.7) | 0 | 0.052 | |
| Nervous system | 10 (40) | 5 (20) | 2 (8) | 7 (30.4) | 6 (26.1) | 0 | ||
| Eye | 9 (36) | 1 (4) | 3 (12) | 10 (43.4) | 0 | 0 | NS | |
| Gastrointestinal | ||||||||
| N/V/D/C | 8 (32) | 6 (24) | 2 (8) | 8 (34.8) | 5 (21.7) | 0 | NS | |
| Oral pain/mucositis | 6 (24) | 2 (8) | 1 (4) | 3 (13.0) | 3 (13.0) | 1 (4.3) | NS | |
| Abdominal pain | 1 (4) | 0 | 0 | 1 (4.3) | 0 | 1 (4.3) | NS | |
| Liver transaminase abnormality | 5 (20) | 2 (8) | 3 (12) | 6 (26.1) | 6(26.1) | 1 (4.3) | NS | |
| General | ||||||||
| Fever, chills, flu-like | 7 (28) | 1 (4) | 0 | 4 (17.4) | 1 (4.3) | 1 (4.3) | NS | |
| Edema | 9 (36) | 2 (8) | 0 | 3 (13.0) | 0 | 0 | 0.018 | |
| Fatigue | 7 (28) | 7 (28) | 1 (4) | 4 (17.4) | 8 (34.8) | 2 (8.7) | NS | |
| Weight change | 0 | 1 (4) | 0 | 1 (4.3) | 1 (4.3) | 0 | NS | |
| Anorexia | 3 (12) | 2 (8) | 0 | 4 (17.4) | 0 | 0 | NS | |
| Injection site reaction | 2 (8) | 14 (56) | 0 | 0 | 0 | 0 | <0.0001 | |
URI, upper respiratory infection; N/V/D/C, nausea/vomiting/diarrhea/constipation. There was no grade 4 or 5 adverse event seen on study. There was no grade 3 or greater adverse event attributed to vaccine seen on study.
Clinical Outcomes
The study was powered to detect a trend toward improvement in PFS. The median PFS in Arm A was 7.9 months compared with 3.9 months in Arm B (one-sided p = 0.09, HR = 0.65 (95% CI: 34 – 1.14)), indicating a trend toward improvement with the combination arm (Figure 2). The overall confirmed partial response rate (PR) on Arm A was 16% compared with 13% in Arm B. There was a notably higher confirmed PR rate at NCI (24%) when compared with MDACC (4%). The median number of vaccines administered in Arm A was 8 (range 2–20). Only 4 patients were eligible and opted to cross over to vaccine after progression docetaxel alone (Arm B). PFS on Arm B was measured to the date of first progression and further clinical outcome data on the patients who crossed-over is not reported due to a small sample size.
Figure 2.
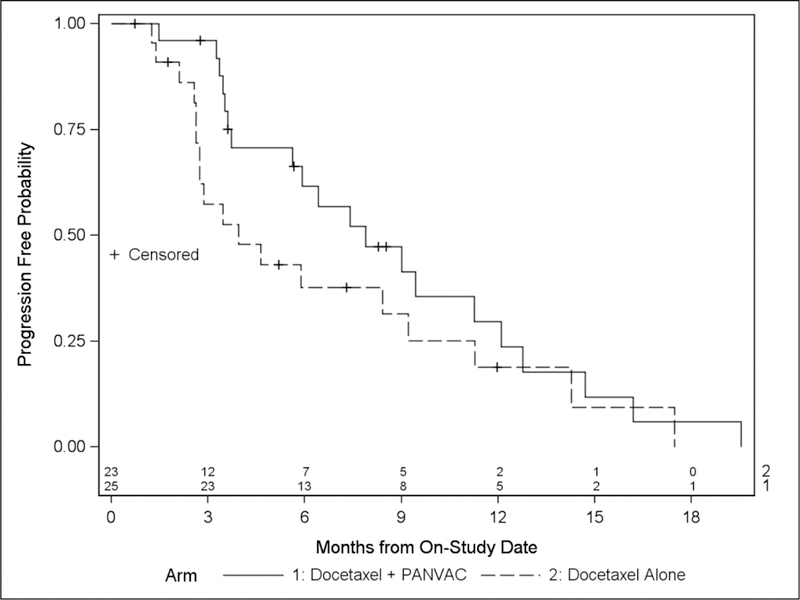
Comparison of progression-free survival (PFS). Medians: Arm A (median 5 cycles docetaxel given) 7.9 months versus Arm B (median 3 cycles given) 3.9 months (p = 0.09, 1-sided, meets predefined statistical goal), HR = 0.65 (95% CI: 0.34 – 1.14). Median potential follow-up 42.8 months.
Since the overall result demonstrated a trend toward benefit with vaccine, we undertook an analysis to determine if this association would remain if clinical and immune response parameters were included in a Cox model. The following parameters were all considered for evaluation: Time since last chemotherapy (<30 vs. >=30 days, detailed below), pre-treatment sCD27, CD4/Treg ratio, Tregs, age at on study, age at diagnosis, ECOG status, gender, ER/PR status, HER2 status, time from diagnosis to enrollment, time from diagnosis to metastases, metastatic disease at diagnosis. By univariate analyses, time since last chemo (<30 vs. >=30 days; p=0.016), CD4/Treg ratio (<0.20 vs. >0.20; p=0.024 unadjusted; p=0.048 adjusted), T regs (<20 vs. >20; p=0.019 unadjusted; p=0.058 adjusted), and ER/PR – status (p<0.0001) were considered for evaluation in a Cox model along with treatment arm to determine the association of treatment arm with progression free survival after taking into consideration potential prognostic factors. After backward selection, treatment (HR=0.60; 95% CI: 0.30–1.19; two-tailed p=0.14; one-tailed p=0.07) retained its trend toward association with PFS after adjusting for higher Tregs (HR=0.30; 95% CI: 0.13–0.72; two-tailed p=0.0065) and not having ER-/PR- status (HR=0.11; 95% CI: 0.04–0.27; two-tailed p<0.0001).
Effect of Time Since Last Chemotherapy
Patients treated at MDACC had a shorter time since last chemotherapy (median 34.0 days; range 15–198 days) compared to those treated at the NCI (median 192; range 25–2800; p = 0.0006). The median (range) time since last chemotherapy in the docetaxel alone arm was 199.5 (25–2800) days for patients enrolled at NCI vs 43 (15–198) days at MDACC (p=0.067). For the combination arm, the median time since last chemotherapy was 192 (27–1607) days for patients enrolled at NCI vs. 30 (20–175) days at MDACC (p=0.0025). There was a trend toward improvement in median PFS in Arm A for patients (n=13) with greater than 30 days since prior chemotherapy compared with those patients (n=6) with less than 30 days since last chemotherapy (9.4 months vs. 5.8 months, p=0.12). We have previously reported21 that sCD27 plays a role in T-cell activation, and the levels of sCD27 in sera in healthy donors are greater than those in prostate cancer patients. Here we have found (Figure 3A) that sCD27 in sera is also lower in breast cancer patients as compared with healthy donors (p < 0.001). Moreover, the pretreatment levels of sCD27 were somewhat lower in patients treated at MDACC vs. the NCI (p =0.082; Figure 3A). This may be due to differences in the 2 centers in the time since last chemotherapy in patients prior to entering this trial, described above.
Figure 3.
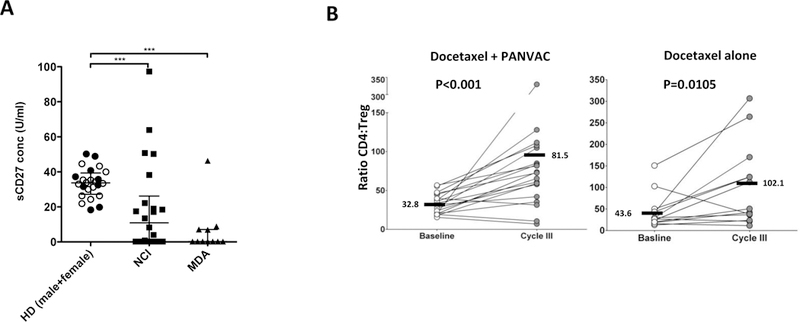
Immune changes pre- and post-therapy. A, Comparison of serum levels of soluble CD27 (sCD27) in healthy donors (female = black dots, male = white dots) vs. pre-treatment samples from enrolled patients, divided by the 2 centers. Dot plots show medians and interquartile range. ***p<0.001. B, Analysis of the changes in the ratio between CD4+ T lymphocytes and regulatory T cells (Tregs) at baseline vs. cycle 3 of docetaxel (28-day cycles, first restaging). Bars represent mean. P values calculated by Wilcoxon signed rank test.
Immune Assays
Sufficient PBMCs were available from patients in the docetaxel alone arm (n=15) and the docetaxel plus vaccine arm (n=16) to analyze antigen-specific immune responses before vs. during (at first restaging, approximately day 85 from start of docetaxel) therapy using ICS. Eleven of 16 (69%) patients in the docetaxel plus vaccine arm developed T-cell responses to 15-mer peptide pools of CEA, MUC-1 and/or brachyury (tumor-associated cascade antigen) during treatment, while 8/15 (53%) patients in the docetaxel alone arm developed T-cell responses to these TAAs during treatment. In analyses of immune responses to the 2 transgenes (CEA and MUC-1) in the PANVAC vaccine, 6/15 (40%) of patients in the docetaxel alone arm developed responses vs. 9/16 (56%) in the combination arm (Supplemental Table S1 and Supplemental Figure S1). In patients treated at MDACC, 3/5 (60%) in the docetaxel alone arm and 2/6 (33%) in the combination arm developed CEA and/or MUC-1 responses. In the NCI treated patients, 3/10 (30%) in the docetaxel alone arm vs. 7/10 (70%) in the combination arm developed CEA and/or MUC-1 responses. Thus, due to the small number of patients in each cohort, these resulted are descriptive only. There was also no statistical correlation seen between the generation of TAA-specific immune responses in PBMC and time to progression in either arm. The possible reasons for these findings are discussed below.
No differences in Arms A and B were seen at baseline in CD4+, CD8+, or Tregs. The ratios of CD8+/Tregs increased similarly in both Arms A and B post-3 cycles of chemotherapy. Both CD4+ T cells and Tregs decreased in both arms post-treatment. Because of a greater decrease in Tregs vs. CD4+ T cells, there was an increase in the CD4+/Treg ratios in the both arms. However, post-3 cycles of chemotherapy, there was a clear trend in a greater CD4+/Treg increase in the combination arm vs. the docetaxel alone arm (Figure 3B; p <0.001 vs. p=0.01).
Discussion
This trial was designed to show a trend in improved PFS in the docetaxel plus vaccine arm vs. the docetaxel alone arm. The trend of improved PFS in the combination arm (7.9 vs 3.9 months; Figure 2) thus informs a potential larger multi-center randomized trial. The trial reported here was a dual center study conducted at NCI and MDACC. Both centers entered approximately the same number of patients in each arm. Some interesting observations can be made concerning patient baseline characteristics. Patients in both arms treated at MDACC had a shorter interval since last chemotherapy. For those patients who received chemotherapy greater than 1 month prior to entering this trial, there was a trend toward longer PFS (p=0.12). This observation could be due to at least 2 non-mutually exclusive factors: (a) patients at MDACC had more advanced/progressive disease and thus required a subsequent therapy sooner, and/or (b) the short interval since the last chemotherapy reduced the capacity of the patient to respond to vaccine therapy. This is supported by the sCD27 results seen in Figure 3A, in which patients enrolled at MDACC had a statistically significant lower level of serum sCD27 compared to patients enrolled at NCI. sCD27 has been previously shown21 to be a stimulator of effector T cells, and sCD27 in sera of prostate cancer patients was reduced compared to levels in sera of healthy subjects. These findings reported here are also in support of findings previously reported22 using another vaccine (rF-CEA-TRICOM) in patients with advanced carcinoma; in that study there was a direct correlation between the length of time since last chemotherapy and immune response to vaccine.
Another factor in the interpretation of the results of the trial reported was that patients at NCI received GM-CSF along with vaccine while patients at MDACC did not. GM-CSF has been associated with both enhancing and suppressing immune responses in both preclinical and clinical studies. These prior findings may be due to the dose and schedule of GM-CSF used and the vaccine with which it was combined. A prior small phase II study13 attempted to answer the question of the use of GM-CSF in combination with a similar vaccine platform, i.e., PROSTVAC in patients with prostate cancer, with non-conclusive results. Consequently, an international phase III study of PROSTVAC vaccine in patients (n=1,200) with asymptomatic prostate cancer has recently finished accruing. In that trial, patients (n=400 per arm) will receive PROSTVAC vaccine, PROSTVAC vaccine plus GM-CSF, or placebo.
T-cell immune responses in PBMC to the 2 transgenes (CEA and MUC-1) in the vaccine and a cascade antigen not in the vaccine (brachyury) were seen post-therapy in both arms. Eleven of 16 (69%) patients developed immune responses post-therapy to CEA, MUC-1, or brachyury pools of 15-mer peptides in the combination arm, and 8 of 15 (53%) patients developed these responses in the docetaxel alone arm; these results were not statistically significant. Similar responses to brachyury 15-mer peptides were seen post-therapy in both arms. Although there is no clear relationship between the evaluated T-cell specific response to CEA and MUC-1 and PFS, this finding is not without precedent in the field of cancer immunotherapy.23 One possible explanation for this finding is that tumor-associated T cells observed in PBMC do not necessarily reflect those at the site of the tumor. Unfortunately, it was not feasible to obtain tumor biopsies in these patients with metastatic breast cancer. Another possibility is that, after vaccination against target antigens (CEA and MUC-1) T-cell mediated killing results in antigen cascade and more ideal candidate antigens are selected, resulting in expansion of a population of T cells against an unknown antigen, which was not measured in this analysis. Because of these limitations, we explored other markers of immune activation and response as correlatives of clinical benefit. Patients in the combination arm also had a greater increase in the ratio of CD4 to Tregs post-therapy than patients in the docetaxel arm alone (Figure 3B). It has previously been shown that docetaxel alone has immune modulatory effects. Preclinical in vivo murine studies and in vitro studies involving murine and human T cells have shown that docetaxel can alter the phenotype of tumor cells to render them more susceptible to T-cell lysis. In the trial reported here, this could have initiated a cascade of immune responses mediated by T cells at the tumor site.
We demonstrated in this trial the ability to safely combine vaccine therapy with a standard-of-care chemotherapy. This study was powered to detect a trend toward improvement in PFS. The results suggest the combination of PANVAC with docetaxel in metastatic breast cancer may provide a clinical benefit. The clear separation of the curves indicates potential benefit, which while it met the pre-specified parameters set out in the trial design is not statistically significant, likely due to the small number of subjects enrolled. This study has a number of limitations including a small size, heterogeneous patient population, and an unknown impact of GM-CSF. However, the intriguing findings in this hypothesis-generating study provide both rationale and statistical assumptions for a larger appropriately powered and designed definitive randomized study in a more uniform patient population, such patients with ER/PR-positive tumors who have not previously received cytotoxic chemotherapy.
Supplementary Material
Acknowledgments
The authors thank Debra Weingarten for her editorial assistance in the preparation of the manuscript.
Grant Support
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. [DOI] [PubMed] [Google Scholar]
- 2.Gulley JL, Arlen PM, Tsang KY, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14(10):3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohebtash M, Tsang KY, Madan RA, et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011;17(22):7164–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morse MA, Niedzwiecki D, Marshall JL, et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann Surg. 2013;258(6):879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madan RA, Arlen PM, Gulley JL. PANVAC-VF: poxviral-based vaccine therapy targeting CEA and MUC1 in carcinoma. Expert Opin Biol Ther. 2007;7(4):543–554. [DOI] [PubMed] [Google Scholar]
- 6.Garnett CT, Schlom J, Hodge JW. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: effects of docetaxel on immune enhancement. Clin Cancer Res. 2008;14(11):3536–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodge JW, Garnett CT, Farsaci B, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. 2013;133(3):624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arlen PM, Gulley JL, Parker C, et al. A randomized phase II study of concurrent docetaxel plus vaccine versus vaccine alone in metastatic androgen-independent prostate cancer. Clin Cancer Res. 2006;12(4):1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115(16):3670–3679. [DOI] [PubMed] [Google Scholar]
- 10.Higano CS, Small EJ, Schellhammer P, et al. Sipuleucel-T. Nat Rev Drug Discov. 2010;9(7):513–514. [DOI] [PubMed] [Google Scholar]
- 11.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. [DOI] [PubMed] [Google Scholar]
- 12.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gulley JL, Arlen PM, Madan RA, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59(5):663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein WD, Gulley JL, Schlom J, et al. Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res. 2011;17(4):907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madan RA, Bilusic M, Heery C, Schlom J, Gulley JL. Clinical evaluation of TRICOM vector therapeutic cancer vaccines. Semin Oncol. 2012;39(3):296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon RM, Steinberg SM, Hamilton M, et al. Clinical trial designs for the early clinical development of therapeutic cancer vaccines. J Clin Oncol. 2001;19(6):1848–1854. [DOI] [PubMed] [Google Scholar]
- 18.Jochems C, Tucker JA, Vergati M, et al. Identification and characterization of agonist epitopes of the MUC1-C oncoprotein. Cancer Immunol Immunother. 2014;63(2):161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsang KY, Zaremba S, Nieroda CA, et al. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995;87(13):982–990. [DOI] [PubMed] [Google Scholar]
- 20.Tucker JA, Jochems C, Boyerinas B, et al. Identification and characterization of a cytotoxic T-lymphocyte agonist epitope of brachyury, a transcription factor involved in epithelial to mesenchymal transition and metastasis. Cancer immunology, immunotherapy : CII. 2014;63(12):1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Jochems C, Anderson AM, et al. Soluble CD27-pool in humans may contribute to T cell activation and tumor immunity. J Immunol. 2013;190(12):6250–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Mehren M, Arlen P, Gulley J, et al. The influence of granulocyte macrophage colony-stimulating factor and prior chemotherapy on the immunological response to a vaccine (ALVAC-CEA B7.1) in patients with metastatic carcinoma. Clin Cancer Res. 2001;7(5):1181–1191. [PubMed] [Google Scholar]
- 23.Butterfield LH, Disis ML, Fox BA, Khleif SN, Marincola FM. Preamble to the 2015 SITC immunotherapy biomarkers taskforce. J Immunother Cancer. 2015;3:8 Published online 3/24/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.