

Abstract
Diabetic neuropathic pain imposes a huge burden on individuals and society, and represents a major public health problem. Despite aggressive efforts, diabetic neuropathic pain is generally refractory to available clinical treatments. A structure–function link between maladaptive dendritic spine plasticity and pain has been demonstrated previously in CNS and PNS injury models of neuropathic pain. Here, we reasoned that if dendritic spine remodeling contributes to diabetic neuropathic pain, then (1) the presence of malformed spines should coincide with the development of pain, and (2) disrupting maladaptive spine structure should reduce chronic pain. To determine whether dendritic spine remodeling contributes to neuropathic pain in streptozotocin (STZ)-induced diabetic rats, we analyzed dendritic spine morphology and electrophysiological and behavioral signs of neuropathic pain. Our results show changes in dendritic spine shape, distribution, and shape on wide-dynamic-range (WDR) neurons within lamina IV–V of the dorsal horn in diabetes. These diabetes-induced changes were accompanied by WDR neuron hyperexcitability and decreased pain thresholds at 4 weeks. Treatment with NSC23766 (N6-[2-[[4-(diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine trihydrochloride), a Rac1-specific inhibitor known to interfere with spine plasticity, decreased the presence of malformed spines in diabetes, attenuated neuronal hyperresponsiveness to peripheral stimuli, reduced spontaneous firing activity from WDR neurons, and improved nociceptive mechanical pain thresholds. At 1 week after STZ injection, animals with hyperglycemia with no evidence of pain had few or no changes in spine morphology. These results demonstrate that diabetes-induced maladaptive dendritic spine remodeling has a mechanistic role in neuropathic pain. Molecular pathways that control spine morphogenesis and plasticity may be promising future targets for treatment.
Introduction
Diabetic neuropathic pain affects >50% of people with diabetes. In many patients, the neuropathic pain that accompanies diabetes is severe and, in some cases, detracts substantially from quality of life. Unfortunately, despite aggressive efforts, diabetic neuropathic pain is largely refractory to available clinical treatments (Setacci et al., 2009). Because of its global impact and burden on individuals and society, diabetic neuropathic pain is a major public health problem.
Mechanisms that have been suggested to contribute to diabetic neuropathic pain include chronic nerve damage (i.e., peripheral neuropathy) due to secondary vascular disease (Beisswenger, 1976; Bays and Pfeifer, 1988), microglial inflammation in the CNS (Tsuda et al., 2008; Pabreja et al., 2011), dysregulation of potassium–chloride cotransporter 2 (KCC2) activity (Morgado et al., 2011), and altered growth factor and sodium channel expression in DRG neurons (Craner et al., 2002a,b). Only a few studies have investigated the possibility of morphological alterations of postsynaptic structures within second-order sensory neurons in diabetes as an underlying mechanism for diabetic neuropathic pain (Yamano et al., 1986; Malone et al., 2008). Our previous work has shown that structural alterations of synaptic spines within dorsal horn neurons can have adverse functional implications (Tan et al., 2009a), which may contribute neuropathic pain in spinal cord injury (SCI) and peripheral nerve injury (Tan et al., 2008, 2011).
Here, we hypothesize that dendritic spine remodeling contributes to neuropathic pain in diabetic animals. We reasoned that if dendritic spine remodeling contributes to diabetic neuropathic pain, then the presence of malformed spines should coincide with pain, and disrupting maladaptive spine structure should reduce chronic pain. We report here that, at 4 weeks after diabetes induction by streptozotocin (STZ) injection, targeted inhibition of Rac1, a GTPase that is known to be important in dendritic spine plasticity, reduces diabetes-induced changes in morphology of dendritic spines in the dorsal horn, decreases neuronal excitability and attenuates spontaneous background firing activity from WDR neurons, and improves nociceptive mechanical pain thresholds. In hyperglycemic animals with diabetes before the development of neuropathic pain (i.e., 1 week after STZ injection), spine morphologies appeared normal. These results suggest that dendritic spine remodeling of dorsal horn nociceptive neurons contributes to diabetic neuropathic pain.
Materials and Methods
Animals/STZ-induced diabetes.
Experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All animal protocols were approved by the Yale University Institutional Animal Use Committee. Adult male Sprague Dawley rats (225–300 g; Harlan) were divided into two treatment arms (see study design in Fig. 1): the first group was injected with STZ (50 mg/kg body weight, i.p.; Sigma-Aldrich), a pancreatic beta cell toxin that produces type I diabetes (n = 48) (Morrow, 2004). Sham animals were injected with equal volumes of saline vehicle (n = 20). Glucose levels were measured from blood samples drawn from the ventral tail vein 4 d after STZ injection and immediately at the end of the experiment (OneTouch Ultra glucose monitoring system; LifeScan). Diabetes mellitus was defined as hyperglycemia with blood glucose of >250 mg/dl (Craner et al., 2002b). STZ-injected animals were removed from the study if they did not display hyperglycemia 4 d after injection. In our previous experience (Fischer et al., 2009), diabetic animals tended to lose body weight over the course of 4 weeks. All animals were housed under a 12 h light/dark cycle in a pathogen-free area with water and food provided ad libitum. To maintain body mass in diabetic animals, supplemental liquid food was provided at a level (Ensure; 150 ml daily; Abbott Laboratories) that permitted ad libitum feeding. Because we were interested in studying the relationship between diabetic neuropathic pain and dendritic spine remodeling, we performed two interrelated investigations: (1) in the loss-of-function experiments, at 4 weeks after STZ injection (Fig. 1), we studied STZ-induced diabetic animals that exhibited significant tactile neuropathic allodynia in our analyses (see below). Thus, for the 4 week endpoint, 34 of 38 animals reached the pretreatment threshold for allodynia and were studied pretreatment and posttreatment, and 4 of 38 animals that failed to reach the threshold for tactile allodynia were excluded. Based on our previous studies (Craner et al., 2002b; Fischer et al., 2009), an exclusion rate of 10–15% was expected for the 4 week endpoint. (2) To assess dendritic spine structure in diabetic animals with hyperglycemia without neuropathic pain, we assessed a cohort of STZ-injected animals before the development of significant pain (Fischer et al., 2009) at 1 week after STZ injection (Fig. 1). All animals underwent blood glucose and behavioral testing, and subpopulations of animals from each treatment group were randomly assigned to undergo histological analysis or terminal electrophysiological assessment.
Figure 1.

Study design. In week 1, weight-matched rats were randomly assigned to Sham (i.e., saline) or STZ treatment groups. To validate STZ induction of diabetes, blood glucose testing was performed 4 d after STZ injection, and just before experimental endpoints. A subpopulation of STZ-injected rats received NSC23766 (a Rac1 inhibitor) or vehicle via intrathecal catheter and were analyzed at 1 week after STZ induction of diabetes (dashed arrow). Another subpopulation of STZ-injected animals and Sham animals received intrathecal NSC23766 in week 3. All animals were tested for behavior at baseline, before STZ or vehicle injection, and at four time points: 1, 2, 3, and 4 weeks. All animals underwent glucose and behavioral testing, and subpopulations of animals from each treatment groups were randomly assigned to undergo histological analysis or terminal electrophysiological assessment.
Behavioral assays.
A blinded experimenter performed behavioral experiments in a dedicated quiet room under invariant conditions. Baseline behavioral testing was performed before STZ or vehicle injections, and also at four time points: 1, 2, 3, and 4 weeks (Fig. 1). After acclimation to the testing area (60 min), mechanical sensory thresholds were determined by paw withdrawal away from a series of von Frey filaments (Stoelting) applied to the glabrous surface of the right hindpaw. To determine the applied force value, we used a modification of the Dixon “up-down” method, which interprets paw withdrawal occurring 50% of the time as mechanical nociceptive threshold (Dixon, 1980; Chaplan et al., 1994). Heat nociceptive thresholds were assessed by measuring the latency of paw withdrawal in response to a radiant heat source (Dirig et al., 1997). Rats were placed in a Plexiglas box situated on an elevated glass plate. The glass plate directly under the glabrous surface of the paw was heated by a radiant heat source (4.7 A). Upon paw withdrawal, a photocell automatically turned off the heat source and measured withdrawal latency. If no response was detected, the heat source was automatically shut off at 20.5 s to prevent tissue damage. Three trials were performed and averaged for each animal. Five minutes of rest was allowed between trials.
Intrathecal catheterization and drug delivery.
All animals included in the final analysis received catheter implants. Animals were deeply anesthetized with ketamine/xylazine anesthesia. A small slit was made in the atlanto-occipital membrane, between the base of the skull and spinal vertebra C1 and a 32 gauge catheter was threaded caudally to the lumbar enlargement and secured by suturing. To prevent CSF leakage and infection, the rostral opening was heat sealed by pinching the exposed tip with a sufficiently heated forcep (Tan et al., 2008). The catheter tip location was confirmed within the intrathecal space after the animals were killed. For the week 1 animal cohort (STZ week 1; Fig. 1), drug vehicle (0.9% sterile saline; n = 10) was infused through the catheter for 3 d (5 μl volume; twice daily) by injection with a Hamilton syringe using a 32 gauge needle (Hamilton Co.). For the remaining animal population of week 3, Sham or STZ-injected animals, drug vehicle (0.9% saline; n = 16) or N6-[2-[[4-(diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine trihydrochloride (NSC23766) (1 mg/ml; n = 28), a Rac1 GTPase-specific inhibitor (EMD Chemicals) (Gao et al., 2004), were infused (5 μl volume; twice daily), beginning 3 d after catheter implantation (days 23–24), through the catheter by Hamilton syringe injection, followed by sterile saline flush (10 μl) for 3 d. By the end of the experiment, this produced five treatment groups (Fig. 1): STZ (week 1), Sham, Sham plus NSC23766, STZ, and STZ plus NSC23766.
Histology.
Rats were killed without fixation by decapitation for Golgi-Cox staining using a FD Rapid GolgiStain Kit (FD NeuroTechnologies) [STZ (week 1), n = 5; Sham, n = 4; Sham plus NSC, n = 4; STZ plus Veh, n = 5; STZ plus NSC, n = 5], as described previously (Tan et al., 2008). Briefly, unfixed spinal cord tissue (between spinal segments L3 and L6) was rapidly removed (<5 min), washed in distilled water, and immersed in the impregnation solutions according to manufacturer's instructions. The 200-μm-thick sections were cut on a vibratome (DTK-1000 microslicer; Ted Pella) and mounted on gelatinized glass slides. Sections were stained, rinsed in distilled water, dehydrated, cleared, and coverslipped with Permount medium.
Electrophysiology.
All animal groups underwent terminal electrophysiological experiments. Extracellular single-unit recording methods and identification of WDR neurons have been described previously (Tan et al., 2008, 2009b). Animals were anesthetized with sodium pentobarbital (40 mg/kg, i.p.), and a laminectomy was performed to expose the lumbar spinal enlargement. The overlying dura was carefully excised and warm mineral oil was applied over the recording area. Core body temperature was monitored with a rectal thermometer and maintained (34 ± 2°C) using a circulating water heat pad. Recordings were obtained with a low-impedance 5 mΩ tungsten insulated microelectrode (A-M Systems) that was positioned near the L4/L5 dorsal root insertion zone. Electrical signals were amplified and filtered at 300–3000 Hz, processed by a data collection system (CED 1401+; Cambridge Instruments). Stored recordings were analyzed off-line with Spike2 software (version 5.09; Cambridge Electronic Design). WDR neurons were first identified by their general response to a range of low- and high-threshold peripherally evoked stimuli (i.e., light brushing with a cotton swab and pinching with sharp forceps) applied to their cutaneous receptive fields. Following WDR neuron identification, we determined the presence or absence of spontaneous background activity (defined as firing activity in the absence of any peripheral stimulation). We mapped the cutaneous receptive field surface area of each neuron on an outline of the dorsal surface of the rat hindquarters (shown in Fig. 6) by lightly brushing, pinching, and probing with von Frey filaments (Fischer et al., 2009; Chang et al., 2010). The areas of each receptive field were measured using NIH ImageJ (software download at http://rsbweb.nih.gov/ij/). An arbitrary standard unit for area was assigned and used for quantitative comparison between treatment groups (Chang et al., 2010). The following stimuli were then applied: (1) phasic brush (PB) stimulation of the skin with a cotton applicator, (2) compressive pressure (144 g/mm2), (3) compressive pinch (583 g/mm2), and (4) calibrated von Frey filaments of increasing force (0.04, 0.16, 0.4, 0.6, 1.0, 4.0, 6.0, 15.0, 26.0 g). These stimulus modalities are reflective of human nociceptive testing (Baumgärtner et al., 2002; Pitcher and Henry, 2004). We confirmed that responses were maximal by stimulating the primary receptive field of each unit and ensured that isolated units remained stable during recording by using software wave template matching routines. The activities of 4–6 WDR units/animal were recorded from the lumbar enlargement (∼1000 μm deep), which yielded 17–27 units/treatment group.
Figure 6.
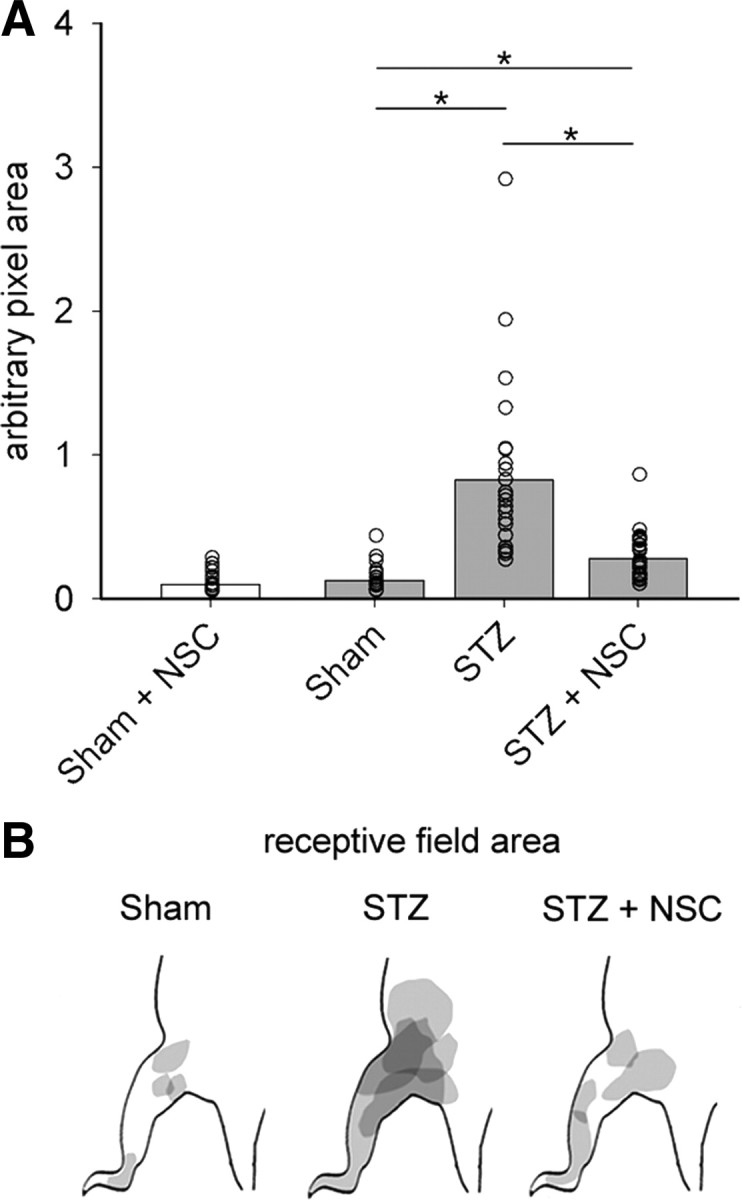
Receptive field areas expand in diabetic neuropathic pain. A, Quantification of WDR unit receptive field areas demonstrate that diabetic animals had receptive fields that were significantly larger, by more than sixfold, compared with Sham receptive field areas (one-way ANOVA with Bonferonni's post hoc, *p < 0.05). Treatment with NSC23766 decreased the size of receptive fields compared with untreated diabetic animals (one-way ANOVA with Bonferonni's post hoc, *p < 0.05), but not back to Sham levels (one-way ANOVA with Bonferonni's post hoc, *p < 0.05). NSC23766 treatment in Sham animals did not change receptive field size (white bar). B, A representative map shows that the receptive field areas in diabetic animals, STZ, increases compared with Sham and STZ animals treated with NSC23766, STZ plus NSC. In A, the open circles show individual animal means. The bar graph is mean ± SEM.
Dendritic spine/reconstruction analysis.
Golgi-stained coronal spinal cord tissue sections were examined under transmitted light microscopy (Nikon Eclipse 80i). Images shown in Figure 2 were captured with a Nikon Eclipse E800 microscope with a HQ Coolsnap camera (Roper Scientific). To identify whole cells in dorsal horns, we used five inclusion criteria that allowed us to sample cells with morphologies similar to those observed for WDR neurons (Woolf and King, 1987; Tan et al., 2008): (1) neurons were located within Rexed lamina 4 and 5; (2) Golgi-stained neurons must have had dendrites and spines that were completely impregnated, appearing as a continuous length; (3) at least one dendrite extended into an adjacent lamina relative to the origin of the cell body; (4) at least one-half of the primary dendritic branches remained within the thickness of the tissue section, such that their endings were not cut and instead appeared to taper into an ending; and (5) the cell body diameter fell between 20 and 50 μm (see example shown in Fig. 2). To determine whether there were any significant morphological differences between neurons, cell diameter, total dendritic length, number of primary dendrites, length of primary dendrites, and the percentage of primary dendrites with secondary dendritic branches were measured and compared across animals and treatment groups post hoc. Together, these measures control for the morphological diversity of spinal cord dorsal horn neurons. We identified a total of 116 neurons [STZ (week 1), 20; Sham, 20; Sham plus NSC, 21; STZ plus Veh, 23; STZ plus NSC, 31] for inclusion in our analysis.
Figure 2.

Golgi-stained coronal sections of spinal cord tissue reveal dendritic spine remodeling on dorsal horn neurons. A, A representative image of the dorsal horn with a WDR neuron located in lamina 5. B, A high-power field of the neuron shown in A (inset). C, D, A blinded observer analyzed dendritic spines from 10–13 randomly sampled neurons from each treatment arm for spine length (C) and spine head diameter (D). Dendritic spines in the STZ arm displayed longer spine lengths compared with Sham. NSC23766 treatment decreased spine length in STZ-induced diabetes (one-way ANOVA with Bonferonni's post hoc test, **p < 0.001). D, STZ-induced diabetes significantly increased spine head diameters by >50% compared with Sham. Administration of NSC23766 decreased spine head diameter compared with STZ animals (one-way ANOVA with Bonferonni's post hoc test, **p < 0.001). Treatment with NSC23766 in nondiabetic Sham animals (C, D, white bars; Sham plus NSC) decreased spine length (one-way ANOVA with Bonferonni's post hoc test, *p < 0.05) but had no effect on spine head diameter. By week 4, after saline or STZ injection, qualitative assessment of high magnification images (E–G) demonstrates differences in dendritic spine morphological profiles between the treatment arms with the presence of more mushroom-shaped spines in the STZ group (F′) compared with Sham (E′) or STZ plus NSC (G′). Treatment with NSC23766 appeared to restore thin-shaped spine structures (G′) with similar profiles comparable with Sham. Scale bars: A, 500 μm; B, 100 μm; E–G, 10 μm; E′, F′, G′, 1 μm. Graphs are mean ± SEM.
To identify dendritic spines, we used specific morphological characteristics as reported previously (Kim et al., 2006; Tan et al., 2008). We considered a protrusion without a visible neck structure (from the main dendrite shaft) a spine only if there was a visible indentation on either side of the junction of the protrusion from the dendrite branch. A spine neck was defined as the structure between the base of the spine, the interface between the parent dendrite branch, and the base of the spine head where the appearance of the spine began to swell distally. Spine head structure varied greatly, but we defined these as the visible bulb-like structures located at the ends of protrusions (Tan et al., 2008). Thin- and mushroom-shaped spines were classified as follows: thin spines have head diameters that are less than or equal to the length of the spine neck. Mushroom spines have head diameters that are greater than the length of the neck. There are three rationales for using these geometric categories for spines: first, two spine shapes allowed us to use simple but strict rules in classifying spine morphology; second, this approach, by precluding discrimination of subtle variations in spine shape, allows collection of a large sample size; third, there is a large body of literature describing the different physiological characteristics associated with the morphologies of thin- and mushroom-shaped spines (Calabrese et al., 2006).
To reconstruct WDR neurons, we used Neurolucida software (version 9.0; MicroBrightField). We analyzed the complete three-dimensional reconstructions of dorsal horn neurons for spine density and distribution. Two Neurolucida traces were made for each neuron. (1) An outline trace of the spinal cord section upon which the location of identified WDR neurons was marked. (2) A three-dimensional reconstruction of each WDR cell, which was created by tracing through the x-, y-, and z-axes. Dendritic spine type (thin spines, blue; mushroom spines, red) and location were marked on these three-dimensional reconstructions. Dendritic spine density was expressed as spine number per 10 μm dendritic length. To determine spatial distribution of spines (relative to the cell body), we used modified Sholl's analysis using Neurolucida software. Seven 50-μm-wide spherical bins were formed around the cell body, and spine density within each bin was averaged for each treatment group. Mean data were compared against equivalent bins across treatment groups.
Statistical analysis.
All statistical tests were performed at the α-level of significance of 0.05 by two-tailed analyses using parametric or nonparametric tests, if appropriate. We used repeated-measures ANOVA and Kruskal–Wallis one-way ANOVA on ranks, followed by Bonferonni's post hoc analysis. Data management and statistical analyses were performed using SigmaStat (version 3.0.1a; Aspire Software International) and Microsoft Office Excel (2011). Data in the text are described as mean ± SD. All graphs were plotted as mean ± SEM using SigmaPlot (version 8.02a).
Results
Tactile allodynia develops rapidly following induction of diabetes
The injection of STZ is a well established procedure for producing type I diabetes mellitus in adult rats (Morrow, 2004). Four days after administration of STZ, injected animals had a significant increase in blood glucose levels compared with vehicle-injected controls (p < 0.0001; group mean for STZ: 460 ± 155.3 vs 105.6 ± 22.5 mg/dl) (Table 1). Blood glucose remained at elevated levels for the duration of the experiment compared with controls (at 4 weeks after STZ injection: p < 0.0001; group mean for STZ: 504.2 ± 167.7 vs 106.6 ± 12.0 mg/dl). To maintain the health of all experimental animals, supplementary food was provided ad libitum. Although there were fluctuations in body weight over the course of the experiment, we found no significant differences in body weights when compared across treatment group or at any time point measured, weeks 1 and 4 shown in Table 1. By the fourth week, the weight of STZ (294.2 ± 26.8 g) and STZ plus NSC23766-treated animals (304 ± 48.5 g) was similar to nondiabetic animals: Sham (293.8 ± 13.3 g) or Sham plus NSC23766 (289.2 ± 13.3 g). As we expected from our previous study (Fischer et al., 2009), not all STZ-injected animals develop neuropathic pain by the week 4 endpoint. Because we were interested in studying the relationship between diabetic neuropathic pain and dendritic spine remodeling, we only included STZ-induced diabetic animals that exhibited significant tactile neuropathic allodynia in our analyses for the week 4 endpoint (see below). We used the criterion for tactile allodynia as an observed withdrawal threshold of <4.0 g at any point during behavioral testing (for von Frey tests; see Materials and Methods). Thirty-four of 38 animals met this criterion and were included in this analysis. A proportion of 10–15% animals that failed to reach the 4.0 g threshold for tactile allodynia was expected based on earlier data (Craner et al., 2002b; Fischer et al., 2009).
Table 1.
Weight and glucose levels of rats
| Sham | Sham + NSC | STZ (week 1) | STZ | STZ + NSC | |
|---|---|---|---|---|---|
| Week 1 | |||||
| Weight (g) | 283.8 ± 43.9 | 303.6 ± 19.2 | 287.2 ± 14.0 | 311.9 ± 42.1 | 301.4 ± 27.8 |
| Glucose level (mg/dl) | 105.6 ± 22.5 | 105.2 ± 10.3 | 537.4 ± 39.8 | 534.3 ± 53.8 | 531 ± 65.4 |
| Week 4 | |||||
| Weight (g) | 293.8 ± 13.3 | 289.2 ± 17.0 | N/A | 294.2 ± 26.8 | 304.8 ± 48.5 |
| Glucose level (mg/dl) | 106.6 ± 12.0 | 89.6 ± 11.1 | N/A | 547 ± 77.9 | 580 ± 29.8 |
Data are shown as mean ± SD.
Dendritic spines remodel on dorsal horn neurons in experimental diabetes
Dendritic spine remodeling in the dorsal horn has been observed following SCI and peripheral nerve injury (Kim et al., 2006; Tan et al., 2008, 2011). In particular, we have shown that maladaptive dendritic spine plasticity occurs on WDR neurons, which can contribute to adversely altered sensory processing associated with chronic pain (Tan et al., 2008, 2009a). Neuropathic pain develops progressively over time in diabetic animal models and chronic diabetes in humans (Malcangio and Tomlinson, 1998; Marchettini et al., 2004; Singleton and Smith, 2007; Fischer et al., 2009). To determine whether diabetic neuropathic pain associated with changes in dendritic spine morphology, we first analyzed dendritic spines from spinal cord tissue collected 4 weeks after STZ or control injection (Fig. 2A,B). Wide-dynamic-range neurons (WDR) were identified in Golgi-stained coronal sections of spinal cord at lumbar segments L3–L5 on the basis of five criteria (see Materials and Methods). All WDR neurons were located in the intermediate zone of the dorsal horn (lamina IV–V, ∼400–1200 μm deep) (Fig. 2A). To control for possible variation in cell morphologies, we compared the dimensions of sample cells for the following: cell body diameter, total dendrite length, number of primary dendrites (dendrites projecting directly from the cell body), length of primary dendrites, and the percentage of primary dendrites with secondary branches (Table 2). These morphological parameters were not significantly different when compared across treatment groups (for all comparisons, p > 0.20). Thus, any changes we observed in dendritic spine morphologies were not due to differences in neuronal sampling. A representative neuron located in lamina 5 is shown in Figure 2, A and B. We categorized all observed spines into two categories: thin- and mushroom-shaped (see Materials and Methods).
Table 2.
Cell body dimensions and dendritic branch morphology comparison
| Maximum cell diameter (μm) | Total dendrite length (μm) | No. of primary dendrites | Length of primary dendrite (μm) | Primary dendrites with secondary branches (%) | |
|---|---|---|---|---|---|
| Sham | 33.2 ± 7.6 | 1025.8 ± 436.0 | 3.6 ± 1.1 | 97.3 ± 50.7 | 53.5 ± 27.4 |
| Sham + NSC | 35.1 ± 8.4 | 1117.1 ± 567.8 | 3.9 ± 0.7 | 82.8 ± 43.5 | 53.4 ± 24.8 |
| STZ (week 1) | 36.7 ± 7.9 | 1162.8 ± 533.8 | 3.6 ± 0.7 | 105.8 ± 84.6 | 58.6 ± 26.6 |
| STZ (week 4) | 36.1 ± 11.4 | 985.2 ± 387.5 | 3.6 ± 0.9 | 90.3 ± 50.9 | 54.6 ± 30.6 |
| STZ + NSC | 34.4 ± 7.1 | 1190.4 ± 465.4 | 3.7 ± 0.8 | 120.4 ± 118.2 | 50.2 ± 28.2 |
Data are shown as mean ± SD.
To quantify the effects of STZ-induced diabetes on spine length and spine head diameter at the 4 week endpoint, we analyzed 2518–2870 spines from each treatment arm (from randomly selected 10–13 WDR neurons per arm; Fig. 1) and a blinded observer measured spine dimensions from each neuron. For statistical comparison, we first pooled spine data within each animal, and then averaged individual animal data within treatment groups. Final comparisons were performed across treatment groups. As shown in Figure 2C, gray bars, spines in the STZ animal group (n = 2870 spines) had significantly longer spine lengths compared with spines in the Sham group (n = 2518 spines) (p < 0.001; 1.55 ± 0.06 μm vs 1.22 ± 0.02 μm). Treatment with NSC23766 (STZ plus NSC) reduced spine lengths (n = 2430 spines) compared with the STZ group (p < 0.001; 1.19 ± 0.16 vs 1.55 ± 0.06 μm). There was no difference between STZ plus NSC and Sham treatment groups (p > 0.05). To determine whether diabetes changes spine head morphology at the week 4 endpoint, we also measured spine head diameter (Fig. 2D). Dorsal horn neurons in the STZ group had spines with larger spine head diameters compared with neurons in the Sham group (p < 0.001; 2.09 ± 0.67 vs 0.88 ± 0.008 μm). NSC23766 treatment in STZ animals (STZ plus NSC) reduced spine head diameter compared with STZ animals without drug treatment (p < 0.001; 1.00 ± 0.19 vs 2.09 ± 0.67 μm). STZ animals treated with NSC23766 had similar spine head diameters with Sham (p = 1.0). These results demonstrate that dendritic spine length and head diameter increase in diabetic neuropathy, but these structural changes can be reversed to close-to-normal by Rac1 inhibition.
As a control for the Rac1 inhibitor, NSC23766, in nondiabetic animals, we treated a population of Sham animals with NSC23766. NSC23766 treatment in Sham animals (Sham plus NSC; n = 2053 spines) reduced spine length compared with untreated Sham animals (p < 0.05; 0.96 ± 0.11 vs 1.22 ± 0.02 μm) (Fig. 2C). NSC23766 treatment of nondiabetic Sham animals did not affect spine head diameter (p = 1.0) (Fig. 2D). This suggests that NSC23766 has the potential to effect spine morphology, regardless of any changes caused by diabetes. This, however, is not surprising since studies by others (Tashiro and Yuste, 2004; Choi et al., 2005) have shown that Rac GTPase is involved in spine plasticity and given that we have also shown that NSC23766 can affect dendritic spines in vitro (Tan and Waxman, 2011; Tan et al., 2011).
Dendritic spine density increases with STZ-induced diabetic neuropathic pain
We observed qualitative differences in the density and morphology of dendritic spines on Golgi-stained WDR neurons (Fig. 2E–G). To obtain an accurate count of dendritic spines from spinal cord tissue, we used Neurolucida software to reconstruct identified WDR neurons (see Materials and Methods) (Fig. 3). To indicate the location of sampled WDR neurons, we first mapped the contour outline of each spinal cord section and the gray matter borders (see representative traces from three week 4 groups; Fig. 3A–C). WDR neurons were marked on each of these maps. On traced neurons (Fig. 3A′,B′,C′), visible spines were color-coded as thin (blue dot) or mushroom-shaped (red dot) (Fig. 3A″,B″,C″). We measured the total number of spines on each neuron and calculated spine density as the number of spines per 10 μm dendrite lengths. Dendritic spine densities from neurons within each treatment group were pooled and compared across groups. As shown in Figure 4A, 4 weeks after STZ-induced diabetes, total spine density was similar compared with Sham (p = 0.07; 3.52 ± 0.81 vs 2.83 ± 0.86 spines/10 μm dendrite). Treatment of diabetic animals with NSC23766, however, significantly decreased total spine densities compared with both STZ (p < 0.05; 1.97 ± 0.73 vs 3.52 ± 0.81 spines/10 μm dendrite) and Sham (p < 0.05; 1.97 ± 0.73 vs 2.83 ± 0.86 spines/10 μm dendrite). Similarly, thin-shaped spine density did not change in diabetic animals compared with Sham (p = 0.56; Sham, 2.63 ± 0.83, vs STZ plus Veh, 2.80 ± 0.98 spines/10 μm dendrite) (Fig. 4B). Treatment of diabetic animals with NSC23766 did decrease thin-shaped spine density below that of Sham (p < 0.05; 1.93 ± 0.74 vs 2.63 ± 0.83 spines/10 μm dendrite) and STZ animals (p < 0.05; 1.93 ± 0.74 vs 2.80 ± 0.98 spines/10 μm dendrite). In contrast, we observed a qualitatively dramatic increase in the number of mushroom spines in diabetic animals compared with neurons in either Sham or STZ plus NSC animals (Fig. 2D). Quantification demonstrated a nearly threefold increase in mushroom spine density in diabetic animals compared with Sham (p < 0.01; 0.63 ± 0.23 vs 0.21 ± 0.13 spines/10 μm dendrite) (Fig. 4C). Importantly, inhibiting Rac1 with NSC23766 treatment in diabetic animals significantly reduced mushroom spine density (p < 0.01; 0.08 ± 0.05 vs 0.63 ± 0.23 spines/10 μm dendrite). NSC23766 treatment also reduced mushroom spine density below Sham (p < 0.05; 0.08 ± 0.05 vs 0.21 ± 0.13 spines/10 μm dendrite). Sham animals (i.e., no diabetes) treated with NSC23766 (Fig. 4A–C, white bars) did not differ significantly compared with untreated Sham animals in any spine density category (p = 0.06–1.0).
Figure 3.

Three-dimensional Neurolucida reconstructions of representative dorsal horn neurons. Digital reconstructions (A–C) show the locations of sampled neurons (black dot and arrow), with the complete three-dimensional distribution and density of thin- and mushroom-shaped spines on neurons from Sham (A′), STZ (B′), and STZ plus NSC (C′). A ∼50 μm length of the dendritic branch (gray shade) from neurons is shown in A″, B″, and C″. These branches reveal the presence of mushroom spines (red dots) and thin spines (blue dots). Branches from STZ (B″) displayed more spines, particularly mushroom spines compared with Sham (A″) or STZ plus NSC (C″). Scale bars: A–C, 500 μm; A′, B′, C′, 50 μm; A″, B″, C″, 10 μm.
Figure 4.

Quantitative analysis of dendritic spine density and spatial distribution. Analyses of reconstructed neurons reveal differences in spine density (top row panels) and distribution (bottom row panels). A, B, Total spine density, which includes all spine shapes (A), and thin spine density (B) did not change in diabetic animals compared with Sham (one-way ANOVA with Bonferonni's post hoc, p > 0.05). C, Mushroom spine density, however, increased by more than threefold in STZ-induced diabetic animals compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.05). Treatment with NSC23766 significantly reduced spine density for total, thin-shaped, and mushroom-shaped spines compared with STZ (one-way ANOVA with Bonferonni's post hoc or one-way ANOVA on ranks with Dunn's method, *p < 0.05). Dendritic spine distribution for total (D), thin (E), and mushroom (F) spines demonstrate significant changes in STZ diabetic animals. Total (D) spine distribution assessment shows significant redistribution in STZ diabetic animals compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.01; at 250 μm: 2.37 ± 0.93 vs 3.89 ± 1.78 spines/10 μm dendrite). Similarly, for mushroom spines (F), significantly more spines were located more proximal to the soma in the STZ group compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.05; at 50 μm: 0.20 ± 0.22 vs 0.86 ± 1.07 spines/10 μm dendrite; at 100 μm: 0.15 ± 0.12 vs 0.86 ± 0.82 spines/10 μm dendrite; at 150 μm: 0.18 ± 0.17 vs 0.71 ± 0.71 spines/10 μm dendrite). NSC23766 treatment of STZ animals resulted in decreased thin spine (E) densities at multiple regions compared with untreated STZ animals (one-way ANOVA on ranks with Dunn's method, #p < 0.05; at 50 μm: 1.26 ± 0.82 vs 2.35 ± 1.34 spines/10 μm dendrite; at 100 μm: 1.93 ± 0.91 vs 2.77 ± 1.11 spines/10 μm dendrite; at 300 μm: 1.96 ± 0.91 vs 3.32 ± 1.41 spines/10 μm dendrite; at 350 μm: 1.60 ± 1.0 vs 3.27 ± 1.17 spines/10 μm dendrite). NSC23766 treatment in STZ animals restored mushroom spine (F) distribution back to Sham levels by decreasing dendritic spine densities compared with untreated STZ animals (one-way ANOVA on ranks with Dunn's method, #p < 0.05; at 50 μm: 0.08 ± 0.13 vs 0.86 ± 1.07 spines/10 μm dendrite; at 100 μm: 0.09 ± 0.08 vs 0.86 ± 0.82 spines/10 μm dendrite; at 150 μm: 0.08 ± 0.10 vs 0.71 ± 0.71 spines/10 μm dendrite; at 200 μm: 0.10 ± 0.10 vs 0.77 ± 0.85 spines/10 μm dendrite; at 250: 0.10 ± 0.11 vs 0.72 ± 0.78 spines/10 μm dendrite). Treatment with NSC23766 in nondiabetic Sham animals (A–C, white bars; D–F, dashed line) had no significant effect compared with untreated Sham. For spine density graphs, the open circles show individual animal means (for Sham plus NSC, n = 21; Sham, n = 20; STZ, n = 23; STZ plus NSC, n = 31). Distribution graphs are mean ± SEM.
Dendritic spines redistribute in animals with diabetic neuropathic pain
The location of spines relative to the cell body has physiological implications for a neuron. Excitatory inputs that synapse closer to the cell body have a greater weighted contribution to the cumulative output of a neuron compared with those forming synapses farther away (Pongrácz, 1985; Yuste and Urban, 2004; Tan et al., 2009a). To examine whether STZ-induced diabetes results in a spatial redistribution of spines along dendritic branches, we used modified Sholl's analysis (Tan et al., 2008). In STZ-induced diabetic animals, spine densities increased on dendritic branches located at 250 μm from the cell body compared with Sham animals (p < 0.01) (Fig. 4D). Thin-shaped spine distribution did not significantly change in STZ animals compared with Sham (p > 0.05) (Fig. 4E). NSC23766 treatment in STZ animals decreased thin-shaped spine densities at multiple regions compared with untreated STZ animals: at 50, 100, 300, and 350 μm compared with STZ (p < 0.05). In STZ-induced diabetic animals, mushroom-shaped spines increased in regions proximal to the soma at three regions between 50 and 150 μm from the cell body compared with Sham (p < 0.05). (Fig. 4F). In five proximal regions, NSC23766 treatment significantly reduced mushroom-shaped spine densities in STZ diabetic animals (p < 0.05). NSC23766 treatment in Sham animals (Fig. 4D–F, dashed gray line) did not significantly affect spine densities across any region analyzed (p > 0.05) compared with untreated Sham animals.
Dendritic spine remodeling is accompanied by electrophysiological signs of neuropathic pain in diabetes
To test the response of WDR neurons in animals with diabetic neuropathic pain, we sampled single units from dorsal horn lamina IV–V. We assessed three physiological signs attributed to chronic neuropathic pain: (1) the presence of spontaneous activity (defined as firing activity in the absence of any peripheral stimulation), (2) hyperresponsiveness to peripheral stimuli, and (3) expansion of cutaneous receptive fields. We identified WDR neurons by their responsiveness to both low- and high-threshold peripheral stimuli applied to their cutaneous receptive fields (Gjerstad et al., 2001; Hains and Waxman, 2006). Of the WDR neurons sampled from animals at the week 4 endpoint, 2 of 17 (11.7%) neurons in Sham animals exhibited spontaneous activity, whereas in diabetic animals there was a greater proportion of sampled neurons with spontaneous activity, 12 of 27 (44%). Disruption of dendritic spines by inhibiting Rac1 with NSC23766 decreased the proportion of sampled WDR neurons, to 4 of 25 (16%) sampled units, with spontaneous activity. Sham animals treated with NSC23766 displayed a similar fraction of sampled neurons, 2 of 17 (11.7%), with spontaneous activity compared with untreated Sham animals.
Compared with Sham, STZ animals had significantly increased peripherally evoked unit activity to PB, press (144 g/mm2), and pinch (538 g/mm2) stimuli, indicative of dynamic mechanical allodynia (p < 0.01) (Fig. 5A). Similarly, compared with Sham, STZ animals had significantly increased evoked single-unit activity in response to increasing von Frey filament intensities (gradient from 0.04 to 26 g) applied to hindlimb receptive fields, demonstrating punctate hyperalgesia (p < 0.01) (Fig. 5B). Inhibiting Rac1 activity with NSC23766 treatment in diabetic animals significantly decreased mechanical allodynia for all tactile modalities tested: phasic brush, press, pinch, and von Frey filament tests (p < 0.01). NSC23766 treatment in Sham animals had no significant effect on evoked firing response of single units in any stimulus modality tested compared with untreated Sham animals. We observed no significant differences for peripherally evoked spiking rates between Sham and STZ plus NSC animal groups, suggesting that NSC23766 treatment restored close-to-normal WDR neuron excitability. Figure 5C shows a representative peristimulus histogram of responses (spikes/1 s bin) for WDR neurons in Sham, STZ, and STZ plus NSC groups.
Figure 5.

Assessments for electrophysiological signs of diabetic neuropathic pain. All sampled WDR neurons responded to low- and high-threshold stimuli. A, STZ-induced diabetes significantly increased spike-firing rate compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.01; for PB: 8.71 ± 5.16 vs 33.1 ± 15.50; for press: 12.7 ± 9.23 vs 33.5 ± 21.7; for pinch: 14.4 ± 9.9 vs 42.7 ± 23.2). NSC23766 treatment significantly reduced the firing activity in STZ-induced diabetic animals (one-way ANOVA on ranks with Dunn's method, *p < 0.01; for PB: 9.8 ± 6.27 vs 33.1 ± 15.50; for press: 13.18 ± 8.93 vs 33.5 ± 21.7; for pinch: 16.7 ± 10.6 vs 42.7 ± 23.2). B, In all animals, stimulation with progressively more intense von Frey filaments produced a directly corresponding increase in spike-firing rates. In STZ diabetic animals, however, there was a significantly greater slope in spike-firing rates compared with Sham, demonstrating the presence of punctate mechanical allodynia (one-way ANOVA on ranks with Dunn's method, *p < 0.01; for 0.04 g: 2.04 ± 2.35 vs 5.7 ± 3.70 Hz; for 0.16 g: 3.18 ± 2.35 vs 9.52 ± 6.9 Hz; for 0.4 g: 4.29 ± 2.71 vs 14.0 ± 11.8 Hz; for 0.6 g: 4.73 ± 2.70 vs 14.03 ± 11.8 Hz; for 1.0 g: 5.1 ± 3.37 vs 15.3 ± 14.8 Hz; for 4.0 g: 6.65 ± 4.36 vs 20.9 ± 15.3 Hz; for 6.0 g: 7.54 ± 5.3 vs 22.0 ± 13.8 Hz; for 15.0 g: 8.08 ± 6.41 vs 26.6 ± 17.8 Hz; for 26.0 g: 9.8 ± 5.61 vs 31.4 ± 17.6 Hz). NSC23766 treatment in STZ plus NSC animals attenuated diabetic-induced neuronal hyperexcitability, and spike-firing rates were similar to Sham (one-way ANOVA on ranks with Dunn's method, #p < 0.01; for 0.04 g: 2.63 ± 3.87 vs 5.7 ± 3.70 Hz; for 0.16 g: 3.99 ± 4.41 vs 9.52 ± 6.9 Hz; for 0.4 g: 5.59 ± 6.04 vs 14.0 ± 11.8 Hz; for 0.6 g: 6.19 ± 7.36 vs 14.03 ± 11.8 Hz; for 1.0 g: 6.98 ± 7.82 vs 15.3 ± 14.8 Hz; for 4.0 g: 8.93 ± 7.14 vs 20.9 ± 15.3 Hz; for 6.0 g: 9.86 ± 8.69 vs 22.0 ± 13.8 Hz; for 15.0 g: 12.3 ± 10.3 vs 26.6 ± 17.8 Hz; for 26.0 g: 11.9 ± 8.31 vs 31.4 ± 17.6 Hz). C, A representative peristimulus histogram shows the peripherally evoked response of WDR single units to PB, compressive press (144 g/mm2), and pinch (583 g/mm2) of cutaneous receptive fields. Representative unit spike waveforms are shown (right side; C). STZ WDR units peristimulus histogram responses (gray) are overlaid with STZ plus NSC peristimulus histogram responses (black). Electrophysiological properties of WDR single units in Sham plus NSC were similar to untreated Sham for all assessments (A, white bar; B, dashed line). All graphs are mean ± SEM.
The receptive fields of dorsal horns units expand in animal models of diabetic neuropathic pain (Fischer et al., 2009). To test whether there are changes in receptive fields of WDR neurons (Fig. 6), we mapped receptive fields in sampled neurons by light brushing, pinching, and gentle probing with von Frey filaments (see Materials and Methods). As shown in representative maps (Fig. 6B), at the week 4 endpoint, the receptive field areas in diabetic animals increased compared with Sham animals, and NSC23766 treatment decreased receptive field size. Quantification demonstrated that neurons in diabetic animals had receptive fields that were significantly larger, by more than sixfold compared with Sham receptive field areas (Fig. 6A) (p < 0.001; 0.82 ± 0.62 vs 0.12 ± 0.09 arbitrary unit area). Treatment of diabetic animals with NSC23766 significantly decreased the size of receptive fields compared with untreated diabetic animals (p < 0.001; 0.27 ± 0.16 vs 0.82 ± 0.62 arbitrary unit area). However, the receptive field areas in NSC23766-treated diabetic animals did not fully return to sham levels, and still remained significantly larger (p < 0.001; 0.27 ± 0.16 vs 0.12 ± 0.09 arbitrary unit area) than those in sham animals. NSC23766 treatment in nondiabetic Sham animals did not change receptive field size (Fig. 6A, white bar).
Neuropathic pain behavior accompanies dendritic spine remodeling in diabetes
For all animals, a blinded observer gathered behavioral data for mechanical and heat pain thresholds at the following: baseline (before STZ or control injections; see study design in Fig. 1) and each week until the end of the experiment at 4 weeks. To demonstrate the progression and establishment of neuropathic pain exhibited in STZ-induced diabetic animals, behavioral data gathered within three treatment arms (Fig. 1) in week 4 was pooled, and compared two ways: (1) against baseline (data not graphed) and (2) across the treatment arms (Fig. 7A). As expected from previous work (Chen and Pan, 2002; Craner et al., 2002b; Fischer et al., 2009), STZ-induced diabetes gradually decreased mechanical pain thresholds over the 4 week period compared with baseline, reaching significance by the second week after STZ injection (week 2: p < 0.05; 10.2 ± 5.95 vs 19.53 ± 4.6 g; week 3: p < 0.01; 5.19 ± 2.2 vs 19.53 ± 4.6 g; week 4: p < 0.01; 6.89 ± 5.6 vs 19.53 ± 4.6 g). Compared with Sham controls, diabetic animals also had decreased mechanical pain thresholds (week 2: p < 0.01; 21.8 ± 2.8 vs 10.16 ± 5.9 g; week 3: p < 0.01; 17.3 ± 6.8 vs 5.19 ± 2.2 g; week 4: p < 0.01; 22.5 ± 7.5 vs 6.89 ± 5.6 g) (Fig. 7A,C). Before NSC23766 administration (in animals assigned to the STZ plus NSC treatment arm), mechanical pain thresholds also decreased compared with baseline (week 2: p < 0.01; 11.6 ± 7.6 vs 20.78 ± 5.17 g; week 3: p < 0.01; 3.83 ± 0.66 vs 20.78 ± 5.17 g) and Sham (week 2: p < 0.01; 11.6 ± 7.6 vs 25.9 ± 0.22 g; week 3: p < 0.01; 3.83 ± 0.66 vs 16.8 ± 4.9 g). After 3 d of intrathecal NSC23766 treatment, there was a restorative increase in mechanical pain thresholds compared with untreated diabetic animals in week 4 (p < 0.01; 17.3 ± 8.1 vs 7.56 ± 6.98 g). At week 4, following NSC23766 treatment, mechanical pain thresholds were similar to sham threshold levels (p = 0.25) and comparable with baseline (p = 0.28). This demonstrates that NSC23766 rapidly and significantly reduces mechanical allodynia in animals with STZ-induced diabetic neuropathic pain. In contrast, we observed no significant development of heat hyperalgesia (Fig. 7B,D). By week 4, both STZ treatment arms, STZ and STZ plus NSC, showed no significant difference in heat sensitivity between baseline (p = 0.39–0.97) and Sham control animals (p = 0.051–0.92). To determine the effects of NSC23766 on pain-related behavior, apart from diabetic induced neuropathic pain, we had also treated Sham animals with intrathecal NSC23766. Treatment with NSC23766 did not significantly affect mechanical allodynia or heat hyperalgesia readouts for nondiabetic Sham animals at the week 4 endpoint or naive animals at baseline.
Figure 7.

Testing for tactile allodynia and heat hyperalgesia. A blinded observer gathered behavioral data for mechanical allodynia (A, C) and heat hyperalgesia (B, D) at the following: baseline (before STZ or vehicle injections) (Fig. 1) and each week until week 4. STZ-induced diabetes progressively decreased mechanical pain thresholds, reaching significance by week 2 and maintaining decreased thresholds until the end of the experiment compared with Sham (one-way ANOVA with Bonferonni's post hoc, *p < 0.05). Similarly, before NSC23766 treatment, animals assigned to the STZ plus NSC treatment arm also had decreased mechanical pain thresholds compared with Sham (one-way ANOVA with Bonferonni's post hoc, §p < 0.01). After NSC23766 treatment over 3 d in week 3 (Fig. 1), there was a restorative increase in mechanical pain thresholds in STZ-injected animals compared with pain thresholds in STZ animals without drug treatment (one-way ANOVA with Bonferonni's post hoc, #p < 0.05). B, NSC23766 treatment had no effect on thermal paw withdrawal latency compared with Sham or STZ animals at any time point. The graphs show mechanical threshold (C) and thermal paw withdrawal latency (D) data at 4 weeks from the three main comparator groups (gray bars; *p < 0.01) and NSC23766 treatment in Sham animals (white bars). There was no significant different in pain thresholds for Sham plus NSC compared with Sham (p > 0.05). Graphs are mean ± SEM.
Diabetic hyperglycemia precedes maladaptive dendritic spine remodeling
The findings reported here and our previous work suggest that morphologic changes in dendritic spines are mechanistically linked to neuropathic pain (Tan et al., 2008, 2009a, 2011). Thus, an important follow-up question is whether or not diabetic animals with hyperglycemia exhibit dendritic spine abnormalities in the absence of pain. Hyperglycemia induced by STZ injection occurs rapidly, within 4 d (Table 1), but neuropathic pain generally begins to appear in the second or third week after diabetes induction (Fig. 7) (Malcangio and Tomlinson, 1998; Fischer et al., 2009). To determine whether diabetic animals with hyperglycemia analyzed before the development of pain-related behaviors exhibit dendritic spine abnormalities, we investigated a subpopulation of diabetic animals 1 week after STZ injection. We first analyzed dendritic spine morphological characteristics (see Materials and Methods), including spine shape, density, and distribution.
As shown in Figure 8, A and B, dendritic spine shape did not change at 1 week after STZ injection: dendritic spine length and head diameter, respectively, were comparable with Sham animals [p = 1.0; STZ (week 1); n = 2139 spines]. In contrast, by week 4, STZ animals had significantly increased dendritic spine length and head diameter compared with STZ animals in week 1 (p < 0.001) and Sham animals (p < 0.001). Similarly, dendritic spine density did not change in hyperglycemic animals 1 week after STZ injection compared with nondiabetic Sham animals (Fig. 8C) (p = 0.32, for total spines; p = 0.58, for thin spines; p = 0.12, for mushroom spines). After 4 weeks, total and mushroom spine density increased for hyperglycemic STZ animals compared with STZ animals at 1 week (p < 0.001, for total spines; p < 0.001, for mushroom spines). Compared with Sham, week 4 STZ animals had increased mushroom spine density (p < 0.05). Dendritic spine spatial distribution generally appeared similar to Sham animals (Fig. 8), with the exception of a change in spine spatial distribution within a single dendrite compartment at 100 μm in diabetic animals 1 week after STZ injection compared with Sham animals (p < 0.01). By 4 weeks after STZ injection, diabetic animals had a significantly greater density of dendritic spines located closer to the cell body compared with STZ animals at 1 week in all spine shape categories: for total spines (p < 0.001; Fig. 8D), for thin spines (p < 0.01; Fig. 8E), and for mushroom spines (p < 0.01; Fig. 8F). Together, these results demonstrate that diabetic animals with hyperglycemia do not exhibit robust changes in dendritic spine morphology before the development of neuropathic pain.
Figure 8.

Dendritic spine morphology 1 and 4 weeks after STZ induction of diabetes. Graphs of spine shape measurements show data for spine length (A) and spine head (B) diameter from neurons in nondiabetic Sham, 1 and 4 weeks after STZ induction of diabetes. There was no difference in spine length or spine head diameter 1 week after STZ injection compared with Sham (p = 1.0, for spine length: 1.11 ± 0.09 vs 1.22 ± 0.02 μm; p = 1.0, for head diameter: 0.69 ± 0.19 vs 0.88 ± 0.008 μm). In contrast, by week 4, STZ animals had increased spine length and head diameter compared with STZ animals in week 1 (one-way ANOVA with Bonferonni's post hoc test, *p < 0.001; for spine length: 1.11 ± 0.09 vs 1.55 ± 0.06 μm; for head diameter: 0.69 ± 0.19 vs 2.08 ± 0.67 μm) and Sham (one-way ANOVA with Bonferonni's post hoc test, *p < 0.001; for spine length: 1.22 ± 0.02 vs 1.55 ± 0.06 μm; for head diameter: 0.88 ± 0.008 vs 2.08 ± 0.67 μm). Analysis of hyperglycemic diabetic animals 1 week after STZ induction demonstrated no change in spine density (C) for total, thin-shaped, and mushroom-shaped spines compared with Sham (p = 0.32, for total spines: 2.26 ± 0.08 vs 2.83 ± 0.86 spines/10 μm; p = 0.58, for thin spines: 2.13 ± 0.78 vs 2.63 ± 0.82 spines/10 μm; p = 0.12, for mushroom spines: 0.15 ± 0.14 vs 0.21 ± 0.13 spines/10 μm). For total spine density, there was a significant increase in STZ animals at 4 weeks, compared with STZ animals at 1 week (one-way ANOVA with Bonferonni's post hoc test, *p < 0.001; for total spines: 3.52 ± 0.81 vs 2.26 ± 0.80 spines/10 μm; for mushroom spines: 0.63 ± 0.24 vs 0.15 ± 0.14 spines/10 μm). Mushroom spine density in STZ animals at 4 weeks was greater than STZ animals at 1 week and nondiabetic Sham (one-way ANOVA with Bonferonni's post hoc test, *p < 0.05; 0.63 ± 0.24 vs 0.22 ± 0.13 spines/10 μm). Total (D), thin (E), and mushroom (F) spines redistributed significantly in STZ diabetic animals at 1 week compared with STZ animals in week 4 (one-way ANOVA on ranks with Dunn's method, §p < 0.01; for total spines, at 50 μm: 3.00 ± 1.35 vs 1.7 ± 0.81 spines/10 μm; at 100 μm: 3.5 ± 1.08 vs 2.1 ± 0.69 spines/10 μm; at 150 μm: 3.41 ± 1.08 vs 2.5 ± 0.86 spines/10 μm; at 250 μm: 2.37 ± 0.93 vs 2.43 ± 1.18 spines/10 μm; at 300 μm: 2.71 ± 1.54 vs 2.06 ± 1.31 spines/10 μm; for thin spines, at 300 μm: 3.32 ± 1.4 vs 1.6 ± 1.33 spines/10 μm; for mushroom spines, at 50 μm: 0.86 ± 1.07 vs 0.12 ± 0.12 spines/10 μm; at 100 μm: 0.85 ± 0.82 vs 0.09 ± 0.08 spines/10 μm; p < 0.001, at 150 μm: 0.71 ± 0.72 vs 0.13 ± 0.08 spines/10 μm; at 200 μm: 0.76 ± 0.85 vs 0.12 ± 0.09 spines/10 μm; at 250 μm: 0.71 ± 0.78 vs 0.15 ± 0.16 spines/10 μm). In the total spine distribution analysis (D), STZ animals at 1 week had a single region at 100 μm with decreased spine density compared with Sham (one-way ANOVA on ranks with Dunn's method, #p < 0.01; at 100 μm: 3.05 ± 1.25 vs 2.1 ± 0.69 spines/10 μm). STZ animals in week 4 had significantly redistributed total dendritic spines and thin-shaped spines compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.01). All graphs are mean ± SEM.
Normal dendritic spine profiles in early diabetes coincide with the absence of neuropathic pain phenotype
To determine whether the normal spine morphology we observed 1 week after STZ injection predicted the absence of neuropathic pain, we performed electrophysiological and behavioral assessment of this subpopulation of animals with hyperglycemia (Fig. 9). Of the WDR neurons sampled from hyperglycemic animals 1 week after STZ injection, 3 of 20 (15%) neurons exhibited spontaneous activity. This was a qualitatively subtle increase compared with nondiabetic Sham animals, which had 2 of 17 (12%) sampled WDR neurons with spontaneous activity.
Figure 9.

Electrophysiological and behavioral pain assessment 1 and 4 weeks after STZ induction of diabetes. A, Dynamic mechanical allodynia of sampled WDR neurons in STZ animals at 1 week demonstrated no difference compared with Sham (p > 0.05). By week 4, STZ animals displayed significantly increased peripherally evoked activity of WDR single units compared with STZ animals at 1 week and Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.05; for PB: 6.57 ± 2.71 vs 33.1 ± 15.5 Hz; for 144 g/mm2: 13.1 ± 8.1 vs 33.5 ± 21.7 Hz; for 538 g/mm2: 13.6 ± 6.9 vs 42.7 ± 23.2 Hz). B, In all animals, stimulation with progressively more intense von Frey filaments produced a corresponding increase in spike-firing rates. In STZ animals at 1 week, at the lowest stimulus intensities (0.04 and 0.16 g), there was a significant increase in evoked response of WDR units compared with Sham, demonstrating a subtle presence of punctate mechanical allodynia (one-way ANOVA on ranks with Dunn's method, #p < 0.05; for 0.04 g: 2.04 ± 2.35 vs 5.9 ± 3.14 Hz; for 0.16: 3.18 ± 2.36 vs 8.51 ± 5.27 Hz). There was no significant difference between STZ animals at 1 week compared with those in week 4 until the strongest von Frey stimuli intensity (26.0 g) (one-way ANOVA on ranks with Dunn's method, §p < 0.05). For all stimulus intensities, STZ animals in week 4 had significantly greater spike-firing rates compared with Sham (one-way ANOVA on ranks with Dunn's method, *p < 0.05). C, Receptive field area of STZ animals at 1 week was not significantly different compared with Sham (p > 0.05). STZ animals in week 4 had receptive fields that were significantly larger compared with STZ at week 1 and Sham (one-way ANOVA with Bonferonni's post hoc, *p < 0.05). Behavioral assessment for pain for mechanical allodynia (D) demonstrated no difference between STZ animals at 1 week compared with Sham (p = 0.59; 17.2 ± 5.42 vs 19.6 ± 4.6 g for von Frey). In contrast, STZ animals in week 4 displayed significantly decreased mechanical pain threshold compared with STZ animals at 1 week (one-way ANOVA with Bonferonni's post hoc, *p < 0.05; 6.89 ± 5.63 vs 17.2 ± 5.42 g) and Sham (one-way ANOVA with Bonferonni's post hoc, *p < 0.05) (Fig. 7). E, There was no significant difference across Sham, STZ (week 1), and STZ (week 4) groups in behavioral tests for heat hyperalgesia (p > 0.05). Graphs are mean ± SEM.
STZ animals 1 week after STZ injection did not exhibit dynamic mechanical allodynia; these hyperglycemic animals had peripherally evoked unit activity following responses to PB, press (144 g/mm2), and pinch (538 g/mm2) stimuli that were not significantly different from the responses in Sham (Fig. 9A) (for all sensory modalities, p > 0.05). In addition, 1 week diabetic animals had lower single-unit activity rates compared with diabetic animals 4 weeks after STZ injection (p < 0.05, for PB, press, and pinch). In von Frey testing, week 1 STZ animals demonstrated evidence of punctate hyperalgesia when compared with Sham (Fig. 9B) (p < 0.05, for von Frey filament intensity of 0.04 and 0.16 g). There was no difference in peripherally evoked single WDR unit activity between week 1 STZ and Sham at von Frey filament intensities >0.16 g (p > 0.05). As reported earlier (see above), STZ animals at week 4 demonstrated significantly increased peripherally evoked unit activity for all sensory modalities compared with Sham. The mean receptive field size of WDR single units in 1 week diabetic animals was statistically equivalent to Sham animals (Fig. 9C) (p > 0.05).
Our previous work (Fischer et al., 2009) and data reported here demonstrate that diabetic animals with hyperglycemia progressively develop significant neuropathic pain-related behavior. As expected, 1 week following STZ-induced hyperglycemia, animals do not present with significant mechanical allodynia (Fig. 9D), demonstrated by equivalent stimulus withdrawal threshold compared with nondiabetic Sham animals (p = 0.59). In contrast, STZ animals in week 4 had significantly reduced mechanical withdrawal threshold compared with animals 1 week after STZ-induced diabetes (p < 0.05). STZ animals at week 4 demonstrated significant mechanical allodynia compared with Sham (see behavioral data presented earlier). We observed no significant development of heat hyperalgesia across any treatment group or experimental endpoint (Fig. 9E).
Discussion
Diabetes mellitus occurs in global epidemic numbers, affecting >50 million persons worldwide. Chronic neuropathic pain represents a major complication of diabetes. Although much research has been devoted to understanding the underlying mechanisms of diabetic neuropathic pain, current clinical treatments have had limited effectiveness in reducing this type of pain. Our data demonstrate for the first time a mechanistic role for postsynaptic dendritic spine remodeling within the spinal cord in diabetic neuropathic pain. Dendritic spines on second-order nociceptive neurons showed changes in morphological architecture, increased in density, and redistributed along dendritic branches. These diabetes-induced structural alterations would be expected to have adverse functional implications for sensory neurons and may underlie the pathophysiological properties of these cells associated with neuropathic pain (Tan et al., 2009a). In agreement with this, dendritic spine remodeling in diabetic animals was accompanied by WDR neuron hyperexcitability in response to low- and high-threshold stimuli; and these animals also demonstrated decreased mechanical pain thresholds. Importantly, diabetic animals with hyperglycemia in the absence of neuropathic pain exhibited near-normal spine profiles, comparable with nondiabetic animals (Table 3). Together, these observations support a structure–function link between maladaptive dendritic spine remodeling and neuropathic pain in an animal model of diabetic neuropathic pain.
Table 3.
Summary findings of treatment arm comparisons with Sham
| Treatment arm | Malformed spinesa |
Paina |
|||
|---|---|---|---|---|---|
| Shape | Density | Distribution | Electrical | Behavioral | |
| STZ (week 1) | — | — | *† | * | — |
| STZ (week 4) | *** | *** | *** | *** | * |
| STZ + NSC | * | * | — | * | — |
| Sham + NSC | *† | — | — | — | — |
| Sham | Control | ||||
All STZ animals exhibited significant hyperglycemia at the time of testing. For dendritic spine morphology, outcome measures include shape, density, and distribution. Pain outcome measures include assessments for WDR neuron excitability and behavioral symptoms of neuropathic pain.
aDefined by Sham control values.
—, No significant change in any tests within the outcome measure.
*Significant change in less than or one-half of tests within an outcome measure.
***Significant change in more than one-half of tests within an outcome measure.
†Significant change below Sham control values.
Rac1, a small kinase (∼21 kDa) in the family of Rho GTPases involved in multiple cellular functions (Ridley, 2006), can regulate dendritic spine morphology and function (Tashiro and Yuste, 2004, 2008). Interestingly, emerging evidence has suggested that ROCK inhibitors (inhibitors of RhoA/Rho kinases) are promising candidates for the treatment of painful diabetic neuropathies, as demonstrated by experiments in STZ-induced diabetic mice (Ohsawa and Kamei, 2010; Ohsawa et al., 2011). While the exact mechanism of action for Rac1-inhibition in attenuating neuropathic pain is not well understood, the activity of Rac1 is known to regulate dendritic spine morphology through its action on filamentous actin, a building block of dendritic spines, and promotes the clustering of excitatory AMPA receptors in spines (Nakayama and Luo, 2000). We have previously shown that the Rac1 inhibitor, NSC23766, can disrupt dendritic spine morphology in vitro (Tan et al., 2011). Here, we treated nondiabetic Sham animals with NSC23766, which resulted in decreased spine length compared with untreated Sham animals. Treatment did not affect spine head diameter, spine density, or distribution, and did not significantly change electrical or behavioral outcomes compared with untreated Sham, suggesting that, although NSC23766 treatment may subtly affect spine structure, its action does not significantly affect normal function. To determine the contributory role of structural plasticity of dendritic spines in diabetic neuropathic pain, we administered NSC23766 in animals with diabetic neuropathic pain. We show for the first time that treatment with a Rac1 inhibitor results in a change in spine structure toward normal that is paralleled by a significant reduction in mechanical neuropathic allodynia, and show that it mitigates hyperexcitability of WDR neurons in diabetic animals. In addition, we show that treatment with Rac1 inhibitor NSC23766 is fast-acting and effective within 3 d of administration.
Our results support the interpretation that morphologic changes in dendritic spines are mechanistically linked to injury- or disease-induced neuropathic pain (Tan et al., 2008, 2011; Tan and Waxman, 2011). Thus, we hypothesized that diabetic animals with hyperglycemia would not exhibit dendritic spine abnormalities in the absence of pain. Hyperglycemia induced by STZ injection occurs rapidly, within 4 d, but significant neuropathic pain only occurs by the second week after induction, as demonstrated previously (Malcangio and Tomlinson, 1998; Fischer et al., 2009). Our findings show that before the development of neuropathic pain in diabetic animals, at 1 week after STZ induction, dendritic spines appear similar to nondiabetic control animals. These results have two implications: first, within the context of the hyperglycemic condition, the presence of maladaptive dendritic spines predicts the manifestation of neuropathic pain phenotype. Second, there may be a therapeutic window in the early stages of diabetes for preventing the establishment of intractable neuropathic pain, by targeting disease-induced dendritic spine remodeling.
We observed significant mechanical allodynia within 2 weeks after STZ injection, in agreement with others (Courteix et al., 1993; Chen and Pan, 2002). A trend for mechanical allodynia was observed at 1 week, but was not statistically significant, raising the possibility that, in some animals, there may be early development of diabetic-induced pain. Without treatment, tactile pain thresholds in diabetic animals continued to decline, demonstrating significant allodynia at 3 weeks and remaining severely below normal until the end of the experiment at 4 weeks. In these animals, however, we observed no significant change in withdrawal latency to noxious heat stimuli. Treatment with NSC23766 also produced no change in behavioral responses to heat stimulation. Within the literature, some studies report that STZ-induced diabetic animals develop heat hyperalgesia (Courteix et al., 1993), whereas other studies report no change or decreased thermal pain sensitivity (Raz et al., 1988; Malcangio and Tomlinson, 1998; Pertovaara et al., 2001). This disparity in heat hyperalgesia in diabetic models of neuropathic pain has several possible explanations: First, it has been suggested that the use of radiant heat stimuli to detect thermal hyperalgesia could have limitations as it only measures the noxious heat detection threshold (Chen and Pan, 2002); it is not known whether thermal sensitivity at suprathreshold heat intensities or the response to prolonged noxious heat stimulation is altered in the diabetic rat model of neuropathic pain. Second, in studies showing absence of abnormal heat sensitivity, it is possible that unmyelinated C-fiber afferents, which carry thermal nociceptive information, remain unaffected; whereas in studies that demonstrate thermal hyperalgesia, C-fibers may become more excitable in this rat model of diabetic pain (Ahlgren and Levine, 1994). This latter interpretation is in agreement with evidence supporting multiple pathological mechanisms originating in the periphery (i.e., peripheral neuropathy, vascular dysfunction, hypoxic tissue damage, and altered sodium channel expression, each of which can contribute to diabetic neuropathic pain) (Benbow et al., 1994; Craner et al., 2002b; Boucek, 2006; Jain, 2008; Veves et al., 2008).
Alternatively, emerging evidence supports a central contribution to diabetic neuropathic pain (Fischer et al., 2009; Fischer and Waxman, 2010). Here, we show that dendritic spine morphologies in dorsal horn neurons change in STZ-induced diabetic neuropathic pain. In particular, our results show a significant nearly threefold increase of mushroom-shaped dendritic spines. As we have demonstrated previously, the geometry of mushroom-shaped dendritic spines can promote the transmission of, and amplify high-frequency tactile input (Tan et al., 2009a). Because thermal inputs travel at lower frequency with slower conduction velocities through unmyelinated or thinly myelinated afferents C- or Aδ-afferents (Ossipov et al., 1999), an increase in mushroom spines could have positive bias toward influencing tactile transmission, with a lesser effect on thermal transmission.
Our findings are supported by literature showing that, in many pathological states, spines change shape, size, and number (Calabrese et al., 2006). In the cortex, malformed or altered spines have been observed in a spectrum of diseases, including epilepsy, stroke, schizophrenia, mental retardation, dementia, and chronic substance abuse (Halpain et al., 2005). Although few functional studies have been performed to link these pathological spine structures with dysfunction, evidence is growing for a contribution of dendritic spines in pathological function. In a model of fragile X mental retardation, mice with Fmr1 protein knock-out show impaired long-term potentiation (Wilson and Cox, 2007) and malformed dendritic spines due to dysfunctional Rac1 signaling (Chen et al., 2010). Interestingly, these mice display decreased neuropathic allodynia (Price et al., 2007). Spinal cord injury also results in dendritic spine alterations on motor cortex (Kim et al., 2006), and induces Rac1-regulated spine remodeling on nociceptive dorsal horn neurons that contributes to neuropathic pain (Tan et al., 2008, 2009a).
Relevant to the present study, previous work has shown decreases in dendritic spine density in the prefrontal cortex of diabetic rats 2 months after STZ induction (Joghataie et al., 2007). Another study showed decreases in dendritic spine density in the parietal neocortex in the STZ model of diabetes associated with impaired performance in a water maze memory test at 2 months after induction of diabetes (Malone et al., 2008). These previous studies demonstrate that STZ-induced diabetes is associated with changes in dendritic spines in the brain and impaired long-term memory. Importantly, none of these studies definitively address the question of whether changes in spines are a direct consequence of hyperglycemia or, alternatively, are triggered by changes in presynaptic elements. Similarly, our experiments do not permit us to determine whether spine changes in the dorsal horn in STZ-induced diabetes are triggered early by changes in primary afferents, or as a direct consequence of hyperglycemia or other metabolic alterations.
Dendritic spine architecture could directly affect WDR neuron hyperexcitability through signal amplification, increased signal fidelity, and/or reduced noise-filtering capabilities (Rall, 1962; Tan et al., 2009a). It is intriguing that diabetes can induce structural changes in neurons within the spinal cord, which suggests that metabolic disease may engage synaptic remodeling mechanism that can chronically and adversely alters the central sensory processing system (Ji and Woolf, 2001). Our results show that inhibition of Rac1 activity in animals with diabetic neuropathic pain can attenuate injury-induced changes in dendritic spines and partially restore normal pain function; in agreement, Rac1 inhibition had similar effects after SCI and peripheral nerve injury (Tan et al., 2008, 2011). Together with our earlier observation that dendritic spine remodeling occurs in both SCI and peripheral nerve injury models of neuropathic pain, the present results suggest that molecular pathways that control spine morphogenesis may be promising future targets for treatments in neuropathic pain of multiple etiologies, including diabetic neuropathic pain.
Footnotes
This work was supported in part by grants from the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America and the United Spinal Association with Yale University.
The authors declare no competing financial interests.
References
- Ahlgren SC, Levine JD. Protein kinase C inhibitors decrease hyperalgesia and C-fiber hyperexcitability in the streptozotocin-diabetic rat. J Neurophysiol. 1994;72:684–692. doi: 10.1152/jn.1994.72.2.684. [DOI] [PubMed] [Google Scholar]
- Baumgärtner U, Magerl W, Klein T, Hopf HC, Treede RD. Neurogenic hyperalgesia versus painful hypoalgesia: two distinct mechanisms of neuropathic pain. Pain. 2002;96:141–151. doi: 10.1016/s0304-3959(01)00438-9. [DOI] [PubMed] [Google Scholar]
- Bays HE, Pfeifer MA. Peripheral diabetic neuropathy. Med Clin North Am. 1988;72:1439–1464. doi: 10.1016/s0025-7125(16)30716-7. [DOI] [PubMed] [Google Scholar]
- Beisswenger PJ. Neuropathic and vascular complications occurring in diabetes. Postgrad Med. 1976;59:169–174. doi: 10.1080/00325481.1976.11716534. [DOI] [PubMed] [Google Scholar]
- Benbow SJ, Chan AW, Bowsher D, MacFarlane IA, Williams G. A prospective study of painful symptoms, small-fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet Med. 1994;11:17–21. doi: 10.1111/j.1464-5491.1994.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Boucek P. Advanced diabetic neuropathy: a point of no return? Rev Diabet Stud. 2006;3:143–150. doi: 10.1900/RDS.2006.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (Bethesda) 2006;21:38–47. doi: 10.1152/physiol.00042.2005. [DOI] [PubMed] [Google Scholar]
- Chang YW, Tan A, Saab C, Waxman S. Unilateral focal burn injury is followed by long-lasting bilateral allodynia and neuronal hyperexcitability in spinal cord dorsal horn. J Pain. 2010;11:119–130. doi: 10.1016/j.jpain.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci. 2010;30:10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Pan HL. Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J Neurophysiol. 2002;87:2726–2733. doi: 10.1152/jn.2002.87.6.2726. [DOI] [PubMed] [Google Scholar]
- Choi J, Ko J, Racz B, Burette A, Lee JR, Kim S, Na M, Lee HW, Kim K, Weinberg RJ, Kim E. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci. 2005;25:869–879. doi: 10.1523/JNEUROSCI.3212-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courteix C, Eschalier A, Lavarenne J. Streptozocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain. 1993;53:81–88. doi: 10.1016/0304-3959(93)90059-X. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Klein JP, Black JA, Waxman SG. Preferential expression of IGF-I in small DRG neurons and down-regulation following injury. Neuroreport. 2002a;13:1649–1652. doi: 10.1097/00001756-200209160-00016. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol. 2002b;52:786–792. doi: 10.1002/ana.10364. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fischer TZ, Waxman SG. Neuropathic pain in diabetes—evidence for a central mechanism. Nat Rev Neurol. 2010;6:462–466. doi: 10.1038/nrneurol.2010.90. [DOI] [PubMed] [Google Scholar]
- Fischer TZ, Tan AM, Waxman SG. Thalamic neuron hyperexcitability and enlarged receptive fields in the STZ model of diabetic pain. Brain Res. 2009;1268:154–161. doi: 10.1016/j.brainres.2009.02.063. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerstad J, Tjølsen A, Hole K. Induction of long-term potentiation of single wide dynamic range neurones in the dorsal horn is inhibited by descending pathways. Pain. 2001;91:263–268. doi: 10.1016/S0304-3959(00)00448-6. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S, Spencer K, Graber S. Dynamics and pathology of dendritic spines. Prog Brain Res. 2005;147:29–37. doi: 10.1016/S0079-6123(04)47003-4. [DOI] [PubMed] [Google Scholar]
- Jain R. Pain and the brain: diabetic neuropathic pain. J Clin Psychiatry. 2008;69:e22. doi: 10.4088/jcp.0808e22. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Joghataie MT, Roghani M, Jalali MR, Baluchnejadmojarad T, Sharayeli M. Dendritic spine changes in medial prefrontal cortex of male diabetic rats using Golgi-impregnation method. Arch Iran Med. 2007;10:54–58. [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol. 2006;198:401–415. doi: 10.1016/j.expneurol.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Tomlinson DR. A pharmacologic analysis of mechanical hyperalgesia in streptozotocin/diabetic rats. Pain. 1998;76:151–157. doi: 10.1016/s0304-3959(98)00037-2. [DOI] [PubMed] [Google Scholar]
- Malone JI, Hanna S, Saporta S, Mervis RF, Park CR, Chong L, Diamond DM. Hyperglycemia not hypoglycemia alters neuronal dendrites and impairs spatial memory. Pediatr Diabetes. 2008;9:531–539. doi: 10.1111/j.1399-5448.2008.00431.x. [DOI] [PubMed] [Google Scholar]
- Marchettini P, Teloni L, Formaglio F, Lacerenza M. Pain in diabetic neuropathy case study: whole patient management. Eur J Neurol. 2004;11(Suppl 1):12–21. doi: 10.1111/j.1471-0552.2004.00795.x. [DOI] [PubMed] [Google Scholar]
- Morgado C, Pereira-Terra P, Cruz CD, Tavares I. Minocycline completely reverses mechanical hyperalgesia in diabetic rats through microglia-induced changes in the expression of the potassium chloride co-transporter 2 (KCC2) at the spinal cord. Diabetes Obes Metab. 2011;13:150–159. doi: 10.1111/j.1463-1326.2010.01333.x. [DOI] [PubMed] [Google Scholar]
- Morrow TJ. Animal models of painful diabetic neuropathy: the STZ rat model. Curr Protoc Neurosci Chapter. 2004;9 doi: 10.1002/0471142301.ns0918s29. Unit 9.18. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Luo L. Intracellular signaling pathways that regulate dendritic spine morphogenesis. Hippocampus. 2000;10:582–586. doi: 10.1002/1098-1063(2000)10:5<582::AID-HIPO8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Ohsawa M, Kamei J. RhoA/Rho kinase signaling in the spinal cord and diabetic painful neuropathy. Eur J Pharmacol. 2010;644:1–4. doi: 10.1016/j.ejphar.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Ohsawa M, Aasato M, Hayashi SS, Kamei J. RhoA/Rho kinase pathway contributes to the pathogenesis of thermal hyperalgesia in diabetic mice. Pain. 2011;152:114–122. doi: 10.1016/j.pain.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- Pabreja K, Dua K, Sharma S, Padi SS, Kulkarni SK. Minocycline attenuates the development of diabetic neuropathic pain: possible anti-inflammatory and anti-oxidant mechanisms. Eur J Pharmacol. 2011;661:15–21. doi: 10.1016/j.ejphar.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Wei H, Kalmari J, Ruotsalainen M. Pain behavior and response properties of spinal dorsal horn neurons following experimental diabetic neuropathy in the rat: modulation by nitecapone, a COMT inhibitor with antioxidant properties. Exp Neurol. 2001;167:425–434. doi: 10.1006/exnr.2000.7574. [DOI] [PubMed] [Google Scholar]
- Pitcher GM, Henry JL. Nociceptive response to innocuous mechanical stimulation is mediated via myelinated afferents and NK-1 receptor activation in a rat model of neuropathic pain. Exp Neurol. 2004;186:173–197. doi: 10.1016/j.expneurol.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Pongrácz F. The function of dendritic spines: a theoretical study. Neuroscience. 1985;15:933–946. doi: 10.1016/0306-4522(85)90244-1. [DOI] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W. Theory of physiological properties of dendrites. Ann N Y Acad Sci. 1962;96:1071–1092. doi: 10.1111/j.1749-6632.1962.tb54120.x. [DOI] [PubMed] [Google Scholar]
- Raz I, Hasdai D, Seltzer Z, Melmed RN. Effect of hyperglycemia on pain perception and on efficacy of morphine analgesia in rats. Diabetes. 1988;37:1253–1259. doi: 10.2337/diab.37.9.1253. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Setacci C, de Donato G, Setacci F, Chisci E. Diabetic patients: epidemiology and global impact. J Cardiovasc Surg (Torino) 2009;50:263–273. [PubMed] [Google Scholar]
- Singleton JR, Smith AG. Neuropathy associated with prediabetes: what is new in 2007? Curr Diab Rep. 2007;7:420–424. doi: 10.1007/s11892-007-0070-y. [DOI] [PubMed] [Google Scholar]
- Tan AM, Waxman SG. Spinal cord injury, dendritic spine remodeling, and spinal memory mechanisms. Exp Neurol. 2011 doi: 10.1016/j.expneurol.2011.08.026. [DOI] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28:13173–13183. doi: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Choi JS, Waxman SG, Hains BC. Dendritic spine remodeling after spinal cord injury alters neuronal signal processing. J Neurophysiol. 2009a;102:2396–2409. doi: 10.1152/jn.00095.2009. [DOI] [PubMed] [Google Scholar]
- Tan AM, Zhao P, Waxman SG, Hains BC. Early microglial inhibition preemptively mitigates chronic pain development after experimental spinal cord injury. J Rehabil Res Dev. 2009b;46:123–133. [PubMed] [Google Scholar]
- Tan AM, Chang YW, Zhao P, Hains BC, Waxman SG. Rac1-regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp Neurol. 2011;232:222–233. doi: 10.1016/j.expneurol.2011.08.028. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Role of Rho GTPases in the morphogenesis and motility of dendritic spines. Methods Enzymol. 2008;439:285–302. doi: 10.1016/S0076-6879(07)00421-1. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Ueno H, Kataoka A, Tozaki-Saitoh H, Inoue K. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia. 2008;56:378–386. doi: 10.1002/glia.20623. [DOI] [PubMed] [Google Scholar]
- Veves A, Backonja M, Malik RA. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med. 2008;9:660–674. doi: 10.1111/j.1526-4637.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A. 2007;104:2454–2459. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, King AE. Physiology and morphology of multireceptive neurons with C-afferent fiber inputs in the deep dorsal horn of the rat lumbar spinal cord. J Neurophysiol. 1987;58:460–479. doi: 10.1152/jn.1987.58.3.460. [DOI] [PubMed] [Google Scholar]
- Yamano T, Shimada M, Fujizeki Y, Kawasaki H, Onaga A. Quantitative synaptic changes on Purkinje cell dendritic spines of rats born from streptozotocin-induced diabetic mothers. Brain Dev. 1986;8:269–273. doi: 10.1016/s0387-7604(86)80080-8. [DOI] [PubMed] [Google Scholar]
- Yuste R, Urban R. Dendritic spines and linear networks. J Physiol Paris. 2004;98:479–486. doi: 10.1016/j.jphysparis.2005.09.014. [DOI] [PubMed] [Google Scholar]