

Abstract
Reexposure to trauma reminders is an integral element of trauma-focused cognitive behavioral therapy (Roberts et al., 2009), but little is known about the physiological processes underlying the therapeutic progress. While it is well established that amygdala, prefrontal cortex and hippocampus are key brain structures in fear memory processing (McGaugh, 2004; Herry et al., 2008; Likhtik et al., 2008), it is not well known which neurotransmitters or neuromodulators are involved. Here with a translational approach we investigated the role of dynorphins in the formation and extinction of fear memories in mice and in humans. Mice lacking dynorphin showed an enhanced cue-dependent fear conditioning, as well as delayed extinction in contextual conditioning/extinction paradigms. The pharmacological blockade of κ-opioid receptors before the extinction trials but not before or after the conditioning produced a similar effect. Analysis of neuronal activity, using the immediate early gene c-fos, demonstrated a reduced neuronal activity in key limbic structures during extinction in the absence of dynorphin. Translating these findings into the human domain, fear conditioning and extinction, coupled with functional MRI was then performed in volunteers preselected for a functionally relevant polymorphism in the dynorphin gene. Human volunteers bearing the (T) allele of PDYN (prodynorphin) at rs1997794 showed reduced fear extinction and a significantly diminished functional connectivity between amygdala and ventromedial prefrontal cortex. Our findings establish a role of dynorphin κ-opioid receptor signaling in fear extinction.
Introduction
The formation of fear memories after stress exposure is an adaptive phenomenon that helps to avoid similar situations and to improve future coping strategies. Fear learning is a highly effective and fast process: a single exposure to strong stress often leads to the formation of long-lasting fear memories. However, adaptation to a changing environment also requires the extinction of the fear memories. Extinction learning is slower than fear learning and usually requires multiple nonreinforced expositions to the fear associated cues or context (Myers and Davis, 2002, 2007). Fear learning is essentially a classical Pavlovian conditioning, whereas extinction learning is a more complex process whereby recall of the fear memory and detection of the prediction error are necessary for the acquisition and consolidation of a new safety memory (Myers and Davis, 2007).
The amygdalar nuclei play a pivotal role in the formation of fear memories through a Pavlovian conditioning process (Maren and Quirk, 2004). The infralimbic region of the prefrontal cortex (Milad and Quirk, 2002; Peters et al., 2010) and the hippocampus (Radulovic and Tronson, 2010), areas exerting inhibitory control over the amygdala (Herry et al., 2010), activate the inhibitory circuits of the amygdala during extinction learning (Herry et al., 2008). Thus, the activity of these regions and the communication between them are crucial for successful fear extinction, thus for the efficacy of the exposure therapy.
The endogenous opioid peptide dynorphin and its receptor, the κ-opioid receptor (KOR) (Schwarzer, 2009) are abundantly expressed in areas involved in fear learning (Akil et al., 1984; Ma et al., 2003). In stressful, aversive situations dynorphins are released (Palkovits, 2000; Shirayama et al., 2004) and they mediate acute stress responses influencing the intensity and duration of behavioral and hormonal stress reactivity (Bilkei-Gorzo et al., 2008). Several lines of evidences suggest that dynorphin signaling also contributes to the formation and extinction of aversive memories. It was shown that activation of KORs by dynorphins mediates the aversive component of the stress situation (Land et al., 2009) and KOR signaling encodes the aversive emotional component of the stress-related event by recalling stress memories (Bruchas et al., 2007). Moreover, dynorphins contribute to attention focusing (Iordanova et al., 2006), which is a characteristic feature of the acquisition of stress-related memories (Phelps, 2006). Paradoxically, dynorphin exerts its effect on attention through blocking the formation of memories unrelated to the stress situation. In good accordance with the proposed proamnesic role of dynorphins it was shown that dynorphins contribute to stress-induced learning and memory deficits (Carey et al., 2009).
Materials and Methods
Animals and drugs.
All experiments were conducted with 3- to 5-month-old male wild-type and null-mutant mice lacking either dynorphin (Pdyn−/−) (Zimmer et al., 2001), proenkephalin (Penk1−/−) (König et al., 1996), or β-endorphin (B-end.−/−) (Rubinstein et al., 1996). Animals were kept under a reversed light/dark cycle (lights on: 7:00 P.M.; lights off: 9:00 A.M.) in groups of 3–5. For the pharmacological treatments the opioid receptor blocker nor-binaltorphimine (nor-BNI) was purchased from Sigma-Aldrich. The freshly prepared solution was injected intraperitoneally in a volume of 10 ml/kg.
Context-dependent fear conditioning and extinction.
On day 1 of the preconditioning phase, the animals were placed into an open-field arena with a grid floor (Experimetria Ltd) and time spent with freezing was registered for 5 min (Bilkei-Gorzo et al., 2007). In the immediately following fear-conditioning phase, they received 10 inescapable footshocks (2 s, 0.5 mA) with 30 s intervals. The animals were then returned to their home cage. In the extinction phase, 24, 48, 72, and 96 h after fear conditioning, the animals were placed back to the same arena and freezing time was registered for 5 min. For statistical analysis one-way repeated-measures ANOVA was used followed by Dunnett's test, separately for the groups.
To assess the effect of κ receptor antagonism on the formation and extinction of fear memory three groups of animals were treated with nor-BNI (10 mg/kg, i.p.) or vehicle (saline). Mice from group A were injected 30 min before, from group B just after the fear conditioning phase and the group C was treated just after the extinction trial.
Cue-dependent fear conditioning and extinction.
For fear conditioning, animals were placed into a Plexiglas and wire mesh cage, with metal bars on the floor on a vibration-sensitive platform of a startle response apparatus (TSE Systems GmbH). Each cage was located in a ventilated, sound- and light-isolated chamber. The animals were allowed to habituate for 5 min and time spent with freezing was measured in this preconditioning period. Freezing was assessed as periods spending motionless i.e., producing no signal on the vibration sensor at least 3 s (Marsicano et al., 2002). After habituation the animals received a signal tone (9 kHz, 80 dB, 20 s), which coterminated with an electric footshock (1 s, 0.5 mA). Warning signal with the footshock was presented 10 times with 30 s intervals. In the extinction phase, 24 h and 6 d after the fear conditioning the animals were placed into the startle apparatus, but in a glass cylinder (6 cm diameter). After a 5 min habituation period the warning signal was presented 3 min long without applying footshock. Mean and SE of time spent with freezing during the preconditioning phase and in the two extinction phases was calculated and groups were compared using two-way ANOVA (genotype and phase as main factor). Consecutively, freezing time in 20 s intervals was calculated and the data were analyzed using repeated two-way ANOVA (between factor: genotype, within factor: time) followed by Bonferroni test, separately for the two extinction trials.
c-fos IR in the brain.
A separate group of animals (5 per group) was used to test the effect of fear conditioning and extinction on immediate early gene induction, a surrogate marker for neuronal activation (Bilkei-Gorzo et al., 2008; Kovács, 2008). The brains were removed after transcardial fixation with 4% paraformaldehyde (PFA) solution 2 h after the fear conditioning at day 1, or after the first extinction trial at day 2. The brains were postfixed at 4°C in PFA solution for 90 min and equilibrated in 10% sucrose solution for 24 h. Subsequently, they were shock frozen and stored at −80°C until further processing. Brains were sliced in a cryostat at 14 μm thickness. The sections were labeled using rabbit anti-c-fos primary antibody and a biotinylated donkey anti-rabbit-IgG secondary antibody. Staining was performed with the ABC-Kit (Vector Labs). The number of labeled cells in the infralimbic and prelimbic regions of the prefrontal cortex, basolateral amygdala and the CA3 region of the hippocampus was counted. Density of c-fos-positive nuclei was calculated (count/mm2) using 4–6 slices per animal. The size of the counting frames was identical between the groups but differed between the brain areas. To obtain a baseline c-fos expression under stress-free conditions, control Pdyn-null mutant and wild-type animals were killed just after removing them from their home cage and their brains were prepared, sliced and stained as described above. Groups were compared using two-way ANOVA (main factors: genotype, footshock) genotypes were compared by Bonferroni test.
Human subjects.
Thirty-three healthy German volunteers with parents and grandparents of European origin were recruited in Bonn. All subjects gave written informed consent to the study. No participant had lifetime or family history of affective disorder. To rule out effects of altered mood states in this study, subjects had to complete the Beck Depression Inventory and the Allgemeine Depressionsskala (German version of CES-D-Scale). Eight subjects were rs1997794 CC homozygotes, 13 CT heterozygotes and 12 TT homozygotes. The allele frequencies were in Hardy-Weinberg equilibrium (χ2 = 0.008, p = 0.93). Groups did not differ with respect to age (mean age = 26.7 (SD 6.8); F(2,30) = 0.63; p = 0.54) and gender (χ2 = 2.49; p = 0.29). The study was approved by the local ethics committee of the University of Bonn.
DNA-extraction and genotyping.
EDTA anti-coagulated venous blood samples were collected from all individuals. Lymphocyte DNA was isolated by a Chemagic Magnetic Separation Module I (Chemagen) according to the manufacturer's recommendations. Rs1997794 was genotyped using the ABI Assays-On-Demand SNP Genotyping products (Applied Biosystems) according to the manufacturer's instructions.
Functional imaging task.
The paradigm used for acquisition and extinction of fear responses was based on a partial reinforcement paradigm previously published (Phelps et al., 2004). Conditioned stimuli (CSs) were colored squares (blue and green) and the unconditioned stimulus (UCS) was a mildly painful laser shock to the foot. All CSs were presented for 4 s with an intertrial interval of 12 s. One of the colored squares was designated as the CS+, which was paired with the UCS, and the other was designated as the CS−, which was never paired with a UCS. Subjects were informed about these contingencies before the start of the study. The study was conducted on two consecutive days and consisted of three phases. During acquisition (phase 1) on day 1 subjects were exposed to 20 presentations each of the CS+ and CS− in randomized order. Ten additional CS+ presentations coterminated with a UCS. After the acquisition phase subjects rated the intensity of the pain stimulus on a 10-point Likert scale (0 = not painful at all, 10 = unbearable pain). This was followed by the early extinction (phase 2), which consisted of 20 unreinforced presentations of the CS+ and 20 presentation of the CS−. On day 2 ∼24 h after the first session, subjects participated in the late extinction (phase 3), which consisted of 22 unreinforced CS+ and 22 CS− stimuli. Subjects were told before each of the phases, that they will see blue and green colored squares and that it may be possible that during presentation of the green square a painful stimulus will be applied on their left foot. All subjects were able to complete all sessions. Additionally, before and after each scanning day, subjects had to complete the state version of the State-Trait-Anxiety-Inventory (STAI) to test for effects of genotype on anxiety levels before and after scanning. Finally, subjects completed the BDI, ADS and the trait version of the STAI.
FMRI acquisition.
BOLD fMRI was performed on a Phillips Achieva 3T scanner at the Department of Radiology, University of Bonn. Scanning parameters were the same for all tasks. Functional images were taken with a gradient echo planar imaging sequence using parallel imaging (sensitivity encoding, SENSE factor 2). Whole brain coverage was obtained with 38 axially tilted slices (slice thickness 2.4 mm + 0.6 mm gap, FOV 192 mm, repetition time 1960 ms, echo time 30 ms, 64 × 64 matrix, reconstructed voxel size 3 × 3 × 3 mm3, flip angle 80°).
Laser stimulation was performed by a Thulium (Tm)-YAG-Laser (Themis, StarMedTec) and stimulation was applied at the dorsal surface of the left foot with 650 mJ. To avoid damage of the epidermis by repetitive application of laser stimuli to the same skin area, the laser stimulation focus was moved around the target area by keeping the angle of the laser beam constant (rectangular to the skin surface).
Skin conductance responses (SCRs) were acquired during fMRI acquisition using Ag/AgCl electrodes attached to the palmar surface of the left index and middle finger, and the signal was amplified and digitized via a skin conductance processing unit (Nexus-16, Mind Media). The filtered analog output of the SCR was displayed online and recorded digitally (sampling rate 100 Hz) using Biotrace software (Mind Media).
Functional image processing.
Image processing and statistical analyses were conducted using statistical parametric mapping methods as implemented in SPM5 (http://www.fil.ion.ucl.ac.uk/spm/software/spm5/) and were similar for all tasks. Briefly, images were realigned to a mean image, slice time corrected, spatially normalized to a standard stereotactic space (a brain template created by the Montreal Neurological Institute), resliced to a spatial resolution of 2 × 2 × 2 mm3, smoothed with an 8 mm FWHM (full-width at half-maximum) Gaussian filter and intensity normalized to the whole-brain global mean. The regression model at the first level consisted of three (CS+, CS-UCS, acquisition) or two (CS+, CS−, extinction) regressors convolved with the hemodynamic response function and six regressors describing residual motion. For each subject, statistical contrast images of CS+ versus CS− were obtained. For the acquisition period we obtained an additional contrast image for UCS versus CS−. To test for genetic association, these contrast images were analyzed using the general linear model in a second-level random effects analysis (two-sample t test (Group 1: CC + CT; Group 2: TT). Because of the small number of CC individuals, we pooled individuals with either one or two copies of the C allele (CT, CC) for subsequent analyses to identify genotype effects on activation or connectivity. To test for effects of decreasing activation (time by event interaction) during early and late extinction, mirroring SCRs, we performed an additional first-level analysis with parametric modulation, including a linear decay over time for the CS+ during early extinction and an exponentially decaying function over time for the CS+ during late extinction. Second level analyses were similar to those described above.
Functional connectivity analyses.
Analyses to measure functional connectivity used a seed region approach (Esslinger et al., 2009; Erk et al., 2010). For each subject, seed time series from the left and right amygdala were extracted using first eigenvariates from all voxels within an amygdala mask. To derive a robust summary measure of activity in the amygdala region of interest (ROI), we excluded white matter by restricting the averaging to voxels related to task at a p < 0.5 level; this was not used for statistical inference. Using SPM5, seed time series were high-pass filtered (128 s) and task related variance was removed to avoid measuring coactivation that is solely due to temporal correlation with the experimental paradigm. To account for unspecific noise, first eigenvariates from masks covering CSF and white matter were extracted for each individual and entered, together with movement covariates and task regressors, into whole brain multiple regression analyses where the respective seed region time series, i.e., the time series from the right or left amygdala was the covariate of interest. Thus, we identified voxels whose activity show significant covariation with the left or with the right amygdala. Here, two brain areas are called functionally connected if their BOLD signal time series covary over time. The resulting maps of partial correlation with the left or right amygdala seed region were then each subjected to a random effects analysis in SPM5 using a two-sample t test as described above.
Statistical inference.
For all imaging methods, the significance threshold was set to p < 0.05, corrected for multiple comparisons across the whole brain, or for anatomical ROI. We used familywise error (FWE), a widely used method, which strongly controls for type I error in imaging genetics (Meyer-Lindenberg et al., 2008). The amygdala ROI for seed voxel extraction and ROI analysis was defined a priori and created using anatomical labels provided by the Wake Forest University PickAtlas (www.fmri.wfubmc.edu/downloads).
Analysis of skin conductance responses.
Data were resampled to 10 Hz, smoothed using a 0.7 s FWHM Gaussian kernel and log (10) transformed. We analyzed amplitudes of responses that occurred in a time window of 0–8 s after CS onset (duration of CS = 4 s). For analysis of CS+ responses we only analyzed responses on unpaired trials, to exclude possible influences of the UCS. Amplitudes were determined as the difference between the maximum response within the analysis interval and a baseline response within a 2 s interval preceding the respective stimulus. We tested for a condition × trial and a group × condition × trial interaction with ANOVA for repeated measures. Additionally we tested for an interaction of group × CS+ trials only.
Results
Fear conditioning and extinction in mice
To investigate the role of dynorphin in the formation and extinction of fear memories, we first studied preprodynorphin deficient animals (Pdyn−/−) and wild-type littermates in a contextual fear conditioning and extinction paradigm. Duration of freezing during the conditioning and extinction trials significantly changed in mice from both genotypes (F(4,49) = 8.552; p < 0.001 for Pdyn+/+ and F(4,49) = 3.997; p < 0.01 for Pdyn−/−). Post hoc analysis of the data with Dunnett's test revealed that animals from both lines displayed a significantly increased freezing behavior in the footshock-associated environment (compare Fig. 1A, PC vs E1), indicative of a conditioned fear response. In consecutive trials, after 24 h intervals, the time spent with freezing gradually returned to the pre-footshock value in Pdyn+/+ animals, indicating an extinction of the conditioned fear response. In contrast, the freezing behavior of Pdyn−/− mice remained constantly high, indicating that these mice show no extinction of the conditioned fear response (Fig. 1A). This effect cannot be attributed to an enhanced habituation in the null mutants, because the time spent with freezing did not differ between the genotypes, when we repeated the experiment with a separate group of animals without applying footshock stress (genotype effect: F(1,90) = 0.354, not significant). These animals therefore showed normal fear conditioning, but a strikingly impaired extinction of fear memory.
Figure 1.
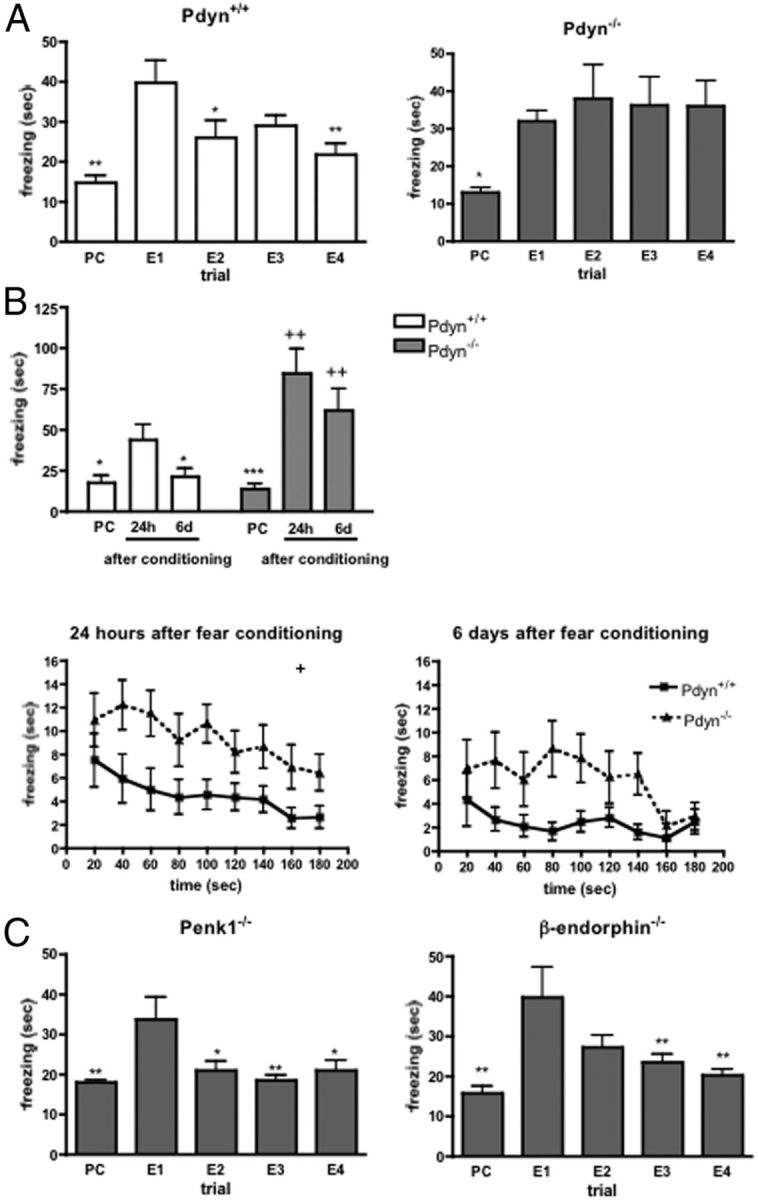
Formation and extinction of stress-induced freezing. A, Normal conditioning, but impaired extinction of context-specific fear responses was observed in Pdyn−/− mice. Enhanced freezing behavior elicited by footshock stress persisted during the four consecutive extinction trials in Pdyn−/− animals, whereas wild-type mice showed an extinction of the fear response in the second extinction trial. PC, Preconditioning trial; E1–E4, extinction trial 1–4. *p < 0.05; **p < 0.01 significantly different from the value measured at the first extinction trial (one-way ANOVA followed by Dunnett's test; n = 10). B, When the animals were tested in a new environment in the presence of a stress associated cue, Pdyn−/− animals showed an enhanced freezing during the tone presentation both 24 h and 6 days after the fear conditioning accompanied by an impaired fear extinction. PC, Preconditioning trial; 24h, 24 h after conditioning: 6d, 6 days after conditioning. *p < 0.05; ***p < 0.001 significantly different from the value measured 24 h after the conditioning. ++p < 0.01 significant difference between the genotypes on the same test day (two-way ANOVA followed by Bonferroni's t test; n = 10–8). C, The time spent with freezing in Penk1 or β-endorphin-null mutant animals was significantly enhanced 24 h after the fear conditioning in the first extinction trial, but not later on. PC, Preconditioning trial; E1–E4, extinction trial 1–4. **p < 0.01 significantly different from the value measured at the first extinction trial (E1) (one-way ANOVA followed by Dunnett's test; n = 10).
We next studied a different group of animals in a cued version of the fear conditioning/extinction paradigm, where mice were conditioned to associate a tone with a footshock stressor (Marsicano et al., 2002). The change in freezing time in the three trials of the experiment (preconditioning, 24 h and 6 d after the conditioning) significantly differed between the genotypes (genotype × trial interaction: F(2,51) = 3.891; p < 0.05; Fig. 1B). Post hoc analysis of the data revealed that the time spent with freezing was significantly elevated 24 h after the fear conditioning in the first extinction trial in wild-type animals, but in the second extinction trial 6 d after the conditioning it returned to the preconditioning values. In contrast, the freezing behavior of Pdyn−/− mice remained constantly high. The freezing behavior significantly reduced within the extinction trial 24 h after the conditioning (time effect: F(8,144) = 4.930; p < 0.001), similarly in both genotypes (genotype × time interaction: F(8,144) = 0.573; p > 0.05). However, Pdyn−/− mice showed more freezing during the whole trial (genotype effect: F(1,144) = 5.944; p < 0.05; Fig. 1B). When animals were reexposed to the tone stimulus 6 d after the conditioning, we still found an enhanced freezing response in Pdyn−/− mice (genotype effect: F(1,120) = 7.217; p < 0.05; Fig. 1B). The extinction of the fear response within the trial was again similar between Pdyn+/+ and Pdyn−/− mice (genotype × time interaction: F(8,144) = 0.814; p > 0.05).
To determine whether other endogenous opioid peptides also contribute to the formation and extinction of fear memories, we studied mice with a genetic invalidation of β-endorphin (Rubinstein et al., 1996) or preproenkephalin (Penk1) (König et al., 1996). As shown in Figure 1C, mice from both strains showed normal contextual fear conditioning and extinction (F(4,49) = 4.589; p < 0.01 for Penk1−/− and F(4,49) = 6.526; p < 0.01 for β-endorphin−/−. Thus, although enkephalin and endorphin opioid peptides are involved in the regulation of emotional behaviors and stress responses (Bilkei-Gorzo et al., 2008), only dynorphin seems to affect extinction of fear memories.
We next determined whether acute pharmacological blockage of dynorphin signaling with the KOR antagonist nor-BNI had similar effects on fear memory extinction. Thus, wild-type mice were treated intraperitoneally with nor-BNI (10 mg/kg, i.p.) 30 min before or immediately after fear conditioning or the extinction trials. Fear conditioning followed by context-specific extinction trials induced a significant change in freezing time in wild-type animals treated before conditioning (vehicle-treated: F(4,49) = 9.071; p < 0.001; norBNI-treated F(4,49) = 8.164; p < 0.001), after conditioning (vehicle-treated: F(4,44) = 13.47; p < 0.001; norBNI-treated F(4,44) = 10.96; p < 0.001) or after the extinction trials (vehicle-treated: F(4,49) = 12.30; p < 0.001; norBNI-treated F(4,49) = 5.055; p < 0.01). As shown by the significant difference between the time spent with freezing in the first extinction trial and freezing time in the preconditioning phase or in the consecutive extinction trials pharmacological blockade of κ-opioid receptors by nor-BNI had no effect on fear memory formation or extinction when applied before or after the conditioning session (Fig. 2A,B). However, it completely blocked the extinction of fear memory when administered after the extinction sessions (Fig. 2C).
Figure 2.

A, B, Intraperitoneal injection of the κ-antagonist nor-BNI (10 mg/kg), 1 h before the fear conditioning (A) or just after it (B), did not alter the formation or extinction of fear memory. C, Animals injected with nor-BNI after the extinction trials did not show a significant decrease in freezing behavior in the extinction trials. PC, Preconditioning trial; E1–E4, extinction trial 1–4. *p < 0.05; **p < 0.01 significantly different from the value measured at the first extinction trial (E1) (one-way ANOVA followed by Dunnett's test; n = 10).
c-fos IR in the brain
To identify brain areas responsible for the extinction deficit in Pdyn−/− mice we compared the neuronal activity in the brains of Pdyn+/+ and Pdyn−/− animals during contextual fear conditioning and extinction. For this purpose, we analyzed induction of the immediate early gene c-fos 1 h after contextual fear conditioning and extinction (Kovács, 2008). In good agreement with the behavioral results, we found a significant difference in the c-fos reactivity between the strains in the basolateral amygdala (genotype × trial interaction: F(2,137) = 15.84; p < 0.001), infralimbic cortex (genotype × trial interaction: F(2,137) = 5.956; p < 0.01), CA3 (genotype × trial interaction: F(2,150) = 6.300; p < 0.01) region of the hippocampus and in the ventrolateral periaqueductal gray area (genotype × trial interaction: F(2,150) = 18.85; p < 0.001). In the prelimbic cortex fear conditioning and extinction induced similar expression change in Pdyn+/+ and Pdyn−/− mice (genotype × trial interaction: F(2,137) = 0.330; not significant) (Fig. 3D). Post hoc analysis of the data using Bonferroni test revealed a similar pattern and intensity of c-fos immunoreactivity in both genotypes in the basolateral amygdala, infralimbic cortex, and in the hippocampus after the conditioning phase. However, after the extinction trial we found a striking genotype difference, with much reduced c-fos intensities in Pdyn−/− animals (Fig. 3A–C). In the ventrolateral periaqueductal gray area fear conditioning induced a lower, whereas fear extinction a higher c-fos expression in Pdyn−/− mice as in the wild-type animals (Fig. 3E).
Figure 3.

Influence of fear conditioning and extinction on c-fos expression in wild-type and Pdyn−/− mice. A, Basolateral amygdala. B, Infralimbic cortex. C, Hippocampus CA3 region. D, Prelimbic cortex. E, Ventrolateral periaqueductal gray. Pdyn−/− animals showed a reduced c-fos expression during the extinction phase in the basolateral amygdala, infralimbic cortex and hippocampus. In the prelimbic cortex fear conditioning and extinction induced similar c-fos expression, whereas in the ventrolateral periaqueductal gray area the expression of c-fos was reduced after fear conditioning and enhanced after extinction training in the Pdyn−/− mice. **p < 0.01; ***p < 0.001 difference between the genotypes (two-way ANOVA followed by Bonferroni test). Scale bars, 50 μm.
Functional imaging study in humans
Analysis of genotype effects on regional brain activation revealed a significantly increased activation of the right amygdala during acquisition and late (day 2) extinction in homozygote carriers of the (T) allele in response to the conditioned stimulus (CS+), when compared with (C) allele carriers (x = 22, y = −4, z = −18, Z = 3.55, p < 0.05 familywise corrected for multiple comparisons across region of interest) (Fig. 4A). We did not find a genotype effect on early (day 1) extinction. Homozygous carriers of the (T) allele also displayed significantly increased skin conductance response compared with (C) allele carriers during late extinction (condition × trial: F = 4.0, p < 0.001; condition × trial × genotype: F = 1.7, p = 0.026; CS+ trial × genotype: F = 2.44, p < 0.001, post hoc tests significant for trial number 5, 9, 13, 14, 16, 20), which might be indicative of prolonged heightened stress reactivity in this group (Fig. 4B). No significant genotype-dependent differences were found during acquisition (condition × trial: F = 3.1, p < 0.001, condition × trial × genotype: F = 0.37, p = 0.99; CS+ trial × genotype F = 0.66, p = 0.86) or early extinction (condition × trial: F = 2.0, p = 0.009, condition × trial × genotype: F = 1.36, p = 0.14; CS+ trial × genotype F = 1.17, p = 0.28).
Figure 4.

A, Regional brain activation. Homozygote carriers of the rs1997794 T allele (n = 12) show significantly stronger activation in the right amygdala (x = 22, y = −4, z = −18, Z = 3.55) during acquisition and late extinction compared with C allele carriers (n = 21, corrected for multiple comparisons across region of interest, FWE p < 0.05). Bar plots indicate size of effect at the maximum activated voxel within the respective region. T, T-value of regional brain activation. B, Compared with C allele carriers T homozygotes exhibit significantly increased skin conductance responses to the CS+ only during late extinction (p < 0.001) but not during conditioning (p = 0.86) or early extinction (p = 0.28).
In addition, during late extinction, we found significantly altered functional coupling between amygdala and ventromedial prefrontal cortex (VMPFC), a region crucially implicated in extinction of conditioned fear responses (Maren and Quirk, 2004): Homozygote carriers of the rs1997794 T allele exhibited significantly diminished functional connectivity between left amygdala and VMPFC (x = 16, y = 24, z = −14, Z = 3.92, p < 0.05 familywise corrected for multiple comparisons within region of interest) and at a lower level of significance also between right amygdala and VMPFC (x = 16, y = 26, z = −14, Z = 3.24, p < 0.001 uncorrected) (Fig. 5A,B).
Figure 5.
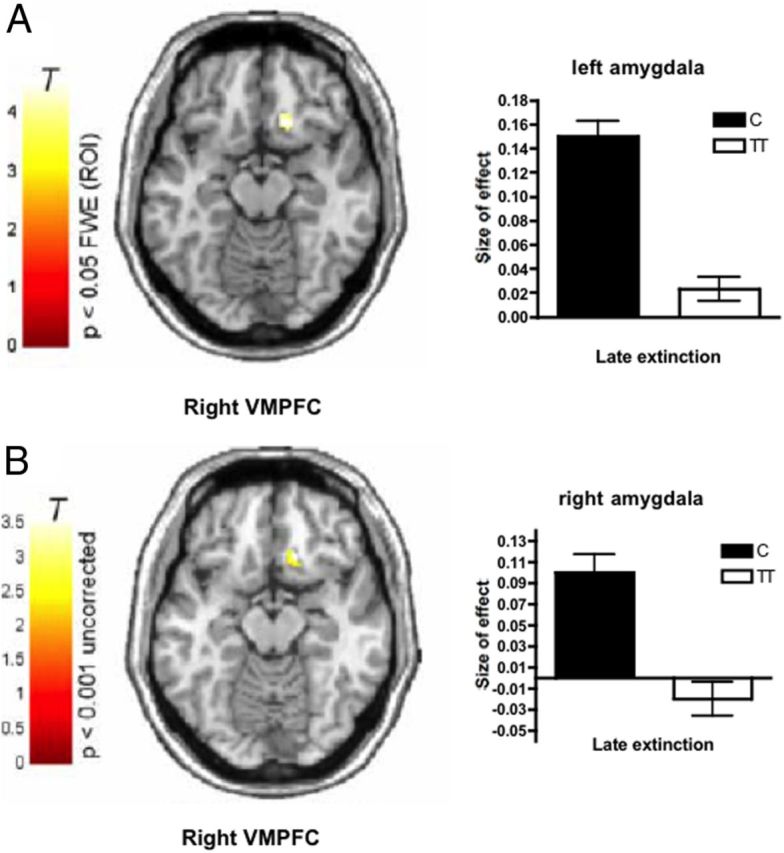
Functional coupling during late extinction. A, B, Homozygote carriers of the rs1997794 T allele (n = 12) exhibit significantly reduced coupling of the VMPFC with the (A) left (VMPFC, x = 16, y = 24, z = −14, Z = 3.92) and (B) right (VMPFC, x = 16, y = 26, z = −14, Z = 3.24) amygdala seed region compared with C allele carriers (n = 21, coupling with left amygdala corrected for multiple comparisons within region of interest, FWE p < 0.05; coupling with right amygdala seed region, uncorrected p < 0.001). Bar plots indicate size of effect at the maximum voxel in the left and right VMPFC. T, T-value of regional brain activation.
Importantly, these pronounced differences in neural activity were not related to increased pain sensitivity in (T) allele carriers: Rs1997794 had no impact on pain intensity ratings or brain activation in response to the UCS. Moreover, no impact of the PDYN variant on anxiety and depression scores was observed (data not shown).
Discussion
This study identifies dynorphin signaling through κ-opioid receptors as a critical mediator of fear extinction in rodents and in humans. Mice lacking dynorphin showed an impaired extinction in the context-dependent paradigm, as well as an increased acquisition of a conditioned fear response in a cue-dependent paradigm. These data indicate that dynorphin signaling contributes to the acquisition of amygdala-driven (Johansen et al., 2010; Pape and Pare, 2010) cue-dependent fear learning, but not to the hippocampus-driven contextual fear conditioning. Furthermore, they demonstrate that dynorphin is required for the extinction of contextual fear memories. The relevance of these findings for humans was assessed by an imaging genetics approach in healthy volunteers. Our data show that a functionally relevant polymorphism in the PDYN gene also affected fear extinction and the functional connectivity between amygdala and ventromedial prefrontal cortex in humans.
We hypothesize that the footshock used for fear conditioning in our study enhances dynorphin signaling in wild-type animals as observed previously using different stressors (Shirayama et al., 2004; Knoll et al., 2011). Dynorphin release helps to fight the stress situation (Knoll and Carlezon, 2010) because it induces analgesia (McLaughlin et al., 2003), regulates the hormonal and behavioral stress responses (McLaughlin et al., 2003; Bilkei-Gorzo et al., 2008; Schwarzer, 2009) and increases the motivation to avoid stress inducing dysphoria (Bruchas et al., 2007). Dynorphin also impairs memory formation (Carey et al., 2009), therefore it may protect against the generation of intensive stress-related memories. Although it was suggested that it improves focusing of attention to stress-relevant cues (Iordanova et al., 2006), our results now show that the amnesic, but not the attention focusing effects of dynorphin predominates during cue-dependent fear conditioning.
Pdyn-null mutant animals continued to show freezing behaviors in the cue und context-dependent fear conditioning paradigms after repeated (in context-dependent) or long lasting (cue-dependent) extinction trials, whereas this behavior was gradually extinguished in wild-type animals. We interpret the behavior of the mutant mice as a deficit in context-dependent fear extinction. The significantly elevated freezing time in the extinction trials of the cued version of the model in Pdyn−/− mice is probably due to an enhanced fear memory formation and not due to an impaired extinction. However, it is not possible to exclude that impaired fear extinction also contributes to the enhanced freezing behavior in the cued version.
It was previously reported that norBNI treatment leads to a long-term blockade (> 3 weeks) of the analgesic effect of dynorphin. However, norBNI did not alter KOR binding characteristics when tested 1 week after the treatment (Bruchas et al., 2007). It is thought that norBNI stabilizes the association between KOR and c-Jun N-terminal kinase (JNK). This leads to a long lasting activation of JNK signaling (Bruchas and Chavkin, 2010). Our data showing that norBNI-treatment before or after the conditioning trial did not alter fear extinction therefore indicates that JNK activation is not involved in KOR-mediated fear extinction (Radulovic and Tronson, 2010).
Interestingly, only the deletion of dynorphin, but not enkephalin or β-endorphin, resulted in delayed fear extinction thus suggesting a unique role of dynorphins within the endogenous opioid family in the termination of stress-related memories. This conclusion contradicts to the results of Fanselow, who reported an enhanced conditioning after pharmacological blockade of MOR, reduced response using κ-antagonist and no effect of δ-antagonists in rats (Fanselow et al., 1991). In mice, acute treatment by DOR agonist attenuated conditioned fear response (Saitoh et al., 2004), and elevation of DOR signaling in the ventrolateral periaqueductal gray (but not outside this area) (McNally, 2005) facilitated extinction. We now show that life-long reduction of enkephalin signaling does not influence fear learning and extinction. The reason of the discrepancy between the results of pharmacological or genetic manipulation of opioid system activity is probably due to the different consequences of short-term pharmacological versus long-term genetic disruption of enkephalin signaling.
c-fos mapping of limbic areas after fear conditioning and extinction supported our hypothesis about the role of dynorphins in fear memory extinction. We found a similar c-fos staining intensity after fear learning and extinction training section in wild-type animals in the basolateral amygdala and in the CA3 region of the hippocampus suggesting the active contribution of these areas both to fear and safety learning. In Pdyn−/− mice, however, the activity of these areas was reduced after the extinction trial. The activity of prelimbic cortex correlates with freezing behavior, whereas infralimbic cortex, an analog area of the human vmPFC (Phelps et al., 2004), is critical for the extinction of fear-related behaviors (Sierra-Mercado et al., 2011). The number of c-fos-positive cells in the prelimbic cortex was lower after extinction trial as after conditioning in both genotypes, showing that the reason of the impaired fear extinction in Pdyn−/− mice is not an enhanced activity of this area after extinction (Burgos-Robles et al., 2009). In the infralimbic cortex, however, the density of c-fos-expressing cells was two times lower in Pdyn−/− as in Pdyn+/+ mice after the extinction training. The low c-fos expression in the infralimbic cortex and hippocampus suggest a reduced inhibitory control over the amygdala during extinction training in the null mutants, which could be responsible for the fear extinction deficit when the KOR signaling is reduced (Kim et al., 2010). In the basal nucleus of the amygdala distinct neuronal populations are active during fear conditioning and extinction (Herry et al., 2008). It is therefore likely that the reduced c-fos expression in this area after extinction training reflects a diminished activity of extinction-related neurons. Our results now suggest an essential role of dynorphin signaling in the activation of this neuronal population during the extinction of fear memories.
It has been shown that cocaine-induced expressions dynorphin and c-fos are inversely related (Steiner and Gerfen, 1993) therefore one would assume an enhanced c-fos staining after footshock or after the extinction trial in Pdyn−/− animals. However, the inverse relation between c-fos and dynorphin expression is probably specific to cocaine-induced expression changes. Neither methylphenidate treatment (Yano and Steiner, 2005), withdrawal from chronic amphetamine (Horner et al., 2009) nor postoperative pain (Campillo et al., 2010) induced opposite change in Pdyn and c-fos expression. Moreover, the expression of c-fos after mild stress was reduced in limbic areas of Pdyn−/− mice (Bilkei-Gorzo et al., 2008), similar to its expression in the present study.
A prerequisite for safety learning, thus for the extinction of a conditioned behavior, is the recall of the fear memory and the detection of the prediction error (Herry et al., 2010). The aversive component of the past stress experience is encoded by dynorphin. Thus, in Pdyn−/− mice or in humans having reduced dynorphin signaling because of a genetic variation in PDYN the recall is incomplete, because the aversive component of the past event is not appropriately represented. It may next impair the prediction error detection and thus delay the extinction of the fear memory. We cannot, however, exclude a direct contribution of dynorphins in prediction error detection: Hippocampus (Radulovic and Tronson, 2010) and the prelimbic cortex (Matsumoto et al., 2007) are key areas for prediction error detection, and the activity of the hippocampus (but not the activity of the prelimbic cortex) was significantly reduced in Pdyn−/− animals after extinction training. It has been suggested that the ventrolateral periaqueductal gray area (vlPAG) is also involved in prediction error detection (McNally and Westbrook, 2003) and danger vicinity (Mobbs et al., 2007). In wild-type animals the c-fos expression in this area was low (Fig. 3E) after extinction training suggesting that the animals did not expect shock under no-shock conditions. The high expression of c-fos in the vlPAG on the other hand in Pdyn−/− mice shows that the vlPAG neurons detected the deviation from the predicted shock, but the animals continued to show freezing response. These results together suggest a role of dynorphins in the fear extinction in the hippocampus and prefrontal cortex but not in prediction error detection in the vlPAG (McNally et al., 2005).
It was previously suggested that combination of the human neuroimaging studies with animal model is an effective strategy (Milad et al., 2006) to overcome the limitations of human functional imaging and animals models alone (Milad and Quirk, 2012). Our result clearly shows the efficacy of this strategy. However, when comparing the results of the human and animals experiments two important differences should be noted: First, in the animal studies the activity of KOR was near to zero after pharmacological KOR blockade or genetic deletion of Pdyn. In contrast, the Pdyn polymorphisms in humans probably produced only moderate differences in KOR activity. Also the experimental condition was different, because the humans were instructed before the conditioning paradigm, which is obviously not possible for animals. Despite these significant differences the fear conditioning and extinction tests in humans and mice provided largely similar results: Pdyn-null mutant mice and humans bearing the TT allele of PDYN at rs1997794 showed stronger fear reactions after conditioning, impaired extinction of the conditioned fear response and altered activity of the amygdala and prefrontal cortex during extinction. The involvement of amygdala and prefrontal cortex (Gottfried and Dolan, 2004) as well as the same role of hippocampus-vmPFC network (LaBar and Phelps, 2005) in fear conditioning and extinction in humans and rodents suggests an evolutionary conserved neuronal base of fear and extinction learning. Previous functional magnetic resonance imaging and anatomical studies also indicated the activation of vmPFC (Milad et al., 2005, 2007), and fMRI studies the activation of amygdala during fear learning and extinction (Bach et al., 2011). These studies revealed that amygdala activation correlates with the autonomic indices of fear (LaBar et al., 1998). We used skin conductance as readout for fear conditioning, similar to the method previously reported (Dunsmoor et al., 2012), because it was shown that it is a sensitive parameter of fear in humans (Bach et al., 2010) and correlates well with the recall and extinction of fear memory (Linnman et al., 2012). We did not find decreased vmPFC activation during late extinction in homozygote T-allele carriers—what should be expected from the literature and our results of increased amygdala activation and heightened SCR—might be due to the small number of subjects. However, diminished connectivity between amygdala and vmPFC in this group can well explain our results and is in accordance with previous evidence, suggesting that connectivity between these regions is in some way a better outcome predictor than activity of one region alone (Kim et al., 2011).
The rs1997794 single nucleotide polymorphism is located in the promoter region of the Pdyn gene (Kölsch et al., 2009), therefore it may influence the expression of dynorphins (Babbitt et al., 2010). Previously, in the 3′region of the gene located polymorphisms were associated with altered Pdyn expression in the human brain, where the rare allele was associated with lower Pdyn levels (Yuferov et al., 2009). However, no direct functional effect, such as conferring differences in gene expression or splicing, has yet been attributed to this variant. At present it is unclear whether the observed effects are due to rs1997794 or another genetic variant in linkage disequilibrium (LD). There even remains the possibility that variation at a different gene locus than PDYN, which is in LD with rs1997794, is responsible for the observed effect. However, the most straightforward interpretation of our data suggests that PDYN is the gene causally implicated in the observed changes: our results suggest that bearers of the (T) allele of PDYN at rs1997794 probably have lower dynorphin levels, because they show alterations in fear-related behaviors and brain area activities similar to those of animals with genetic deletion of Pdyn.
These results point to a diminished extinction of the amygdala response as a consequence of reduced coupling between amygdala and VMPFC in humans bearing the (T) allele of PDYN rs1997794. It was previously shown that the neuronal activity in the prefrontal cortex is increased during extinction (Milad and Quirk, 2002), which in turn leads to a reduced amygdalar activity (Quirk et al., 2003) and thus to lower fear reactivity (Milad et al., 2004). Diminished coupling between the prefrontal cortex and amygdala could thus be one mechanism contributing to the delayed fear extinction observed in these individuals—and also in animals having reduced KOR signaling.
Footnotes
This work was supported by grants from the National Institutes of Health (Grants RO1, DA016768) and the Federal Ministry for Education and Research (NGFN Plus, FKZ: 01GS08144).
References
- Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, Walker JM. Endogenous opioids: biology and function. Annu Rev Neurosci. 1984;7:223–255. doi: 10.1146/annurev.ne.07.030184.001255. [DOI] [PubMed] [Google Scholar]
- Babbitt CC, Silverman JS, Haygood R, Reininga JM, Rockman MV, Wray GA. Multiple functional variants in cis modulate PDYN expression. Mol Biol Evol. 2010;27:465–479. doi: 10.1093/molbev/msp276. [DOI] [PubMed] [Google Scholar]
- Bach DR, Daunizeau J, Friston KJ, Dolan RJ. Dynamic causal modelling of anticipatory skin conductance responses. Biol Psychol. 2010;85:163–170. doi: 10.1016/j.biopsycho.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach DR, Weiskopf N, Dolan RJ. A stable sparse fear memory trace in human amygdala. J Neurosci. 2011;31:9383–9389. doi: 10.1523/JNEUROSCI.1524-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Michel K, Noble F, Roques BP, Zimmer A. Preproenkephalin knockout mice show no depression-related phenotype. Neuropsychopharmacology. 2007;32:2330–2337. doi: 10.1038/sj.npp.1301370. [DOI] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Racz I, Michel K, Mauer D, Zimmer A, Klingmüller D. Control of hormonal stress reactivity by the endogenous opioid system. Psychoneuroendocrinology. 2008;33:425–436. doi: 10.1016/j.psyneuen.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci. 2009;29:8474–8482. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillo A, González-Cuello A, Cabañero D, Garcia-Nogales P, Romero A, Milanés MV, Laorden ML, Puig MM. Increased spinal dynorphin levels and phospho-extracellular signal-regulated kinases 1 and 2 and c-Fos immunoreactivity after surgery under remifentanil anesthesia in mice. Mol Pharmacol. 2010;77:185–194. doi: 10.1124/mol.109.059790. [DOI] [PubMed] [Google Scholar]
- Carey AN, Lyons AM, Shay CF, Dunton O, McLaughlin JP. Endogenous kappa opioid activation mediates stress-induced deficits in learning and memory. J Neurosci. 2009;29:4293–4300. doi: 10.1523/JNEUROSCI.6146-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunsmoor JE, Martin A, LaBar KS. Role of conceptual knowledge in learning and retention of conditioned fear. Biol Psychol. 2012;89:300–305. doi: 10.1016/j.biopsycho.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erk S, Meyer-Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P, Grimm O, Arnold C, Haddad L, Witt SH, Cichon S, Nöthen MM, Rietschel M, Walter H. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry. 2010;67:803–811. doi: 10.1001/archgenpsychiatry.2010.94. [DOI] [PubMed] [Google Scholar]
- Esslinger C, Walter H, Kirsch P, Erk S, Schnell K, Arnold C, Haddad L, Mier D, Opitz von Boberfeld C, Raab K, Witt SH, Rietschel M, Cichon S, Meyer-Lindenberg A. Neural mechanisms of a genome-wide supported psychosis variant. Science. 2009;324:605. doi: 10.1126/science.1167768. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Kim JJ, Young SL, Calcagnetti DJ, DeCola JP, Helmstetter FJ, Landeira-Fernandez J. Differential effects of selective opioid peptide antagonists on the acquisition of pavlovian fear conditioning. Peptides. 1991;12:1033–1037. doi: 10.1016/0196-9781(91)90056-u. [DOI] [PubMed] [Google Scholar]
- Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7:1144–1152. doi: 10.1038/nn1314. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Lüthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Horner KA, Noble ES, Lauterbach EC. Differential regulation of prodynorphin, c-fos, and serotonin transporter mRNA following withdrawal from a chronic, escalating dose regimen of d-amphetamine. Synapse. 2009;63:257–268. doi: 10.1002/syn.20606. [DOI] [PubMed] [Google Scholar]
- Iordanova MD, McNally GP, Westbrook RF. Opioid receptors in the nucleus accumbens regulate attentional learning in the blocking paradigm. J Neurosci. 2006;26:4036–4045. doi: 10.1523/JNEUROSCI.4679-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Tarpley JW, LeDoux JE, Blair HT. Neural substrates for expectation-modulated fear learning in the amygdala and periaqueductal gray. Nat Neurosci. 2010;13:979–986. doi: 10.1038/nn.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Loucks RA, Palmer AL, Brown AC, Solomon KM, Marchante AN, Whalen PJ. The structural and functional connectivity of the amygdala: from normal emotion to pathological anxiety. Behav Brain Res. 2011;223:403–410. doi: 10.1016/j.bbr.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Jo YS, Kim IH, Kim H, Choi JS. Lack of medial prefrontal cortex activation underlies the immediate extinction deficit. J Neurosci. 2010;30:832–837. doi: 10.1523/JNEUROSCI.4145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA., Jr Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry. 2011;70:425–433. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kölsch H, Wagner M, Bilkei-Gorzó A, Toliat MR, Pentzek M, Fuchs A, Kaduszkiewicz H, van den Bussche H, Riedel-Heller SG, Angermeyer MC, Weyerer S, Werle J, Bickel H, Mösch E, Wiese B, Daerr M, Jessen F, Maier W, Dichgans M. Gene polymorphisms in prodynorphin (PDYN) are associated with episodic memory in the elderly. J Neural Transm. 2009;116:897–903. doi: 10.1007/s00702-009-0238-5. [DOI] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Kovács KJ. Measurement of immediate-early gene activation—c-fos and beyond. J Neuroendocrinol. 2008;20:665–672. doi: 10.1111/j.1365-2826.2008.01734.x. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Phelps EA. Reinstatement of conditioned fear in humans is context dependent and impaired in amnesia. Behav Neurosci. 2005;119:677–686. doi: 10.1037/0735-7044.119.3.677. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454:642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnman C, Zeidan MA, Furtak SC, Pitman RK, Quirk GJ, Milad MR. Resting amygdala and medial prefrontal metabolism predicts functional activation of the fear extinction circuit. Am J Psychiatry. 2012;169:415–423. doi: 10.1176/appi.ajp.2011.10121780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ye N, Lange N, Cohen BM. Dynorphinergic GABA neurons are a target of both typical and atypical antipsychotic drugs in the nucleus accumbens shell, central amygdaloid nucleus and thalamic central medial nucleus. Neuroscience. 2003;121:991–998. doi: 10.1016/s0306-4522(03)00397-x. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Matsumoto K, Abe H, Tanaka K. Medial prefrontal cell activity signaling prediction errors of action values. Nat Neurosci. 2007;10:647–656. doi: 10.1038/nn1890. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP. Facilitation of fear extinction by midbrain periaqueductal gray infusions of RB101(S), an inhibitor of enkephalin-degrading enzymes. Behav Neurosci. 2005;119:1672–1677. doi: 10.1037/0735-7044.119.6.1672. [DOI] [PubMed] [Google Scholar]
- McNally GP, Westbrook RF. Opioid receptors regulate the extinction of Pavlovian fear conditioning. Behav Neurosci. 2003;117:1292–1301. doi: 10.1037/0735-7044.117.6.1292. [DOI] [PubMed] [Google Scholar]
- McNally GP, Lee BW, Chiem JY, Choi EA. The midbrain periaqueductal gray and fear extinction: opioid receptor subtype and roles of cyclic AMP, protein kinase A, and mitogen-activated protein kinase. Behav Neurosci. 2005;119:1023–1033. doi: 10.1037/0735-7044.119.4.1023. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Nicodemus KK, Egan MF, Callicott JH, Mattay V, Weinberger DR. False positives in imaging genetics. Neuroimage. 2008;40:655–661. doi: 10.1016/j.neuroimage.2007.11.058. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ. Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci. 2004;118:389–394. doi: 10.1037/0735-7044.118.2.389. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quinn BT, Pitman RK, Orr SP, Fischl B, Rauch SL. Thickness of ventromedial prefrontal cortex in humans is correlated with extinction memory. Proc Natl Acad Sci U S A. 2005;102:10706–10711. doi: 10.1073/pnas.0502441102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Mobbs D, Petrovic P, Marchant JL, Hassabis D, Weiskopf N, Seymour B, Dolan RJ, Frith CD. When fear is near: threat imminence elicits prefrontal-periaqueductal gray shifts in humans. Science. 2007;317:1079–1083. doi: 10.1126/science.1144298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Palkovits M. Stress-induced expression of co-localized neuropeptides in hypothalamic and amygdaloid neurons. Eur J Pharmacol. 2000;405:161–166. doi: 10.1016/s0014-2999(00)00549-5. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA. Emotion and cognition: insights from studies of the human amygdala. Annu Rev Psychol. 2006;57:27–53. doi: 10.1146/annurev.psych.56.091103.070234. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Paré D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Tronson NC. Molecular specificity of multiple hippocampal processes governing fear extinction. Rev Neurosci. 2010;21:1–17. doi: 10.1515/revneuro.2010.21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts NP, Kitchiner NJ, Kenardy J, Bisson JI. Systematic review and meta-analysis of multiple-session early interventions following traumatic events. Am J Psychiatry. 2009;166:293–301. doi: 10.1176/appi.ajp.2008.08040590. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG, Low MJ. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci U S A. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. J Pharmacol Sci. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Schwarzer C. 30 years of dynorphins—new insights on their functions in neuropsychiatric diseases. Pharmacol Ther. 2009;123:353–370. doi: 10.1016/j.pharmthera.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. 1993;13:5066–5081. doi: 10.1523/JNEUROSCI.13-12-05066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M, Steiner H. Topography of methylphenidate (ritalin)-induced gene regulation in the striatum: differential effects on c-fos, substance P and opioid peptides. Neuropsychopharmacology. 2005;30:901–915. doi: 10.1038/sj.npp.1300613. [DOI] [PubMed] [Google Scholar]
- Yuferov V, Ji F, Nielsen DA, Levran O, Ho A, Morgello S, Shi R, Ott J, Kreek MJ. A functional haplotype implicated in vulnerability to develop cocaine dependence is associated with reduced PDYN expression in human brain. Neuropsychopharmacology. 2009;34:1185–1197. doi: 10.1038/npp.2008.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Valjent E, Konig M, Zimmer AM, Robledo P, Hahn H, Valverde O, Maldonado R. Absence of delta-9-tetrahydrocannabinol dysphoric effects in dynorphin-deficient mice. J Neurosci. 2001;21:9499–9505. doi: 10.1523/JNEUROSCI.21-23-09499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]