

Abstract
Upon illumination several phototransduction proteins translocate between cell body and photosensory compartments. In Drosophila photoreceptors arrestin (Arr2) translocates from cell body to the microvillar rhabdomere down a diffusion gradient created by binding of Arr2 to photo-isomerized metarhodopsin. Translocation is profoundly slowed in mutants of key phototransduction proteins including phospholipase C (PLC) and the Ca2+-permeable transient receptor potential channel (TRP), but how the phototransduction cascade accelerates Arr2 translocation is unknown. Using real-time fluorescent imaging of Arr2-green fluorescent protein translocation in dissociated ommatidia, we show that translocation is profoundly slowed in Ca2+-free solutions. Conversely, in a blind PLC mutant with ∼100-fold slower translocation, rapid translocation was rescued by the Ca2+ ionophore, ionomycin. In mutants lacking NINAC (calmodulin [CaM] binding myosin III) in the cell body, translocation remained rapid even in Ca2+-free solutions. Immunolabeling revealed that Arr2 in the cell body colocalized with NINAC in the dark. In intact eyes, the impaired translocation found in trp mutants was rescued in ninaC;trp double mutants. Nevertheless, translocation following prolonged dark adaptation was significantly slower in ninaC mutants, than in wild type: a difference that was reflected in the slow decay of the electroretinogram. The results suggest that cytosolic NINAC is a Ca2+-dependent binding target for Arr2, which protects Arr2 from immobilization by a second potential sink that sequesters and releases arrestin on a much slower timescale. We propose that rapid Ca2+/CaM-dependent release of Arr2 from NINAC upon Ca2+ influx accounts for the acceleration of translocation by phototransduction.
Introduction
Photoreceptors are highly polarized cells with membrane-rich photosensory compartments separated from the rest of the cell body. It has recently become widely recognized that several phototransduction proteins translocate between these compartments in response to light, representing a form of long-term light and dark adaptation (for review, see Frechter and Minke, 2006; Slepak and Hurley, 2008). One of the best-studied examples is arrestin, which terminates the light response by binding to photo-isomerized rhodopsin (metarhodopsin). In dark-adapted photoreceptors most arrestin localizes to the cell body in both vertebrate and insect photoreceptors, but on illumination translocates to the photosensory compartment (Broekhuyse et al., 1985; Alloway et al., 2000; Kiselev et al., 2000; Lee et al., 2003; Peterson et al., 2003; Calvert et al., 2006; Slepak and Hurley, 2008; Satoh et al., 2010). In fly (Drosophila) photoreceptors the photosensory compartment is represented by the rhabdomere, a light-guiding, rod-like stack of ∼30,000 densely packed apical microvilli loaded with rhodopsin and proteins of a phototransduction cascade mediated by heterotrimeric Gq protein, phospholipase C (PLC), and Ca2+-permeable “transient receptor potential” (TRP) channels (for review, see Wang and Montell, 2007; Katz and Minke, 2009; Yau and Hardie, 2009; Hardie, 2012).
Although the possible role of active transport by molecular motors remains debated (Lee and Montell, 2004; Calvert et al., 2006; Strissel et al., 2006; Orisme et al., 2010), recent evidence in both vertebrate rods and Drosophila microvillar photoreceptors supports an essentially passive diffusional model of arrestin translocation, down gradients established by light-regulated “sinks” (Nair et al., 2005; Slepak and Hurley, 2008; Satoh et al., 2010). We recently provided evidence that metarhodopsin (M) is the major light-activated sink in fly rhabdomeres by showing that the dominant arrestin isoform (Arr2) translocated in a 1:1 stoichiometric relationship to the number of rhodopsin photo-isomerizations (Satoh et al., 2010). We also showed that Arr2 translocation was very rapid (τ ∼10 s), but profoundly slowed in mutants of various phototransduction proteins including Gq, phospholipase C (PLC) (norpA), and the major Ca2+-permeable TRP channel (Satoh et al., 2010). Our evidence suggested that Ca2+ influx via the light-sensitive channels was required to accelerate Arr2 translocation, possibly by releasing Arr2 from a Ca2+-dependent cytosolic sink (Satoh et al., 2010). However, direct evidence for the role of Ca2+ was lacking, while the identity of the putative cytosolic sink and the mechanism(s) mediating the acceleration remained unresolved.
Here, we show directly that Ca2+ is both necessary and sufficient to accelerate Arr2 translocation and provide evidence that the Ca2+-regulated cytosolic sink is the cytosolic isoform of NINAC, a calmodulin (CaM) binding myosin III. Our evidence also suggests the existence of another potential Ca2+ dependent cytosolic sink, which sequesters and releases arrestin on a much slower timescale, and that NINAC protects Arr2 from sequestration and immobilization by this site. The data support a mechanism for the Ca2+-dependent translocation of Arr2 that is remarkably similar to a previously proposed disinhibitory mechanism of Ca2+-dependent inactivation of M required for rapid termination of the light response (Liu et al., 2008).
Materials and Methods
Flies.
Flies (Drosophila melanogaster) were reared in the dark on the standard cornmeal/agar diet at 25°C. Both male and females were used. Mutants included norpAP24 (null mutant of PLC) (Bloomquist et al., 1988; Pearn et al., 1996); trp343 (null mutant of light-sensitive TRP channel) (Scott et al., 1997); ninaCP235 (null mutant of NINAC MyoIII) (Montell and Rubin, 1988), ninaCΔ174, and ninaCΔ132 (transgenic lines expressing only cytosolic p132 or rhabdomeric p174 NINAC isoforms, respectively) (Porter et al., 1992); and ninaCΔC1, a transgenic line expressing a NINAC construct lacking the CaM binding domain common to both NINAC p132 and p174 (Porter et al., 1995). Null rac2Δ mutants (Ng et al., 2002) were obtained from a stock maintained in Cambridge, UK, and the mutation confirmed by sequencing. Green fluorescent protein (GFP) imaging was performed using flies expressing Arr2-GFP driven by the Rh1 promoter as previously described (Satoh et al., 2010). Double mutant combinations and mutants expressing Rh1-Arr2-GFP were generated by genetic crosses using standard balancers. Rhodopsin content measured by M fluorescence was near maximal (compared, e.g., to “wild-type” w1118 flies fed on a β-carotene-enriched diet) except in ninaCP235 (where levels were ∼2-fold reduced). All fly stocks were on a white (w1118) background to eliminate compound eye-screening pigments, except weak eye pigmentation from the mini-white marker gene in Rh1-Arr2-GFP. Controls using cn,bw to eliminate even this weak pigmentation showed no significant differences in Arr2-GFP translocation (Satoh et al., 2010).
Live GFP imaging.
Fluorescence from the deep pseudopupil (DPP) of intact flies was measured by a photomultiplier tube (PMT), essentially as previously described (Satoh et al., 2010). Briefly, transgenic flies expressing Arr2-GFP in photoreceptors R1–R6 were fixed with low melting point wax in truncated Eppendorf pipettes, mounted on a micromanipulator on the stage of a Nikon Eclipse TE300 microscope and observed with a 20×/0.35 NA Fluor objective. The DPP was cropped via a rectangular diaphragm and sampled at up to 500 Hz using a PMT (Cairn Research) collecting fluorescence excited by a blue (470 nm peak) ultrabright light-emitting diode (LED) and imaged/measured via 515 nm dichroic and OG515 long-pass filters. Data were recorded and analyzed using pClamp10 software (Molecular Devices). Photoreisomerization of M to rhodopsin (R) was achieved by long wavelength light delivered by an ultrabright orange LED (Thorlabs) via the front port of the microscope. In some experiments a 75W Xe arc lamp and a monochromator (Photon Technology Instruments) were used to deliver precisely defined wavelengths.
It is important to know that fly M is thermostable and absorbs maximally at ∼570 nm, while the R state absorbs maximally at 480 nm. The two states are photo-interconvertible; hence, the fraction of pigment in the M state is determined by the spectral content of illumination, reaching a photo-equilibrium (after sufficiently long and intense illumination) determined by the ratio of the R and M photosensitivity spectra (Minke and Kirschfeld, 1979; Belusic et al., 2010). Photo-equilibration was typically achieved within 100–200 ms for the blue excitation LED (generating ∼70% M, 30% R) and 1–2 s for the orange LED (generating ∼2%M, 98% R) (Satoh et al., 2010). Thus the blue illumination used to excite Arr2-GFP fluorescence rapidly (∼100 ms) generates a large number of M molecules, which bind Arr2, creating the gradient that drives its translocation. Following orange light, virtually all M is photoreisomerized to R (which no longer binds Arr2) resulting in rapid (<10 s) reverse translocation back to the cell body. Unless otherwise stated, translocation measurements using blue excitation were made after photo-equilibration with orange light, followed by a 30–60 s dark-adaptation period, which is sufficient time to allow full reverse translocation of Arr2 out of the rhabdomere (Satoh et al., 2010).
Imaging of dissociated ommatidia.
Fluorescence from dissociated ommatidia was measured on the same microscope using freshly dissociated ommatidia prepared as previously described for electrophysiological patch-clamp experiments (Hardie, 1991; Hardie et al., 2002). Ommatidia were imaged with a 40× oil-immersion objective through a glass coverslip forming the floor of a Perspex recording chamber. Images were captured using 125 ms exposures at up to three frames s−1 by a Scion Firewire camera. To minimize exposure to potentially damaging light levels, single translocation runs were typically restricted to only 12 s. The average intensity was measured in regions of interest in rhabdomere and cytosol using ImageJ. Standard bath contained the following (in mm): 120 NaCl, 5 KCl, 10 N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulfonic acid, 4 MgCl2, 1.5 CaCl2, 25 proline, and 5 alanine, pH 7.15. Ca2+-free bath was identical except for the omission of CaCl2 and the addition of 1.6 mm EGTA. Ionomycin (14 μm) was dissolved from a 14 mm dimethylsulfoxide stock in (Ca2+-containing) bath solution, in which NaCl had been substituted for LiCl to inactivate extrusion of Ca2+ by the Na+/Ca2+ exchanger. Both Ca2+-free and ionomycin-containing solutions were applied by pressure ejection from a closely positioned puffer pipette. All chemicals were ordered from Sigma-Aldrich.
Electrophysiology.
Electroretinograms (ERGs) were recorded as described previously (Satoh et al., 2010). Briefly, intact flies were immobilized with low melting point wax in truncated Eppendorf tips. Recordings were made using low resistance (∼10 MΩ) glass microelectrodes filled with fly Ringer (140 mm NaCl, 5 KCl, 1.5 CaCl2, 4 MgCl2) inserted into the eye. The indifferent electrode was a similar electrode inserted into the head capsule near the ocelli. Signals were amplified by a Neurolog NL102 DC preamplifier (Digitimer) and sampled and analyzed using pClamp software (Molecular Devices). Light was provided by a 75 W Xenon arc lamp and monochromator, delivered to within 5 mm of the eye by a liquid light guide (diameter 5 mm).
Indirect immunohistochemistry.
Fixation and staining methods were as previously described (Satoh and Ready, 2005; Satoh et al., 2010). Primary antisera were rabbit anti-NinaCp132 (1:100; a gift from Dr. Montell) and chicken anti-Arr2 antibody (1:100). Samples were examined and images recorded using a Bio-Rad MRC1024 or Olympus FV1000 confocal microscope. Flies were either dark adapted and dissected using infrared illumination and infrared-sensitive eyepieces or illuminated with saturating blue light before fixation and dissection as previously described (Satoh and Ready, 2005).
Results
Ca2+ dependence of Arr2 translocation in dissociated ommatidia
The blue excitation required to excite Arr2-GFP fluorescence rapidly (within ∼100 ms) establishes a photo-equilibrium with a high proportion (∼70%) of M, which binds Arr2 thus creating a gradient that drives translocation (Satoh et al., 2010). We previously monitored Arr2-GFP translocation in real time in vivo by imaging its fluorescence in the rhabdomeres using the DPP in completely intact flies (see Materials and Methods). To allow direct manipulation of Ca2+, we used instead a preparation of freshly dissociated ommatidia. This ex vivo preparation has been extensively used in electrophysiological patch-clamp experiments (Hardie, 1991; Hardie et al., 2002), and the photoreceptors remain viable and fully functional for at least 1 h in simple Ringer solutions. Importantly, for the present experiments, it allows rapid perfusion of the extracellular solution and resolution of subcellular detail.
In wild-type flies expressing Arr2-GFP, arrestin translocation induced by blue excitation light could be readily imaged in dissociated ommatidia as a rapid decrease in cytosolic fluorescence mirrored by an increase in rhabdomere fluorescence (Fig. 1). To quantify the signal, we measured the average fluorescence intensity in a region of interest in the rhabdomere and divided this by that of a region in the cell body—a metric that minimizes any effect of bleaching during the experiment. Under control conditions, this ratio rapidly increased by ∼40% within 12 s, with a time constant of 5–10 s, similar to that measured in vivo using fluorescence of the DPP. Translocation could be rapidly reversed by subsequent photoreisomerization of M to R with long wavelength (orange/red) light; following which translocation could be repeated by another blue stimulus (typically after ∼1 min in the dark). To test whether translocation required Ca2+ influx, the ommatidia were perfused with Ca2+-free, EGTA buffered solution from a puffer pipette. Strikingly, under these conditions there was virtually no change in fluorescence over the same time course. However, when normal Ca2+-containing bath was restored, and following M→R photoreconversion by orange light, robust translocation could again be elicited by R→ M conversion with blue excitation. These results indicate that Ca2+ influx was indeed required to facilitate rapid translocation.
Figure 1.

Arr2-GFP translocation in dissociated ommatidia is Ca2+ dependent. A, Frames from movies of Arr2-GFP fluorescence in otherwise wild-type ommatidium (125 ms exposures, sampled at 3 Hz). In the first frame (t = 0 s) fluorescence is seen in both cell body (c) and rhabdomeres (r), but not in nucleus (n). By 12 s under control conditions (above), fluorescence in the cell body had decreased and increased in the rhabdomere. In the same ommatidium perfused with Ca2+-free EGTA-buffered solution, there was little change after 12 s. Ai, Time course of fluorescence increases (ratio of average fluorescence in rhabdomere and cytosol) under the two conditions (mean from n = 6 ommatidia). Aii, Bar graph summarizing data, expressed as relative increase in the rhab/cyto ratio over 12 s (mean ± SEM n = 6). B, Similar measurements made in null norpAP24 mutant lacking PLC. In normal bath (control), there was now no obvious change in the fluorescence distribution; however, in measurements made ∼30 s after brief application of a Ca2+ ionophore (ionomycin, 14 μm dotted trace), robust translocation was observed. Bi, Bii, Summary of time course and relative increase in the rhab/cyto ratio over the 12 s measurement period as in A (n = 6). Scale bar, 5 μm. Significance levels (paired, two-tailed t test): **p < 0.005; ***p < 10−6 with respect to same cell controls.
We previously found that translocation in vivo was profoundly slowed in norpA mutants, which lack PLC activity and have no response to light. Correspondingly, in ommatidia prepared from norpAP24 flies expressing Arr2-GFP, virtually no translocation (change in fluorescence) was detected during 12 s of blue excitation. However, in measurements made after brief (∼10 s) puffer pipette application of the Ca2+ ionophore, ionomycin (14 μm), rapid translocation was rescued to at least wild-type performance with rapid reversible clearance of cytosolic Arr2-GFP and an increase in rhabdomeric signal (Fig. 1B). Together, these experiments demonstrate that after R→M conversion by blue light, Ca2+ is both necessary and sufficient to accelerate Arr2 translocation without any products of PLC activity.
Role of NINAC in Ca2+-dependent acceleration of translocation
One mechanism by which Ca2+ influx might accelerate translocation is if Arr2 is bound to a target in the cell body and Ca2+ promotes its release from this cytosolic sink. A candidate for such a sink was suggested by a previous study, which indicated that Arr2 can bind to the NINAC myosin III in the microvilli in a Ca2+/CaM-dependent manner (Liu et al., 2008). NINAC exists in two isoforms: a 174 kDa protein (p174), which localizes exclusively to the rhabdomere, and a shorter (132 kDa) cytosolic protein (Porter et al., 1992). Because both share an identical CaM binding site, we wondered whether NINAC p132 might represent a similar Ca2+/CaM-dependent cytosolic binding partner for Arr2. As a first test we asked whether NINAC p132 and Arr2 colocalized in dark-adapted retina. Indeed, in double immunolabeling experiments we found that NINACp132 was extensively colocalized with Arr2 in dark-adapted ommatidia, with both showing a similar textured distribution throughout most of the cell body (Fig. 2E).
Figure 2.
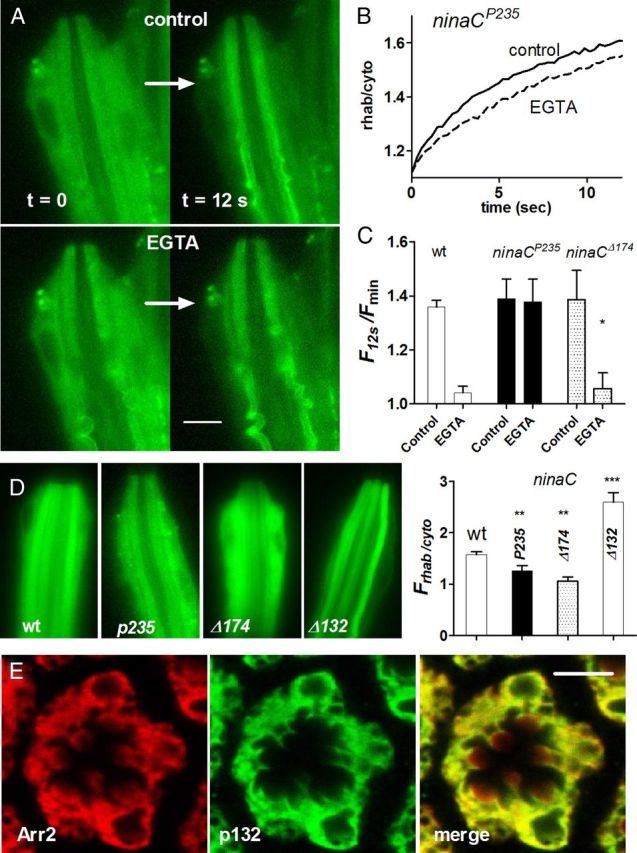
ninaC mutants lacking cytosolic MyoIII do not require Ca2+ influx for rapid translocation. A, Frames from movies of Arr2-GFP fluorescence in ommatidium of ninaCP235-null mutant. In the first frame (t = 0 s) fluorescence is evenly distributed throughout the cell body and rhabdomeres, but by 12 s the rhabdomere is brighter in both control (1.5 mm Ca2+) solution (top) and in the same cell perfused with Ca2+-free, EGTA- buffered solution (bottom). B, Time courses of fluorescence increase (ratio of average fluorescence in rhabdomere and cytosol). C, Bar graph summarizing data, expressed as relative increase in the rhab/cyto ratio over 12 s. In wild-type (data from Fig. 1) and ninaCΔ174 mutants lacking just the rhabdomeric p174 isoform, translocation was suppressed in EGTA-buffered solution, but not in ninaCP235 (mean ± SEM n = 4–7 ommatidia; *p < 0.005). D, Representative images of Arr2-GFP fluorescence at t = 0 s (i.e., dark adapted) in dissociated ommatidia from wild-type ninaCp235 ninaCΔ174 (expressing only cytosolic NINAC) and ninaCΔ132 (expressing only rhabdomeric NINAC) flies. Right, Quantification showing initial dark-adapted ratio of rhabdomere to cytosol fluorescence (mean ± SEM, n = 4–14 ommatidia). All ninaC alleles were significantly increased/decreased with respect to wild-type (**p < 0.001; ***p < 10−5 unpaired, two-tailed t test). E, Transverse section of dark-adapted wild-type (w1118) ommatidium immunolabeled with Arr2 (left) and NINAC p132 antibodies (center) merged image (right). Apart from the presence of Arr2 in the rhabdomeres, there is tight colocalization (representative of n = 5 flies). Scale bar, 5 μm.
If NINAC is a Ca2+-dependent binding target responsible for tethering Arr2 in the dark-adapted cell body, we predicted that under Ca2+-free conditions Arr2 would be free to diffuse and translocate in ninaC mutants. Indeed, in marked contrast to wild-type flies, the time course and extent of Arr2-GFP translocation monitored in dissociated ninaCP235 ommatidia was very similar, whether measured in normal Ca2+-containing bath or when perfused with Ca2+-free solution buffered with EGTA (Fig. 2). The Ca2+ dependence of Arr2 translocation appeared to be due to the cytosolic NINAC p132 isoform, because ninaCΔ174 mutants lacking rhabdomeric p174, but still expressing cytosolic p132, again showed strong inhibition of translocation by EGTA perfusion (Fig. 2C). Consistent with a role of NINAC as an Arr2 binding target, the initial dark-adapted distribution of Arr2 was strongly dependent upon the distribution of NINAC. In null ninaCP235 mutants and ninaCΔ174 mutants lacking rhabdomeric p174 NINAC, Arr2-GFP-relative fluorescence levels in the rhabdomere were initially lower than in wild-type, while in ninaCΔ132 mutants lacking only the cytosolic p132 isoform, the initial rhabdomeric fluorescence was much higher (Fig. 2D). While not systematically analyzed, it was clear that the absolute fluorescence of Arr2-GFP in ninaCP235 was considerably weaker than in a wild-type background, consistent with the reduced level of Arr2 expression previously reported in ninaCP235 (Liu et al., 2008).
NINAC-dependent biphasic fluorescence increase
Arr2-GFP translocation in ninaC-null mutants, measured in vivo from the DPP, is similar in time course to that observed in wild-type flies (Satoh et al., 2010). Nevertheless, measurements made with high time resolution reveal a subtle difference. On a wild-type background the Arr2-GFP fluorescence increase follows a distinctive biphasic time course, with a very rapid increase in fluorescence over a period of ∼500 ms, followed by a more gradual increase (τ ∼10s). Although the overall time course of translocation in ninaCP235-null mutants was similar, this initial rapid phase was always conspicuously absent. The rapid phase was specifically dependent upon NINAC in the microvilli as it was still observed in ninaCΔ132 mutants but not in ninaCΔ174 flies lacking the rhabdomeric isoform (Fig. 3). By fitting a double exponential function to the first 5 s of the traces in otherwise wild-type flies, the time constant of the rapid phase was estimated at 265 ± 16 ms (mean ± SEM n = 11). A similar rapid phase was also detected in wild-type (but not ninaC) dissociated ommatidia (Figs. 1, 2).
Figure 3.

Rapid fluorescence increase, dependent on rhabdomeric NINAC. A, Arr2-GFP fluorescence measured in vivo from the DPP in an otherwise wild-type fly and a ninaCP235-null mutant. The wild-type (wt) trace is characterized by an early rapid phase lasting ∼0.5 s that is absent in ninaC. B, Same traces on a faster timescale, plus traces from ninaCΔ132, which retains the fast phase and ninaCΔ174, which lacks this component. Representative of n ≥ 20 (wild-type or ninaCP235) and n ≥ 6 ninaCΔ132 or ninaCΔ174 flies. Traces normalized (between Fmin at t = 0, and F after 10 s) for comparison of kinetics. C, Arr2-GFP fluorescence in a wild-type fly measured 30 s after 10s photo-equilibrating green (540 nm) illumination (G1). A second 4 s green stimulus (G2) was delivered at variable delays (DA) before the blue (B) excitation triggered translocation. When excited 0.05 s after termination of the second pulse, the fast phase was almost eliminated, but recovered rapidly (arrowheads) as the dark-adaptation period (DA) was increased to 10 s. D, Using the same protocol in ninaCP235 the second pulse made no difference (0.05 and 10 s only shown). E, Time course of the recovery of the fast phase (as in C) measured as the relative incremental increase in fluorescence averaged between 0.3 and 0.5 s (ΔF0.5/Fmin) after onset of blue excitation. Mean ± SEM n = 5, data fitted with single exponential (τ = 2.6 s).
Because the initial rapid fluorescence increase required the presence of the rhabdomeric form of NINAC, we hypothesized that, rather than representing an improbably rapid phase of translocation, this initial phase represents a change in the fluorescence efficiency of Arr2-GFP dependent upon binding to NINAC in the rhabdomere. Thus previously, we proposed that in the dark-adapted state, Arr2 is bound to NINAC, and that upon illumination, Ca2+ influx into the rhabdomeric microvilli releases Arr2 from NINAC via a Ca2+/CaM-dependent action (Liu et al., 2008). We suggest that when bound to NINAC in the rhabdomere, Arr2-GFP fluorescence is partially quenched, and that the rapid fluorescence increase represents unquenching as Arr2 is released from NINAC following Ca2+ influx.
If this is the case, long wavelength illumination, which itself causes no net change in the M fraction, but which still elicits large light-induced Ca2+ influx, should also be able to release Arr2 from NINAC. We therefore predicted that the rapid phase should no longer be detected if measured immediately after such a long wavelength stimulus, because Arr2 should already have been released. To test this we first established a photo-equilibrium with most pigment in the R state with a bright 540 nm stimulus, waited 20 s (sufficient for full reverse translocation), and then delivered a second 540 nm stimulus at varying times before inducing and measuring translocation as usual with blue 470 nm excitation (Fig. 3). As predicted, when measured immediately (50 ms) after the end of the 540 nm stimulus, the rapid phase was essentially abolished, but then recovered rapidly as the dark period between the two flashes was increased, with a time constant of ∼3 s (Fig. 3C,E). This was strictly dependent upon NINAC as the kinetics of fluorescence increase in ninaCP235-null mutants was unaffected by the timing of the second light pulse (Fig. 3D). We suggest that this behavior represents the rapid Ca2+-dependent release of Arr2 from rhabdomeric NINAC p174, followed by rapid rebinding as Ca2+ falls in the dark.
A second cytosolic sink with slow sequestration and release kinetics
We previously reported that Arr2 translocation measured after 10 min dark adaptation is somewhat slower than after 30 s to 1 min dark adaptation (Satoh et al., 2010). Here, we confirmed this finding and explored the change in translocation kinetics in more detail. By varying the dark adaptation time we found that translocation in otherwise wild-type flies gradually slowed over a time course of 20–30 min; however, the rapid increase in fluorescence, which we attribute to unbinding of Arr2 from rhabdomeric NINAC p174, appeared unaffected (Fig. 4A). Interestingly, we found an even more pronounced time-dependent slowing of translocation in ninaCP235 photoreceptors, which was also more conspicuous because of the absence of the initial rapid phase (Fig. 4B). To quantify this behavior we estimated the time taken to reach 50% maximum increase in fluorescence (t1/2), discounting the initial rapid phase in wild type (by zeroing the baseline after the rapid phase ∼500 ms after light onset). On this measure, the slowing of translocation in ninaC was significantly more pronounced than in wild-type, and could be fitted by a single exponential time constant of ∼8 min dark-adaptation time. A very similar behavior was also observed in ninaCΔC1 mutants lacking the CaM binding site that is common to both NINAC p132 and p174 isoforms (Fig. 4C).
Figure 4.

Evidence for an alternative cytosolic sink in ninaC mutants. A–C, Effect of dark adaptation (DA). A, Arr2-GFP translocation measured from the DPP in a control fly (expressing Arr2-GFP on arr2 mutant background), after 30 s, 5 min, and 30 min in the dark following photo-equilibrating orange illumination. Although the rapid phase (arrow) was unaffected, translocation was slower after prolonged dark adaptation. Inset, Shows detail of first 5 s. B, Similar protocol in ninaCP235-null mutant. The fast phase was absent, but the slowing of translocation with dark adaptation was more pronounced. C, t1/2 of translocation as a function of dark adaptation time in ninaC mutant (ninaCP235, solid symbols; ninaCΔC1, open symbols) and wild-type backgrounds. Mean ± SEM (n = 5–6 flies). ninaCP235 data fitted with a single exponential (τ = 7.6 min). D–F, Effect of pre-illumination. D, Translocation time course of Arr2-GFP in wild-type background measured (from DPP) after 0.5 and 15 min DA (solid traces) and after 15 min DA, but with either 0.1 or 30 s orange pre-illumination immediately (∼2 s) before measurement (gray traces). As in A, translocation was slowed following 15′ DA; orange pre-illumination resulted in a reduced initial fluorescence signal, whereafter translocation was initially faster, finally approximately overlapping the 15 min DA “control” trace. Pre-illumination with even 0.1 s orange light was sufficient to achieve most of the effect (representative of n = 5). Inset, Shows first 5 s. E, Similar protocol in ninaCP235: translocation was even slower, now after only 10 min DA. Orange pre-illumination after 10 min DA accelerates translocation, but at least 60 s were required to accelerate translocation to rates seen after only 0.5 min DA. F, t1/2 of Arr2-GFP translocation in 10 min dark-adapted ninaCP235 flies as a function of the duration of orange pre-illumination delivered immediately before measuring translocation (mean ± SEM n = 4). Data fitted with single exponential (14.9 s). Open symbol: control, t1/2 for translocation measured after only 0.5 min DA in the same flies.
If ninaC mutants were first dark adapted for sufficiently long to induce a pronounced slowing of the response (10–15 min), but then pre-illuminated with bright orange light (causing no net change in M fraction) before measuring (blue-induced) translocation, the time course of translocation could be restored to values almost as rapidly as when tested after only 30 s dark-adaptation time (Fig. 4E). The extent of acceleration increased with the duration of the pre-illumination, and could be fit with a time constant of ∼15 s pre-illumination time (Fig. 4F). This suggests that the orange pre-illumination resulted in the gradual release of Arr2 from some binding target(s) or compartment(s). We suggest the most likely mechanism for this behavior in ninaC mutants is release from a second Ca2+-dependent site/compartment with binding/sequestration kinetics (under low Ca2+ conditions in the dark) on the order of minutes and release kinetics (following light-induced Ca2+ influx) on the order of tens of seconds.
In wild-type photoreceptors, a rather different behavior was observed (Fig. 4D): following prolonged dark adaptation, brief orange pre-illumination resulted in a reduced initial fluorescence on subsequent blue excitation, presumably representing movement of Arr2 out of the rhabdomere (see further below). The initial translocation then appeared accelerated compared with controls kept for the same time in the dark without pre-illumination, but at later stages followed a time course that approximately overlapped the control time course. In further marked contrast to ninaC mutants, only a very brief pre-illumination was required for this effect, with a 100 ms orange flash essentially as effective as 60 s.
We interpret these results as follows: in ninaC mutants, Arr2 slowly (τ ∼ 8 min) becomes sequestered in the dark by one or more binding site(s)/compartment(s), which then release Arr2 on a relatively slow timescale of several seconds (τ ∼15 s) during orange pre-illumination. Without pre-illumination, translocation in ninaC in response to blue light after several minutes in the dark is substantially slowed for the same reason (i.e., slow sequestration in the dark followed by slow release from this second sink). We suggest that in wild-type cells, Arr2 is protected from sequestration by this potential sink because it binds with higher affinity to NINAC under the low Ca2+ conditions prevailing in the dark. Following light-induced Ca2+ influx, however, NINAC's affinity for Arr2 drops dramatically resulting in rapid release on a subsecond timescale (Fig. 3). The less severe slowing of translocation with dark adaptation still seen in wild-type flies might represent the gradual movement of Arr2 (presumably still bound to NINAC) into regions of the cell, on average more distant from the rhabdomere. The effect of pre-illumination in wild-type photoreceptors can be interpreted as rapid release of Arr2 from NINAC in the microvilli, resulting in a net redistribution of Arr2 into the cell body thus accounting for the reduced initial fluorescence (see further below). Translocation measured immediately after this then starts more rapidly as there is now a new pool of cytosolic Arr2, which has just vacated the rhabdomere.
Electrophysiological correlates of slowed translocation
We previously reported that the time course of translocation could be resolved in the decay of ERG responses to carefully calibrated stimuli (Satoh et al., 2010). In fully dark-adapted cells with high R (and low M) fraction, ∼25% of the cell's Arr2 is already in the rhabdomere in the dark (equivalent to ∼100 molecules per microvillus). This “first responder” pool of Arr2 rapidly binds to and inactivates M until it is all bound (i.e., until ∼100 rhodopsin molecules per microvillus are photo-isomerized to M equivalent to an M fraction of ∼10%). Until this happens, the ERG recovers rapidly to baseline (t1/2 ∼2–3 s); however, any M generated in excess of ∼10% can only be inactivated by Arr2 from the cytosolic pool translocating from the cell body, and hence with stimulation generating >10% M, the ERG decays more slowly reflecting the time course of translocation (Satoh et al., 2010). We therefore predicted that the time course of the ERG decay to wavelengths generating >10% M should show a similar dependence on the preceding dark-adaptation period, with a more pronounced slowing expected in ninaC mutants. As shown in Figure 5 this was indeed observed, with decay time course to a 537 nm stimulus (generating ∼20% M), increasing as the dark-adaptation period was increased, but with a significantly more pronounced slowing measured in ninaC mutants (Fig. 5B). Note that the wavelength for these experiments was carefully chosen individually in each fly to achieve a similar translocation-limited slowing of the ERG decay, but without inducing the “prolonged depolarizing afterpotential,” which ensues when the amount of M generated out titrates all available arrestin (Dolph et al., 1993). It was necessary to select slightly shorter wavelengths (Fig. 5B, 532 nm) for this experiment in ninaC mutants because of their lower rhodopsin/Arr2 ratio (Satoh et al., 2010).
Figure 5.
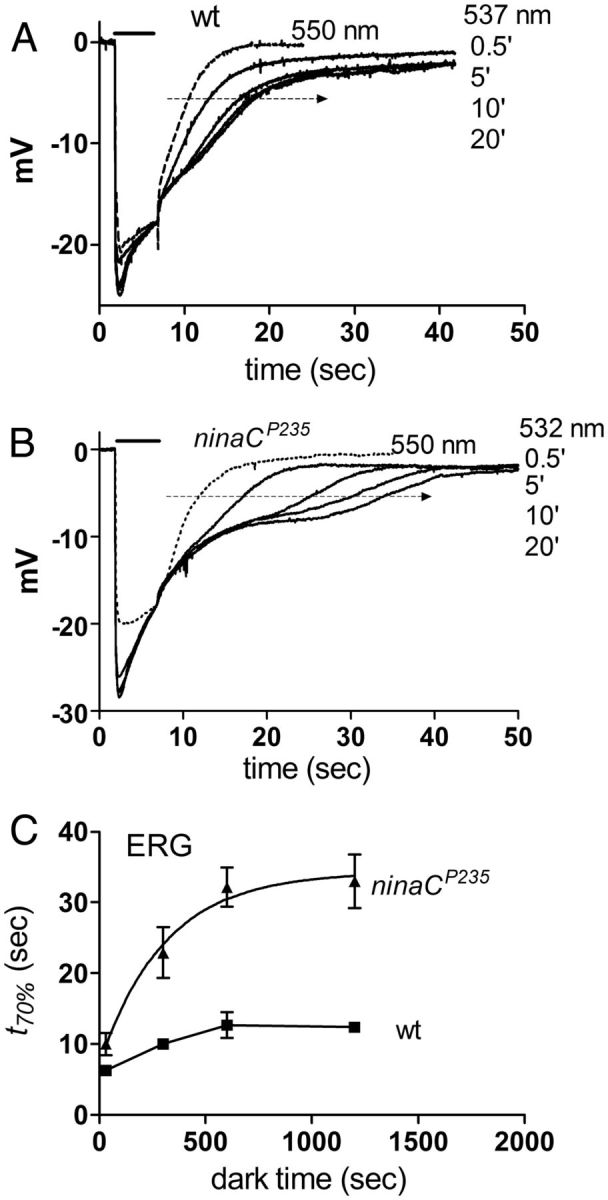
Electrophysiological correlates of slowed translocation. A, ERG recording from wild-type (w1118) in response to 5 s photo-equilibrating 537 nm monochromatic light (bar) after varying times in the dark. A response to 550 nm (dotted trace), which elicited no translocation is also shown. There was a significant slowing of the decay as dark-adaptation time (in each case following photo-equilibrating orange illumination) was increased from 0.5 to 20 min. B, Similar protocol in a ninaCP235-null mutant. The decay of the ERG was now more obviously prolonged with increasing dark adaptation. The wavelength was adjusted individually for each fly (in this case 532 nm) such that the ERG decay was limited by translocation time course (see text and Satoh et al., 2010, for further details). C, Decay time of the ERG (t70%: measured from the end of the 5 s light pulse to 70% decay—i.e., the level indicated by arrows in A and B) as a function of dark adaptation time in wild-type (wt) and ninaCP235 flies (mean ± SEM n = 4–5). ninaC data are fitted with a single exponential (τ = 4.5 min). The slowing of the response with increasing dark time closely reflects the effect of dark adaptation on the translocation time course (compare Fig. 4C).
NINAC asymmetry allows Ca2+-dependent translocation without net M generation
The rapid, albeit slight, reduction in Arr2-GFP fluorescence seen following orange pre-illumination in a wild-type background, but not in ninaC mutants (Fig. 5), was interpreted as rapid Ca2+/CaM-dependent release of Arr2 from rhabdomeric NINAC, resulting in a net redistribution of Arr2 to the cytosol, despite no net change in M fraction. However, we also propose that Ca2+ influx releases Arr2 from the cytosolic NINAC p132 isoform in a similar manner. Hence, if this interpretation is correct, it presumably relies upon an imbalance between the overall binding capacity of rhabdomeric and cytosolic NINAC isoforms. To explore this further we investigated the consequence of generating much larger imbalances by using flies expressing only the cytosolic or rhabdomeric NINAC isoform, respectively. Specifically, in ninaCΔ174 mutants lacking rhabdomeric p174 we predicted that Ca2+ influx induced by long wavelength light should release cytosolic Arr2 from p132 resulting in net redistribution of Arr2GFP into the rhabdomere. Conversely, in ninaCΔ132 photoreceptors lacking the p132 cytosolic isoform, we predicted that Ca2+ influx alone would result in a net movement of Arr2-GFP out of the rhabdomere.
To test these predictions, we measured rhabdomeric Arr2-GFP fluorescence in the DPP of intact flies. Before blue excitation the eye was stimulated with two 4 s pulses of photo-equilibrating orange (O) illumination, which sets the M fraction to low levels (<2% M). The flashes were either delivered together, followed by 30 s dark adaptation before the blue (B) test excitation (30 s DA), or separated by 30 s, with the second pulse terminating only 1 s before the blue excitation (Fig. 6, 1 s DA). Under these conditions, Ca2+ levels should have returned to low basal levels after 30 s, while the second 4 s orange flash would generate massive Ca2+ influx, but without any net change in the M fraction.
Figure 6.

Arr2 translocation driven by NINAC asymmetry. Arr2-GFP fluorescence in rhabdomeres measured from DPP with 1 s blue (B) excitation. Before excitation the eye was stimulated with two 4 s pulses of photo-equilibrating orange (O) illumination delivered either together but allowing 30 s dark adaptation before the blue (B) test excitation (30 s DA) or separated by 30 s, with the second pulse terminating only 1 s before the blue excitation (1 s DA). A, In wild-type controls, expressing both rhabdomeric and cytosolic forms of NINAC, there was a slight decrease in Arr2-GFP fluorescence (movement out of rhabdomere) when the orange light was delivered only 1 s before measurement (two repeated traces for each condition). B, In contrast, in ninaCΔ174 flies lacking rhabdomeric NINAC but with p132 in the cell body there was a significant increase in Arr2-GFP fluorescence in the rhabdomere (note also absence of the rapid phase). C, In ninaCΔ132 flies expressing rhabdomeric NINAC, but none in the cell body, the same stimulus resulted in a large movement of Arr2-GFP out of the rhabdomere. D, Bar graph summarizing data expressed as relative fluorescence increase (movement into rhabdomere) or decrease (movement out of rhabdomere into cell body) in measurements made when the second orange flash was delivered 1 s before measurement compared with measurement made with 30 s DA. Data normalized with respect to total range of translocation (Fmax − Fmin), where Fmin is the fluorescence at time 0 for whichever condition gave the lower fluorescence and Fmax determined after 60 s full translocation (data not shown).
In control flies (which included flies expressing Arr2-GFP on wild-type, ninaCP235/+ heterozygotes, and arr23 mutant backgrounds) when the second flash was delivered 1 s before testing (1 s DA), there was a slight (7% n = 12), but highly significant (two-tailed t test p < 10−9) reduction in the level of Arr2-GFP in the rhabdomere (Fig. 6A) as also noted above (Fig. 4D). However, in ninaCΔ174 flies lacking the rhabdomeric NINAC isoform, there was a small (6%), but highly significant (p < 10−7) increase in Arr2-GFP fluorescence indicative of net translocation into the rhabdomere as predicted. In ninaCΔ132 flies lacking cytosolic p132, the effect of delivering the second flash only 1 s before measurement was yet more pronounced, with a ∼40% reduction in normalized fluorescence, indicating a large net reverse translocation out of the rhabdomere into the cell body (Fig. 6C). This reflected abnormally high levels of Arr2 in the dark-adapted rhabdomere, presumably sequestered by binding to rhabdomeric p174 under low Ca2+ conditions, now unopposed by a p132 sink in the cytosol (Fig. 2D).
Rescue of rapid translocation in trp and norpA by ninaC mutation
Arr2 translocation is slowed by at least an order of magnitude in phototransduction mutants such as trp and norpA, where there is little or no Ca2+ influx (Satoh et al., 2010). In trp mutants (lacking the major light-sensitive channel), Ca2+ influx is reduced, both because the remaining TRP-like (TRPL) channels have a reduced Ca2+ permeability, and also because the electrophysiological response to light decays to baseline within seconds of bright illumination (Cosens and Manning, 1969; Hardie and Minke, 1992); while in null norpA mutants there is no response to light at all. If, the impaired translocation in norpA and trp is due to failure of Ca2+-dependent release of Arr2 from NINAC, we predicted that translocation might be rescued in ninaCP235;trp343 and norpAP24;ninaCP235 double mutants. To test this we expressed Arr2-GFP in ninaCP235;trp343 and norpAP24;ninaCP235 double mutants and monitored translocation in vivo using the DPP.
As previously reported (Satoh et al., 2010), in single trp mutant controls translocation initially starts normally, but stalls after ∼5 s (presumably due to the decay of the light response) and then proceeds with a profoundly slowed time course. As predicted, however, in the ninaCP235;trp343 double mutant, full translocation was observed with a time course almost as fast as that in wild-type or ninaC single mutant backgrounds (Fig. 7A,B), even though the electrophysiological light response decays in a similar manner to single trp mutants (Fig. 7C).
Figure 7.
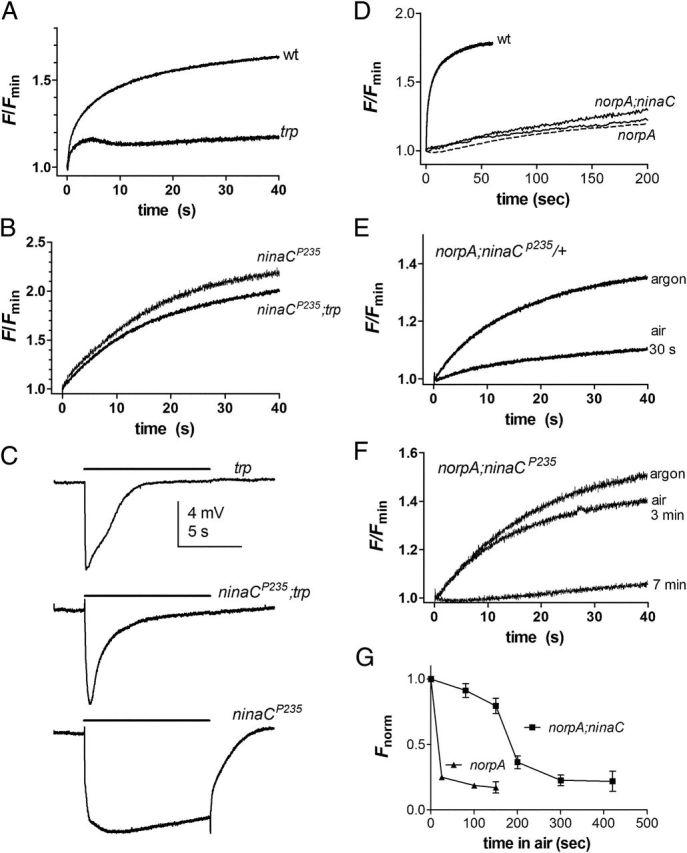
Translocation in ninaC;trp and norpA;ninaC double mutants. A, Arr2-GFP fluorescence increase measured from DPP in an otherwise wild-type fly and a trp343-null mutant. As previously reported, translocation in trp mutants “stalls” after a few seconds and then progresses very slowly. A fast phase is, however, still apparent. B, In contrast, Arr2-GFP translocation in ninaCP235;trp343 double mutant is rescued and similar to ninaC single mutant control (n = 4). C, ERG responses to the 10s bright yellow illumination (same intensity) in trp, ninaCP235;trp, and ninaCP235 (representative of n ≥ 3 flies). D, Arr2-GFP translocation in norpAP24;ninaCP235 double mutants (two examples) and norpAP24 control (dotted trace). In both cases translocation was extremely slow compared with wild type. E, As previously reported, rapid translocation in a norpAP24 control (actually norpA;ninaC/+) was rescued by anoxia induced by streaming argon over the fly; but largely suppressed in the first measurement 30 s after return to air. F, In contrast, in norpAP24;ninaCP235 double mutants, near normal translocation was still observed 3 min after return to air, finally stalling after 7 min. G, Time course of suppression of Arr2-GFP translocation on return to air after rescue by argon in norpAP24;ninaCP235 and norpA controls (norpAP24;ninaCP235/+ heterozygotes from same vials). Fnorm is the fluorescence increase after 40 s blue excitation (as in E and F) normalized to maximum increase seen under argon (Mean ± SEM n = 3–5).
In norpAP24-null mutants, as previously described, translocation is even more severely impaired than in trp with only a relatively small increase in fluorescence with a time course ∼10-fold to 100-fold slower than in wild type (Satoh et al., 2010). In contrast to the rescue just described for trp mutants, in norpAP24;ninaCP235 double mutants, there seemed to be no rescue of Arr2 translocation, and Arr2-GFP translocated very slowly with a time course similar to that seen in norpA controls (Fig. 7C). This would, however, be consistent with the existence of the proposed second Ca2+-dependent sink. Thus, in norpAP24;ninaCP235 flies there is never any light-dependent Ca2+ influx, and in the absence of NINAC, we would therefore predict that Arr2 should have become firmly bound or sequestered through this second Ca2+-dependent sequestration mechanism.
To test this further we exploited our recent demonstration that it is possible to rescue Arr2-GFP translocation in norpA by anoxia, which leads to Ca2+ influx by spontaneous activation of the light-sensitive TRP channels (Satoh et al., 2010). In single norpA mutant controls (in this case norpAP24;ninaCP235/+ flies from the same vials), rapid translocation was rescued by anoxia, but stalled within only a few seconds of return to air (Fig. 7D) as previously reported (Satoh et al., 2010). On our model, this is because Arr2 rapidly binds to cytosolic NINAC as soon as the channels close and Ca2+ is extruded from the cell on re-oxygenation. In norpAP24;ninaCP235 double mutants, however, the results were clearly different. Once again, translocation was dramatically rescued by anoxia; however, now on return to air, rapid reversible translocation could still be induced for several minutes before finally stalling, presumably because of the much slower immobilization by the second putative Ca2+-dependent sequestration mechanism (Fig. 7F,G).
Translocation is normal in rac2 mutants
The results presented here are consistent with, and interpreted within, a model of translocation whereby Arr2 is proposed to move between cell body and rhabdomere by diffusion along gradients established by cytosolic and rhabdomeric sinks (see Fig. 9). This mechanism contrasts with an earlier model in which NINAC was proposed as an obligatory molecular motor (Lee and Montell, 2004). Our repeated demonstration of robust translocation in ninaC mutants is clearly inconsistent with this earlier model, while indicating a novel role for NINAC in Ca2+-dependent regulation. However, a recent report that Arr2 translocation was severely impaired in mutants of the small GTP-ase Rac2 (Elsaesser et al., 2010) was difficult to understand on the relatively simple diffusional model we propose. We therefore reinvestigated Arr2 translocation in null rac2Δ mutants using both immunocytochemistry of endogenous Arr2 and Arr2-GFP expressed in the rac2Δ mutant background. In both cases, we found that Arr2 translocated normally, with no significant differences in either extent or time course from wild-type controls (Fig. 8). We also measured the spectral dependence of Arr2-GFP translocation in rac2Δ mutants and found that Arr2-GFP translocated in a wavelength-dependent manner indicative of a stoichiometric relationship with photo-isomerized rhodopsin (M) as previously shown in wild-type photoreceptors (Satoh et al., 2010).
Figure 9.

Multisink model for Ca2+-dependent Arr2 translocation. Left, In the dark-adapted state the visual pigment is in the rhodopsin state (R; blue) with negligible affinity for Arr2 (green). Most Arr2 is bound to either the cytosolic (p132) or microvillar (p174) isoform of NINAC, a CaM binding myosin III. NINACp174 is attached to a central actin filament found in each microvillus (Hicks et al., 1996). In the cell body, Arr2 and NINAC p132 are shown associated with endomembrane, because cytosolic Arr2 was previously reported to colocalize with an endomembrane marker (Satoh et al., 2010), but this is not critical for the model. In ninaCP235-null mutants, Arr2 becomes bound or sequestered to an alternative cytosolic site with slow kinetics (not shown). The identity of this site is unknown: one candidate may be a negatively charged phosphoinositide species on the endomembrane. By analogy with vertebrate rods, microtubules are another candidate. Center, Bright long wavelength (orange) illumination leads to activation of the light-sensitive TRP channels, resulting in massive Ca2+ influx, which rapidly (<1 s) releases Arr2 from both p174 and p132: any M (red) formed is rapidly inactivated by high-affinity binding to Arr2, which is now freely diffusible. In wild-type photoreceptors orange light induces minimal translocation of Arr2 because only a tiny fraction (∼1–2%) of the visual pigment is photo-isomerized to M. However, in mutants lacking p132 or p174, the asymmetry in NINAC distribution results in translocation of Arr2 from rhabdomere to cell body or vice versa in response to orange light (Fig. 6). Right, Bright blue illumination again activates Ca2+ influx, thereby releasing Arr2 from NINAC; however, now the majority (∼70%) of visual pigment is converted to M, which acts as a high-affinity sink driving translocation to the rhabdomere (Satoh et al., 2010).
Figure 8.

Normal Arr2 translocation in rac2 Δ mutants. A, Arr2-GFP translocation measured from DPP of wild-type and rac2Δ mutant. Both fast (putatively representing dissociation of Arr2 from NINAC) and slow (translocation) phases were similar. B, Bar graphs showing time constant (tau) of translocation, time constant of rapid phase, and extent of translocation (Fmax/Fmin) in rac2Δ (mean ± SEM n = 4) and wild-type controls (n = 11–12). No significant difference was observed in any of these parameters (t test p > 0.5). C, Translocation as a function of wavelength in rac2Δ mutants (mean ± SEM n = 2); each point represents Arr2-GFP fluorescence measured immediately (<100 ms) after onset of blue excitation following photo-equilibration to a different wavelength of monochromatic light, normalized to Fmax (Satoh et al., 2010). Dotted line wild-type data replotted from Satoh et al. (2010). D, Ommatidia labeled with a-Arr2 antibody in flies fixed in dark under infrared illumination (left) and after 3 min in the dark following 2 min exposure to bright blue light (right) in wild-type (top) and rac2Δ mutants (bottom). Both show similar translocation to rhabdomeres (representative of n = 3 dark and 4 blue-adapted rac2Δ flies; and 4–7 wild type). Scale bar, 5 μm.
Discussion
We previously proposed that Ca2+ influx via the light-sensitive TRP channels is required for rapid Arr2 translocation, because the slow translocation in trp mutants could be rescued by genetic elimination of Na+/Ca2+ exchanger activity (Satoh et al., 2010). In the present study, we confirmed the role of Ca2+ directly by imaging Arr2-GFP translocation in dissociated ommatidia, and showing that extracellular Ca2+ is required for rapid Arr2 translocation. Ca2+ was not only required, but also sufficient to enable rapid translocation without any products of PLC activity, since the Ca2+ ionophore, ionomycin, fully rescued translocation in blind norpA mutants lacking PLC. Significantly, we found that the requirement of Ca2+ for rapid translocation was obviated in null mutants of NINAC (CaM binding MyoIII), with the cytosolic p132 isoform of NINAC alone being sufficient to slow down translocation in Ca2+-free conditions. This suggests that cytosolic NINAC p132 acts as a Ca2+/CaM-dependent “brake” on translocation by binding Arr2, releasing it in response to Ca2+ influx associated with the photoresponse. This conclusion was further supported by finding that the ninaC mutation rescued rapid translocation in trp mutants (in ninaC;trp double mutants). However, the failure to rescue translocation in norpA;ninaC mutants, except under special conditions, and the demonstration of significant slowing of translocation following prolonged dark adaptation in ninaC mutants also indicated the existence of a second Ca2+-dependent cytosolic sink.
ninaC phenotypes
Despite an earlier study reporting that Arr2 translocation was impaired in ninaC mutants (Lee and Montell, 2004), as shown here and previously (Satoh and Ready, 2005; Satoh et al., 2010), Arr2 translocation, whether of endogenous Arr2 or GFP-tagged Arr2, appears essentially intact in ninaC-null mutants. In fact, far from being impaired, our results indicate that translocation can be rescued by ninaC mutations under conditions where translocation is slowed down by reduced Ca2+ influx. Although translocation was significantly slower in ninaC mutants following prolonged dark adaptation, it was never prevented and full translocation was always achieved within ∼2–3 min of appropriate illumination.
A novel phenotype of ninaCP235-null mutants, and also ninaCΔ174 mutants lacking only the rhabdomeric p174 isoform, was the complete absence of an early rapid increase in fluorescence routinely observed during the first ∼500 ms of measurements of Arr2-GFP fluorescence from the DPP of wild-type photoreceptors or dissociated ommatidia. With a time constant of ∼260 ms, this rapid phase was ∼40× faster than the overall translocation (τ ∼ 10 s) itself the fastest protein translocation reported in a photoreceptor to date, and probably diffusionally limited (Satoh et al., 2010). It therefore seems unlikely that the fast phase represents a 40× faster, ninaC-dependent movement of Arr2 from cell body into the rhabdomere. Instead we suggest that it represents a change in the fluorescence efficiency of Arr2-GFP as it is released (via Ca2+ influx) from the rhabdomeric p174 NINAC isoform. A lower fluorescence when bound to NINAC might reflect crowding of the GFP-fluorophore, or could be due to some other feature of the nano-environment of the fluorophore when Arr2 is bound to NINAC in the microvilli. This interpretation was supported by the ability to eliminate the rapid phase by pre-illumination with long wavelength light, which induces Ca2+ influx without net change in M. The rapid phase then re-emerged with a time constant of ∼3 s in the dark, presumably representing rapid rebinding of Arr2-GFP to NINAC (Fig. 3).
A second Ca2+-dependent cytosolic sink
After more than a few minutes in the dark, Arr2 translocation into the rhabdomere became progressively slower, with clear functional consequences in a parallel slowing of the decay of the ERG (Figs. 4, 5). This gradual slowing was considerably more pronounced in ninaC-null mutants, where we propose that it represents binding or sequestration of Arr2 via one or more NINAC-independent target(s) or compartment(s). Release from such sites also requires activation of the phototransduction cascade, and translocation could be accelerated back to levels typical of short dark-adaptation times by pre-illumination with bright orange light, which itself does not generate a net increase in M. It seems likely that the rise in Ca2+ is also responsible for release from this site; however, we cannot exclude the involvement of other products of the phototransduction cascade. The identity of this second site or compartment remains a subject for future investigation. Given previous reports that Arr2 can bind to phosphoinositides (Lee et al., 2003; Lee and Montell, 2004), negatively charged phosphoinositide species on endomembranes, which could be screened by Ca2+, might represent promising candidates. Drosophila Arr2 is an unusually basic (positively charged) protein (Alloway and Dolph, 1999) and may thus have a strong tendency to bind to such sites. The finding that the slowing of translocation with dark adaptation was more pronounced in ninaC mutants suggests that one of the functions of cytosolic NINAC may be to prevent immobilization of Arr2 by this alternative potential sink. Because the Ca2+-dependent release of Arr2 from NINAC occurs on a subsecond timescale, this then allows more rapid translocation (and hence recovery of the electrical response) after a period in the dark.
A common mechanism for Ca2+-dependent regulation of response termination and translocation
Our results demonstrate that Arr2 translocation is accelerated by Ca2+ influx, and suggest that this is mediated by a disinhibitory mechanism, whereby NINAC p132 binds to Arr2 under low Ca2+ conditions in the dark, rapidly releasing it in response to Ca2+ influx associated with the photoresponse (Fig. 9). Although inferred from essentially independent experiments, this mechanism is strikingly similar to one previously proposed for the rapid, Ca2+-dependent inactivation of M during the light response itself (Liu et al., 2008). In that study the time constant of M inactivation by Arr2 was found to be accelerated from ∼200 ms under Ca2+-free conditions to ∼20 ms following Ca2+ influx. This Ca2+ dependence was eliminated in both ninaC-null mutants, and in ninaCΔ174 mutants lacking the rhabdomeric p174 (but not in ninaCΔ132 mutants lacking cytosolic p132). The results also indicated a disinhibitory mechanism, leading us to propose that Arr2 in the microvilli was bound to rhabdomeric NINAC p174 under low Ca2+ conditions in the dark, thus hindering its diffusional access to activated M. Ca2+ influx via the first activated TRP channels, then rapidly releases Arr2, allowing it to diffuse, bind to, and inactivate M.
NINAC p132 and p174 share a common CaM binding site (CBS) and although p174 has a second CBS not found in p132 (Porter et al., 1993, 1995), the pronounced slowing of translocation with dark adaptation in null ninaCP235 mutants was recapitulated in mutants lacking the common CBS (Fig. 4E). We therefore suggest that essentially the same mechanism underlies the Ca2+-dependent rapid translocation of Arr2, but now acting via NINAC p132 rather than p174 and working over much larger distances (several micrometers as opposed to the nanometer dimensions of single microvilli) and hence slower timescales.
The proposed interaction between NINAC and Arr2 finds some support from biochemical data reporting coimmunoprecipitation of Arr2 and NINAC in extracts from whole heads (Lee and Montell, 2004). However, these authors also reported that both Arr2 and NINAC had significant in vitro affinity for phosphoinositides. They proposed that the NINAC/Arr2 association was indirect and mediated by both Arr2 and NINAC binding to phosphoinositide-rich membrane. Although our results clearly indicate that Ca2+-dependent modulation of Arr2 binding to M (Liu et al., 2008) and Ca2+-dependent translocation of Arr2 are both dependent upon NINAC, we cannot exclude the possibility that the interaction is mediated indirectly via a NINAC-dependent target. Ultimate verification will require direct demonstration of NINAC/Arr2 binding and its dependence upon Ca2+/CaM.
Concluding remarks
The regulated multisink model proposed here (Fig. 9) differs fundamentally from an earlier model in which NINAC was proposed as a molecular motor transporting Arr2 in phosphoinositide-rich vesicles (Lee and Montell, 2004). By contrast it shows strong parallels with current models for arrestin translocation in vertebrate rods (Calvert et al., 2006; Slepak and Hurley, 2008). Here, phosphorylated rhodopsin represents the light-activated sink in the outer segments, while microtubules have been proposed as the cytosolic sink in the inner segments (Nair et al., 2005). There is also evidence indicating light-regulated acceleration of translocation in vertebrate rods (Strissel et al., 2006). The mechanism is unclear; however, intriguingly a recent study has implicated roles for PLC and protein kinase C possibly stimulating release of arrestin from its cytosolic sink (Orisme et al., 2010).
Such regulated-sink models have the advantage of simplicity: directed translocation requires no more than diffusion coupled with regulated binding, can rapidly transport virtually unlimited quantities of protein, and per se consumes essentially no energy (Nair et al., 2005; Slepak and Hurley, 2008) (but see Orisme et al., 2010). While it can be conveniently studied in photoreceptors with their distinctive polarized morphologies and high concentrations of transduction machinery, translocation according to the same general principles may represent a general and elegant solution to the problem of directed movements of signaling proteins.
Footnotes
This research was supported by grants from the Biotechnology and Biological Sciences Research Council (BB/G0068651) to R.C.H. and the Naito Foundation (25-040920) and KAKENHI (21687005 and 23113712) to A.K.S. We thank Drs. M. Postma and D.F. Ready for critical comments on earlier versions of the manuscript.
References
- Alloway PG, Dolph PJ. A role for the light-dependent phosphorylation of visual arrestin. Proc Natl Acad Sci U S A. 1999;96:6072–6077. doi: 10.1073/pnas.96.11.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Belusic G, Pirih P, Stavenga DG. Photoreceptor responses of fruitflies with normal and reduced arrestin content studied by simultaneous measurements of visual pigment fluorescence and ERG. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2010;196:23–35. doi: 10.1007/s00359-009-0489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist BT, Shortridge RD, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak WL. Isolation of putative phospholipase C gene of Drosophila, norpA and its role in phototransduction. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- Broekhuyse RM, Tolhuizen EF, Janssen AP, Winkens HJ. Light induced shift and binding of S-antigen in retinal rods. Curr Eye Res. 1985;4:613–618. doi: 10.3109/02713688508999993. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Strissel KJ, Schiesser WE, Pugh EN, Jr, Arshavsky VY. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006;16:560–568. doi: 10.1016/j.tcb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Cosens DJ, Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- Dolph PJ, Ranganathan R, Colley NJ, Hardy RW, Socolich M, Zuker CS. Arrestin function in inactivation of G protein-coupled receptor rhodopsin in vivo. Science. 1993;260:1910–1916. doi: 10.1126/science.8316831. [DOI] [PubMed] [Google Scholar]
- Elsaesser R, Kalra D, Li R, Montell C. Light-induced translocation of Drosophila visual Arrestin2 depends on Rac2. Proc Natl Acad Sci U S A. 2010;107:4740–4745. doi: 10.1073/pnas.0906386107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frechter S, Minke B. Light-regulated translocation of signaling proteins in Drosophila photoreceptors. J Physiol Paris. 2006;99:133–139. doi: 10.1016/j.jphysparis.2005.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC. Whole-cell recordings of the light-induced current in Drosophila photoreceptors: evidence for feedback by calcium permeating the light sensitive channels. Proc Roy Soc Lond B Biol Sci. 1991;245:203–210. [Google Scholar]
- Hardie RC. Phototransduction mechanisms in Drosophila microvillar photoreceptors. WIRES Membr Transp Signal. 2012;1:162–187. [Google Scholar]
- Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- Hardie RC, Martin F, Cochrane GW, Juusola M, Georgiev P, Raghu P. Molecular basis of amplification in Drosophila phototransduction. Roles for G protein, phospholipase C, and diacylglycerol kinase. Neuron. 2002;36:689–701. doi: 10.1016/s0896-6273(02)01048-6. [DOI] [PubMed] [Google Scholar]
- Hicks JL, Liu X, Williams DS. Role of the NinaC proteins in photoreceptor cell structure: ultrastructure of ninaC deletion mutants and binding to actin filaments. Cell Motil Cytoskeleton. 1996;35:367–379. doi: 10.1002/(SICI)1097-0169(1996)35:4<367::AID-CM8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Katz B, Minke B. Drosophila photoreceptors and signaling mechanisms. Front Cell Neurosci. 2009;3:2. doi: 10.3389/neuro.3303.3002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev A, Socolich M, Vinós J, Hardy RW, Zuker CS, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Montell C. Light-dependent translocation of visual arrestin regulated by the NINAC myosin III. Neuron. 2004;43:95–103. doi: 10.1016/j.neuron.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Xu H, Kang LW, Amzel LM, Montell C. Light adaptation through phosphoinositide-regulated translocation of Drosophila visual arrestin. Neuron. 2003;39:121–132. doi: 10.1016/s0896-6273(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Liu CH, Satoh AK, Postma M, Huang J, Ready DF, Hardie RC. Ca2+-dependent metarhodopsin inactivation mediated by Calmodulin and NINAC myosin III. Neuron. 2008;59:778–789. doi: 10.1016/j.neuron.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B, Kirschfeld K. The contribution of a sensitizing pigment to the photosensitivity spectra of fly rhodopsin and metarhodopsin. J Gen Physiol. 1979;73:517–540. doi: 10.1085/jgp.73.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C, Rubin GM. The Drosophila ninaC locus encodes two photoreceptor cell specific proteins with domains homologous to protein kinases and the myosin heavy chain head. Cell. 1988;52:757–772. doi: 10.1016/0092-8674(88)90413-8. [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Nardine T, Harms M, Tzu J, Goldstein A, Sun Y, Dietzl G, Dickson BJ, Luo L. Rac GTPases control axon growth, guidance and branching. Nature. 2002;416:442–447. doi: 10.1038/416442a. [DOI] [PubMed] [Google Scholar]
- Orisme W, Li J, Goldmann T, Bolch S, Wolfrum U, Smith WC. Light-dependent translocation of arrestin in rod photoreceptors is signaled through a phospholipase C cascade and requires ATP. Cell Signal. 2010;22:447–456. doi: 10.1016/j.cellsig.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearn MT, Randall LL, Shortridge RD, Burg MG, Pak WL. Molecular, biochemical, and electrophysiological characterization of Drosophila norpA mutants. J Biol Chem. 1996;271:4937–4945. doi: 10.1074/jbc.271.9.4937. [DOI] [PubMed] [Google Scholar]
- Peterson JJ, Tam BM, Moritz OL, Shelamer CL, Dugger DR, McDowell JH, Hargrave PA, Papermaster DS, Smith WC. Arrestin migrates in photoreceptors in response to light: a study of arrestin localization using an arrestin-GFP fusion protein in transgenic frogs. Exp Eye Res. 2003;76:553–563. doi: 10.1016/s0014-4835(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Porter JA, Hicks JL, Williams DS, Montell C. Differential localizations of and requirements for the two Drosophila ninaC kinase/myosins in photoreceptor cells. J Cell Biol. 1992;116:683–693. doi: 10.1083/jcb.116.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JA, Yu M, Doberstein SK, Pollard TD, Montell C. Dependence of calmodulin localization in the retina on the NINAC unconventional myosin. Science. 1993;262:1038–1042. doi: 10.1126/science.8235618. [DOI] [PubMed] [Google Scholar]
- Porter JA, Minke B, Montell C. Calmodulin binding to Drosophila NinaC required for termination of phototransduction. EMBO J. 1995;14:4450–4459. doi: 10.1002/j.1460-2075.1995.tb00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh AK, Ready DF. Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol. 2005;15:1722–1733. doi: 10.1016/j.cub.2005.08.064. [DOI] [PubMed] [Google Scholar]
- Satoh AK, Xia H, Yan L, Liu CH, Hardie RC, Ready DF. Arrestin translocation is stoichiometric to rhodopsin isomerization and accelerated by phototransduction in Drosophila photoreceptors. Neuron. 2010;67:997–1008. doi: 10.1016/j.neuron.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K, Sun Y, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–383. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- Slepak VZ, Hurley JB. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: interaction-restricted diffusion. IUBMB Life. 2008;60:2–9. doi: 10.1002/iub.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. 2007;454:821–847. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- Yau KW, Hardie RC. Phototransduction motifs and variations. Cell. 2009;139:246–264. doi: 10.1016/j.cell.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]