

Abstract
Neurotrophin-3 (NT-3) plays numerous important roles in the CNS and the elevation of intracellular Ca2+ ([Ca2+]i) is critical for these functions of NT-3. However, the mechanism by which NT-3 induces [Ca2+]i elevation remains largely unknown. Here, we found that transient receptor potential canonical (TRPC) 5 protein and TrkC, the NT-3 receptor, exhibited a similar temporal expression in rat hippocampus and cellular colocalization in hippocampal neurons. Stimulation of the neurons by NT-3 induced a nonselective cation conductance and PLCγ-dependent [Ca2+]i elevation, which were both blocked when TRPC5, but not TRPC6 channels, were inhibited. Moreover, the Ca2+ influx through TRPC5 induced by NT-3 inhibited the neuronal dendritic growth through activation of calmodulin-dependent kinase (CaMK) IIα. In contrast, the Ca2+ influx through TRPC6 induced by NT-4 promoted the dendritic growth. Thus, TRPC5 acts as a novel and specific mediator for NT-3 to regulate dendrite development through CaMKIIα.
Introduction
Neurotrophin-3 (NT-3) is largely enriched and has multiple roles in the CNS (Huang and Reichardt, 2003). It functions mainly through the TrkC, its high-affinity receptor with intrinsic receptor tyrosine kinase (RTK) activity (Ip et al., 1993). NT-3 also binds to two other high-affinity receptors, TrkA and TrkB, as well as a low-affinity receptor, p75NTR. Activation of TrkC receptors can stimulate three classical signaling cascades, whose components include phospholipase C (PLC)-Ca2+, Ras-Raf-extracellular signal-regulated kinase, and phosphatidylinositol 3 (PI3) kinase-Akt (Huang and Reichardt, 2003). Activation of these cascades is correlated with intracellular Ca2+ ([Ca2+]i) changes (Xia et al., 1996; Furukawa and Mattson, 1998; Clapham, 2007) and implicated in the varied actions of NT-3, ranging from modulation of neuronal morphology (McAllister et al., 1997), synaptic plasticity (Lohof et al., 1993; Yang et al., 2001), neuronal survival (Schecterson and Bothwell, 1992), and differentiation (Averbuch-Heller et al., 1994), to neurotransmitter release (Bonni et al., 1999; Poo, 2001). Furthermore, the [Ca2+]i changes due to extracellular Ca2+ influx are important for NT-3 to exert its physiological functions. For example, NT-3-induced Ca2+ influx following TrkC activation is required for modulation of the excitatory GABAergic synaptic transmission (Gao and van den Pol, 1999), differentiation of calbindin-D28k-positive pyramidal neurons (Boukhaddaoui et al., 2001), and activation of Ca2+-dependent K+ channels (Holm et al., 1997). However, the mechanism that enables NT-3 to induce Ca2+ influx remains largely unknown.
The transient receptor potential canonical (TRPC) channels, a subfamily of nonselective cation channels, emerge as a new route responsible for [Ca2+]i elevation when receptor tyrosine kinase (RTK) or G-protein-coupled receptors (GPCRs) are activated (Clapham, 2007). The TRPC subfamily contains seven members (TRPC1–TRPC7). Based on amino acid similarities, the TRPCs are divided into three subgroups: (1) TRPC1; (2) TRPC3, TRPC6, and TRPC7; and (3) TRPC4 and TRPC5. Meanwhile, TRPC2 is classified as a human pseudogene. TRPC activation includes both receptor-operated and store-operated mechanisms. The stimulation of GPCRs or RTKs leads to PLC activation, which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG could directly activate TRPCs and IP3 binds to IP3 receptors, leading to store-operated TRPC activation. Unlike the relatively wide distribution of other TRPCs, the distribution of TRPC5 is mainly restricted to the CNS. However, the physiological agonist for TRPC5 in the CNS remains largely unknown.
It has been reported that brain-derived neurotrophic factor (BDNF) can activate TRPC3 and TRPC6 channels via the TrkB-PLC pathway (Li et al., 1999; Amaral and Pozzo-Miller, 2007; Jia et al., 2007). Because NT-3 can also stimulate PLC, we hypothesized that NT-3 might induce Ca2+ influx through TRPC channels. Indeed, the RTK is involved in the activation of nonselective cation channels formed by TRPC4 and TRPC5 independent of internal Ca2+ stores in human embryonic kidney (HEK) 293 cells (Schaefer et al., 2000). Furthermore, both NT-3 and TRPC5 are implicated in the inhibition of neuronal dendritic growth (McAllister et al., 1997; Greka et al., 2003; Puram et al., 2011). Therefore, we asked whether NT-3 inhibits dendritic growth through activation of TRPC5. We showed here that TRPC5 was activated by NT-3 via the TrkC-PLCγ pathway and that TRPC5 mediated the effect of NT-3 on dendritic growth via CaMKIIα in rat hippocampal neurons.
Materials and Methods
Reagents and antibodies.
Neurotrophin-3, neurotrophin-4, and brain-derived neurotrophic factor were purchased from Millipore; TrkC-IgG from R&D Systems; K252a from Merck; Fura-2 AM and Mitotracker from Invitrogen; and nifedipine, SKF96365, and Hoechst33342 from Sigma-Aldrich. The 3-nitrocoumarin (3-NC) was a gift from R.T. Martegani (Milano-Bicocca University, Italy). The 7-OH-3-NC was synthesized according to the previous report and confirmed by mass spectrometry and nuclear magnetic resonance spectroscopy (Perrella et al., 1994; Ward et al., 2002). All the other reagents were from Sangon Biotech unless otherwise specified.
We purchased anti-TRPC4, anti-TRPC5, and anti-TRPC6 antibodies from Alomone Labs; anti-HSP60 and anti-TrkC from R&D Systems (for immunostaining) and Santa Cruz Biotechnology (for immunoprecipitation); anti-TrkA, anti-PLCγ1, anti-CaMKIV, and anti-pan-CaMKII from Cell Signaling Technology; anti-TrkB from Becton Dickinson; anti-α-tubulin and anti-CaMKIIα from Sigma-Aldrich; anti-IP3R, anti-pCaMKI (Thr177), anti-pCaMKIIα (Thr286), and anti-pCaMKIV (Thr196), anti-rabbit IgG, HRP-conjugated anti-goat IgG from Santa Cruz Biotechnology; anti-CaMKIγ from Abcam; HRP-conjugated anti-mouse, rabbit IgG from GE Healthcare; and Alexa fluor 488, 546 conjugated anti-mouse, rabbit IgG from Invitrogen. Anti-TRPC3 was produced in our laboratory (Ding et al., 2010).
Cell culture and transfection.
Primary hippocampal neurons were isolated and cultured as described previously (Shi et al., 2004). Briefly, the neurons were obtained by dissociating the hippocampus of embryonic day 18 Sprague Dawley rat (Shanghai Slack Laboratory Animal) brains of either sex, and seeded onto coverslips (No.1 Glass, Warner Instruments) coated with 50 μg/ml poly-d-lysine (Sigma-Aldrich) at a density of 100,000 neurons per coverslip and cultured in Neurobasal A supplemented with B-27 and 0.5 mm glutamine (Invitrogen). All the rats were anesthetized and treated according to animal welfare guidelines of the Institute of Neuroscience, Chinese Academy of Sciences. The HEK293 cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Biochrom).
For Ca2+ imaging assay and biochemical experiments, 4 × 106 neurons were electroporated using Nucleofector Kit (Lonza Cologne) with 4 μg of indicated siRNA or plasmids plus 1 μg of Discosoma red (dsRed) as an indicator for the transfected neurons. Parts of the transfected neurons were seeded on the coverslips for Ca2+ imaging and the rest of the transfected neurons were cultured for biochemical experiments. Additionally, 2 μg of each indicated plasmid was transfected in HEK293 cells using lipofectamin 2000 (Invitrogen). DsRed was cotransfected with the plasmids at a ratio of 1:6 to indicate the transfected HEK293 cells. For the morphology experiments, we transfected neurons at 3–4 d in vitro (DIV) using Ca2+ phosphate as reported previously (Ramos et al., 2007). The yellow fluorescent protein (YFP) was used at the ratio of 1:5 (YFP/target plasmids) to indicate the transfected cells.
Immunoblotting.
Total proteins extracted from male rat brain hippocampal tissues using the radioimmunoprecipitation assay (RIPA) buffer (in mm, 150 NaCl, 5 EDTA, 1% Triton X-100, 1 Na3VO4, 50 NaF, 1 PMSF, 1 aprotinin, 1 leupeptin, 5 DTT, protease inhibitors, and 10 Tris-Cl, pH 7.4) or total proteins extracted from cultured cells using SDS lysis buffer (2% SDS, 10% glycerol, 0.1 mm dithiothreitol, and 0.2 m Tris-HCl, pH 6.8) were Western-blotted with the indicated primary and secondary antibodies. Bound antibodies were detected by the enhanced chemiluminescence detection system (GE Healthcare). The band optical densities were quantified by ImageQuant software (GE Healthcare). The relative amount of proteins was determined by normalizing the densitometry value of interest bands to that of the internal loading control and the external control.
Immunostaining.
The cultured neurons at 7 DIV were fixed with paraformaldehyde buffer (4% paraformaldehyde in 0.1 m phosphate buffer supplemented with 0.1% picric acid and 0.05% glutaraldehyde, pH 7.4) for 20 min and then permeabilized with 0.1% Triton X-100 for 2 min. After blocking in 10% goat serum, the interesting proteins were probed with the primary antibodies at 4°C overnight and with fluorescent secondary antibodies at room temperature for 50 min. For morphology assay, we stained the transfected neurons with the antibody against YFP to observe the details of neuronal branches. The fluorescent signals were examined using an A1R laser-scanning confocal microscope (Nikon).
Electrophysiology.
Whole-cell currents were recorded using a MultiClamp patch 700A amplifier (Molecular Devices). Patch pipettes were made from borosilicate glass and had resistances of 5–7 MΩ when filled with high-cesium intracellular solutions (in mm, 140 CsCl, 10 EGTA, 0.3 Mg2ATP, 0.3 CaCl2, and 10 HEPES, pH 7.25). Before recording, the cells were transferred into the normal extracellular solution (in mm, 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4). Drugs were applied to cells by the eight-channel perfusion system (ALA Scientific Instruments). The flow rate was adjusted to 1–2 ml/min. Currents were filtered at 5 kHz, digitized using a Digidata 1322 Interface (Molecular Devices), and analyzed with pClamp 9.0 software (Molecular Devices). Experiments were performed at room temperature (22–24°C). Cells were recorded with a holding potential at −60 mV.
RNAi construct.
The sequences (5′-3′) of siRNA synthesized to specifically target indicated rat genes by GenePharma were as follows: negative control sense: UUCUCCGAACGUGUCACGUTT; negative control antisense: ACGUGACACGUUCGGAGAATT; siTrkA_1402 sense: GGAGCAAAUUUGGGAUCAATT; siTrkA_1402 antisense: UUGAUCCCAAAUUUGCUCCTT; siTrkB_1766 sense: GGACGAGAGACAGAUUUCUTT; siTrkB_1766 antisense: AGAAAUCUGUCUCUCGUCCTT; siTrkC_1425 sense: GGUCCAAAUUUGGAAUGAATT; siTrkC_1425 antisense: UUCAUUCCAAAUUUGGACCTT; siTRPC5_1686 sense: AACGCCUUCUCCACGCUCUUU; siTRPC5_1686 antisense: AAAGAGCGUGGAGAAGGCGUU; siTRPC6_2066 sense: CGGUGGUCAUCAACUACAATT; siTRPC6_2066 antisense: UUGUAGUUGAUGACCACCGTT.
Short hairpin RNA (shRNA) were designed and cloned into the pPRIME vector (Stegmeier et al., 2005). The shRNA sequences (5′-3′) targeting rat TRPC5, TRPC6, or scramble were as follows: shTRPC5i: GCACTCTATGTGGCAGGACAT; shTRPC6i: CCTCTCCATATGGTATGAGAA; scramble: TTCTCCGAACGTGTCACGAAG.
Immunoprecipitation.
Whole brain lysates extracted from postnatal day 8 rats of either sex using the RIPA buffer were precipitated with 2 μg of rabbit anti-TrkC antibody or rabbit IgG at 4°C overnight. Protein A beads (20 μl, GE Healthcare) were then added to the supernatant and incubated at 4°C overnight. The samples were centrifuged to pellet the beads and washed three times using the RIPA buffer and resuspended using 2 × SDS buffer for Western blotting.
Cytosolic Ca2+ measurement.
Changes in [Ca2+]i concentration were measured using Fura-2-AM. Briefly, a total of 7 × 104 primary cultured neurons were seeded on coverslips and at 6–8 DIV incubated with 2 μm Fura-2-AM at 37°C for 25 min. Cells were washed three times with normal extracellular solution (in mm, 120 NaCl, 6 KCl, 2 CaCl2, 2 MgCl2, 12 glucose, 12 sucrose, and 10 HEPES-free acid, pH 7.4) or with 0 Ca2+ extracellular solution (in mm, 120 NaCl, 6 KCl, 2 MgCl2, 12 glucose, 5 EGTA, and 10 HEPES-free acid, pH 7.4) and imaged using a Eclipse Ti microscope (Nikon) with dual excitation wavelengths for Fura-2-AM at 340 and 380 nm and detection of fluorescent emission at 500 nm. For the Ca2+ imaging in neurons with gene manipulation, only the neurons with red fluorescence were measured and included in calculations. For the experiment of reconstructing NT-3-induced Ca2+ change in the heterologous expression system, only the cells with red fluorescence were measured. Also, to make sure the exogenously expressed TRPC5 channels were functional, only the cells responding to LaCl3 in the groups of TRPC5 and TRPC5 plus TrkC were included in calculations. The ratios (R) of the emission at 500 nm induced by 340 and 380 nm excitation were captured at 6 s intervals and presented. The ΔR/R0 was calculated as following: (R−R0)/R0 where R0 was the mean value of R during the pretreatment time frame (baseline) in each experiment (Jia et al., 2007). The ΔR/R0 area was defined as the cumulative area of the [Ca2+]i elevation curve within the stimulating period, which indicated the changes of total amount of intercellular Ca2+ within a given time.
Image analysis.
Confocal images of the neurons were obtained using A1R laser-scanning confocal microscope (Nikon) with sequential acquisition settings at the resolution of 1024 × 1024 pixels. Each image was a z-series of seven images at 1 μm depth interval when 40× oil objective was used. The z-series images were “flattened” into a single image using the maximal projection. Using Neurolucida software (MicroBrightField), the dendritic morphology was quantified in two ways: (1) total dendritic tip numbers and total dendritic length per neuron; and (2) Sholl analysis. Transfection experiments were performed in duplicate wells and all experiments were repeated at least three times. More than 10 neurons per group were obtained each time. The colocalizations between Trk and TRPC proteins were estimated using Fiji ImageJ JACoP (NIH) (Bolte and Cordelières, 2006). To access colocalization, we used both Pearson's correlation coefficient (PCC) and Mander's overlap coefficient (MOC). PCC is a parameter used to quantify correlation across pixels of all intensities in each channel with a value close to 1 indicating reliable colocalization. MOC calculates the percentage of pixel intensities of the TRPC merged with Trk relative to the total intensities of TRPC, with 1 being high colocalization and 0 being low colocalization.
Statistics.
Statistical analysis for protein levels, Ca2+ imaging, and dendritic morphology was performed using a two-sided Student's t test. p Values <0.05 were considered statistically significant. Data were presented as mean ± SEM.
Results
The expression pattern of TRPC5 was similar to that of TrkC
We first examined the expression of TRPC5 and Trks in the developing rat hippocampus by Western blot. The specificity of the commercial antibodies against TRPC5 and TRPC6 was examined and confirmed. The expression of TrkA started from the embryonic stage and decreased four weeks after birth. The expression of TrkB and TrkC was similar to that of TRPC6 and TRPC5, with a low level at early developmental stages and peak level between postnatal day 7 and 14, remaining high up to adult (Fig. 1A). We then examined the subcellular localization of TrkB, TrkC, TRPC5, and TRPC6 by immunostaining on cultured hippocampal neurons. As shown in Figure 1, B–D, both TrkC and TRPC5 were localized on the dendrites and soma, with most TRPC5 staining merged with TrkC staining (PCC, 0.856 ± 0.009; MOC, 0.895 ± 0.032). In contrast, most staining of TRPC6 merged well with that of TrkB (PCC, 0.882 ± 0.014; MOC, 0.936 ± 0.041), which is consistent with the previous report (Jia et al., 2007). Together, TRPC5 and TrkC showed a similar developmental expression pattern in rat hippocampus and they had a subcellular colocalization in cultured hippocampal neurons. These results provide anatomic evidence to indicate a possible physiological relationship between TRPC5 channels and TrkC.
Figure 1.
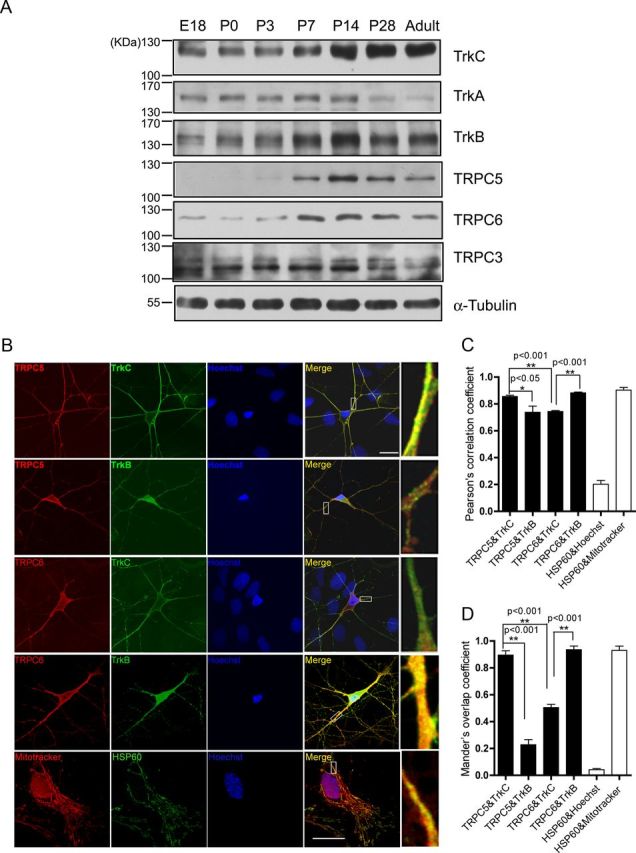
The expression of Trks and TRPCs in rat hippocampus. A, Western blots of total lysates extracted from rat hippocampus at different developmental stages with indicated antibodies. α-Tubulin served as a loading control. B, Representative staining of cultured hippocampal neurons at 7 DIV with the indicated antibodies or dyes. Right, Ninefold magnification of the white rectangle areas in “Merge.” Bottom, Representative staining of culture astrocytes at 7 DIV labeled with Mitotracker anti-HSP60 for mitochondria and Hoechst33342 for nucleus. Scale bar: 20 μm. C, D, Quantification of the colocalization of TRPCs and Trks shown in B. Lower limit: HSP60 and Hoechst33342, PCC, 0.202 ± 0.028; MOC, 0.042 ± 0.008. Upper limit: HSP60 and Mitotracker, PCC, 0.903 ± 0.020; MOC, 0.931 ± 0.032. In each group, 4–11 pictures were analyzed, *p < 0.05; **p < 0.01.
NT-3 induced Ca2+ influx sensitive to SKF96365 in cultured hippocampal neurons
Using Ca2+ imaging analysis, we then determined whether NT-3 could induce [Ca2+]i changes in cultured hippocampal neurons (Fig. 2A). NT-3 induced a slow and small [Ca2+]i elevation in neurons in a dose-dependent manner and the time of the delay to onset or the rising time of the [Ca2+]i elevation was 1.326 ± 0.190 min or 10.090 ± 0.582 min, respectively (Fig. 2B). Further quantitative analysis in [Ca2+]i by measuring the area under the [Ca2+]i elevation curve within the treatment period revealed that NT-3 induced the [Ca2+]i elevation with a threshold concentration of 25 ng/ml (10 ng/ml, 0.025 ± 0.008; 25 ng/ml, 0.102 ± 0.043; 50 ng/ml, 0.513 ± 0.075 normalized to 100 ng/ml. Fig. 2C,D). Because 50 ng/ml NT-3 was able to induce a repeatable [Ca2+]i elevation, we performed the following experiments with this concentration. We next used an NT-3 scavenger TrkC-IgG, a chimeric recombinant protein known to specifically neutralize NT-3 at an equimolar ratio (Shelton et al., 1995), to determine the minimal concentration of NT-3 needed to evoke Ca2+ signals. We estimated that 300 ng/ml TrkC-IgG was able to completely neutralize the action of 50 ng/ml NT-3. Indeed, as shown in Figure 2, E and F, 300 ng/ml TrkC-IgG completely blocked NT-3-induced [Ca2+]i elevation (0.192 ± 0.033, p < 0.001 compared with 0 ng/ml TrkC-IgG). However, in the presence of 150 ng/ml TrkC-IgG, NT-3 still induced [Ca2+]i elevation (0.402 ± 0.159 compared with 0 ng/ml TrkC-IgG), indicating that the minimal concentration of NT-3 needed to induce Ca2+ signals in cultured neurons was ∼22 ng/ml or less. Furthermore, the [Ca2+]i elevation induced by NT-3 was prevented in the absence of extracellular Ca2+ (0 mm Ca2+, 0.160 ± 0.044, p < 0.001 vs 1 mm Ca2+, Fig. 2G,I) and suppressed by SKF96365, a nonspecific TRPC channel inhibitor (SKF, 0.179 ± 0.041, p < 0.001 vs vehicle. Fig. 2H,I). Together, these results suggested that TRPC channels may be involved in the NT-3-induced Ca2+ influx.
Figure 2.
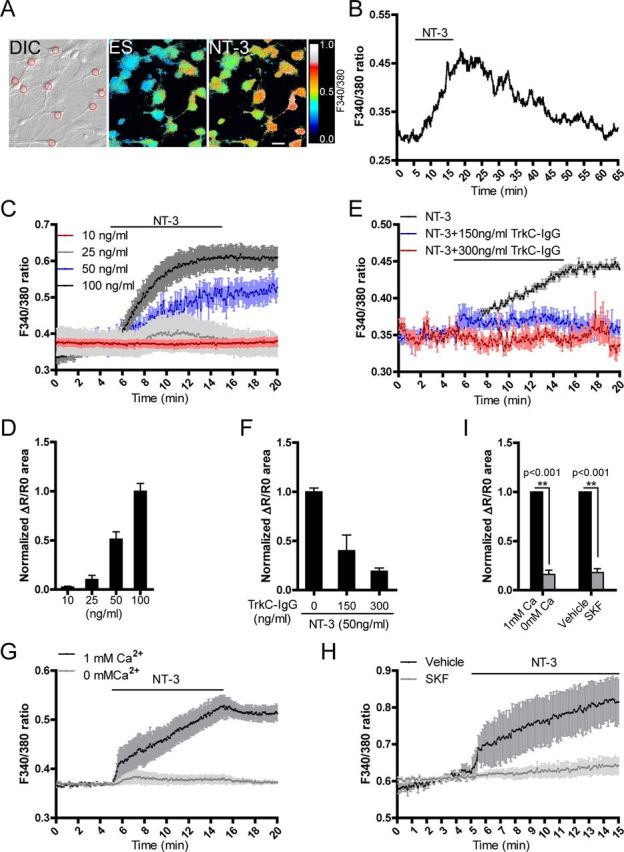
NT-3 induced [Ca2+]i elevation in cultured hippocampal neurons. A, An example of Fura-2-AM-loaded hippocampal neurons. Red circle in “DIC” panel indicates the regions from where the 340/380 ratios were obtained. The pseudocolor panels show the 340/380 ratio changes of the cells before and after applying NT-3. DIC, differential interference contrast; ES, extracellular solution. Scale bar: 20 μm. B, A representative trace showing the F340/380 ratio values of neurons in response to NT-3. Delay time was the period from applying NT-3 to the initial Ca2+ rising. Rise time was the period from the initial Ca2+ rising to the peak level. C, Dose-effect of NT-3 on [Ca2+]i elevation. D, Quantification of C. E, Effective concentration of NT-3 needed to evoke Ca2+ signals. F, Quantification of E. G, H, Effects of extracellular Ca2+ (G) or SKF96365 (10 μm) (H) on NT-3-induced [Ca2+]i changes. I, Statistics of the normalized area of ΔR/R0 curve in G and H. Cultured hippocampal neurons were measured at 7–9 DIV. NT-3, 50 ng/ml unless otherwise specified. Data were presented as means ± SEM from at least three independent experiments with >60 cells in each group.
TRPC5 was required for NT-3-induced Ca2+ influx in the hippocampal neurons
We next examined whether NT-3 stimulates TRPC5 to induce [Ca2+]i elevation in the neurons transfected with constructs and dsRed to indicate the neurons analyzed (Fig. 3A). As shown in Figure 3, B and H, the NT-3-induced [Ca2+]i change in the neurons transfected with a dominant-negative form of TRPC5 (dnTRPC5), in which three residues' mutations in the pore region were generated to inhibit its channel activity (Strübing et al., 2003), was 34.56 ± 11.76% of that in the neurons transfected with the control vector (p = 0.005 vs control). Moreover, expression in the neurons of a small interfering RNA against TRPC5 (siTRPC5), which downregulated TRPC5 without affecting TRPC4 or TRPC6 (Fig. 3C), greatly inhibited NT-3-induced [Ca2+]i elevation [siTRPC5, 0.580 ± 0.118, p = 0.012 vs negative control siRNA (N.C.), Fig. 3C,H]. In contrast, in the neurons transfected with dnTRPC6, in which three mutations in a pore region were made to block the channel activity (Hofmann et al., 2002), NT-3-induced [Ca2+]i elevation was similar to that in the neurons transfected with control vectors (dnTRPC6, 1.139 ± 0.196, p = 0.510 vs control, Fig. 3D,H). Similarly, expression in the neurons of a small interfering RNAi against TRPC6 (siTRPC6), which downregulated TRPC6 without affecting TRPC5 or TRPC3, did not affect the NT-3-induced [Ca2+]i elevation (siTRPC6, 1.092 ± 0.074, p = 0.344 vs N.C., Fig. 3E,H). Furthermore, dnTRPC5 did not affect the [Ca2+]i elevation induced by NT-4, the receptor of which is TrkB (Ip et al., 1993) (dnTRPC5, 0.940 ± 0.195, p = 0.788 vs control, Fig. 3F,H), whereas dnTRPC6 (dnTRPC6, 0.243 ± 0.074, p = 0.009 vs control, Fig. 3F,H) or siTRPC6 (siTRPC6, 0.370 ± 0.167, p = 0.019 vs N.C., Fig. 3G,H) suppressed NT-4-induced [Ca2+]i elevation.
Figure 3.
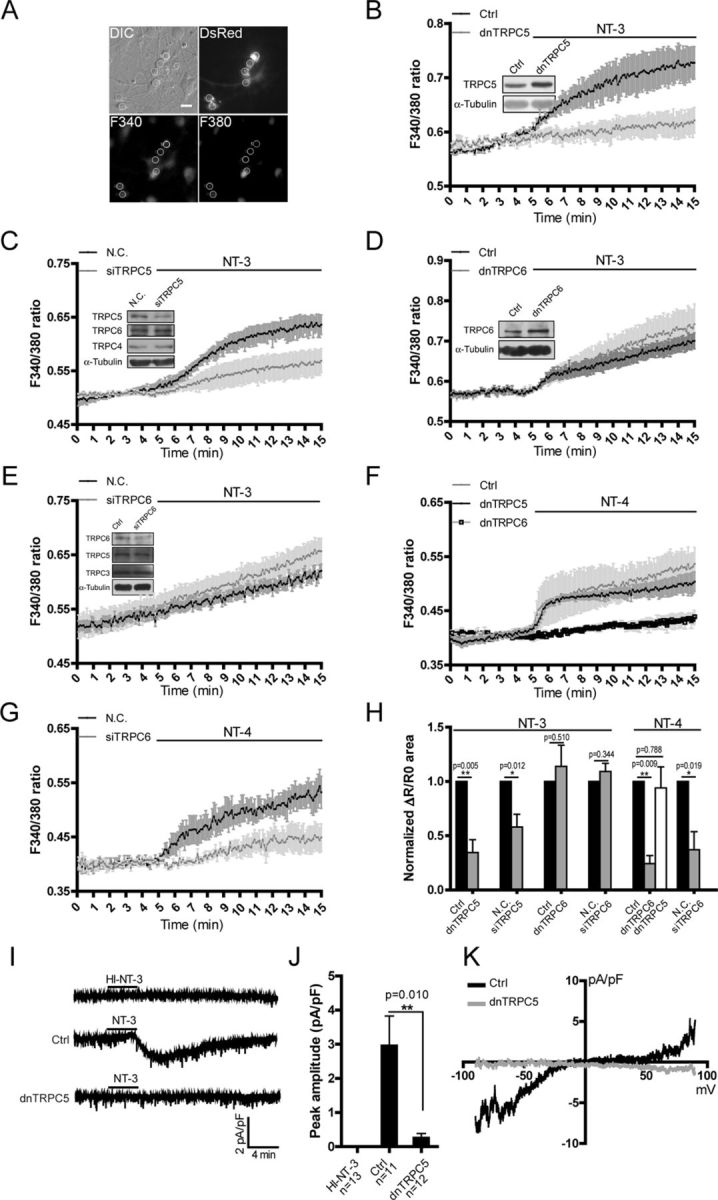
TRPC5 mediated the NT-3-induced [Ca2+]i elevation in cultured hippocampal neurons. A, An example showing how the transfected neurons with red fluorescence were identified and measured. Scale bar: 20 μm. B–E, NT-3-induced [Ca2+]i elevation in the neurons transfected with (B) the control vector or dnTRPC5; (C) N.C. or siTRPC5; (D) control vector or dnTRPC6; (E) N.C. or siTRPC6. Insets, representative immunoblots showing the effect of gene manipulation in the measured neurons. F, G, NT-4-induced [Ca2+]i elevation in the neurons transfected with (F) control vector (gray dot), dnTRPC5 (black dot), or dnTRPC6 (open squares); (G) N.C. or siTRPC6. H, Statistics of the normalized area of ΔR/R0 curve from B to G. *p < 0.05; **p < 0.01. I, Representative whole-cell recording traces with active or heat-inactivated NT-3 (HI-NT-3) in the neurons transfected with control vector (n = 11) or dnTRPC5 (n = 12). Control (Ctrl), 2.973 ± 0.854 pA/pF; dnTRPC5, 0.269 ± 0.116 pA/pF. NT-3, 100 ng/ml. J, Statistic results of the peak amplitude of the current shown in I. K, Representative current–voltage curves of the currents shown in I. A ramp potential holding from −90 to +90 mV was applied before and after NT-3 stimulation to obtain purified NT-3 conductance. Data in Ca2+ imaging were means ± SEM from at least three independent experiments with >40 cells in each group. Cultured hippocampal neurons were electroporated with indicated siRNA or plasmids and measured at 7–9 DIV. For electrophysiology experiment, cultured hippocampal neurons were transfected with control vector or dnTRPC5 plasmids at 3 DIV and recorded at 7–9 DIV. α-Tubulin served as a loading control. NT-3 and NT-4, 50 ng/ml unless otherwise specified.
To further verify that NT-3 indeed activates TRPC5, we performed whole-cell patch-clamp recording. In the control neurons, heat-inactivated NT-3 did not evoke a membrane current, whereas applying active NT-3 induced a small inward current (the peak current density was 2.973 ± 0.854 pA/pF, n = 11). In the neurons transfected with dnTRPC5, the current density was greatly suppressed (the peak current density was 0.269 ± 0.116 pA/pF, n = 12, p = 0.010 vs control, Fig. 3I,J). The current–voltage relationship analysis showed a double rectifying conductance with a reverse potential near 0 mV, which is a characteristic of the TRPC-like current (Fig. 3K). Together, these results suggest that TRPC5 mediated the NT-3-induced [Ca2+]i elevation in the hippocampal neurons.
TrkC receptors mediated the activation of TRPC5 by NT-3
To identify the receptor that mediates the activation of TRPC5 by NT-3, we examined the effect of K252a, an agent known as a nonspecific Trk receptor inhibitor, on [Ca2+]i elevation induced by NT-3. As shown in Figure 4, A and H, treatment with K252a greatly inhibited the [Ca2+]i elevation (K252a, 0.260 ± 0.069, p = 0.009 vs vehicle), indicating that the high-affinity Trk receptors, but not P75NTR, mediated NT-3-induced [Ca2+]i elevation. To specify which Trk receptor mediated this process, we designed three siRNA molecules (siTrkA, siTrkB, and siTrkC) specifically against TrkA, TrkB, and TrkC (Fig. 4B–D). In the neurons transfected with either siTrkA or siTrkB, the NT-3-elevated [Ca2+]i was not changed compared with that in the neurons transfected with negative control siRNA (N.C.) (siTrkA, 0.966 ± 0.217, p = 0.890 vs N.C.; siTrkB, 1.229 ± 0.238, p = 0.438 vs N.C., Fig. 4B,C,H). In contrast, the NT-3-elevated [Ca2+]i in the neurons transfected with siTrkC was reduced to 40.96 ± 10.58% of that in the neurons transfected with negative control siRNA (p = 0.005 vs N.C., Fig. 4D,H). The [Ca2+]i elevation induced by NT-4 in the neurons treated with K252a or siTrkB was 23.29 ± 6.68% or 56.67 ± 11.99%, respectively, of that in the neurons treated with vehicle or N.C. siRNA (p = 0.007 vs vehicle, p = 0.022 vs N.C., Fig. 4E,F,H). These results indicated that TrkC mediated the [Ca2+]i elevation induced by NT-3, whereas TrkB mediated the [Ca2+]i elevation induced by NT-4 in the neurons.
Figure 4.

TrkC mediated the activation of TRPC5 by NT-3. Fura-2 AM imaging showed [Ca2+]i changes in the neurons. A–D, Effect of NT-3 on [Ca2+]i elevation in the presence or absence of 200 nm K252a (A) or in the neurons transfected with N.C. or siTrkA (B), siTrkB (C), or siTrkC (D). E, F, Effect of NT-4 on [Ca2+]i elevation in the presence or absence of 200 nm K252a (E) or in the neurons transfected with N.C. siRNA or siTrkB (F). Insets, representative immunoblots showing the specific knockdown of endogenous TrkA (B), TrkB (C, F) or TrkC (D) in the neurons. Cultured hippocampal neurons were electroporated with indicated siRNA and imaged at 7–9 DIV. G, Effect of NT-3 or LaCl3 on [Ca2+]i elevation in HEK293 cells expressing TRPC5 and TrkC either individually or in combination. Upper line, Time scale with indicated stimulations. ES, extracellular solution. Inset, representative immunoblots showing the expression of exogenous TrkC and TRPC5 in the cells. H, Statistics for A–G, *p < 0.05; **p < 0.01. Corresponding bar charts show the normalized area of ΔR/R0 curves within stimulation. Data were means ± SEM from at least three independent experiments with >40 cells in each group.
To provide further evidence that TRPC5 was indeed activated by NT-3 through TrkC, we tested whether NT-3 could induce [Ca2+]i elevation after expressing TRPC5 and TrkC in HEK293 cells. As shown in Figure 4, G and H, NT-3-induced [Ca2+]i elevation level in the cells expressing both TrkC and TRPC5 was nearly fivefold higher than that in the cells expressing either TrkC or TRPC5 (TRPC5 plus TrkC, 5.083 ± 0.343, p < 0.001 vs TRPC5; TrkC, 0.836 ± 0.220, p < 0.001 vs TRPC5 plus TrkC). Together, these results suggest that TrkC mediated the activation of TRPC5 by NT-3.
NT-3 activated TRPC5 through PLCγ
Activation of TrkC induces the phosphorylation and activation of PLCγ, which plays important roles in TRPC activation (Venkatachalam and Montell, 2007). To study whether PLCγ mediated the NT-3-induced [Ca2+]i elevation, we used 3-nitrocoumarin, a compound known to inhibit PLCγ activity without affecting PLCβ (Perrella et al., 1994; Ward et al., 2002). As shown in Figure 5, A and C, 3-NC, but not the inactive analog 7-OH-3-NC, greatly suppressed the NT-3-induced [Ca2+]i elevation (3-NC, 0.250 ± 0.083, p = 0.012 vs 7-OH-3-NC). Similarly, NT-4-induced [Ca2+]i elevation was also blocked by 3-NC (3-NC, 0.341 ± 0.046, p = 0.004 vs 7-OH-3-NC, Fig. 5B,C). These results indicate that PLCγ, via activation of TRPC5 or TRPC6, mediated the effects of NT-3 or NT-4 on [Ca2+]i elevation (Figs. 3, 4, 5A–C). We then asked how TrkC specifically activates TRPC5. We examined whether TrkC was in the complex containing PLCγ and TRPC5. Immunoprecipitation of the neonatal rat brain lysates using the anti-TrkC antibody revealed that PLCγ1 and TRPC5, but not CaMKII, CaMKIγ, TRPC6, or TRPC3, were found in the precipitated complexes (Fig. 5D), suggesting that TrkC was specifically associated with TRPC5, but not TRPC6 or TRPC3. Together, these results suggest that NT-3 induced [Ca2+]i elevation through the TrkC-PLCγ-TRPC5 pathway.
Figure 5.

NT-3 activated TRPC5 through PLCγ. Fura-2 AM imaging showed [Ca2+]i changes in the neurons. A, C, NT-3-induced [Ca2+]i elevation in the presence of 30 μm 3-NC or the inactive analog 7-OH-3-NC. p = 0.012 vs 7-OH-3-NC. B, C, NT-4-induced [Ca2+]i elevation in the presence of 30 μm 3-NC or 7-OH-3-NC. p = 0.004 vs 7-OH-3-NC. C, Statistics of the normalized area of ΔR/R0 curves induced by indicated neurotrophins shown in A and B. Data were means ± SEM from at least three independent experiments with >60 cells in each group. D, Representative immunoblot shows that TrkC was immunoprecipitated (IP) with TRPC5 and PLCγ1, but not TRPC6, TRPC3, CaMKIIα, CaMKIIβ, and CaMKIγ. IgG served as a negative control. Asterisk indicates the IgG heavy chain.
TRPC5 mediated the effect of NT-3 on dendritic growth
To explore the physiological roles of the Ca2+ influx through TRPC5 activated by NT-3, we examined the effect of NT-3 on dendritic growth in the hippocampal neurons, in which the critical period for dendritic development is after day 4 in culture (Dotti et al., 1988). We measured the total dendritic tips and total dendritic length to assess the dendritic growth. As shown in Figure 6, A–E, treatment with NT-3 for 48 h decreased the total dendritic tips and length by 21% (p = 0.002 vs vehicle) and 36% (p < 0.001 vs vehicle), respectively. Knocking down TRPC5 with a vector-based short hairpin RNA (shTRPC5i) increased the total dendritic tips and length by 21% (p = 0.020 vs scramble) and 24% (p = 0.009 vs scramble), and abolished the inhibitory effect of NT-3 on the total dendritic tips (p = 0.163 vs vehicle) and length (p = 0.839 vs vehicle). However, knocking down TRPC6 with shTRPC6i decreased the total number of dendritic tips by 20% (p = 0.017 vs scramble) and total dendritic length by 34% (p < 0.001 vs scramble), and did not reverse the inhibitory effect of NT-3 on dendrite growth. In contrast, NT-4 increased both total dendritic tips by ∼47% and length by ∼41% (p = 0.003 for total dendritic tips and p < 0.001 for total dendritic length vs vehicle, Fig. 6F–J), which is similar to the BDNF effect on dendrite growth. The NT-4-increased dendritic growth was eliminated when the TRPC6 was knocked down (p = 0.142 for total dendritic tips and p = 0.636 for total dendritic length vs vehicle, Fig. 6G,H). These results suggest that TRPC5 mediates the effect of NT-3 on dendritic growth and that TRPC6 mediates the effect of NT-4 on dendritic growth.
Figure 6.

NT-3 or NT-4 regulated dendritic growth via TRPC5 or TRPC6, respectively. A, Representative images for the neurons transfected with shTRPC5i or shTRPC6i and stimulated with NT-3. B, Quantification of total dendritic tips for the neurons shown in A. Scramble vehicle: 27.50 ± 1.35; scramble NT-3: 21.73 ± 1.22; shTRPC5i vehicle: 33.21 ± 1.93; shTRPC5i NT-3: 29.59 ± 1.67; shTRPC6i vehicle: 22.07 ± 1.76; shTRPC6i NT-3: 18.00 ± 2.48. C, Quantification of total dendritic length for the neurons shown in A. In μm, scramble vehicle: 1231.00 ± 64.36; scramble NT-3: 785.90 ± 50.85; shTRPC5i vehicle: 1529.00 ± 86.70; shTRPC5i NT-3: 1504.00 ± 85.68; shTRPC6i vehicle: 818.08 ± 43.00; shTRPC6i NT-3: 595.70 ± 76.82. *p < 0.05; **p < 0.01. D, E, Sholl analyses of the neurons shown in A. F, Representative images for the neurons transfected with shTRPC6i and stimulated with NT-4. G, Quantification of total dendritic tips for the neurons shown in F. Scramble vehicle: 27.50 ± 1.93; scramble NT-4: 40.30 ± 3.54; shTRPC6i vehicle: 17.02 ± 1.00; shTRPC6i NT-4: 20.60 ± 1.41. H, Quantification of total dendritic length for the neurons shown in F. In μm, scramble vehicle: 1231.43 ± 97.46; scramble NT-4: 1735.08 ± 85.61; shTRPC6i vehicle: 722.05 ± 35.50; shTRPC6i NT-4: 746.78 ± 38.08. *p < 0.05; **p < 0.01. I, J, Sholl analyses of the neurons shown in F. Culture hippocampal neurons were transfected with indicated shRNAi at 3 DIV, stimulated with neurotrophin at 5 DIV for 48 h and observed at 7 DIV. Scale bar: 50 μm. Data were means ± SEM from three independent experiments with >30 neurons in each group. The numbers of neurons analyzed are shown inside the bars.
NT-3 regulated dendrite growth through the TRPC5-CaMKIIα pathway
Next we explored how the Ca2+ influx through TRPC5 induced by NT-3 affected dendritic growth. As the Ca2+ effectors, CaMKs are widely involved in Ca2+ signaling. Studies from different groups consistently reported that CaMKIIα can inhibit dendritic development while CaMKIV can promote dendritic development (Wu and Cline, 1998; Redmond et al., 2002). Specifically, CaMKIγ (Davare et al., 2009) or CaMKIIβ (Puram et al., 2011) was found downstream from TRPC5. To examine whether CaMKs were involved in NT-3 regulation of dendritic growth, we investigated the activity of CaMKIγ, CaMKIIα, CaMKIIβ, and CaMKIV in response to NT-3 by detecting the phosphorylation at Thr177 (Fujimoto et al., 2011), Thr286/287 (Barkai et al., 2000), or Thr196 (Means, 2000) of the kinases. As shown in Figure 7, A–D, time course experiments revealed that NT-3 increased the phosphorylation of CaMKIIα and CaMKIIβ, but not CaMKIγ and CaMKIV, whereas NT-4 increased the phosphorylation of CaMKIV, but not CaMKIγ, CaMKIIα, and CaMKIIβ. Because the amount of CaMKIγ is low in young cultures (Davare et al., 2009) and because CaMKIγ promotes dendritic arborization (Wayman et al., 2006; Takemoto-Kimura et al., 2007), we speculated that CaMKIγ does not mediate the NT-3-TRPC5 inhibition of dendrite growth. The NT-3-induced phosphorylation of CaMKII was abolished when TRPC5 was knocked down (Fig. 7E,F), indicating that TRPC5 was required for the activation of CaMKII induced by NT-3. Moreover, overexpressing TRPC5 did not enhance CaMKIIβ activity, but greatly enhanced CaMKIIα activity, although downregulating TRPC5 suppressed both CaMKIIα and CaMKIIβ activity (Fig. 7G,H), suggesting that activation of TRPC5 was necessary and sufficient for the activation of CaMKIIα in the neurons. Because CaMKIIα is predominantly in the forebrain and CaMKIIβ is predominantly in the cerebellum (McGuinness et al., 1985; Miller and Kennedy, 1985), and because mRNA of CaMKIIα is selectively found in dendrites, in contrast to mRNA of CaMKIIβ, which is not selectively found in dendrites (Schulman, 2004), we next examined whether CaMKIIα was involved in TRPC5 regulation of dendrite development in hippocampal neurons. In the dendrite development assay, overexpressing TRPC5 reduced the number of dendritic tips by 34% and dendritic length by 29% compared with control (p < 0.001 vs control). However, expression of a dominant-negative form of CaMKIIα (Abraham et al., 1997) eliminated the TRPC5 inhibitory effect on dendritic growth (Fig. 7I–M). Responding to reports of the possible involvement of L-type voltage-gated Ca2+ channels (L-VGCCs) in NT-3-elevated [Ca2+]i (Baldelli et al., 2000; Kang and Schuman, 2000), we examined whether L-VGCCs played a role in NT-3 inhibition of dendritic growth. However, inhibition of L-VGCCs with nifedipine did not change the NT-3 inhibitory effect on dendritic growth in the neurons (total dendritic length in μm: vehicle, 991.90 ± 112.00; NT-3, 641.80 ± 77.76; nifedipine, 665.70 ± 60.04; NT-3 plus nifedipine, 508.60 ± 43.65. Data not shown). Together, these results suggest that NT-3 regulated dendritic growth via the TRPC5-CaMKIIα pathway.
Figure 7.

NT-3 inhibited dendritic growth through TRPC5-CaMKIIα pathway. A, NT-3 enhanced the CaMKIIα or CaMKIIβ phosphorylation at Thr286 or Thr287, respectively, in a time-dependent manner. B, Statistics for A. C, NT-4 enhanced CaMKIV phosphorylation at Thr196. D, Statistics for C. Cell lysates were extracted from cultured hippocampal neurons at 7 DIV. E, Knocking down TRPC5 abolished NT-3-induced CaMKII activation. Cell lysates extracted from the neurons electroporated with siTRPC5 and treated with NT-3 for 15 min were Western-blotted with indicated antibodies. F, Statistics for E. G, TRPC5 was necessary and sufficient to activate CaMKIIα. Immunoblots showed that increasing TRPC5 expression raised the CaMKIIα phosphorylation level in the neurons and that, conversely, decreasing TRPC5 expression lowered both the CaMKIIα and the CaMKIIβ phosphorylation levels in the neurons. Cell lysates were extracted from the neurons electroporated with TRPC5 or siTRPC5 at 4 DIV. H, Statistics for G. α-Tubulin served as a loading control. I, Representative images for the neurons transfected with TRPC5 or the dnCaMKIIα individually or in combination at 4–5 DIV and observed at 9–10 DIV. Scale bar: 50 μm. J, Quantification of total dendritic tips of the neurons shown in I. Control (Crtl), 35.80 ± 1.67; TRPC5, 23.48 ± 1.54; dnCaMKIIα, 42.87 ± 1.70; TRPC5 plus dnCaMKIIα, 38.70 ± 1.69. K, Quantification of total dendritic length of the neurons shown in I. In μm, control (Ctrl), 1481.71 ± 46.05; TRPC5, 1058.63 ± 62.71; dnCaMKIIα, 1882.03 ± 69.00; TRPC5 plus dnCaMKIIα, 1788.68 ± 45.06. L, M, Sholl analyses of the neurons shown in I. The numbers of neurons analyzed shown within the bar. Data were means ± SEM from at least three independent experiments. *p < 0.05; **p < 0.01.
Discussion
In this study, we showed that TRPC5 channels were important for NT-3-induced [Ca2+]i elevation and the Ca2+ influx through TRPC5 via activation of CaMKIIα mediated the inhibitory effect of NT-3 on dendritic growth in the hippocampal neurons. In contrast, Ca2+ influx through TRPC6 was important for NT-4-induced CaMKIV activation and subsequent stimulation of dendritic growth. Our results thus suggest that NT-3 is a physiological activator for TRPC5 channels and that different neurotrophins stimulate distinct signaling modules to activate different channels and the downstream Ca2+ signaling effectors, leading to opposing effects on dendritic development.
TRPC5 activation mechanism
TRPC5, as one of the predominant members of TRPC family expressed in the CNS, plays important roles in axon growth (Greka et al., 2003; Davare et al., 2009), fear emotion (Riccio et al., 2009), and neuronal differentiation (Shin et al., 2010). Although the activation mechanism of TRPC5 has been extensively investigated, the physiological factors that lead to its activation in the CNS remain unidentified. Two endogenous factors reported so far for TRPC5 activation are nitric oxide (NO) in endothelial cells (Yoshida et al., 2006) and reduced thioredoxin in synoviocytes from patients with rheumatoid arthritis (Xu et al., 2008). Reduced thioredoxin is produced pathologically and endothelial NO is not specifically present in the CNS. Additionally, carbachol (Andrade, 1991) or histamine (Schaefer et al., 2000), agonists for Gq/11 protein-coupled muscarinic or histamine 1 receptors, respectively, were used to activate overexpressed TRPC5 and other TRPCs (Vazquez et al., 2003; Kwon et al., 2007) in cell lines. Meanwhile, reducing extracellular pH (Semtner et al., 2007; Kim et al., 2008) or elevating [Ca2+]i (Blair et al., 2009) were reported to potentiate the activation of TRPC5 in HEK293 cells. Our results here suggest that NT-3 is a physiological activator for TRPC5 in the CNS. Further, unlike the activation by redox molecules through a direct modification on TRPC5 proteins, the activation for TRPC5 in neurons by NT-3 was through a receptor-operated mechanism. Activation of TrkC by NT-3 leads to the activation of PLCγ, which hydrolyzes PIP2 into DAG and IP3. DAG may modify membrane curvature and stretch, leading to TRPC5 opening (Allan and Michell, 1978; Gomis et al., 2008; Beech et al., 2009). As TRPC5 channels can be inhibited by PIP2 (Estacion et al., 2001; del Pilar Gomez and Nasi, 2005; Otsuguro et al., 2008), it is also possible that PLCγ releases TRPC5 from inhibition by hydrolyzing PIP2, leading to its opening. Further, TRPC5 and TrkC shared the same subcellular localization and TrkC, PLCγ, and TRPC5 were found in the same complex. In contrast, TrkB and TRPC6 shared the same subcellular localization and might coexist in a separate complex. Due to lack of a TrkB antibody able to perform immunoprecipitation, we could not test whether TrkB forms a complex with TRPC6 rather than TRPC5. Collectively, it is likely that subcellular localization and interacting partners play critical roles in the specification of the activation for TRPC5 or TRPC6.
NT-3 induced Ca2+ influx in hippocampal neurons
It is important to note that in Xenopus spinal neurons, the NT-3-induced [Ca2+]i elevation was due to internal Ca2+ release (He et al., 2000; Yang et al., 2001). Our results here and those of others (Marsh and Palfrey, 1996) suggest that in rat hippocampal neurons, the NT-3-induced [Ca2+]i elevation is mainly due to Ca2+ influx. The reason for the difference is not entirely clear, but may stem from the differences in species and cell types. Indeed, in Xenopus motoneurons, NT-3 activates all three classical signaling pathways and potentiates transmitter release (Yang et al., 2001). However, in the hippocampal slices, NT-3 does not activate MAP (mitogen-activated protein) kinase and PI3 kinase pathways (Gottschalk et al., 1999) and has no effect on the transmitter release in dentate granule cells (Kokaia et al., 1998). Thus, it is possible that NT-3 may use the PLC-IP3 pathway to stimulate IP3 receptors to release Ca2+ in the motoneurons (Berridge, 1998; He et al., 2000). However, in the hippocampal neurons, NT-3 may use the PLC-DAG pathway to couple Ca2+ channels, such as TRPs and VGCCs, to induce Ca2+ influx.
The signaling pathway underlying NT-3 regulation on dendrite growth
The roles of NT-3 in cell survival, differentiation, synaptic potentiation, and neuronal morphogenesis have been widely studied. However, the results related to NT-3's role in neuronal morphogenesis have been controversial. Some groups reported that NT-3 promotes neurite growth in neocortex pyramidal neurons (Baker et al., 1998), cerebellum granule neurons (Ramos et al., 2009), or inhibitory neurons (Vicario-Abejón et al., 1998), whereas other studies reported that it inhibits the dendritic growth in other types of neurons (McAllister et al., 1997; Schwyzer et al., 2002; Liu et al., 2009). In this study, we found that NT-3 inhibited dendritic growth by stimulating the TRPC5-CaMKIIα pathway in cultured hippocampal neurons. Consistently, CaMKIIα is involved in the inhibitory regulation of neuronal dendrite development (Wu and Cline, 1998; Redmond et al., 2002). Moreover, CaMKIIα is important for the activity-dependent secretion of NT-3 (Kolarow et al., 2007) and the secreted NT-3 in turn induces [Ca2+]i elevation and CaMKIIα activation (He et al., 2000). It is thus likely that the Ca2+ influx, through TRPC5 induced by NT-3, activates CaMKIIα, and then initiates a signaling cascade to inhibit dendritic development.
A Ying Yang regulation on dendrite growth
One new finding in the current study was that NT-4, one of the ligands of TrkB, promoted dendritic growth through activation of TRPC6. It is possible that NT-4-induced Ca2+ influx through TRPC6 specifically activates CaMKIV and that the activated CaMKIV translocates into the nucleus to activate CREB (Redmond et al., 2002; Tai et al., 2008) and induce gene expression (Shieh et al., 1998), leading to the promotion effect on dendritic growth (McAllister et al., 1997; Horch et al., 1999). On the other hand, it is also possible that the Ca2+ influx through TRPC5 induced by NT-3 activates CaMKIIα and that the activated CaMKIIα regulates the activity of some small GTPases, which are repressed by CaMKII in the postsynapse (Chen et al., 1998; Li, 2000), leading to repression of dendritic growth. Collectively, a Ying Yang regulation on neuronal morphogenesis was explored (Fig. 8). In the CNS, a couple of neurotrophins, NT-3 or NT-4/BDNF, could respectively activate TRPC5 or TRPC6 via TrkC or TrkB receptors, leading to the specific activation of CaMKII or CaMKIV. The activation of the kinases results in negative or positive regulation of dendrite development, which might be important for the proper development of neuronal dendrites and the formation of correct neuronal circuitry.
Figure 8.

Schematic diagram of the signaling pathways underline the opposing roles of TRPCs in dendrite development. The Ca2+ influx through TRPC5 triggered by NT-3-TrkC-PLCγ activates CaMKII and initiates a negative regulation on dendrite development. In contrast, the Ca2+ influx through TRPC6 triggered by NT-4/BDNF-TrkB-PLCγ activates CaMKIV and initiates a positive regulation on dendrite development.
Footnotes
This work was supported by the 973 Program (2011CB00400) and a grant (81130081) from National Natural Science Foundation of China. We thank M.X. Zhu, T. Gudermann, J. Yang, and X. Yu for constructs; R.T. Martegani for 3-nitrocoumarin; L. Birnbaumer for samples of TRPC5 knock-out mice; and Q. Hu for imaging analysis.
The authors declare no competing financial interests.
References
- Abraham ST, Benscoter HA, Schworer CM, Singer HA. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circ Res. 1997;81:575–584. doi: 10.1161/01.res.81.4.575. [DOI] [PubMed] [Google Scholar]
- Allan D, Michell RH. A calcium-activated polyphosphoinositide phosphodiesterase in the plasma membrane of human and rabbit erythrocytes. Biochim Biophys Acta. 1978;508:277–286. doi: 10.1016/0005-2736(78)90330-9. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci. 2007;27:5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R. Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res. 1991;548:81–93. doi: 10.1016/0006-8993(91)91109-e. [DOI] [PubMed] [Google Scholar]
- Averbuch-Heller L, Pruginin M, Kahane N, Tsoulfas P, Parada L, Rosenthal A, Kalcheim C. Neurotrophin 3 stimulates the differentiation of motoneurons from avian neural tube progenitor cells. Proc Natl Acad Sci U S A. 1994;91:3247–3251. doi: 10.1073/pnas.91.8.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RE, Dijkhuizen PA, Van Pelt J, Verhaagen J. Growth of pyramidal, but not non-pyramidal, dendrites in long-term organotypic explants of neonatal rat neocortex chronically exposed to neurotrophin-3. Eur J Neurosci. 1998;10:1037–1044. doi: 10.1046/j.1460-9568.1998.00118.x. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Forni PE, Carbone E. BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur J Neurosci. 2000;12:4017–4032. doi: 10.1046/j.1460-9568.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- Barkai U, Prigent-Tessier A, Tessier C, Gibori GB, Gibori G. Involvement of SOCS-1, the suppressor of cytokine signaling, in the prevention of prolactin-responsive gene expression in decidual cells. Mol Endocrinol. 2000;14:554–563. doi: 10.1210/mend.14.4.0437. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bahnasi YM, Dedman AM, Al-Shawaf E. TRPC channel lipid specificity and mechanisms of lipid regulation. Cell Calcium. 2009;45:583–588. doi: 10.1016/j.ceca.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Blair NT, Kaczmarek JS, Clapham DE. Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J Gen Physiol. 2009;133:525–546. doi: 10.1085/jgp.200810153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Boukhaddaoui H, Sieso V, Scamps F, Valmier J. An activity-dependent neurotrophin-3 autocrine loop regulates the phenotype of developing hippocampal pyramidal neurons before target contact. J Neurosci. 2001;21:8789–8797. doi: 10.1523/JNEUROSCI.21-22-08789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Davare MA, Fortin DA, Saneyoshi T, Nygaard S, Kaech S, Banker G, Soderling TR, Wayman GA. Transient receptor potential canonical 5 channels activate Ca2+/calmodulin kinase Iγ to promote axon formation in hippocampal neurons. J Neurosci. 2009;29:9794–9808. doi: 10.1523/JNEUROSCI.1544-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pilar Gomez M, Nasi E. A direct signaling role for phosphatidylinositol 4,5-bisphosphate (PIP2) in the visual excitation process of microvillar receptors. J Biol Chem. 2005;280:16784–16789. doi: 10.1074/jbc.M414538200. [DOI] [PubMed] [Google Scholar]
- Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D, Cai R, Jin Y, Dong B, Xu Y, Wang Y. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J Nat Cancer Inst. 2010;102:1052–1068. doi: 10.1093/jnci/djq217. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Sinkins WG, Schilling WP. Regulation of Drosophila transient receptor potential-like (TrpL) channels by phospholipase C-dependent mechanisms. J Physiol. 2001;530:1–19. doi: 10.1111/j.1469-7793.2001.0001m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, Hatano N, Nozaki N, Yurimoto S, Kobayashi R, Tokumitsu H. Identification of a novel CaMKK substrate. Biochem Biophys Res Commun. 2011;410:45–51. doi: 10.1016/j.bbrc.2011.05.102. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP. The transcription factor NF-kappaB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-alpha in hippocampal neurons. J Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Neurotrophin-3 potentiates excitatory GABAergic synaptic transmission in cultured developing hypothalamic neurones of the rat. J Physiol. 1999;518:81–95. doi: 10.1111/j.1469-7793.1999.0081r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis A, Soriano S, Belmonte C, Viana F. Hypoosmotic- and pressure-induced membrane stretch activate TRPC5 channels. J Physiol. 2008;586:5633–5649. doi: 10.1113/jphysiol.2008.161257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk WA, Jiang H, Tartaglia N, Feng L, Figurov A, Lu B. Signaling mechanisms mediating BDNF modulation of synaptic plasticity in the hippocampus. Learn Mem. 1999;6:243–256. [PMC free article] [PubMed] [Google Scholar]
- Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- He X, Yang F, Xie Z, Lu B. Intracellular Ca(2+) and Ca(2+)/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J Cell Biol. 2000;149:783–792. doi: 10.1083/jcb.149.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm NR, Christophersen P, Olesen SP, Gammeltoft S. Activation of calcium-dependent potassium channels in mouse [correction of rat] brain neurons by neurotrophin-3 and nerve growth factor. Proc Natl Acad Sci U S A. 1997;94:1002–1006. doi: 10.1073/pnas.94.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, Krüttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559–567. doi: 10.1038/nn1870. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Intracellular Ca(2+) signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci Lett. 2000;282:141–144. doi: 10.1016/s0304-3940(00)00893-4. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Jeon JP, Kim HJ, Kim BJ, Lee YM, Choe H, Jeon JH, Kim SJ, So I. Molecular determinant of sensing extracellular pH in classical transient receptor potential channel 5. Biochem Biophys Res Commun. 2008;365:239–245. doi: 10.1016/j.bbrc.2007.10.154. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Asztely F, Olofsdotter K, Sindreu CB, Kullmann DM, Lindvall O. Endogenous neurotrophin-3 regulates short-term plasticity at lateral perforant path-granule cell synapses. J Neurosci. 1998;18:8730–8739. doi: 10.1523/JNEUROSCI.18-21-08730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson ML, Schreiber AM, Wu V, Lavail MM, Cang J, Copenhagen DR. Regulation of neonatal development of retinal ganglion cell dendrites by neurotrophin-3 overexpression. J Comp Neurol. 2009;514:449–458. doi: 10.1002/cne.22016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Marsh HN, Palfrey HC. Neurotrophin-3 and brain-derived neurotrophic factor activate multiple signal transduction events but are not survival factors for hippocampal pyramidal neurons. J Neurochem. 1996;67:952–963. doi: 10.1046/j.1471-4159.1996.67030952.x. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McGuinness TL, Lai Y, Greengard P. Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J Biol Chem. 1985;260:1696–1704. [PubMed] [Google Scholar]
- Means AR. Regulatory cascades involving calmodulin-dependent protein kinases. Mol Endocrinol. 2000;14:4–13. doi: 10.1210/mend.14.1.0414. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J Biol Chem. 1985;260:9039–9046. [PubMed] [Google Scholar]
- Otsuguro K, Tang J, Tang Y, Xiao R, Freichel M, Tsvilovskyy V, Ito S, Flockerzi V, Zhu MX, Zholos AV. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2008;283:10026–10036. doi: 10.1074/jbc.M707306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrella FW, Chen SF, Behrens DL, Kaltenbach RF, 3rd, Seitz SP. Phospholipase C inhibitors: a new class of cytotoxic agents. J Med Chem. 1994;37:2232–2237. doi: 10.1021/jm00040a016. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Puram SV, Riccio A, Koirala S, Ikeuchi Y, Kim AH, Corfas G, Bonni A. A TRPC5-regulated calcium signaling pathway controls dendrite patterning in the mammalian brain. Genes Dev. 2011;25:2659–2673. doi: 10.1101/gad.174060.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B, Gaudillière B, Bonni A, Gill G. Transcription factor Sp4 regulates dendritic patterning during cerebellar maturation. Proc Natl Acad Sci U S A. 2007;104:9882–9887. doi: 10.1073/pnas.0701946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B, Valín A, Sun X, Gill G. Sp4-dependent repression of neurotrophin-3 limits dendritic branching. Mol Cell Neurosci. 2009;42:152–159. doi: 10.1016/j.mcn.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, Gapon S, Yao GL, Tsvetkov E, Rodig SJ, Van't Veer A, Meloni EG, Carlezon WA, Jr, Bolshakov VY, Clapham DE. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137:761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- Schulman H. Activity-dependent regulation of calcium/calmodulin-dependent protein kinase II localization. J Neurosci. 2004;24:8399–8403. doi: 10.1523/JNEUROSCI.3606-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwyzer L, Mateos JM, Abegg M, Rietschin L, Heeb L, Thompson SM, Lüthi A, Gähwiler BH, McKinney RA. Physiological and morphological plasticity induced by chronic treatment with NT-3 or NT-4/5 in hippocampal slice cultures. Eur J Neurosci. 2002;16:1939–1948. doi: 10.1046/j.1460-9568.2002.02259.x. [DOI] [PubMed] [Google Scholar]
- Semtner M, Schaefer M, Pinkenburg O, Plant TD. Potentiation of TRPC5 by protons. J Biol Chem. 2007;282:33868–33878. doi: 10.1074/jbc.M702577200. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Cox DN, Wang D, Jan LY, Jan YN. Control of dendrite arborization by an Ig family member, dendrite arborization and synapse maturation 1 (Dasm1) Proc Natl Acad Sci U S A. 2004;101:13341–13345. doi: 10.1073/pnas.0405370101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Shin HY, Hong YH, Jang SS, Chae HG, Paek SL, Moon HE, Kim DG, Kim J, Paek SH, Kim SJ. A role of canonical transient receptor potential 5 channel in neuronal differentiation from A2B5 neural progenitor cells. PloS One. 2010;5:e10359. doi: 10.1371/journal.pone.0010359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- Tai Y, Feng S, Ge R, Du W, Zhang X, He Z, Wang Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J Cell Sci. 2008;121:2301–2307. doi: 10.1242/jcs.026906. [DOI] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T, Hoshino M, Suzuki S, Kojima M, Mishina M, Okuno H, Bito H. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIγ. Neuron. 2007;54:755–770. doi: 10.1016/j.neuron.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Trebak M, St John Bird G, Putney JW., Jr Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem. 2003;278:21649–21654. doi: 10.1074/jbc.M302162200. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PD, Klein RR, Troutman MD, Desai S, Thakker DR. Phospholipase C-gamma modulates epithelial tight junction permeability through hyperphosphorylation of tight junction proteins. J Biol Chem. 2002;277:35760–35765. doi: 10.1074/jbc.M203134200. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A, Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451:69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He X, Feng L, Mizuno K, Liu XW, Russell J, Xiong WC, Lu B. PI-3 kinase and IP3 are both necessary and sufficient to mediate NT3-induced synaptic potentiation. Nat Neurosci. 2001;4:19–28. doi: 10.1038/82858. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]