

Abstract
The blood–brain barrier (BBB), which is formed by adherens and tight junctions (TJs) of endothelial cells, maintains homeostasis of the brain. Disrupted intracellular Ca2+ homeostasis and breakdown of the BBB have been implicated in the pathogenesis of Alzheimer's disease (AD). The receptor for advanced glycation end products (RAGE) is known to interact with amyloid β-peptide (Aβ) and mediate Aβ transport across the BBB, contributing to the deposition of Aβ in the brain. However, molecular mechanisms underlying Aβ-RAGE interaction-induced alterations in the BBB have not been identified. We found that Aβ1–42 induces enhanced permeability, disruption of zonula occludin-1 (ZO-1) expression in the plasma membrane, and increased intracellular calcium and matrix metalloproteinase (MMP) secretion in cultured endothelial cells. Neutralizing antibodies against RAGE and inhibitors of calcineurin and MMPs prevented Aβ1–42-induced changes in ZO-1, suggesting that Aβ-RAGE interactions alter TJ proteins through the Ca2+-calcineurin pathway. Consistent with these in vitro findings, we found disrupted microvessels near Aβ plaque-deposited areas, elevated RAGE expression, and enhanced MMP secretion in microvessels of the brains of 5XFAD mice, an animal model for AD. We have identified a potential molecular pathway underlying Aβ-RAGE interaction-induced breakage of BBB integrity. This pathway might play an important role in the pathogenesis of AD.
Introduction
Alzheimer's disease (AD) is characterized by the accumulation of amyloid β-peptide (Aβ) in the CNS and cerebrovascular changes that lead to cerebral amyloid angiopathy (CAA) (Ellis et al., 1996). Changes of cerebral microvasculature have been reported in the brains of AD subjects and are the major event of AD (Claudio, 1996; Banks et al., 1997; Heyman et al., 1998). However, the deposition of Aβ aggregates in cerebral blood vessels and the brain is poorly understood, and the mechanisms that underlie the response to changes in permeability are unknown.
The blood—brain barrier (BBB) regulates the transport of various molecules and restricts permeability across brain endothelium (Hawkins and Davis, 2005). Tight junctions (TJs) are the most prominent feature of brain endothelium and are responsible for the BBB integrity. As a component of the TJ, zonula occludin-1 (ZO-1) was initially identified in the BBB (Watson et al., 1991), and associated with TJ integrity. ZO-1 binds directly to a wide variety of cellular proteins (Fanning et al., 1998) and orchestrates the formation of TJ complexes. Aβ peptide impairs TJ integrity and increases the paracellular permeability in bovine brain capillary endothelial cell cultures (Strazielle et al., 2000).
The BBB controls the entry of plasma-derived Aβ into the CNS by RAGE (Yan et al., 1996; Deane et al., 2003)and clears brain-derived Aβ in the plasma by LRP-1 (Shibata et al., 2000). Indeed, increased levels of free Aβ in plasma have been reported in AD patients as well as AD mouse models (Matsubara et al., 1999; Kawarabayashi et al., 2001). We hypothesize that an initial small increase in luminal Aβ can trigger a vicious cycle of BBB damage and efflux of abluminal Aβ to blood vessels in the course of AD pathology. The accumulation of Aβ peptides is believed to be an early and causative event in cerebrovascular alterations (Selkoe, 2001). All of these are important clues, suggesting that Aβ may disrupt the TJ of BBB via interaction with RAGE as a specific mediator. Aβ induces calcium influx in the cells either directly or indirectly (Kawahara et al., 2000; Kagan et al., 2002). Also, enhanced intracellular calcium leads to a change of TJs (Stuart et al., 1996) as well as induces expression of matrix metalloproteases (MMPs) (Bond et al., 1998). For example, Aβ increases MMP-9 activity in murine cerebral endothelial cell cultures (Lee et al., 2003). Based on these reports, we hypothesized that TJs are altered by changes in Aβ-triggered cytosolic calcium influx and MMP expression.
In this study, using neutralizing anti-RAGE and specific inhibitors of calcineurin and MMPs, we observed that Aβ-induced TJ disruptions are mediated by RAGE through intracellular Ca2+-calcineurin signaling and MMP secretion. We propose that alteration of cerebral capillaries, RAGE expression, and TJ structural changes have a causal relationship in 5XFAD mouse brains, AD animal model, and supporting in vitro results. Together, we have identified a mechanism by which Aβ-RAGE interaction mediates the attenuation of BBB integrity and the pathogenesis of AD.
Materials and Methods
Reagents.
Aβ1–42 peptide (American Peptide) was dissolved in hexafluoroisopropanol for 4 d at room temperature (RT), and the lyophilized peptide was dissolved in dimethylsulfoxide (DMSO) (Dahlgren et al., 2002). Most of Aβ1–42 forms for this study are monomers. A23187 (Sigma), as a calcium ionophore, was dissolved in DMSO. FK506 was provided by Chong Kun Dang Pharm. GM6001 was purchased from Millipore. The following antibodies were used: anti-ZO-1, anti-claudin-5, and anti-occludin (Zymed Laboratories); anti-RAGE (Millipore); neutralizing anti-RAGE (R & D Systems); and anti-tubulin and anti-β-actin (Sigma). Lipofectamine LTX for cDNA transfection was purchased from Invitrogen.
Cell culture.
The murine endothelial cell line bEnd.3 (ATCC) was cultured in DMEM (HyClone), supplemented with 10% fetal bovine serum (Hyclone), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma), at 37°C in 5% CO2. Before the drug treatments, the media was replaced with Opti-MEM (Invitrogen).
Fluorescein isothiocyanate-dextran permeability assay.
bEnd.3 cells were plated in 6.5 mm diameter Transwell polycarbonate membrane inserts (0.4 μm pore size, 6 × 104 cells/cm2; Costar, Corning) that were assembled into 24-well plates. The cells achieved confluence within 3 d of seeding. The permeability was measured using fluorescein isothiocyanate-conjugated dextran (FD-40, MW 40,000; Sigma-Aldrich) (Gonzalez-Velasquez et al., 2008). FD-40 was added to the apical compartment at 0.1 μg/ml. After 30 min, the samples were collected from the basolateral compartment. The fluorescence (A.U.) was measured on a fluorescence luminometer (Tecan) at the wavelengths of 490 (excitation) and 520 nm (emission).
Western blot analysis.
bEnd.3 cells homogenate was separated 7% SDS-PAGE gels, and then transferred to a PVDF membrane. Membrane was incubated with antibodies against indicated proteins. Enhanced chemiluminescence (GE Healthcare Pharmacia Biotech) was used for visualization. The images were captured using a bioimaging analyzer (LAS-3000; Fuji) and analyzed with a Multi-Gauge program (Fuji).
Immunocytochemistry and immunohistochemistry.
bEnd.3 cells were seeded in a 24-well plate (2 × 104 cells/cm2; SPL) and cultured for 3 d. After being washed in PBS, pH 7.4, the cells were fixed in 4% paraformaldehyde (PFA) for 20 min at RT. After permeabilization with 0.5% Triton X-100 followed by blocking with 5% bovine serum albumin (BSA), cells were incubated with anti-ZO-1 (1:200; Zymed Laboratories) in PBS with 5% BSA at 4°C overnight and Cy3-labeled donkey anti-rabbit IgG (1:1000; Invitrogen) in 5% BSA for 1 h at RT. The cells were counterstained with DAPI (4′, 6′-diamidino-2-phenylindole dihydrochloride) and analyzed on a fluorescence microscope (Olympus DP50). For immunohistochemistry, wild-type (n = 3) and 5XFAD (n = 3) were killed at 8 months of age. Mice were anesthetized with a mixture of Zoletil 50 (Virbac) and Rompun (Bayer) solution (3:1 ratio, 1 ml/kg, i.p.) and perfused transcardially with a freshly prepared solution of 4% PFA in PBS. After mice were decapitated, brains were dissected from the skull. Serial 30-μm-thick coronal tissue sections were cut using a freezing microtome (Leica). Free-floating sections were incubated with the following primary antibodies: biotin-labeled 4G8 (1:700; Covance), rabbit anti-GLUT-1 (1:1000; Millipore), and goat anti-RAGE (1:20; R & D Systems) overnight at 4°C. After washes in PBS, the sections were incubated with the following secondary antibodies: Alexa Fluor 488-conjugated streptavidin (1:1000; Invitrogen), goat anti-rabbit Alexa 594 (1:1000; Invitrogen), and biotinylated anti-goat IgG antibody (1:500; Vector Laboratories) for 1 h and then donkey anti-goat Alexa 594 for 1 h for RAGE detection at RT.
Image analysis.
Images were acquired with a fluorescence microscope (Olympus DP50). For morphological analyses of ZO-1 and fluorescence intensity of Fluo-4 in bEnd.3 cells, acquired images were analyzed by ImageJ (NIH). Under the same threshold line, ZO-1 and Fluo-4 were measured, and the averages were determined (n = 8 for ZO-1 measurement and n = 4 for Fluo-4 intensity).
Visualization of intracellular calcium.
The intracellular calcium in bEnd.3 cells was stained using a fluorescent calcium indicator dye (Fluo-4; Invitrogen). bEnd.3 cells were plated in 96-well plates (5 × 104 cells/cm2; SPL). After 3 d, the cells were incubated with 5 mm Fluo-4 and diluted in Opti-MEM solution at 37°C for 30 min. The cells were treated with 5 μm Aβ1–42 or 1 μm A23187, and intracellular calcium levels were observed. Live images were taken using a fluorescence microscope (Olympus DP50) with a fluorescein isothiocyanate (FITC) filter set.
Gelatin zymography.
Culture media was incubated at 25°C for SDS sample buffer free of reducing agents and then electrophoresed on 10% polyacrylamide gels including 2.65 mg/ml gelatin at 20 mV. After electrophoresis, gels were washed in 2.5% Triton X-100 to remove SDS, and then incubated with at 37°C in 20 mm Tris-HCl, pH7.6, 10 mm CaCl2, 1 mm ZnCl2, and 0.04% NaN3 for 12 h and then stained with 0.1% Coomassie blue in 30% ethanol and 10% acetic acid for 30 min and destained until there were clear gelatinolytic bands. The images were captured using a bioimaging analyzer (LAS-3000; Fuji).
Transfection.
Transfection of the bEnd.3 cells were performed by Lipofectamine LTX according to the manufacturer's protocols (Invitrogen). Cells were plated in six wells (12 × 105 cells/cm2; SPL) and cultured for 2 d. The cyan fluorescent protein cDNA was used as a control for normalizing transfection efficiency using Lipofectamine Plus (Invitrogen) and human full-length RAGE cDNA constructs were mixed with Lipofectamine LTX in Opti-MEM (Invitrogen). After 30 min of incubation, the mixture was then added to the cell culture medium for 24 h and treated with 5 μm Aβ1–42.
Animals and tissue collection.
Female transgenic mice with five familial AD mutations (5XFAD) were purchased from The Jackson Laboratory. These mice express both mutant human amyloid precursor protein (APP695) with the Swedish mutation (K670N, M671L), Florida mutation (I716V), and London mutation (V717I) and human presenilin 1 harboring two FAD mutations (M146L and L286V). Both transgenes are expressed under the control of mouse Thy1 promoter to induce overexpression in the brain. Animal treatment and maintenance were performed in accordance with the Principle of Laboratory Animal Care (NIH publication No.85–23, revised 1985) and the Animal Care and Use Guidelines of Seoul National University, Seoul, Korea. All efforts were made to minimize animal suffering and to reduce the number of mice used.
3D-structured illumination microscopy image acquisition.
To identify the modification of cerebral capillary by senile plaque, we used the super-resolution structured illumination microscopy (SIM; Nikon N-SIM). Fixed brain slices were taken each 3D-SIM image by moving the stage in the z-direction with a step size of 0.150 μm. The sequential z-sections were reconstructed to 3D-SIM image (z-axis; brain slice thickness ∼5.0 ± 0.4 μm) and make the 3D-deconvolution with the α blending function using NIS-E software (Nikon). Images were taken by Eclipse Ti-E research inverted microscope with Nikon's legendary CFI Apo TIRF 100× oil objective lens (NA 1.49) and 512 × 512 pixel resolution with iXon DU-897 EMCCD camera (Andor Technology). Multicolor fluorescence was acquired using a diode laser (488 nm, 561 nm) and exposure times were applied 40 ms, electron microscopic (EM) gain 150, conversion gain 1×, and image processed with NIS-E software later exported to the Adobe Photoshop program.
Transmission electron microscope analysis.
Several cortex pieces from both wild-type and 5XFAD were randomly excised, diced (1 mm3), and then fixed overnight at 4°C in a mixture of cold 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.2, and 2% PFA in 0.1 m phosphate or cacodylate buffer, pH 7.2, and embedded with epoxy resin. The epoxy resin-mixed samples were loaded into capsules and polymerized at 38°C for 12 h and 60°C for 48 h. Thin sections were sliced on an ultramicrotome (RMC MT-XL) and collected on a copper grid. Appropriate areas for thin sectioning were cut at 65 nm and stained with saturated 4% uranyl acetate and 4% lead citrate. Then the ultrastructure of TJ of the brain was examined by a transmission electron microscope (JEM-1400). TJs were measured, and the averages were determined (n = 12 for each group).
Statistics.
All data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prizm4. The data were analyzed by one-way ANOVA with post hoc test or unpaired t test, as appropriate (*p < 0.05, **p < 0.01, and ***p < 0.001).
Results
Aβ1–42 alters distribution of ZO-1 and disrupts TJs in bEnd.3 cells
We examined the mechanisms by which Aβ1–42 disrupts TJs and induces structural alterations in ZO-1 in a monolayer culture of bEnd.3 cells. In control cells, ZO-1 was expressed in TJs, as reported previously (Brown et al., 2007). However, after treatment of Aβ1–42, ZO-1 immunoreactivity declined, staining intermittently in the cell-to-cell borders compared with control; this effect was increased in a dose- and time-dependent manner (Fig. 1A,B, arrowheads). Also, by Western blot, ZO-1 levels fell significantly in bEnd.3 cells after 24 h of Aβ1–42 treatment (Fig. 1C,D; n = 3, 1.6-fold; p < 0.05). Moreover, Aβ1–42 also induced structural alteration and reduced the protein level of other TJ proteins such as claudin-5 and occludin (Fig. 1E—G; n = 3 and 1.3- and 1.2-fold, respectively; p < 0.05). Alteration of these TJ proteins by Aβ1–42 occurred without affecting cell death under this experimental condition (data not shown). To evaluate whether the rearrangement and decrease in ZO-1 levels in Aβ1–42-treated bEnd.3 cells reflected the disruption of TJs, bEnd.3 cells were cultured in a Transwell system, and the amount of diffused FITC-dextran 40 (FD-40) was measured; FD-40 is used to measure BBB permeability in human brain endothelial cells and mice (Tai et al., 2010). Aβ1–42 (5 μm) increased FD-40 levels in the basolateral chamber (Fig. 1H; n = 8, 2.8-fold; p < 0.001). These results demonstrate that Aβ1–42 disrupts TJ and opens paracellular pathways in bEnd.3 cell monolayers.
Figure 1.
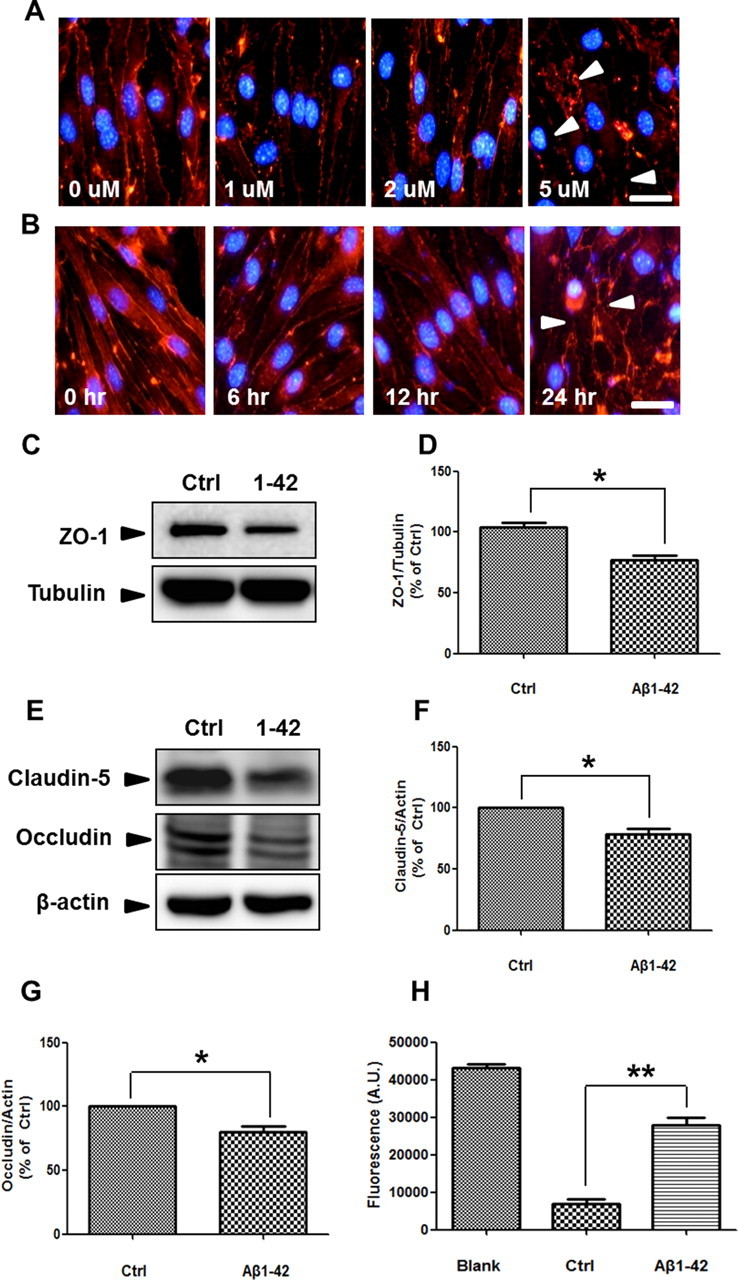
Aβ1–42 alters ZO-1 distribution and disrupts TJs in bEnd.3 cells. A, bEnd.3 cells were incubated with various doses of Aβ1–42 for 24 h (red, ZO-1; blue, DAPI). B, 5 μm Aβ1–42-induced alterations (arrowheads) in ZO-1 distribution in a time-dependent manner. Scale bar, 50 μm. n = 8 for each group. C, D, ZO-1 levels in bEnd.3 cells were decreased significantly with Aβ1–42 (5 μm, 24 h) in total lysate. Tubulin is a loading control. E–G, Claudin-5 and occludin levels in bEnd.3 cells were decreased significantly with Aβ1–42 (5 μm, 24 h) in total lysate. β-actin is a loading control. H, Diffusion of FD-40 through the bEnd.3 monolayer under the same conditions as performed in C was increased significantly after Aβ1–42 (5 μm, 24 h). y-axis is the fluorescence signals from the bottom chamber of Transwells. Blank shows a high value when no cells were on the Transwell plate. Control (Ctrl) shows a low value when bEnd.3 cells were compacted on the Transwell.*p < 0.05, **p < 0.01 versus control sample.
Aβ1–42 increases intracellular calcium level and MMP secretion in bEnd.3 cells
To examine whether Aβ1–42 induces calcium increase and MMP activation in bEnd.3 cells, changes of intracellular calcium and MMP activities were measured. Since A23187 showed similar Aβ1–42-induced ZO-1 alteration (data not shown), we expected that calcium affects alteration of TJs by Aβ1–42 in bEnd.3 cells. By fluorescence microscopy, Aβ1–42 induced a time-dependent increase in intracellular calcium levels in bEnd.3 cells (Fig. 2A). We quantified fluorescence intensity of Fluo-4. Intracellular calcium level exhibited a tendency to increase to 1 h after treatment of Aβ1–42 and A23187 and showed significant increase of Fluo-4 intensity at 6 h (Fig. 2B; n = 4 and 1.4- and 1.5-fold; p < 0.05). Aβ1–42 increased the enzymatic activity at 72 and 92 kDa against gelatin, revealed by zymography using conditioned media from Aβ1–42-treated cell cultures (Fig. 2C). These sizes correspond to MMP-2 and MMP-9 (Yang et al., 2007), respectively, suggesting that Aβ1–42 induces MMP-2 and MMP-9 by increasing intracellular calcium levels in bEnd.3 cells.
Figure 2.
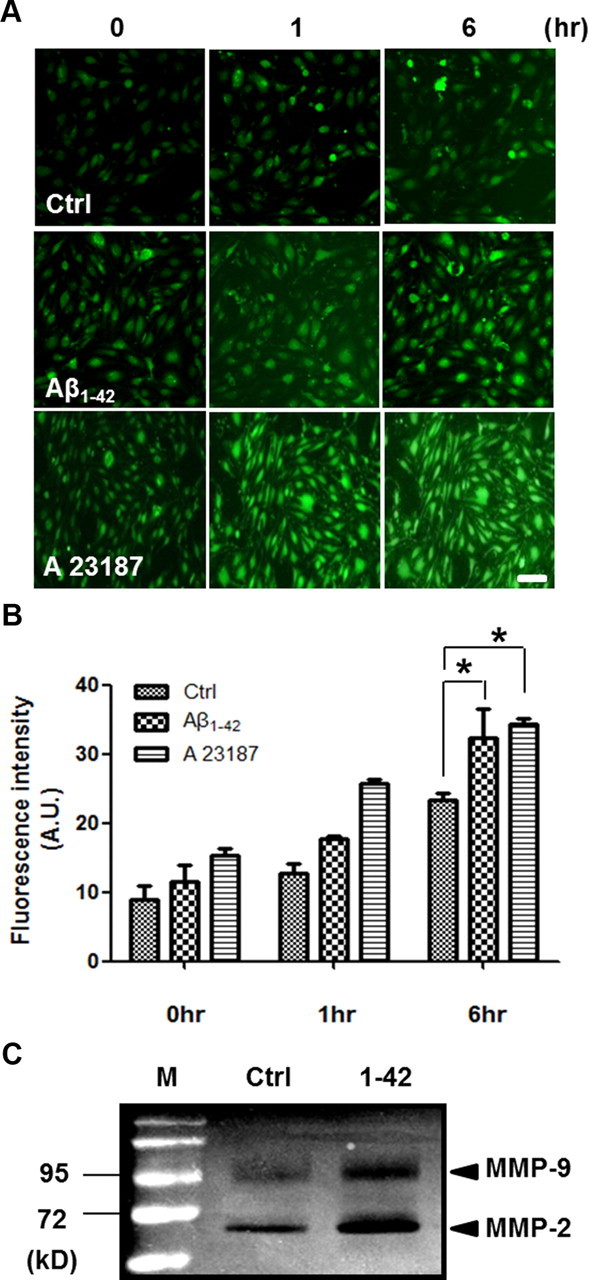
Aβ1–42 increases intracellular calcium levels and secretion of MMPs in bEnd.3 cells. A, Fluo-4-loaded bEnd.3 cells were incubated with 5 μm Aβ1–42 or 1 μm A23187 (calcium ionophore). Live cell images were obtained on a fluorescent microscope at the indicated times. Intensity of Fluo-4-staining in cells was increased in a time-dependent manner after treatment of Aβ1–42 compared with control (DMSO). Scale bar, 50 μm. B, Fluorescence intensity in bEnd.3 cells had a tendency to increase to 1 h with Aβ1–42 and A23187. At 6 h, fluorescence intensity was increased significantly compared with control (Ctrl). *p < 0.05 versus control sample at 6 h. C, Gelatin zymography using conditioned medium from bEnd.3 cells showed an increase of MMP-9 (95 kDa) and MMP-2 (72 kDa) after treatment with 5 μm Aβ1–42 (lane 3) compared with control (DMSO). M, molecular weight marker.
Aβ1–42-induced TJ alterations are attenuated by GM6001 and FK506
To confirm that the increase in MMPs mediated the Aβ-induced disruptions in TJs, we cotreated bEnd.3 cells with Aβ1–42 and the inhibitor GM6001, which prevents the conversion of pro-MMPs to active forms of matrix-degrading MMPs (Restituito et al., 2011). In the presence of GM6001, Aβ1–42-induced dysregulation of ZO-1 distribution was attenuated in a dose-dependent manner (Fig. 3A). Transcription factors, such as NF-κB and AP-1, regulate the transcription of MMP-9 (Bond et al., 1998). These transcription factors are also controlled by calcineurin (CaN), a calcium/calmodulin-dependent protein phosphatase that is sensitive to calcium concentrations. CaN is activated by Aβ and is upregulated in the brains of AD patients (Liu et al., 2005; Agostinho et al., 2008). Doller et al. (2007) have reported that CaN inhibitors suppress MMP transcription that is induced by NF-κB-dependent pathways.
Figure 3.
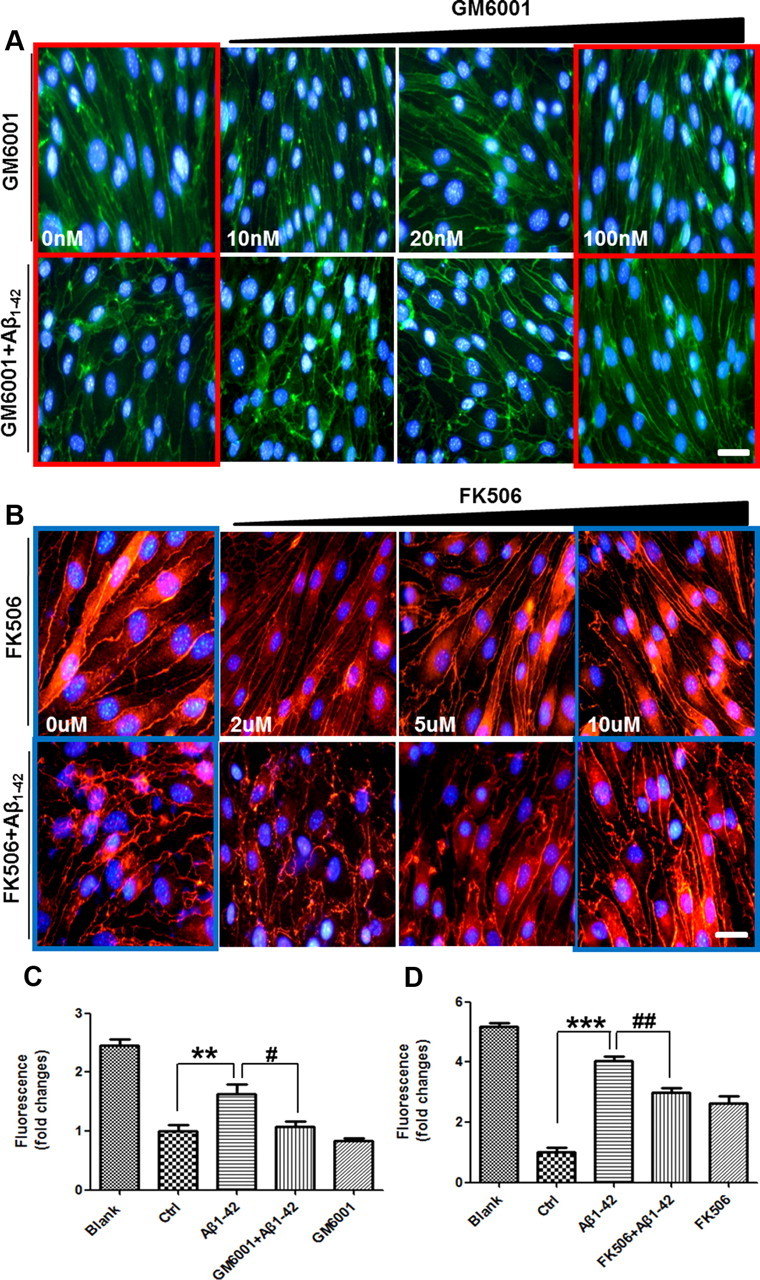
Aβ1–42-induced alterations of TJs in bEnd.3 cells are attenuated by GM6001 and FK506. A, B, bEnd.3 cells were incubated with 5 μm Aβ1–42 for 24 h in the presence of GM6001 or FK506. ZO-1 immunoreactivity (red) in the presence of Aβ1–42 was rescued in a dose-dependent manner by both GM6001 (A) and FK506 (B). Three independent experiments were done for both A and B. Scale bar, 50 μm. C, D, Changes of diffusion for FD-40 through the bEnd.3 monolayer were measured at each dose of GM6001 (100 nm) and FK506 (10 μm). Data are represented as mean ± SEM of three independent experiments performed in triplicate. *p < 0.05, ***p < 0.001 versus control sample; #p < 0.05, ##p < 0.01 versus Aβ1–42-treated sample.
To determine whether intracellular calcium influx/CaN activation and MMP secretion mediates Aβ-induced TJ disruption, we cotreated bEnd.3 cells with Aβ1–42 and FK506, a CaN inhibitor (Snyder et al., 1998). In the presence of FK506, Aβ1–42-induced changes in ZO-1 distribution were mitigated in a dose-dependent manner (Fig. 3B), as with GM6001. However, because the changes of ZO-1 redistribution seem to be different from treatment of GM6001 or FK506, the mechanisms of action to ameliorate Aβ-induced ZO-1 changes might be different. In addition, permeability assay using FD-40 in the basolateral chamber in bEnd.3 cell monolayers showed that cotreatment of 100 nm GM6001 or 10 μm FK506 as a representative concentration with Aβ1–42 significantly attenuated membrane permeability enhanced by Aβ1–42 (Fig. 3C,D; n = 5 and 1.5- and 1.4-fold; p < 0.05 and p < 0.01). Collectively, these data show that intracellular calcium and MMPs are associated with Aβ1–42-induced alterations in TJs in bEnd.3 cells.
RAGE mediates Aβ1–42-induced alterations of TJs in bEnd.3 cells
Endothelial cells express RAGE, a serum Aβ binding partner in the BBB system, transporting Aβ into the brain (Deane et al., 2003). To determine whether RAGE mediates Aβ-induced TJ disruptions, a neutralizing antibody against RAGE, which recognizes its extracellular domain, was used to block Aβ-RAGE interactions (Li et al., 2009). Anti-RAGE antibody attenuated Aβ1–42-induced perturbations in ZO-1 distribution, but normal goat IgG did not (Fig. 4A). It suggested that RAGE plays an important role in ZO-1 alteration by Aβ1–42. ZO-1 levels were measured in bEnd.3 cells that overexpressed full-length human RAGE in the presence or absence of Aβ1–42 for 24 h. In contrast to what was observed with anti-RAGE antibody, Aβ1–42 decreased ZO-1 levels in RAGE-overexpressing bEnd.3 cells to a greater extent than mock-transfected cells (Fig. 4B; n = 3, 1.2-fold; p < 0.05), suggesting that RAGE mediates Aβ1–42-induced changes in ZO-1 localization and protein level.
Figure 4.

RAGE mediates Aβ1–42-induced disruptions in TJs. A, Aβ1–42-induced alterations in ZO-1 distribution are attenuated by an anti-RAGE antibody. Con, DMSO; RAGE-Ab, neutralizing anti-RAGE antibody; goat-IgG, IgG from goat used as a negative control. Red signal is ZO-1 staining. Three independent experiments were performed. Scale bar, 100 μm. B, C, Transiently transfected bEnd.3 cells with mock or full-length human RAGE (hRAGE) were incubated with 5 μm Aβ1–42 for 24 h. Representative Western blot images (B) and the densitometry results (C) are presented. Data are represented as mean ± SEM of three independent experiments performed in triplicate. #p < 0.05 versus Mock Aβ1–42-treated sample of RAGE Aβ1–42.
Cerebral capillaries were impaired in the brains of 5XFAD mice
Since the expression of GLUT-1 is limited to the endothelium-bearing TJs, downregulation of GLUT-1 implies the increased permeability of the BBB (Kalaria, 1999), so, GLUT-1 was used as an endothelial marker. To assess the change of cerebral capillaries in the AD model mouse, we performed costaining with anti-GLUT-1 antibody and the anti-Aβ antibody, 4G8. Since 5XFAD mice show massive amyloid plaques and behavioral changes with neuronal loss at 8 months old (Oakley et al., 2006), the integrity of BBB was examined using GLUT-1 staining. Capillaries stained with GLUT-1 showed long tubular-like form and 4G8 immunoreactivity was not detected in littermates, while capillaries of disconnected short length were observed near the deposited area of amyloid plaques in the cortex of 5XFAD mice (Fig. 5A, yellow dotted circle). To clearly recognize modification of cerebral capillaries by senile plaques in the AD mouse model, we used the super-resolution SIM (Nikon). The reconstructed 3D-SIM image showed that the cerebral capillaries of 5XFAD mice were more damaged compared with that of littermates (Fig. 5B). These results suggest that cerebral capillaries were impaired and increased the BBB permeability in 5XFAD mice compared with littermate mice.
Figure 5.
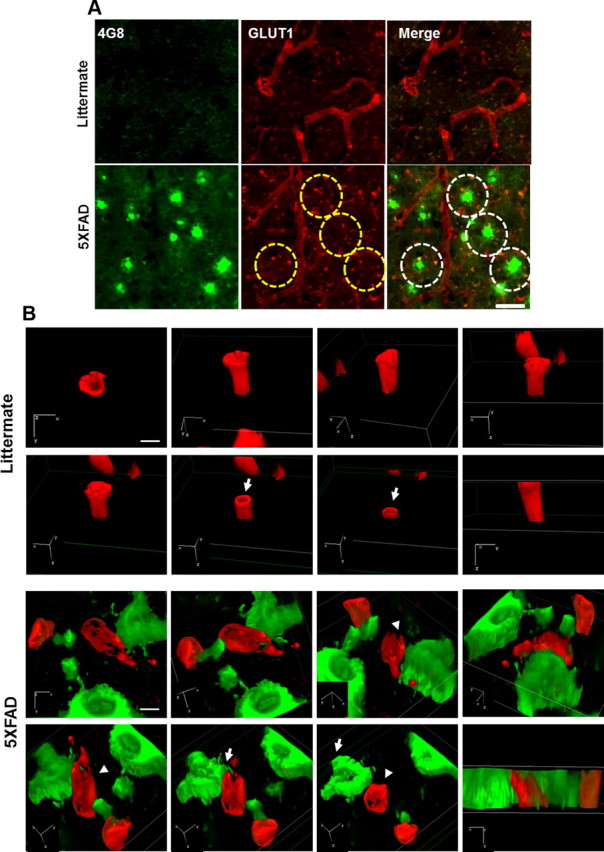
Cerebral capillaries are impaired in the brains of 5XFAD mice. A, Coronal serial sections of brains from 8-month-old mice (n = 3 for each from littermates and 5XFAD mice) were costained with anti-GLUT-1 (red) and anti-Aβ (green; 4G8) antibodies and imaged by confocal microscopy. Capillaries stained with anti-GLUT-1 antibody (red) showed long tubular-like form in littermate mice. 5XFAD mice displayed amyloid plaque deposition (green) and cut capillary forms (the parts shown in the yellow dotted circle). Capillaries adjacent to the amyloid plaques displayed disconnected tubular-like form in 5XFAD in the merged images (the parts shown in the white dotted circle). Scale bar, 40 μm. B, 3D-SIM images of the brains from littermates and 5XFAD mice. Brain slices were each recorded by 3D-SIM images in the z-direction with a thickness of 0.150 μm, reconstructed, and made into a 3D volume image with α blending function. Axial directions were represented on each image. Capillaries stained with anti-GLUT-1 antibody (red) and amyloid plaque stained with anti-Aβ (green; 4G8) antibody. Arrow, sectioned z-axis image; arrowhead, damaged microvessel. Scale bar, 2 μm. 3D depth, 4.65 μm in littermate and 5.45 μm in 5XFAD.
Expressions of RAGE and MMP were increased at the microvessels in cortex of 8-month-old 5XFAD mice
Since the in vitro result from Figure 4 strongly suggests that RAGE mediates Aβ-induced BBB disruption and 5 XFAD mice have massive amyloid depositions in the brain, it is reasonable to examine RAGE expression in the vessels of 5XFAD mouse brains. RAGE expression was increased in cortical layers 1 through 3 compared with that in littermates (Fig. 6A). RAGE and GLUT-1 were colocalized, suggesting RAGE exists in cerebral capillaries. Overlapped RAGE and GLUT-1 staining increased yellow fluorescence in merged images of 5XFAD mice and these images were magnified and quantified thepercentage area occupied by RAGE (Fig. 6B,C; n = 4, 1.6-fold; p < 0.05), suggesting RAGE expression was increased in brain capillaries of 5XFAD mice. To clearly illustrate RAGE expression and localization on cerebral capillaries in the AD mouse model, we used the super-resolution SIM (Nikon). The reconstructed 3D-SIM image showed that 5XFAD mice showed increased RAGE expression on damaged cerebral capillaries compared with that of littermates (Fig. 6D). Since Aβ1–42-induced secretion of MMPs was detected by zymography in bEnd.3 cells (Fig. 2B), immunoreactivity of MMPs was examined. We found that immunoreactivity of MMPs was increased by the surrounding cerebral capillaries of 5XFAD mice (Fig. 6E). These results are consistent with previous reports of increased RAGE expression in the vascular system in AD brain (Yan et al., 1996).
Figure 6.
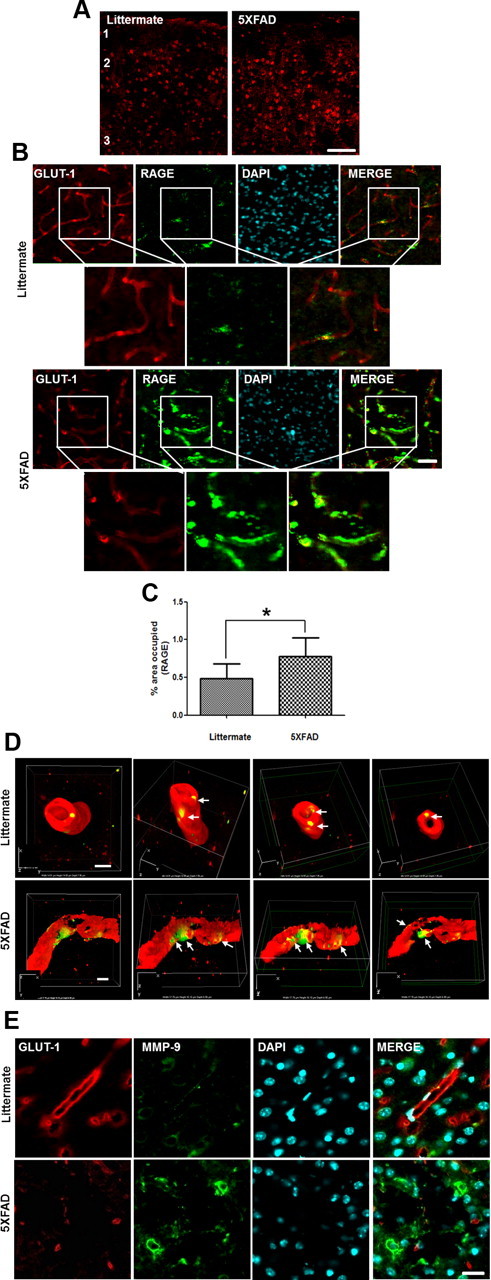
Expressions of RAGE and MMP are increased in the capillary from the brains of 8-month-old 5XFAD mice. A, Representative figure of RAGE expression in the cortex. RAGE-positive cells were increased in the cortex of 5XFAD mice. Red, anti-RAGE antibody staining. 1, 2, and 3 represent cortical layers 1, 2, and 3. Scale bar, 60 μm. B, Immunostainings of both RAGE (green) and GLUT-1 (red). RAGE expression was increased more in GLUT-1-positive capillaries of the brains of 5XFAD than in littermate brains. Red, GLUT-1; green, RAGE; blue, DAPI staining for nucleus. Scale bar, 30 μm. Boxed areas were magnified for each figure. Scale bar, 30 μm. C, Percentage of area occupied by RAGE was quantified. n = 4 for each from littermate and 5XFAD mice. D, 3D-SIM reconstruction images of RAGE expression on the capillaries. RAGE expression on the capillaries (arrows) was increased significantly more in 5XFAD than in littermate. The sectioned z-axis images represented that the capillaries were more impaired in the brains of 5XFAD mice than in the brains of their littermates. Each 3D-SIM image was taken in a z-direction with a thickness of 0.150 μm, reconstructed, and made into a 3D volume image with the α blending function. Axial directions were represented on each of the images. The first frame image showed on top of the reconstructed 3D-SIM image and its scale is represented on the image. Scale bars, 2 μm. 3D depth, 7.95 μm in littermate and 6.90 μm in 5XFAD. E, Immunoreactivity of MMP was increased around the cerebral capillaries of 8-month-old 5XFAD mice brains. n = 3 for each cortex from littermate and 5XFAD. Red, GLUT-1; green, RAGE (D) or MMP (E); blue, DAPI staining for nucleus. Scale bar, 20 μm.
5XFAD mouse brains showed alterations in cerebral TJs
The structure of TJs between adjacent cells was revealed by EM in previous studies (Goodenough and Revel, 1970). Cerebral capillaries from littermates and 5XFAD mouse brains were examined by EM to identify changes of cerebral TJ. Interestingly, TJs in 5XFAD mice appeared to be significantly shorter lengths than TJs seen in littermate mice (Fig. 7A,B, arrows). Fifteen TJs from littermates and 5XFAD mice were measured. The length of TJs from 5XFAD mouse brains (1091.2 ± 217.3 nm) is shorter and is approximately two times the littermate TJs (Fig. 7C; n = 12, 538.1 ± 71.35 nm; p < 0.05).
Figure 7.
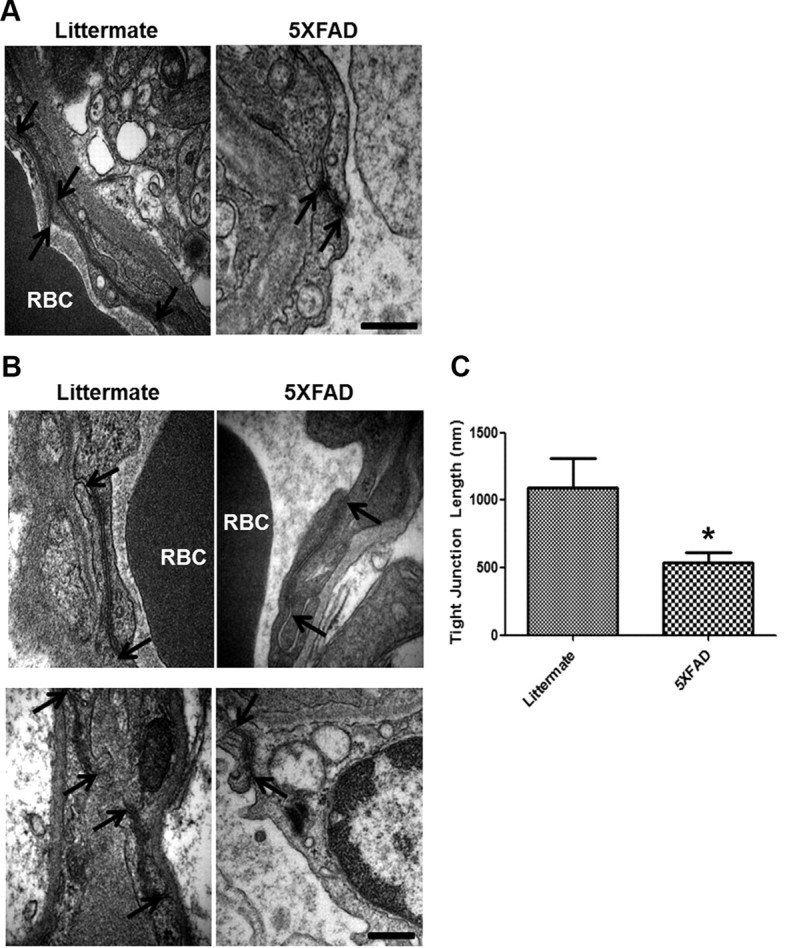
5XFAD mice show alterations in cerebral TJs. A, EM from the brains of 5XFAD showed alterations of TJs compared with littermates. TJs of 5XFAD mice were shorter than the littermate's TJs. n = 3 for each cortex from littermate and 5XFAD. Arrow, TJs; RBC, red blood cells. Scale bar, 500 nm. B, Higher magnification of TJs in the littermate and 5XFAD mice brains. Arrow, TJs. Scale bar, 200 nm. C, Length of 15 TJs from littermate (1091.2 nm) and 5XFAD (538.1 nm) were examined. Graph shows an average of 12 capillaries. *p < 0.05.
Discussion
Despite several lines of evidence that showed the relationship between BBB disruption and AD previously, the precise mechanisms of Aβ-induced TJ alterations are unknown (Wardlaw et al., 2003). Previous reports suggest that Aβ disrupts TJs directly and alters BBB integrity, contributing to AD-related pathogenesis (Claudio, 1996; Marco and Skaper, 2006). Even though it showed the alteration of TJ proteins in a high concentration of 20 μm Aβ1–42 for 3 d (Marco and Skaper, 2006), we confirmed that a low concentration of 5 μm Aβ1–42 also induced structural alteration and reduced the protein level of other TJ proteins such as claudin-5 and occludin as well as ZO-1 (data not shown). FD-40 penetration assay with Transwell plates showed that 5 μm Aβ1–42 increased permeability of FD-40 (Fig. 1E,F), suggesting Aβ alters TJs distribution and breaks BBB integrity.
In this study, we observed that a neutralizing antibody against the extracellular domain of RAGE effectively blocked Aβ1–42-induced disruptions in TJs, suggesting that Aβ-RAGE interactions are critical for BBB integrity. In the brain, RAGE is normally expressed on endothelial cells, microglia, and neurons (Deane et al., 2003; Li et al., 2009). Also, the influx of Aβ across the BBB depends on its interaction with RAGE (Mackic et al., 1998), and RAGE and LRP-1 have been proposed to stabilize Aβ concentrations in the plasma and brain through the BBB (Shibata et al., 2000). Although several pathological functions of RAGE have been reported—including perturbation of neuronal function, amplification of glial inflammatory responses, elevation of oxidative stress, amyloidosis, and increased Aβ influx at the BBB in AD (Yan et al., 1996; Deane et al., 2003)—the mechanism of TJ disruption by RAGE-Aβ interactions has not been documented. We observed that Aβ1–42 increased intracellular calcium levels with RAGE and activated MMPs through the CaN pathway, as demonstrated by treatment with CaN and MMP inhibitors (Fig. 3). In addition, these events were mitigated by a neutralizing antibody against RAGE, suggesting that disruption of the BBB by Aβ1–42 is initiated by an interaction between Aβ1–42 and RAGE, followed by intracellular signaling cascades in bEnd.3 cells.
Dysregulation of intracellular calcium is associated with AD pathogenesis (Mattson et al., 1992). Aβ can bind to NMDA receptors and trigger calcium influx (De Felice et al., 2007). Further, increasing intracellular calcium levels interferes with the formation of TJ (Stuart et al., 1996). We have observed that RAGE affects Aβ-induced calcium influx (Cho et al., 2009) and that the influx is sustained during Aβ treatment (Fig. 2A). In addition, CaN, a calcium-dependent phosphatase, is activated by Aβ in neurons and might influence AD progression (Liu et al., 2005; Agostinho et al., 2008).
FK506, which targets CaN phosphatase activity, is widely used to prevent a CaN upregulation by Aβ. In the APP/PS1 mouse model of AD, FK506 reduces the accumulation of Aβ and mitigates gliosis and CaN activity in the brain (Hong et al., 2010). Little is known about the effect of GM6001 and FK506 on Aβ-induced BBB permeability. Interestingly, both GM6001 and FK506 declined Aβ-induced ZO-1 alteration and BBB permeability, suggesting that both CaN and MMPs are able to affect BBB maintenance. Thus, we suggest that RAGE not only transports Aβ into the brain but also mediates Aβ-induced signaling, implicating a new function of TJ disruption in endothelial cells. Such a phenomenon would allow Aβ and blood-borne substances, including antibodies and other immune-related compounds, to cross the BBB with increased and unregulated influx into the brain.
Warboys et al. (2009) demonstrated that RAGE is required for causing the microvascular permeability to increase, resulting in accumulating advanced glycation end products. Notably, RAGE expression upregulates severalfold in cerebral vessels, microglia, and neurons that are affected in human AD brains and rodent AD model brains (Zlokovic, 2008). Several studies, including our previous report, have shown that RAGE levels rise with age in rodents and humans (Simm et al., 2004; Cho et al., 2009). In addition, RAGE expression in neurons and human brain microvascular endothelial cells is increased on treatment with Aβ (Cho et al., 2009; Li et al., 2009). Figure 4 shows that RAGE-overexpressing bEnd.3 cells experience greater reductions in ZO-1 levels during treatment with Aβ.
To confirm these data in animal models of AD, we used 5XFAD mice, which show massive amyloid plaque deposition with neuronal loss from 2 months old and behavioral abnormality from 6 months old (Oakley et al., 2006). Interestingly, abnormal and disrupted cerebral capillaries were shown in the location where amyloid plaques were deposited by using a cerebral endothelial marker in 5XFAD mice brains (Fig. 5). We used super-resolution microscopy and 3D-SIM image apparently showed that the cerebral capillaries of 5XFAD mice was more damaged and had scratches all over the vessel while littermates showed a whole, enclosed thick and strong vessel. At the same time, upregulation of RAGE expression in cerebral capillaries was detected in 5XFAD mouse brains (Fig. 6), suggesting that the increased Aβ-RAGE interactions could amplify TJ alterations in BBB as confirmed in bEnd.3 cells and disrupted BBB passes many toxic molecules including Aβ itself, following acceleration of neuronal cell death as well as Aβ deposition in the brain. In addition, EM study showed alterations in TJ morphology from 5XFAD mouse brains (Fig. 7). We suggest that shortening of the TJ in 5XFAD mice indicates a few TJ proteins and a lot of difficulty from molecule transport restriction. This result supports a high possibility of BBB breakdown in AD brains. Although we did not perform the experiments with FK506- or GM6001-injected 5XFAD mice, previous reports demonstrated that amyloid plaque burden is reduced in FK506-injected APPswe/PS1dE9 mouse brain (Hong et al., 2010) and GM6001 reduces oxidative stress-associated CAA in the same animal model (Garcia-Alloza et al., 2009).
Together, we believe that Aβ1–42 treatment induces RAGE expression and that the interaction between Aβ and RAGE triggers an intracellular signaling cascade that disrupts TJs, leading to the breakdown of BBB integrity. We confirmed that Aβ-induced TJ breakdown occurred by monitoring increases in intracellular calcium, CaN, and MMPs using potent inhibitors. Also, TJ breakdown was confirmed in in vivo AD animal models. We conclude that the Aβ-RAGE-CaN-MMP cascade is an important mechanism of BBB disruption and AD pathogenesis and is an excellent target for AD therapies.
Footnotes
This work was supported by grants from National Research Foundation (2012R1A2A1A01002881, 2008-05943), Medical Research Center (2011-0030738), World Class University (R32-10084), and Korean National Institute of Health Road R & D program project (A092058; I.M.-J.).
The authors declare no competing financial interests.
References
- Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Maness LM, Banks MF, Shayo M, McLay RN. Interactions of beta-amyloids with the blood-brain barrier. Ann N Y Acad Sci. 1997;826:190–199. doi: 10.1111/j.1749-6632.1997.tb48470.x. [DOI] [PubMed] [Google Scholar]
- Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. FEBS Lett. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- Brown RC, Morris AP, O'Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007;1130:17–30. doi: 10.1016/j.brainres.2006.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer's disease animal model. FASEB J. 2009;23:2639–2649. doi: 10.1096/fj.08-126383. [DOI] [PubMed] [Google Scholar]
- Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol. 1996;91:6–14. doi: 10.1007/s004010050386. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Deane R, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Doller A, Akool el–S, Müller R, Gutwein P, Kurowski C, Pfeilschifter J, Eberhardt W. Molecular mechanisms of cyclosporin A inhibition of the cytokine-induced matrix metalloproteinase-9 in glomerular mesangial cells. J Am Soc Nephrol. 2007;18:581–592. doi: 10.1681/ASN.2006060568. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Prada C, Lattarulo C, Fine S, Borrelli LA, Betensky R, Greenberg SM, Frosch MP, Bacskai BJ. Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathy in vivo in transgenic mice. J Neurochem. 2009;109:1636–1647. doi: 10.1111/j.1471-4159.2009.06096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Velasquez FJ, Kotarek JA, Moss MA. Soluble aggregates of the amyloid-beta protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J Neurochem. 2008;107:466–477. doi: 10.1111/j.1471-4159.2008.05618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, Revel JP. A fine structural analysis of intercellular junctions in the mouse liver. J Cell Biol. 1970;45:272–290. doi: 10.1083/jcb.45.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Heyman A, Fillenbaum GG, Welsh-Bohmer KA, Gearing M, Mirra SS, Mohs RC, Peterson BL, Pieper CF. Cerebral infarcts in patients with autopsy-proven Alzheimer's disease: CERAD, part XVIII. Consortium to Establish a Registry for Alzheimer's Disease. Neurology. 1998;51:159–162. doi: 10.1212/wnl.51.1.159. [DOI] [PubMed] [Google Scholar]
- Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AbetaPP/PS1 double transgenic mice. J Alzheimers Dis. 2010;22:97–105. doi: 10.3233/JAD-2010-100261. [DOI] [PubMed] [Google Scholar]
- Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. The channel hypothesis of Alzheimer's disease: current status. Peptides. 2002;23:1311–1315. doi: 10.1016/s0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. The blood-brain barrier and cerebrovascular pathology in Alzheimer's disease. Ann N Y Acad Sci. 1999;893:113–125. doi: 10.1111/j.1749-6632.1999.tb07821.x. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM, Hsu CY, Xu J. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol. 2003;54:379–382. doi: 10.1002/ana.10671. [DOI] [PubMed] [Google Scholar]
- Li M, Shang DS, Zhao WD, Tian L, Li B, Fang WG, Zhu L, Man SM, Chen YH. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol. 2009;182:5778–5788. doi: 10.4049/jimmunol.0803013. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer's amyloid-beta 1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998;102:734–743. doi: 10.1172/JCI2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco S, Skaper SD. Amyloid beta-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–224. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Matsubara E, Ghiso J, Frangione B, Amari M, Tomidokoro Y, Ikeda Y, Harigaya Y, Okamoto K, Shoji M. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol. 1999;45:537–541. [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restituito S, Khatri L, Ninan I, Mathews PM, Liu X, Weinberg RJ, Ziff EB. Synaptic autoregulation by metalloproteases and gamma-secretase. J Neurosci. 2011;31:12083–12093. doi: 10.1523/JNEUROSCI.2513-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simm A, Casselmann C, Schubert A, Hofmann S, Reimann A, Silber RE. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol. 2004;39:407–413. doi: 10.1016/j.exger.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Lai MM, Burnett PE. Immunophilins in the nervous system. Neuron. 1998;21:283–294. doi: 10.1016/s0896-6273(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF, Ghiso J, Dehouck MP, Frangione B, Patlak C, Fenstermacher J, Gorevic P. In vitro evidence that beta-amyloid peptide 1–40 diffuses across the blood-brain barrier and affects its permeability. J Neuropathol Exp Neurol. 2000;59:29–38. doi: 10.1093/jnen/59.1.29. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem. 1996;271:13636–13641. doi: 10.1074/jbc.271.23.13636. [DOI] [PubMed] [Google Scholar]
- Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14:1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warboys CM, Toh HB, Fraser PA. Role of NADPH oxidase in retinal microvascular permeability increase by RAGE activation. Invest Ophthalmol Vis Sci. 2009;50:1319–1328. doi: 10.1167/iovs.08-2730. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Sandercock PA, Dennis MS, Starr J. Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke. 2003;34:806–812. doi: 10.1161/01.STR.0000058480.77236.B3. [DOI] [PubMed] [Google Scholar]
- Watson PM, Anderson JM, Vanltallie CM, Doctrow SR. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci Lett. 1991;129:6–10. doi: 10.1016/0304-3940(91)90708-2. [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer's disease. Neurotherapeutics. 2008;5:409–414. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]