

Abstract
Mutations in parkin and LRRK2 together account for the majority of familial Parkinson's disease (PD) cases. Interestingly, recent evidence implicates the involvement of parkin and LRRK2 in mitochondrial homeostasis. Supporting this, we show here by means of the Drosophila model system that, like parkin, LRRK2 mutations induce mitochondrial pathology in flies when expressed in their flight muscles, the toxic effects of which can be rescued by parkin coexpression. When expressed specifically in fly dopaminergic neurons, mutant LRRK2 results in the appearance of significantly enlarged mitochondria, a phenotype that can also be rescued by parkin coexpression. Importantly, we also identified in this study that epigallocatechin gallate (EGCG), a green tea-derived catechin, acts as a potent suppressor of dopaminergic and mitochondrial dysfunction in both mutant LRRK2 and parkin-null flies. Notably, the protective effects of EGCG are abolished when AMP-activated protein kinase (AMPK) is genetically inactivated, suggesting that EGCG-mediated neuroprotection requires AMPK. Consistent with this, direct pharmacological or genetic activation of AMPK reproduces EGCG's protective effects. Conversely, loss of AMPK activity exacerbates neuronal loss and associated phenotypes in parkin and LRRK mutant flies. Together, our results suggest the relevance of mitochondrial-associated pathway in LRRK2 and parkin-related pathogenesis, and that AMPK activation may represent a potential therapeutic strategy for these familial forms of PD.
Introduction
Parkinson's disease (PD) is a prevalent neurodegenerative movement disorder characterized pathologically principally by the loss of dopaminergic (DA) neurons in the substantia nigra. Although a subject of intense research, the etiology of PD remains poorly understood. However, during the last decade or so, the identification and functional characterization of several genes whose mutations are causative of familial forms of PD have provided tremendous insights into the molecular events underlying neurodegeneration in PD (Kumar et al., 2010; Corti et al., 2011). Collectively, these studies implicate aberrant mitochondrial homeostasis as one of the key contributors to PD.
Among the disease-associated genes, recessive mutations in Parkin and dominant mutations in LRRK2 (especially the G2019S variant) are currently recognized to be the most prevalent cause of early-onset and late-onset familial PD, respectively (Healy et al., 2008; Kumari and Tan, 2009). Interestingly, Drosophila parkin and LRRK2 mutants could reproduce the essential pathological phenotypes as well as the age of disease onset associated with the two PD-linked genes (Lim and Ng, 2009; Dawson et al., 2010). Further, Drosophila parkin mutants exhibit pronounced mitochondrial abnormalities in their flight musculature, a phenotype that can be rescued by overexpression of wild-type human parkin (Greene et al., 2003; Park et al., 2006; Wang et al., 2007). Importantly, parkin overexpression in flies also protects against LRRK2 G2019S-mediated DA neurotoxicity in the presence or absence of rotenone, a mitochondrial complex I inhibitor (Ng et al., 2009). Together, these results suggest a link between the two PD-linked genes and mitochondrial function. Accordingly, we speculate that mutant LRRK2 expression may affect mitochondrial function in a similar manner brought about by parkin deficiency.
Here, we demonstrated by means of the Drosophila model system that transgenic expression of human LRRK2 G2019S mutant induces marked mitochondrial pathology in vivo, and that parkin coexpression effectively rescued this mutant LRRK2-induced phenotype. Importantly, we found that pharmacological or genetic activation of AMP-kinase (AMPK), a key cellular regulator of energy metabolism (Hardie, 2011), offers significant protection against DA and mitochondrial dysfunction in both LRRK2 and parkin mutant flies. Together, our results suggest that parkin- and LRRK2-related parkinsonism operate through a common pathogenic pathway involving mitochondrial dysfunction and that AMPK activation may represent a viable therapeutic strategy for multiple forms of PD.
Materials and Methods
Fly stocks.
Fly lines for 24B-Gal4 (muscle-specific), ddc-Gal4 (dopaminergic neuron-specific), elav-Gal4 (pan-neuronal), UAS-mito-GFP, and UAS-dAMPK-KA were purchased from Bloomington Drosophila Stock Center. UAS-siAMPK was purchased from Vienna Drosophila RNAi Center (Vienna, Austria). UAS-dAMPK-TD, UAS-parkin, LRRK2-WT, and LRRK2-G2019S transgenic and parkin-null flies were previously described (Lee et al., 2007a; Wang et al., 2007; Ng et al., 2009).
Immunohistochemistry and Western blotting.
Immunohistochemical analysis of whole-mount adult fly brains were prepared according to published protocols (Whitworth et al., 2005) and stained with rabbit anti-TH (1:300, Pel-Freez Biologicals) as primary antibody. The stained samples were viewed using an Olympus Fluoview Upright Confocal Microscope. DA neurons were quantified according to a published method (Whitworth et al., 2005). The size of mito-GFP puncta was measured using the ImageJ program and expressed as mean ± SD (n ≥ 10 DA neurons per experimental group). For Western blotting, 20 fly heads per group were homogenized in PBS containing 1% SDS before centrifugation (16,000 × g × 30 min) to remove the debris. An equivalent amount of the clarified lysate was resolved by SDS-PAGE and subsequently immunoblotted with anti-LRRK2 (Sigma) or anti-tubulin (Developmental Studies Hybridoma Bank).
Climbing assay and drug treatment.
Climbing assays were performed according to a previously described method (Ng et al., 2009). Briefly, 20 female adult flies from each group were randomly selected after anesthetization and placed in a vertical plastic column (length 25 cm; diameter 1.5 cm). Age-matched normal flies were used as controls. After a 2 h recovery period from CO2 exposure, flies were gently tapped to the bottom of the column, and the number of flies that reached the top of column at 1 min was counted. Results are presented as mean ± SEM of the scores obtained from three independent experiments. To study the effect of drugs, flies were fed with cornmeal-agar medium supplemented with 0.5 mm epigallocatechin gallate (EGCG; Sigma), 1 mm 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR; Cayman Chemicals), 10 mm metformin (Sigma), or 0.5 mm Compound C (EMD Biosciences) immediately at posteclosion (for parkin-null flies) or at day 35 onward (for LRRK2 flies) for a period of 25 d.
Dopamine measurement.
Ten fly heads were homogenized in 0.1 m perchloric acid, and the lysates were filtered through 0.1 μm Durapore Ultrafree Centrifugal filters (Millipore) before HPLC measurement of dopamine levels.
Transmission electron microscopy.
Indirect flight muscles were prepared by fixing in 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.2, containing 0.1% Tween-20, before being postfixed in 1% OsO4 for 1 h. Subsequently, they were embedded in Spurr's resin and cut to ultrathin sections before staining with uranyl acetate and lead citrate before they were observed under a JEM-1230 electron microscope (Jeol). Selected specimens were sent for commercial EM analysis at the EM Research Services (Newcastle University, Newcastle, UK) as an added confirmation.
Statistical analysis.
Statistical significance for all the quantitative data obtained were analyzed using one-way ANOVA with Tukey's test HSD post hoc test (*p < 0.05, **p < 0.001).
Results
Disease-associated LRRK2 G2019S mutant induces marked mitochondrial pathology in vivo
To examine the potential relationship between LRRK2 and mitochondrial function in vivo, we expressed human wild-type and G2019S LRRK2 in Drosophila under the direction of a muscle-specific promoter (24B-GAL4) and found that mutant LRRK2 expression in Drosophila flight muscles results in prominent mitochondrial abnormalities that become more severe with age (Fig. 1A). Further, these mutant flies also exhibit significant age-related climbing impairment relative to control (Fig. 1B), suggesting that LRRK2 G2019S-induced mitochondrial pathology in Drosophila flight muscles compromises their locomotion function. In contrast, these mutant LRRK2-associated phenotypes are not evident in flies expressing wild-type LRRK2 (Fig. 1A,B), which is consistent with previous reports by our group and others showing that wild-type LRRK2 overexpression in flies triggers comparatively less toxicity than its disease-associated counterparts (Lee et al., 2007b; Imai et al., 2008; Ng et al., 2009). Given that LRRK2 G2019S-expressing flies seem to phenocopy the reported mitochondrial phenotype of parkin-null flies (Greene et al., 2003), and that parkin protects against LRRK2 G2019S-induced neurotoxicity (Ng et al., 2009), we surmised that parkin overexpression may potentially rescue the mitochondrial pathology induced by mutant LRRK2. Supporting this, double-transgenic flies coexpressing LRRK2 G2019S and wild-type parkin under the direction of 24B-GAL4 exhibit a greater number of normal mitochondria and significantly better climbing scores than single LRRK2 G2019S transgenic flies (Fig. 1B,C). As the above experiments involving Drosophila flight muscles represent more of an assay of toxicity rather than the etiopathogenesis of PD, we also examined the effects of mutant LRRK2 expression in DA neurons. Interestingly, when LRRK2 G2019S expression is confined to DA neurons by the Ddc-GAL4 driver, it results in the appearance of significantly enlarged mitochondria, a phenotype that can also be rescued by Ddc-GAL4-mediated overexpression of parkin (Fig. 1D). Together, our results support an intimate link between parkin and LRRK2 in mitochondrial homeostasis.
Figure 1.

LRRK2-induced mitochondrial pathology can be rescued by parkin overexpression. A, TEM images of indirect flight muscles of 40- and 60-d-old flies expressing 24B-driven wild-type (WT) and G2019S human LRRK2. Boxed regions in the top panels are shown at higher magnification in corresponding bottom panels. B, Climbing score of 24B-driven wild-type, LRRK2 G2019S-expressing, and LRRK2 G2019S/parkin double-transgenic flies (GS>hPK) at different age posteclosion, as indicated. C, TEM images of indirect flight muscles of 40- and 60-d-old 24B-driven LRRK2 G2019S-expressing flies in the absence or presence of human parkin coexpression (GS>hPK). D, Representative confocal microscopy images showing the expression and localization of mito-GFP (under the direction of Ddc-GAL4 promoter) (green) in TH-positive neurons (red) of control and various mutant flies, as indicated. Control refers to yw wild-type fly strain in all cases. Number refers to the average size ± SD of mito-GFP puncta in various LRRK2 flies relative to control (in arbitrary units).
Identification of EGCG as a potent suppressor of parkin- and LRRK2-associated phenotypes
Next, we speculated that compounds that can mitigate parkin dysfunction may potentially suppress LRRK2-associated phenotypes. We therefore used the Drosophila parkin-null mutant as an initial model to evaluate the therapeutic potential of a number of candidate compounds. Among them, we found that EGCG provides the best protection against parkin deficiency (data not shown). Indeed, EGCG-treated parkin-null flies exhibit significantly improved climbing scores compared with untreated mutant flies (Fig. 2A) that is associated with a remarkable recovery of mitochondrial integrity (Fig. 2B). Further, they also show reduced loss of DA neurons in the PPL1 cluster, the degeneration of which is otherwise apparent in parkin-deficient flies (Whitworth et al., 2005; Wang et al., 2007) (Fig. 2C). Thus, EGCG appears capable of compensating for parkin deficiency. We next examined whether EGCG can likewise protect against LRRK2-induced phenotypes and found that EGCG-treated mutant LRRK2 flies also recorded significant improvements in their climbing scores (Fig. 2D) and mitochondrial morphology (Fig. 2E). Similarly, EGCG treatment ameliorates the loss of DA neurons displayed by Ddc-GAL4-LRRK2 G2019S-expressing flies (Fig. 2F). Like their counterparts coexpressing parkin, EGCG-treated LRRK2 G2019S flies also display significantly fewer enlarged mitochondria in their DA neurons (Fig. 1D). Together, we have identified EGCG as a potent suppressor of DA dysfunction and mitochondrial abnormalities in two Drosophila genetic models of PD that represent recessive and dominant parkinsonism, respectively.
Figure 2.

EGCG treatment mitigates dopaminergic and mitochondrial dysfunction in both mutant LRRK2-expressing and parkin-null flies. A, Climbing score of untreated and EGCG-treated parkin-null (pk−/−) flies, as indicated. B, TEM images of indirect flight muscles of pk−/− flies treated with DMSO or EGCG. C, Quantification of DA neurons in the PPL1 cluster of untreated and EGCG-treated pk−/− flies. Inset, Confocal microscopy images of TH-positive neurons in PPL1 cluster of untreated and EGCG-treated pk−/− flies. D–F, same as A–C, respectively, except that pk−/− flies are substituted with Ddc-driven (D, F) or 24B-driven (E) G2019S LRRK2-expressing flies. Boxed regions in top panels of B and E are shown at higher magnification in corresponding bottom panels. G, Climbing score (left) and DA neuronal count (PPL1 cluster) (right) of Ddc-driven untreated or AICAR-treated LRRK2 G2019S-expressing flies relative to control flies. H, As in G, except that LRRK2 G2019S-expressing flies are substituted with pk−/− flies. I, As in H, except that AICAR is replaced by metformin. Data for control and pk−/− (i.e., climbing score and DA neurons) shown in H and I were derived from A and C. Number indicates the number of fly brains used for DA neuronal quantification. J, Survival curves of untreated or compound C-treated pk−/− (left) or LRRK2 mutant flies (right).
Pharmacological activation of AMPK mimics EGCG-mediated protective response
We were next interested to know how EGCG exerts its protective effects. Besides possessing antioxidant properties, EGCG has been reported to activate AMPK, and emerging evidence suggests that AMPK activation may be neuroprotective (Hwang et al., 2009; Spasić et al., 2009). To examine this, we treated Drosophila parkin and LRRK2 mutants with AICAR, a direct pharmacological activator of AMPK. AICAR treatment significantly improves the DA neuronal and climbing phenotypes in Ddc-GAL4-LRRK2 G2019S flies (Fig. 2G) as well as in parkin-null flies (Fig. 2H), suggesting that direct AMPK activation can protect against DA dysfunction in vivo. Similar observations were made when parkin-null flies were treated with metformin, another selective AMPK activator (Fig. 2I). Conversely, when these flies were treated with Compound C, a commonly used AMPK inhibitor, they exhibit markedly increased mortality (Fig. 2J).
EGCG-mediated protection is AMPK dependent
To address whether EGCG-mediated protective response requires AMPK, we knocked down AMPK expression in mutant LRRK2-expressing flies via siRNA strategy and found that reduced AMPK expression aggravates their climbing deficits although without further loss of protocerebral posterior lateral 1 (PPL1) DA neurons (Fig. 3A,B). Importantly, AMPK expression silencing in these flies abolishes the beneficial effects of EGCG treatment (Fig. 3A,B). To verify our finding obtained with siRNA methodology, we also generated bigenic flies coexpressing LRRK2 G2019S and a dominant-negative AMPK-KA mutant (Lee et al., 2007a) under the direction of the Ddc-GAL4 driver. Similar to AMPK expression silencing, AMPK-KA expression exacerbates mutant LRRK2-induced locomotion defects (Fig. 3A,B). Again, EGCG-mediated protective response is blocked in mutant LRRK2 flies in the presence of AMPK-KA coexpression (Fig. 3A,B). Not surprisingly, the dominant-negative AMPK mutant also blocks the protective effects of AICAR in LRRK2 G2019S flies (Fig. 3C,D). Together, our results identified AMPK activation as a potential downstream effector of EGCG-mediated protective pathway in our Drosophila PD models.
Figure 3.
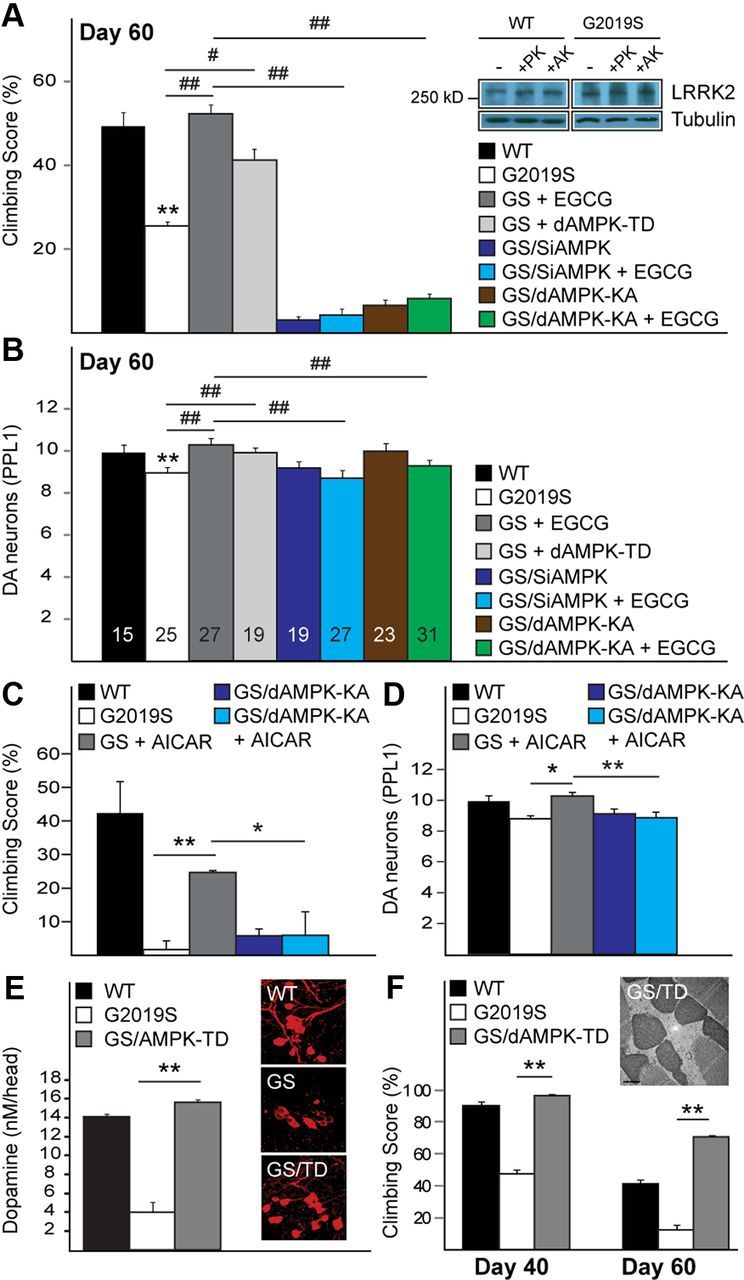
AMPK activation ameliorates Drosophila LRRK2 mutant phenotypes. A, B, Climbing score (A) and DA neuronal count (PPL1 cluster) (B) of untreated or EGCG-treated Ddc-driven G2019S LRRK2-expressing flies in the presence or absence of AMPK expression silencing (siAMPK), dominant-negative (dAMPK-KA), or constitutively active (dAMPK-TD) AMPK mutant, as indicated. Data for WT, G2019S, and G2019S + EGCG were derived from Figure 2, D and F. Inset, LRRK2 expression in the absence or presence of parkin (PK) or dAMPK (AK) coexpression (genotype: elav-Gal4-hLRRK2 or elav-Gal4-hLRRK2;elav-Gal4-hparkin or elav-Gal4-hLRRK2;elav-Gal4-dAMPK). Experiment was repeated at least three times with essentially similar results, C, D, Similar to A and B, except EGCG is replaced by AICAR. E, Dopamine levels in wild-type (WT) LRRK2, LRRK2 G2019S and LRRK2 G2019S/AMPK-TD flies as measured by HPLC. Inset, Confocal microscopy images of TH-positive neurons in PPL1 cluster of WT and G2019S (GS) LRRK2-expressing and G2019S/AMPK-TD (GS/TD) double-transgenic flies. F, Climbing score of 24B-driven G2019S LRRK2-expressing flies in the presence or absence of dAMPK-TD coexpression. Inset, Representative TEM image of indirect flight muscles of dAMPK-TD/G2019S double-transgenic flies (GS/dAMPK-TD).
Genetic activation of AMPK ameliorates parkinsonian phenotypes in mutant LRRK2 and parkin-null flies
To confirm that AMPK activation is indeed protective, we generated double-transgenic flies expressing mutant LRRK2 in the presence of a constitutively active form of AMPK (AMPK-TD) (Lee et al., 2007a). As expected, the climbing deficits and DA neuronal loss associated with mutant LRRK2 expression are significantly ameliorated in these double (Ddc-AMPK-TD/LRRK2 G2019S) transgenic flies (Fig. 3A,B,E). Notably, the level of LRRK2 is not significantly changed in the presence of AMPK (or parkin) coexpression (Fig. 3A). Moreover, HPLC analysis revealed a dramatic increase in dopamine levels in brains of double-transgenic flies relative to flies expressing LRRK2 G2019S alone (Fig. 3E), which we have previously reported to be abnormally low (Ng et al., 2009). When driven by the 24B-GAL4 driver, coexpression of AMPK-TD with LRRK2 G2019S in Drosophila flight muscles results in a significant improvement of the otherwise poor climbing performance of LRRK2 mutant flies (Fig. 3F), and a marked protection against mutant LRRK2-induced mitochondrial abnormalities (Fig. 3F).
Finally, we also generated double-mutant flies expressing the constitutively active AMPK mutant in a parkin-null background and found that AMPK-TD overexpression mitigates the mitochondrial pathology induced by the loss of parkin function to an extent that results in a significant improvement in their climbing ability (Fig. 4A). Accompanying this, AMPK-TD overexpression also leads to a remarkable recovery of the abnormal wing posture that characterized parkin-null flies (Fig. 4B).
Figure 4.
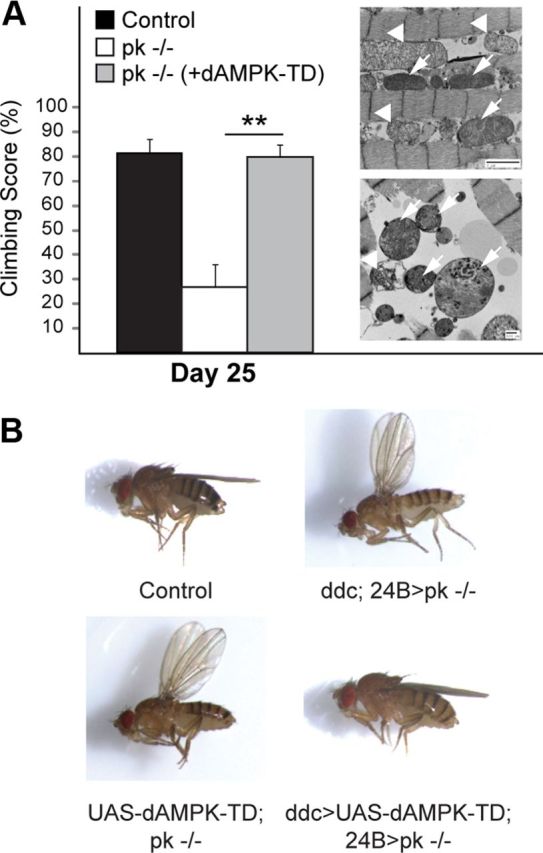
AMPK activation ameliorates Drosophila parkin mutant phenotypes. A, Climbing score of parkin-null (pk−/−) flies in the absence or presence of dAMPK-TD coexpression, as indicated. Inset, Representative TEM images of indirect flight muscles of dAMPK-TD/pk−/− bigenic flies. Note the significantly improved mitochondrial integrity (arrows) amid abnormal mitochondria (arrowheads). B, Representative images showing the wing phenotype of pk−/− flies in the absence or presence of dAMPK-TD coexpression.
Discussion
We have demonstrated in this study that disease-associated LRRK2 G2019S mutation induces mitochondrial pathology in vivo and that parkin coexpression is sufficient to rescue this mutant LRRK2-mediated phenotype. Importantly, we further found that pharmacological or genetic activation of AMPK exerts potent protection against DA and mitochondrial dysfunction in both LRRK2 and parkin mutant flies, suggesting that AMPK activation may represent a viable therapeutic strategy for different forms of PD.
Mitochondrial dysfunction is currently widely accepted to be a key driver of neurodegeneration. Supporting this, several postmortem studies have demonstrated a significant reduction in the activity of mitochondrial complex I as well as ubiquinone (coenzyme Q10) in the substantia nigra of PD brains (Schapira et al., 1989; Shults et al., 1997). Moreover, mitochondrial poisoning recapitulates PD features in humans and represents a popular strategy to model the disease in animals (Dauer and Przedborski, 2003). Despite their seemingly disparate function, it is also becoming clear that the activities of different PD-linked genes all have significant impact on mitochondrial homeostasis (Lim et al., 2012). In particular, a recent landmark paper identified parkin as a key mediator of mitochondrial quality control (Narendra et al., 2008). Consistent with this, abnormalities in mitochondrial structure and/or dynamics were detected in Drosophila parkin mutant and other parkin-related models (Greene et al., 2003; Wang et al., 2011). Comparatively less is known about the relationship between LRRK2 and mitochondrial function, although a Caenorhabditis elegans-based study suggests that LRRK2 may protect against mitochondrial damage (Saha et al., 2009). Here, we show that transgenic overexpression of LRRK2 G2019S in Drosophila flight muscles results in prominent mitochondrial pathology, the toxic effects of which can be rescued by parkin coexpression. Moreover, mutant LRRK2 expression in DA neurons triggers an enlarged mitochondria phenotype, which similarly can be rescued by parkin coexpression. Our results thus suggest a functional link between LRRK2 and parkin in mitochondrial homeostasis. However, an important caveat here is that we have used an overexpression system to assay for LRRK2 toxicity [i.e., whether LRRK2-induced mitochondrial phenotype (particularly in muscles) is relevant to humans or not is debatable]. Notwithstanding this, LRRK2 patient-derived cells reportedly exhibit altered mitochondrial dynamics that is accompanied by a reduction in mitochondrial membrane potential and total intracellular ATP levels (Mortiboys et al., 2010), which lends relevance to our findings.
Given that parkin apparently protects against LRRK2-mediated toxicity, we reasoned that compounds that could compensate for the loss of parkin function would have good potential to mitigate LRRK2-associated phenotypes. In a candidate drug screen, we found that EGCG acts as a potent suppressor of DA and mitochondrial dysfunction in parkin-null flies, and, as expected, in mutant LRRK2-expressing flies. Notably, EGCG treatment has previously been reported to be capable of suppressing the toxic effects of the parkinsonian neurotoxins MPTP and 6-OHDA (Chao et al., 2010). Our current study further elucidated a protective mechanism of EGCG that is dependent on the activation of AMPK. Interestingly, a recent report demonstrated that AMPK is activated in mice treated with MPTP and that inhibition of AMPK function by compound C enhances 1-methyl-4-phenylpyridinium-induced cell death (Choi et al., 2010). More recently, a PD cohort-based study revealed that Metformin-inclusive sulfonylurea therapy reduces the risk for the disease occurring with Type 2 diabetes in a Taiwanese population (Wahlqvist et al., 2012). Together, these findings support our suggestion that AMPK activation may protect against the development of PD.
AMPK is an evolutionarily conserved cellular energy sensor that is activated by ATP depletion or glucose starvation (Hardie, 2011). When activated, AMPK switches the cell from an anabolic to a catabolic mode and, in so doing, helps to regulate cellular energy demands. It is noteworthy that LRRK2-mediated neurodegeneration has been reported to compromise neuronal energy homeostasis due to the chronic activation of protein translation, a highly energy-demanding process (Imai et al., 2008). Accordingly, overexpression of AMPK could restore the energy perturbation induced by mutant LRRK2 and preserve neuronal function. Another attractive explanation regarding AMPK-mediated neuroprotection came from recent studies demonstrating that AMPK, like parkin, can regulate mitophagy (Egan et al., 2011; Kim et al., 2011). Mitophagy induction in this case occurs specifically through AMPK-mediated phosphorylation of the autophagy initiator ATG1. Given this functional convergence between AMPK and parkin, it is tempting to speculate that mitophagy induction may represent a common denominator underlying their respective ability to rescue the phenotypes of LRRK2 G2019S-expressing flies. Thus, in the absence of the parkin or AMPK transgene, LRRK2 mutant expression could significantly retard the clearance of damaged mitochondria, which progressively accumulate with age. This may also explain why the mitochondrial phenotype it induces in the flight muscle is so reminiscent of that brought about by the loss of parkin function.
In sum, our results here suggest that mitochondrial dysfunction may underlie both LRRK2- and parkin-related parkinsonism. Importantly, our results also suggest that pharmacological AMPK activation may represent a viable therapeutic strategy for these forms of PD.
Footnotes
This work was supported by grants from the Singapore Millennium Foundation, A*STAR Biomedical Research Council (K.-L.L.), National Medical Research Council (C.-H.N.), and Ministry of Education (X.Z.). We thank Valerie Tan and Jingjie Chen for technical assistance.
References
- Chao J, Lau WK, Huie MJ, Ho YS, Yu MS, Lai CS, Wang M, Yuen WH, Lam WH, Chan TH, Chang RC. A pro-drug of the green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG) prevents differentiated SH-SY5Y cells from toxicity induced by 6-hydroxydopamine. Neurosci Lett. 2010;469:360–364. doi: 10.1016/j.neulet.2009.12.028. [DOI] [PubMed] [Google Scholar]
- Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson's disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2010;391:147–151. doi: 10.1016/j.bbrc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev. 2011;91:1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetain CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JT, Kwon DY, Yoon SH. AMP-activated protein kinase: a potential target for the diseases prevention by natural occurring polyphenols. N Biotechnol. 2009;26:17–22. doi: 10.1016/j.nbt.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Greggio E, Beilina A, Kaganovich A, Chan D, Taymans JM, Wolozin B, Cookson MR. The Parkinson's disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PLoS One. 2010;5:e8730. doi: 10.1371/journal.pone.0008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari U, Tan EK. LRRK2 in Parkinson's disease: genetic and clinical studies from patients. FEBS J. 2009;276:6455–6463. doi: 10.1111/j.1742-4658.2009.07344.x. [DOI] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, Chung J. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007a;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007b;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Lim KL, Ng CH. Genetic models of Parkinson disease. Biochim Biophys Acta. 2009;1792:604–615. doi: 10.1016/j.bbadis.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Lim KL, Ng XH, Grace LG, Yao TP. Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid Redox Signal. 2012;16:935–949. doi: 10.1089/ars.2011.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–2020. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, Moore L, Goldstein L, Cookson M, Wolozin B. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–264. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- Spasić MR, Callaerts P, Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 2009;15:309–316. doi: 10.1177/1073858408327805. [DOI] [PubMed] [Google Scholar]
- Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord. 2012;18:753–758. doi: 10.1016/j.parkreldis.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]