

Abstract
T-cell recognition of cancer neoantigens is important for effective immune checkpoint blockade therapy, and an increasing interest exists in developing personalized tumor neoantigen vaccines. Previous studies utilizing RNA and long-peptide neoantigen vaccines in pre-clinical and early phase clinical studies have shown immune responses predominantly driven by MHC class II CD4+ T cells. Here, we report on a pre-clinical study utilizing a DNA vaccine platform to target tumor neoantigens. We showed that optimized strings of tumor neoantigens, when delivered by potent electroporation-mediated DNA delivery, were immunogenic and generated predominantly MHC class I–restricted, CD8+ T-cell responses. High MHC class I affinity was associated specifically with immunogenic CD8+ T-cell epitopes. These DNA neoantigen vaccines induced a therapeutic antitumor response in vivo, and neoantigen-specific T cells expanded from immunized mice directly killed tumor cells ex vivo. These data illustrate a unique advantage of this DNA platform to drive CD8+ T-cell immunity for neoantigen immunotherapy.
Keywords: DNA vaccine, neoantigen, personalized medicine, cancer immunotherapy
Introduction
Cancer neoantigens represent epitopes derived from tumor-specific somatic mutations that are presented on MHCs and have emerged as promising targets for personalized cancer immunotherapy. These epitopes are thought to be more robust immunotherapy targets compared to shared, overexpressed tumor-associated self-antigens due to i) their high frequency in human cancers (1), ii) their lack of expression in normal somatic tissues, and iii) their high potential for immunogenicity due to lack of tolerance. Importantly, the same immunogenic neoantigens are rarely shared across multiple patients (2). Therefore, this approach is personalized and requires rapid, efficient, and affordable sequencing, design, and manufacturing. The majority of these mutations are passenger mutations, and thus, a high likelihood of tumor escape exists unless multiple targets are included in the vaccine construct.
Early clinical trials using synthetic long peptides (SLPs) delivered with poly(I:C), dendritic cells loaded with short HLA class I–restricted peptides, or RNA vaccines encoding long neo-epitope peptides have shown immune responses directed against an important fraction of mutated epitopes delivered (3–5). Intriguingly, the vast majority of these responses driven by RNA or SLPs have been MHC class II–restricted, CD4+ T cells, both in early clinical studies and in pre-clinical mouse studies (3,4,6,7). This induction of CD4+ T-cell responses occurs despite the fact that the epitopes were selected in silico for high MHCI binding affinity (3,4,6). Although it has been established that CD4+ T cells are able to recognize tumor neoantigens, the majority of naturally occurring tumor antigen-specific killer T cells identified in patients have been of CD8+ T-cell origin (8).
Newer DNA vaccines are showing efficacy in the clinic (9,10). We have observed induction of CD8+ and CD4+ T cells, as well as anti-neoplastic activity and T-cell infiltration in tumors (9–11). Because of the potential for rapid synthesis of vaccine constructs and delivery of a large number of neo-epitopes simultaneously, we sought to study this DNA vaccine platform as a tool to develop immunity against cancer neoantigens. For this study, we chose tumor models that do not respond to immune checkpoint blockade alone (TC1, LLC, and ID8) (12–14). We sequenced these tumors to identify neoantigens and designed long strings of epitopes (12 epitopes per plasmid) separated by efficient cleavage sites. These synthetic neoantigen DNA vaccines (SNDVs) were then tested for effects on immunity and tumor impact in vivo. We observed that this DNA approach generated robust T-cell immunity against a similar proportion of epitopes compared to other vaccine platforms. However, the SNDVs generated a much larger proportion of CD8+ T-cell responses compared to prior studies. The SNDVs generated 75% of CD8+ only or CD4+/CD8+ T-cell responses, and 25% CD4+ only T-cell responses, showing a significant CD8+ T-cell bias. Inclusion of only high-affinity MHC class I (<500nM) epitopes selected for a larger proportion of immunogenic epitopes and for 100% CD8+ or CD8+/CD4+ T-cell epitopes. These SNDVs encoding neoantigens were able to control tumor growth therapeutically in vivo, and T cells expanded from immunized mice were able to kill tumor cells ex vivo. In summary, SNDVs are a promising approach to develop potent neoantigen-targeted immunity.
Materials and Methods
Animals and cell lines
8–10 week-old C57Bl/6 mice were purchased from Jackson labs. Animal experiments were approved by the Institutional Animal Care and Use Committee at The Wistar Institute. The TC1 cell line was provided by Y. Paterson (University of Pennsylvania) in 2011. B16 melanoma cell line was purchased from ATCC. The LLC cell line was purchased from ATCC in 2017. We generated TC1 and LLC tumors by injecting 100,000 cells subcutaneously in the flank. ID8 cells were provided by J.R. Conejo-Garcia (Moffitt Cancer Center) in 2016. We generated ID8 tumors by injecting 2 million cells intraperitoneally. Cells were maintained at low passage (<10 passages), and thawed directly from a master stock generated upon receipt of the cells for all experiments. Cells were routinely tested for Mycoplasma contamination prior to freezing them for storage, most recently in 2018. Cell lines were not genetically authenticated, but were examined for morphological authenticity in cell culture.
Mice were treated by injecting 25 μg of DNA resuspended in 30 μL of water into the tibialis anterior muscle followed by electroporation with the CELLECTRA®−3P device (Inovio Pharmaceuticals). For each immunization, mice were delivered two 0.1 Amp electric constant current square-wave pulses. For CD8+ and CD4+ T-cell depletion studies, mice were administered 200 μg of each antibody (CD8: YTS 169.4 and CD4: GK1.5, BioXCell) intraperitoneally twice weekly for a total of 13 doses.
Vaccination experiments
Immunogenicity:
Naive mice were vaccinated three times on 3-week intervals, and immune responses were measured a week after final vaccination. This was performed with the following plasmids: TC1 plasmid 1, TC1 plasmid 2, TC1 plasmid 1 without Sgsm2 and Herpud 2, TC1 plasmid 2 without Lta4h, LLC plasmid 1, LLC plasmid 2, ID8 plasmid 1, and ID8 plasmid 2.
TC1 tumor challenge experiments:
We implanted mice with the TC1 cell line (100,000 cells in PBS in the flank), and 7 days later, we vaccinated the mice with weekly 25 μg doses of the dodecamer vaccine TC1 Plasmid 1 (containing the immunogenic neoantigens Sgsm2 and Herpud2), TC1 Plasmid 2 (containing the immunogenic neoantigen Lta4h), or the empty pVax vector for a total of 4 immunizations. Tumors were monitored by caliper measurements twice a week. Mice were euthanized when tumor length reached 15 mm.
To determine if the presence of multiple non-specific plasmids would interfere with the specific antitumor response, we implanted mice with the TC1 cell line (100,000 cells in PBS in the flank), and 7 days later and weekly thereafter for a total of 4 weeks, we immunized mice with 1) a control pVax plasmid (25 μg), 2) the dodecamer vaccine TC1 Plasmid 1 alone (25 μg), 3) 3 LLC plasmids (36 epitopes) plus pVax plasmid (125 μg plasmid total), 4) 2 TC1 plasmids (24 epitopes) plus pVax plasmid (125 μg plasmid total), or 5) 3 LLC plasmids (36 epitopes) plus 2 TC1 plasmids (24 epitopes) (125 μg plasmid total). Tumors were monitored by caliper measurements twice a week. Mice were euthanized when tumor length reached 15mm.
DNA and RNA sequencing
We sequenced TC1, LLC, and ID8 cell lines from in vitro cultures and from in vivo generated tumors after implanting 100,000 TC1 or LLC subcutaneously or ID8 intraperitoneally 3 weeks after tumor implantation (2 mice per tumor). As a control, we used tails from C57Bl/6 mice. The mouse exome and RNA sequencing were performed on the Illumina HiSeq-2500 platform. The SureSelect Mouse All Exon Kit (Agilent Technologies, USA, cat #5190–4642) was used. All samples generated greater than 13 Gb of data, with greater than 98% of the exomes covered at ≥150X. Overall, 99% of the reads aligned to the mouse reference genome (downloaded from ensemble ftp://ftp.ensembl.org/pub/release-78/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz). Mapping quality for 80% of the aligned reads was ≥Q60. Duplicate % was low: 4–6%. Somatic variant calling was performed using Strelka program v1.0.14 (Illumina Inc.). The identified somatic variants were further filtered (using Strelka parameters such as read filtering, indel calling, SNV calling, and other parameters described in https://github.com/Illumina/strelka.), and only passed and on-target variants were considered for further analysis.
The RNA sequencing was done using TrueSeq RNA library prep kit v2 (Illumina, USA, cat# G9641B). All samples generated >100 million reads. Reads mapping to the ribosomal and mitochondrial genome were removed before performing alignment. The reads were aligned using STAR (2.4.1) aligner (open source software distributed under GPLv3). Overall 96–98% of the total pre-processed reads mapped to the reference gene model/genome (Mus musculus GRCm38 DNA). The gene expression was estimated using Cufflinks v2.2.1 (Trapnell et al. Broad Institute of MIT and Harvard).
Design of neoantigen vaccines
We designed the neoantigen vaccines by selecting the predicted neoantigens from the DNA and RNA sequencing data obtained from the TC1, LLC, and ID8 established tumors. Neoepitopes were prioritized from non-synonymous coding missense mutants, where the mutant allele expression was ≥1 FPKM. MHC class I binding analysis was performed for all coding missense mutations. 9-mer epitopes were analyzed using NetMHCons v1.1 (15, http://www.cbs.dtu.dk/services/NetMHCcons/) on the C57Bl/6 MHC alleles (H-2-Kb, H-2-Db). Peptides were further prioritized based on lower proteasomal processing score using NetChop3.1 (http://www.cbs.dtu.dk/services/NetChop/)(16,17). Peptides showing a score ≥10 were selected. Peptides were scored for transporter associated with antigen processing (TAP) binding, and peptides having binding affinities ≤0.5 were prioritized. A list of all predicted epitopes is included in the Supplementary Data. We included 12 epitopes defined as the predicted sequence that would bind to H2-K(b) or H2-D(b), keeping the predicted 9-mer epitope, including the mutation in the central position, and keeping 12 non-mutated amino acids flanking on each side. We concatenated the twelve 33-mers with furin cleavage sites and sub-cloned each construct into the pVax1 plasmid (GenScript). For generating each plasmid, we selected neoepitopes from each cell line that would represent a wide diversity of MHC-I binding. Prediction of binding to MHCII was performed using netMHCII-1.1 (SMM align) and netMHCII-2.2 (NN align) prediction programs (available at www.cbs.dtu.dk/services/NetMHCII-1.1/ and www.cbs.dtu.dk/services/NetMHCII-2.2/).
Flow Cytometry
We used a BD LSRII flow cytometer (BD Biosciences). Mouse antibodies used were directly fluorochrome-conjugated. We used: CD3e (17A2), CD4 (RM4–5), CD8b (YTS156.7.7), Interferon-γ (XMG1.2), TNFα (MP6-XT22), Interleukin-2 (JES6–5H4), and T-bet (4B10), all from Biolegend. Live/dead exclusion was done with the Violet viability kit (Invitrogen).
For the determination of intracellular cytokine production, we cultured 2 million splenocytes from vaccinated mice in the presence of peptides (5 μg/mL) derived from the corresponding wild-type or mutated neoantigen, Golgi-stop protein transport inhibitor (BD biosciences), and CD107a antibody (1D4B, Biolegend) for 4–5 hours prior to surface and intracellular staining. The neoantigen peptides consisted of 15-mer peptides overlapping by 9 amino acids. These peptides spanned the entire 33-mer used for immunization. Mice were vaccinated three times at 3-week intervals and euthanized a week after the last immunization. Spleens were harvested, and splenocyte suspensions obtained using a Stomacher® 80 Biomaster (Thomas Scientific), followed by red blood cell lysis (ThermoFisher).
T-cell expansion and activation
We harvested splenocytes from vaccinated mice and pulsed them with neoantigen-specific peptides (5 μg/mL) and IL2 (30 UI/mL). We refreshed the peptides and IL2 (Peprotech) with irradiated (4000 rad) splenocytes from naïve mice (1:3–10 T-cell:splenocyte ratio) once a week. 4–6 weeks after initiating the T-cell expansion, we cocultured with the tumor cells (TC1 or ID8) for performing in vitro cytotoxicity experiments.
In vitro cytotoxicity
We generated luciferase-transduced TC1 and ID8 cells by culturing them with 5 μl of CMV firefly luciferase lentivirus (Cellomics technology) and polybrene (8 μg/mL; Sigma Aldrich) and performing a spinoculation for 90 minutes at 32°C at 500 x g. Cells were selected using puromycin (2 μg/mL; Takara Bio USA) for 2 weeks and kept in culture with selection media. We plated 10,000 luciferase-transduced TC1 or ID8 cells per well in a 96-well plate, and 18 hours later we co-incubated them for 24 hours with 10,000 or 50,000 in vitro expanded T cells. We measured cytotoxicity using CytoTox-Glo™ Cytotoxicity Assay (Promega), according to the manufacturer’s instructions. We reported cytotoxicity as a ratio of luciferase expression in the T cell–containing study wells divided by luciferase expression in the wells with tumor cells only (no T cells).
ELISPOT
We vaccinated mice three times at 3-week intervals. A week after the final immunization, we harvested splenocytes and co-incubated them with each neoantigen-derived peptide pool comprising 15-mers overlapping by 9 amino acids (5 μg/mL). After a 24-hour incubation, we performed the mouse interferon-γ ELISPOT according to the manufacturer’s instructions (Mabtech, #3321–4APT-10). Spots were read using an ImmunoSpot CTL reader, and spot forming units (SFU) were calculated by subtracting media alone wells from stimulated wells. Concanavalin A was used as a positive control to ensure spot development.
We set the threshold for immunoreactivity to be ≥30 spot forming units per million splenocytes by IFNγ ELISpot using the highest number of spots typically obtained in non-stimulated splenocyte wells. For epitopes to be classified as immunogenic, they required responses to the mutant peptide that were statistically significantly higher than those induced by mice immunized with the control pVax plasmid.
Induction of MHC class II
We cultured 10,000 B16, TC1, LLC, and ID8 cells per well in a 96 well plate in RPMI plus 10% FBS in the presence of different concentrations of interferon gamma or PBS as negative control. 48 hours later, we trypsinized the cells and measured MCH class II by flow cytometry using I-A/I-E antibody (clone M5/114.15.2, Biolegend).
Statistical analysis
Differences between the means of experimental groups were calculated using a two-tailed, unpaired Student’s t test. Comparisons between two groups with repeated measures were done using two-way ANOVA corrected using Bonferroni’s test. Error bars represent standard error of the mean (SEM). For mouse survival analysis, significance was determined using a Gehan-Brelow-Wilcoxon test. All statistical analyses were done using Graph Pad Prism 7.0. p<0.05 was considered statistically significant.
Results
DNA vaccine neo-epitope dodecamers induce frequent immune responses in mice
For our experimental approach, we implanted mice with three different tumor types: LLC, TC1and ID8. After 3 weeks, we harvested tumors and performed DNA and RNA isolation, as well as exome and RNA sequencing. For neoantigen identification, we utilized a prioritization pipeline (Fig. 1A; see methods). We compared sequencing of cell lines cultured in vitro to the same cell lines implanted into mice and observed that a substantial proportion of mutations were differentially expressed, indicating an important influence of the three-dimensional tumor microenvironment (Supplementary Fig. S1)(18).
Figure 1. DNA vaccine neo-epitope dodecamers induce frequent immune responses.
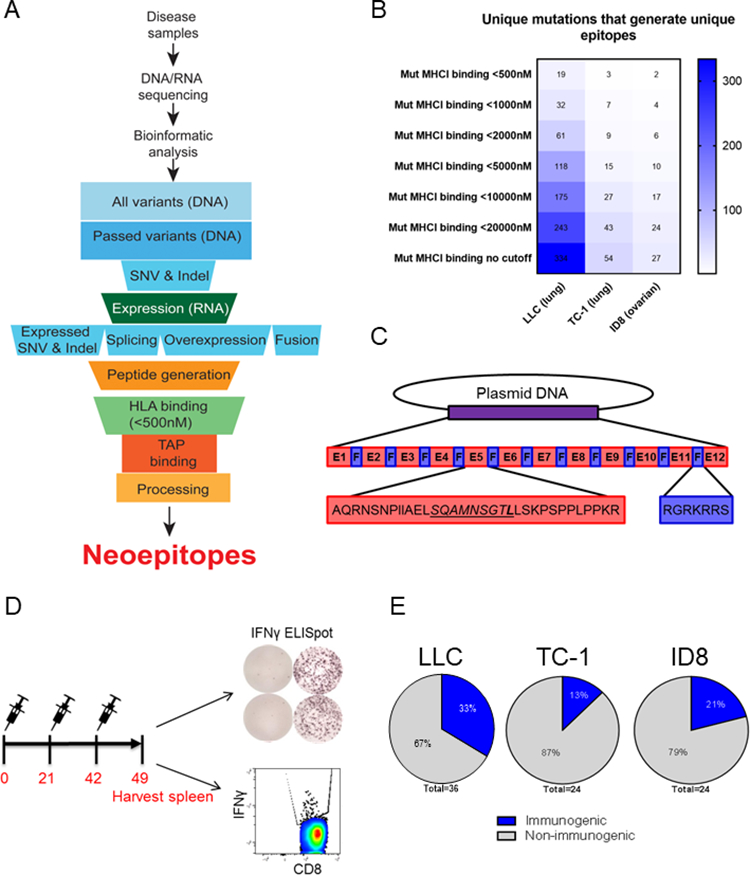
A. Neoantigen prioritization pipeline. Tumor samples and normal tail tissues from 2 mice/tumor type were harvested 3 weeks after implantation for DNA and RNA isolation and sequencing. Identified neoantigens were prioritized according to expression, MHC binding, TAP binding and processing. B. Number of mutations identified for each tumor type with the indicated MHC class I affinity (NetMHCons v1.1). Blue shading: the number of unique mutations having unique epitopes. C. Plasmid DNA design. The predicted MHCI 9-mer epitopes (underlined and italicized) were flanked by 12 amino acids on each side. Each epitope was separated by a furin cleavage site. The mutated amino acid within the 9-mer epitope is shown in bold. D. Schematic of immunization experiments. Mice were immunized with each plasmid with 25μg of DNA followed by electroporation (EP) three times at three-week intervals and euthanized one week after final immunization. E. Percentage of epitopes that generated immune responses (>30 SFU/million splenocytes) for each tumor type.
We identified a total of 334, 54, and 27 non-synonymous, expressed mutations that generated unique neoepitopes in the LLC, TC1, and ID8 tumor models, respectively (Fig. 1B). 19, 3, and 2 epitopes from LLC, TC1, and ID8, respectively, had less than 500nM binding affinity, predicted using NetMHCons v1.1 (Fig. 1B). We chose 36 epitopes from LLC, 24 epitopes from TC1, and 24 epitopes from ID8 to test for immunogenicity using a DNA vaccine platform. We included all the highest affinity epitopes (<500nM), as well as some low affinity epitopes (>500nM), to assess the value of the MHCI prediction programs. We designed 7 total DNA vaccine neo-epitope dodecamers, which included 12 total 33-mer epitopes per plasmid, linked together by furin cleavage sites (Fig. 1C). We tested these 7 plasmidsby immunizing mice at 3-week intervals for a total of three immunizations (Fig. 1D). We sacrificed mice one week following the final immunization, and harvested spleens for IFNγ ELISpot analysis and intracellular cytokine staining (Fig. 1D). We identified 12/36 epitopes from the LLC model, 3/24 epitopes from the TC1 tumor model, and 5/24 epitopes from the ID8 model that were immunogenic when delivered using DNA vaccines (Fig. 1E). Overall, after immunizing naïve mice with an unfiltered group of neoantigens, we observed immune responses to 24% (20/84) of the epitopes. This percentage of responses is similar to what has been reported for RNA and SLP vaccines in pre-clinical studies (6,19).
DNA vaccines generate predominantly CD8+ T-cell responses to neoantigens
We performed flow cytometry to determine which of these epitopes generated CD8+ or CD4+ T-cell responses (Supplementary Fig. S2A–F). 40% of the responses were mediated by CD8+ T cells, 35% of the responses were mediated by both CD8+ and CD4+ T cells, and only 25% of responses were mediated by CD4+ T cells alone (Fig. 2A, Supplementary Fig. S2A–F). The strongest CD8+ (Sgsm2) and CD4+ (Lta4h) T-cell epitopes generated both IFNγ and TNFα cytokine production exclusively in CD8+ and CD4+ T cells, respectively (Fig. 2B,C). Many other epitopes generated polyfunctional responses as well, with expression of IFNγ, TNFα, and IL2 simultaneously, in addition to expression of T-bet and CD107a, indicating cytolytic potential (Supplementary Fig. S3A–F).
Figure 2. DNA vaccines generate predominantly CD8+ T-cell responses to neoantigens.
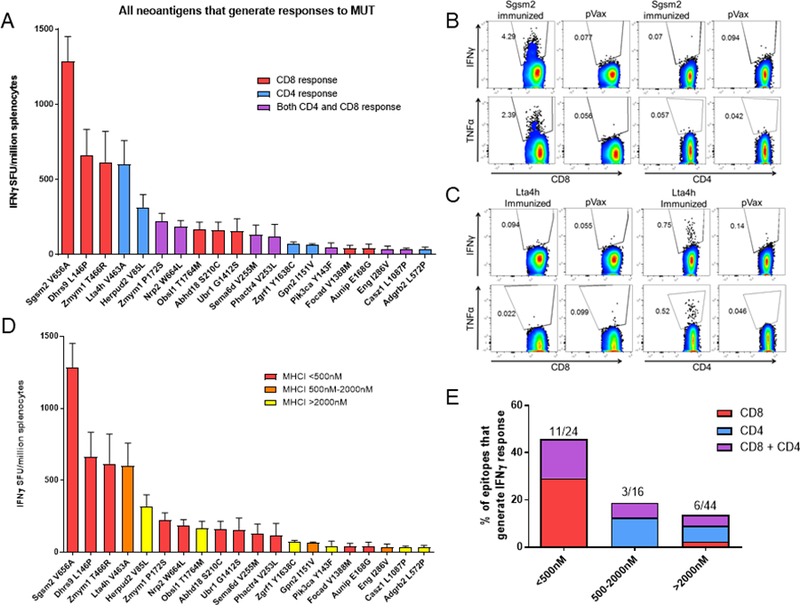
A. Graph of IFNγ ELISpot responses for each epitope that generated >30 SFU/million splenocytes. Red bars: CD8+ T-cell responses; Blue bars: CD4+ T-cell responses; Purple bars: both CD8+ and CD4+ T-cell responses. Control: pVax empty vector. B,C. Example flow cytometry IFNγ and TNFα responses from mice immunized with a (B) TC1 plasmid containing Sgsm2 or (C) Lta4h from (A). D. IFNγ ELISpot responses from (A), displayed according to MHC class I affinity (NetMHCCons v1.1). Red bars: high affinity (<500nM); Orange bars: medium affinity (500nM-2000nM); Yellow bars: low affinity (>2000nM). E. Percentage of epitopes that generate CD4+ versus CD8+ T-cell responses, organized according to MHC class I affinity. Single experiment, N=5 mice/group.
We next assessed the ability of the MHC class I binding affinity to predict immunogenic epitopes. We found that NetMHCcons v1.1 selected for epitopes that were immunogenic: 46% of the epitopes with <500nM binding affinity were immunogenic (Figure 2D,E), and 100% of the high-affinity epitopes generated either CD8+ or CD8+/CD4+ T-cell responses (Fig. 2E). These data suggest that the type of response elicited by the neoantigen may depend on the immunization platform, and that SNDVs can induce robust CD8+ T-cell responses to neoantigens.
We next tested the ability of MHC class II binding affinity to predict immunogenic epitopes (Supplementary Fig. S4). We tested both netMHCII-1.1 (SMM align) and netMHCII-2.2 (NN align) prediction programs. Consistently with previous reports (20), we observed that neither program could accurately predict CD4+ T-cell epitopes (Supplementary Fig. S4A,B).
DNA vaccine–primed T cells selectively kill mutated cells
We next compared immune responses generated from the immunized mice against the corresponding wild-type (non-mutated) epitope. The majority of immune responses were specific to the mutated epitope (Supplementary Fig. S5A,B). 75% of immune responses generated were at least 1.5-fold higher for the mutated epitope compared to the wild-type epitope, with the remaining 25% of responses being similar (Supplementary Fig. 5B). These results are similar to what has been previously reported for neoantigen SLP vaccines, in which 68.8% of responses were specific to the mutated epitope (19).
To determine the cytotoxic functionality and specificity of the T cells primed by our SNDVs, we expanded ex vivo T cells using the TC1 neoantigens that elicited stronger IFNγ responses in vivo (Sgsm2, Herpud2, and Lta4h). After ex vivo expansion, we observed that most T cells expanded with the Herpud2 and Lta4h peptides were CD8+ (Fig. 3A), indicating that these epitopes could generate a CD8+ T-cell response in mice, in addition to a CD4+ T-cell response. Coculture of these expanded T cells with TC1 cells or ID8 cells showed specific cytotoxicity of the TC1 Herpud2-specific and Sgsm2-specific T cells against TC1 cells, but not against ID8 cells (Fig. 3B,C). However, we did not find any cytotoxic activity of Lta4h-specific T cells against TC1 (Fig. 3D). To try to explain this phenomenon, we hypothesized that TC1 cells may not express Lta4h in vitro. We examined the RNA sequencing data generated from the TC1 cultured cells and tumor (Supplementary Fig. S1) and found that Lta4h is only expressed at the RNA level in vivo in tumor tissue (Fig. 3E).
Figure 3. DNA vaccine–primed T cells selectively kill mutated cells.
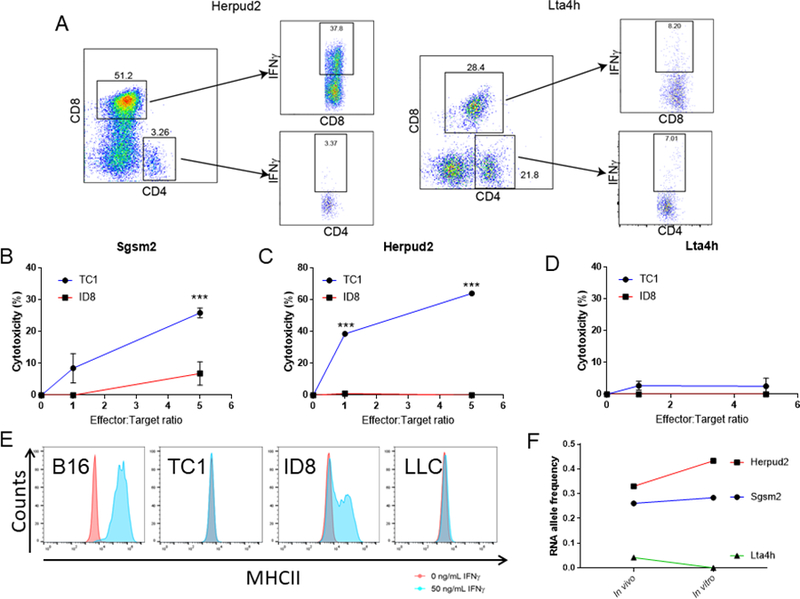
A. Representative flow cytometry plots showing IFNγ-expressing CD4+ and CD8+ T cells resulting from the expansion of T cells from mice immunized with TC1 plasmid 1 or TC1 plasmid 2, stimulated with Herpud2 or Lta4h peptides. Negative control: non-peptide stimulated T cells (n=5 mice/group). B-D. Cytotoxicity of T cells expanded from mice immunized with TC1 plasmids (from A). T cells were expanded with Sgsm2 (B), Herpud2 (C) or Lta4h (D) specific peptides (5μg/mL). After expansion, T cells were cocultured with 10,000 luciferase-tagged TC1 or ID8 tumor cells. Cytotoxicity was measured by luciferase activity after 24 hours of coculture. E. RNA expression Sgsm2, Herpud2 and Lta4h in TC1 tumor cells grown in vivo and in vitro (2 mice/tumor type after 3 weeks). F. Representative flow cytometry histograms showing surface expression of MHC class II on B16, TC1, ID8, and LLC tumor cells in normal conditions or after being exposed to IFNγ (50ng/mL) for 48 hours. (Single experiment). Two-way ANOVA. ***p<0.001.
Although physiological expression of MHC class II is restricted to antigen-presenting cells, some tumors express this presenting protein complex, allowing for direct CD4+ T-cell recognition. To determine if CD4+ T cells could recognize neoantigens directly on tumor cells, we measured if our tumor cells expressed MHC class II. Unlike B16 melanoma or ID8, TC1 and LLC cells did not exhibit MHC class II expression upon incubation with various doses of IFNγ (Fig. 3F, Supplementary Fig. S6), and therefore, tumor cytotoxicity must have occurred through an MHC class I–restricted mechanism.
We next examined the hierarchy of immunodominance for the epitopes within the TC1 neoantigen plasmids. We tested if the responses observed for the Sgsm2, Herpud2, and Lta4h epitopes could be masking potential sub-dominant immune responses from other epitopes within the same plasmid by deleting the immunodominant epitopes from each plasmid (Supplementary Fig. S7). We found that, deleting the immunodominant epitopes did not result in generation of immune responses from sub-dominant epitopes (Supplementary Fig. S7A,B).
DNA neoantigen vaccines delay tumor progression
We next studied the in vivo antitumor effect of the TC1 vaccines (Fig. 4A). We observed a profound delay in tumor progression when we treated with the Plasmid 1 alone (Fig. 4B) and a less intense tumor delay with Plasmid 2 (Fig. 4C). Although the Lta4h epitope was immunogenic, it generated primarily CD4+ T-cell responses (Fig. 2A). These neoantigen-specific CD4+ T cells were likely not as effective at delaying tumor progression in this TC1 tumor model because the TC1 cells do not express MHC class II (Fig. 3F). In addition, the expression of the Lta4h neoantigen was relatively low (Fig. 3E) compared to the neoantigens present in Plasmid 1.
Figure 4. DNA neoantigen vaccines impact tumor growth.
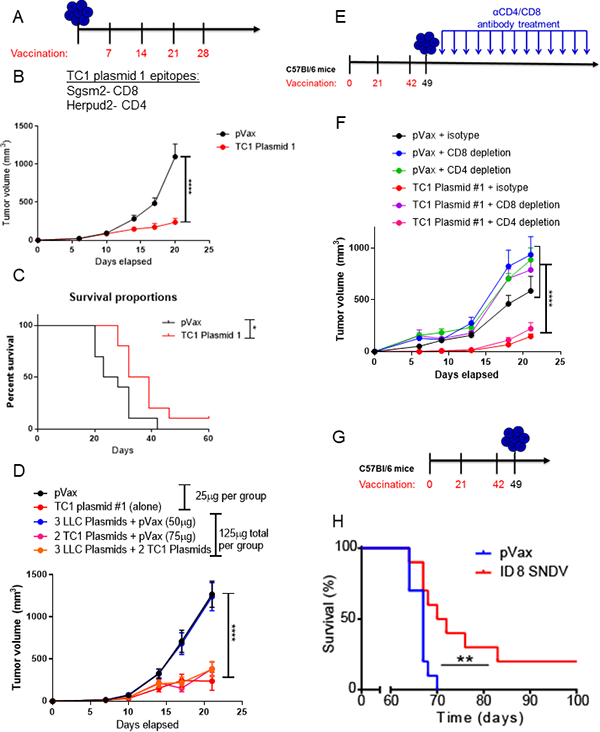
A. Schematic of tumor challenge experiments for panels B-D. Mice were challenged with 100,000 TC1 tumor cells, and immunized weekly starting one week after tumor implantation. B. Tumor volume of mice bearing TC1 tumors treated with 25μg of TC1 plasmid 1 or pVax. C. Tumor volume of mice bearing TC1 tumors treated with 25μg of TC1 plasmid 2 or pVax. D. Tumor volume measurements for mice bearing TC1 tumors treated with indicated vaccine plasmid or plasmid combinations (all diluted in 30μL of water). E. Schematic of tumor challenge experiment for panel F. Mice were immunized three times, at three-week intervals with 25μg of TC1 plasmid 1 or pVax, and challenged with TC1 tumor cells one week following the final immunization. Mice were given 200μg CD4 or CD8 depletion antibodies twice weekly immediately after tumor implantation for a total of 13 injections. F. Tumor volume measurements for mice bearing TC1 tumors treated with the indicated vaccine and antibody combination. G. Schematic of tumor challenge experiment for panel H. Mice were immunized three times, at three-week intervals with 25μg of ID8 plasmids 1&2 or pVax, and challenged with 2×106 ID8 tumor cells injected intraperitoneally one week following the final immunization. H. Survival analysis for mice bearing ID8 tumors treated with ID8 plasmid cocktail (25μg ID8 plasmid 1 and 25μg ID8 plasmid 2 formulated together) or 50μg of pVax control plasmid. For this experiment, mice were euthanized upon development of ascites. For all studies, N=10 mice/group. Two-way ANOVA. Gehan-Brelow-Wilcoxon test. *p<0.05, **p<0.01, ****p<0.0001.
We next determined whether inclusion of high numbers of non-specific neo-epitopes within the vaccine would alter its antitumor impact. Inclusion of additional pVax control plasmid or non-specific neoantigen epitopes within the vaccine formulation did not impact the efficacy of the dodecamer TC1 Plasmid 1 (Fig. 4D), indicating that formulations containing many epitopes is a feasible strategy for incorporation into the SNDV platform.
To verify that the antitumor impact was mediated by CD8+ T cells, we performed a CD4+ and CD8+ T-cell depletion study. We immunized mice prophylactically and depleted mice of CD4+ or CD8+ T cells after tumor implantation (Fig. 4E). The antitumor impact of the TC1 Plasmid 1 vaccine was abrogated upon CD8+ T-cell depletion, but not upon CD4+ T-cell depletion, indicating that CD8+ T cells were the major driver of antitumor immunity (Fig. 4F).
We next studied the in vivo efficacy of the SNDVs in the ID8 ovarian cancer model in a prophylactic setting (Fig. 4G). Previous vaccination studies using peptide vaccines have failed to delay tumor progression, either prophylactically or therapeutically, in this aggressive model of ovarian cancer using neoantigen vaccines, and this model fails to respond to immune checkpoint blockade alone (7,14). We found a significant increase in survival after vaccination in this model using the SNDV platform (Fig. 4H), indicating that the SNDV approach offers a significant advantage over existing immunotherapies for this tumor type.
Discussion
Here, we described the possibility of generating effective antitumor immune responses against cancer neoantigens using synthetic neoantigen DNA based vaccines (SNDVs). An advantage for this approach would be to directly generate in vivo immunity without ex vivo expansion. CD8+ T cells are thought to be the major mediators of antitumor T-cell responses in vivo. As we have previously shown in clinical studies (9–11), engineered DNA vaccines using full length antigens are able to generate robust CD8+ T-cell responses in vivo. Here, we described a strategy for the assembly of multi-epitope strings of neoantigens into plasmid vaccines, which efficiently drive CD8+ T-cell responses. In this study, neoantigens selected based on predictions for high MHCI binding following immunization generated CD8+ or CD8+/CD4+ neoantigen-specific immune responses 75% of the time. Many of the neoantigen-specific CD8+ T cells were polyfunctional, with production of multiple cytokines simultaneously (IFNγ, TNFα, and IL2), and had expression of the degranulation marker CD107a, indicating cytolytic potential.
These data are in contrast to studies of other neoantigen in vivo immunization approaches, which demonstrate predominantly MHC class II–restricted T-cell responses, despite in silico prediction for MHCI responses (3,4). It has been reported that CD4+ T cells can generate antitumor cytotoxicity. CD4+ T cells can exert direct cytotoxicity based on granule exocytosis or by the Fas-Fas ligand pathway upon recognition of MHC class II-peptide complexes (21,22). However, it is not common for solid tumors to express MHCII (23,24). CD4+ T cells have also been suggested to induce killing of tumor cells that do not express MHC class II by activating macrophages (25). It is possible that antigen presentation differences, as well as toll-like receptors (TLR) or PAMP activation pathways, might play a role in the observed CD4+ versus CD8+ T-cell biases (26–29). MHCI presentation of epitopes requires intracellular protein synthesis and proteasomal degradation (29). SLPs are injected directly into tissues, and, therefore, are primarily engulfed and presented by antigen-presenting cells, which will skew them towards a class II presentation. In the case of RNA and DNA, peptide synthesis occurs in the cell and can be cleaved as necessary by the proteasome and enter the MHC class I pathway. However, it appears that RNA may induce more TLR activation (including TLR3,7,8), as well as RIG-I like receptor activation, which could promote pro-inflammatory cytokine production, skewing the T-cell response (26–28). Although more work will be required to understand these differences, these approaches could be considered complimentary due to the divergent T-cell phenotypes induced.
The number of neoantigens in cancers varies widely according to the tumor type but has been defined to be approximately between 33 and 163 expressed, non-synonymous mutations (1). Using this SNDV platform to generate neoantigen based vaccines allowed us to encode a high number of neoantigens in each plasmid. Considering the insert size (33aa), the linker size (7aa), and the capacity of DNA plasmids to include large inserts, it is possible to immunize with all identified neoantigens in each patient with as little as 1–3 plasmids. In our pre-clinical studies, inclusion of a high number of non-specific neo-epitopes did not impair vaccine efficacy. Because immunogenic neoantigens typically occur as passenger mutations, immunizing against a larger number of neoantigens per tumor may prevent or delay tumor immune escape. This approach also eliminates the need to validate each epitope experimentally, shortening valuable time before the vaccine can be produced and administered to the patient. Because humans have six different MHC class I molecules, it is likely that a higher proportion of epitopes will be able to bind to human HLA and generate responses to the vaccine.
In conclusion, we have shown that an engineered DNA vaccine designed to target tumor neoantigens was able to generate potent CD8+ T-cell antitumor–specific responses, which impact tumor progression and survival in mouse models. Further development and possible clinical study of this approach appears warranted.
Supplementary Material
Financial Support:
This work was supported by an NIH/NCI NRSA Individual Fellowship (F32 CA213795 to E.K. Duperret), a Penn/Wistar Institute NIH SPORE (P50CA174523 to D.B. Weiner), the Wistar National Cancer Institute Cancer Center (P30 CA010815), the W.W. Smith Family Trust (to D.B. Weiner), funding from the Basser Foundation (to D.B. Weiner), a grant from Inovio Pharmaceuticals (to D.B. Weiner) and internal research funding support from Geneos Therapeutics.
Footnotes
Competing interests:
N.Y. Sardesai and J. Barlow are employees of Inovio Pharmaceuticals and its subsidiary Geneos Therapeutics, and as such receive salary and benefits, including ownership of stock and stock options. D.B. Weiner receives a commercial research grant from Inovio Pharmaceuticals, has received speakers bureau honoraria from Inovio Pharmaceuticals, GeneOne, AstraZeneca, has ownership interest (including patents) in Inovio Pharmaceuticals and is a consultant/advisory board member for Inovio Pharmaceuticals and Geneos Therapeutics. The other authors declare no competing financial interests.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science (80-). 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rech AJ, Balli D, Mantero A, Ishwaran H, Nathanson KL, Stanger BZ, et al. Tumor Immunity and Survival as a Function of Alternative Neopeptides in Human Cancer. Cancer Immunol Res. 2018;6:276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. Nature Publishing Group; 2017;547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. Nature Publishing Group; 2017;547:222–6. [DOI] [PubMed] [Google Scholar]
- 5.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2015;520:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, et al. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines Ashkar AA, editor. PLoS One. 2016;11:e0155189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front Immunol. Frontiers; 2018;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet (London, England). 2015;386:2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine - Preliminary Report. N Engl J Med. 2017;NEJMoa1708120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aggarwal C, Cohen RB, Morrow MP, Kraynyak K, Bauml J, Weinstein GS, et al. Immunogenicity results using human papillomavirus (HPV) specific DNA vaccine, INO-3112 (HPV16/HPV18 plasmids + IL-12) in HPV+ head and neck squamous cell carcinoma (HNSCCa). Am Soc Clin Oncol. 2017; 10.1200/JC. [DOI] [Google Scholar]
- 12.Duperret EK, Wise MC, Trautz A, Villarreal DO, Ferraro B, Walters J, et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol Ther. 2018;26:435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res. American Association for Cancer Research; 2017;5:767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai M, Wei H, Yip YY, Feng Q, He K, Popov V, et al. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J Immunother. 2013;36:248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–86. [DOI] [PubMed] [Google Scholar]
- 16.Keşmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002;15:287–96. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. [DOI] [PubMed] [Google Scholar]
- 18.Zschenker O, Streichert T, Hehlgans S, Cordes N. Genome-wide gene expression analysis in cancer cells reveals 3D growth to affect ECM and processes associated with cell adhesion but not DNA repair. PLoS One. Public Library of Science; 2012;7:e34279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. American Association for Cancer Research; 2012;72:1081–91. [DOI] [PubMed] [Google Scholar]
- 20.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. BMC Bioinformatics. BMC Bioinformatics. 2008;0101:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang M, Siciliano NA, Hersperger AR, Roscoe F, Hu A, Ma X, et al. Perforin-dependent CD4+ T-cell cytotoxicity contributes to control a murine poxvirus infection. Proc Natl Acad Sci U S A. 2012;109:9983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janssens W, Carlier V, Wu B, VanderElst L, Jacquemin MG, Saint-Remy J-MR. CD4+CD25+ T cells lyse antigen-presenting B cells by Fas-Fas ligand interaction in an epitope-specific manner. J Immunol. 2003;171:4604–12. [DOI] [PubMed] [Google Scholar]
- 23.He Y, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, Yu H, et al. MHC class II expression in lung cancer. Lung Cancer. 2017;112:75–80. [DOI] [PubMed] [Google Scholar]
- 24.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauritzsen GF, Bogen B. The role of idiotype-specific, CD4+ T cells in tumor resistance against major histocompatibility complex class II molecule negative plasmacytoma cells. Cell Immunol. 1993;148:177–88. [DOI] [PubMed] [Google Scholar]
- 26.Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu W, Dietze KK, Gibbert K, Lang KS, Trilling M, Yan H, et al. TLR ligand induced IL-6 counter-regulates the anti-viral CD8(+) T cell response during an acute retrovirus infection. Sci Rep. 2015;5:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86:2900–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock KL, Reits E, Neefjes J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016;37:724–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.