

Abstract
Cerebral energy metabolism is a highly compartmentalized and complex process in which transcellular trafficking of metabolites plays a pivotal role. Over the past decade, a role for lactate in fueling the energetic requirements of neurons has emerged. Furthermore, a neuroprotective effect of lactate during hypoglycemia or cerebral ischemia has been reported. The majority of the current evidence concerning lactate metabolism at the cellular level is based on in vitro data; only a few recent in vivo results have demonstrated that the brain preferentially utilizes lactate over glucose. Using voltage-sensitive dye (VSD) imaging, beta-probe measurements of radiotracer kinetics, and brain activation by sensory stimulation in the anesthetized rat, we investigated several aspects of cerebral lactate metabolism. The present study is the first in vivo demonstration of the maintenance of neuronal activity in the presence of lactate as the primary energy source. The loss of the voltage-sensitive dye signal found during severe insulin-induced hypoglycemia is completely prevented by lactate infusion. Thus, lactate has a direct neuroprotective effect. Furthermore, we demonstrate that the brain readily oxidizes lactate in an activity-dependent manner. The washout of 1-[11C]l-lactate, reflecting cerebral lactate oxidation, was observed to increase during brain activation from 0.077 ± 0.009 to 0.105 ± 0.007 min−1. Finally, our data confirm that the brain prefers lactate over glucose as an energy substrate when both substrates are available. Using [18F]fluorodeoxyglucose (FDG) to measure the local cerebral metabolic rate of glucose, we demonstrated a lactate concentration-dependent reduction of cerebral glucose utilization during experimentally increased plasma lactate levels.
Introduction
For a long time, it was considered that blood-borne glucose is the sole energy substrate in the adult healthy brain. However, several studies have reported a different situation in the developing and diseased brain. For example, in breast-fed newborns and suckling rats, the utilization of monocarboxylates, such as lactate, and ketone bodies is increased (Cremer, 1982; Dombrowski et al., 1989; Nehlig and Pereira de Vasconcelos, 1993). In addition, a neuroprotective role for exogenous lactate in pathologic situations, such as prolonged starvation, diabetes, and cerebral ischemia, has been documented on several occasions (Gjedde and Crone, 1975; Schurr et al., 2001; Mason et al., 2006; Berthet et al., 2009). However, it is still unclear whether the neuroprotective effect of lactate is indirect [e.g., via increased cerebral blood flow (CBF) caused by hyperlactemia (Shackford et al., 1994)] or whether lactate can serve as a direct neuronal energy substrate.
One of the first indications that lactate is not only a product of the metabolic chain but also a potentially important alternative neuronal energy substrate was documented more than two decades ago from the demonstration that lactate is able to support synaptic function in the absence of glucose in rat hippocampal slices (Schurr et al., 1988). For the living brain, the situation is less clear. No information about the capability of lactate to sustain neuronal action in vivo could be provided. Recently, independent groups have reported lactate utilization by the brain in humans (Gallagher et al., 2009; van Hall et al., 2009; Boumezbeur et al., 2010). There is also evidence that lactate oxidation is preferentially located in neurons. Bouzier et al. (2000) used 3-[13C]lactate to investigate cerebral lactate metabolism. From the accumulation of labeled metabolites, they concluded that lactate metabolism is located in a compartment devoid of pyruvate carboxylase, e.g., neurons.
Furthermore, astrocytic production of lactate, which serves as an energy pool for neurons, was proposed (Pellerin and Magistretti, 1994), but neuronal lactate use has still to be demonstrated in situ.
In the present study, we followed a multimodal approach. First, using optical voltage-sensitive dye (VSD) imaging to directly monitor the synaptic activity in the somatosensory cortex, we report for the first time that lactate is capable of sustaining neuronal activity for a time period of hours in the quasi-absence of glucose in the living organism. Second, applying radiotracer kinetic measurements of 1-[11C]l-lactate accumulation and classical cerebral glucose utilization measurements using FDG, we demonstrate that lactate is metabolized by the intact brain in an activity-dependent manner.
Materials and Methods
Animal preparation
All the animal experiments were approved by the local veterinary authorities and were performed by licensed investigators. In total, 64 animals (male adult Sprague Dawley rats; weighing 220–350 g) were included in the study. Before measurements, the animals were fasted overnight to standardize plasma levels of glucose. Surgery was performed under 2.5–3.5% isoflurane anesthesia and involved the placement of an arterial and a venous catheter and tracheotomy for mechanical ventilation. For the cortical beta-probe acquisitions, the cranial bone above somatosensory cortices was thinned to translucency. For the voltage-sensitive dye imaging experiments, a craniotomy was performed. Staining with the voltage-sensitive dye RH1691 (Optical Imaging) was done through the intact dura mater for 90 min. Leaving the dura intact reduces the movement artifact during recording and decreases the risk of cortex irritations (Lippert et al., 2007). After staining, the dye was removed by washing with dye-free Ringer's solution. Plasma glucose and lactate levels were determined periodically (Ektachem DT; Eastman Kodak). The actual experiments were performed under α-chloralose anesthesia (44 mg/kg bodyweight, s.c.). Several blood gas variables (pH, PCO2, PO2) (AVL, Compact 3; Roche Diagnostics) and metabolic measures such as plasma glucose and lactate levels (Ektachem DT; Eastman Kodak), which are relevant parameters influencing lactate uptake in the brain, were measured in all animals before the start of measurements. Ventilation was adjusted to reach physiological blood gas values.
Voltage-sensitive dye imaging
The VSD was excited with 630 nm light from an LED (Aculed; PerkinElmer Life and Analytical Sciences). A custom-built tandem lens system consisting of two 50 mm objectives and dichroic and emission filters was used. Data were collected with a high-speed CMOS-based camera (Micam Ultima; Scimedia). Images were collected with 1 ms temporal resolution. Images were analyzed using custom-written Matlab routines and the software package PMOD (Mikolajczyk et al., 1998). Bleaching of fluorescence was corrected by subtraction of a best-fit double exponential. Time courses of fluorescence changes were quantified as ΔF/F0 from circular regions of interest of constant diameter manually placed over the activated area. To compare VSD signal from different animals, region of interests (ROIs) were centered on the location of the earliest responses. In addition, to estimate the spatial extent of the neuronal activation at the time point of the highest activation (peak amplitude), the area containing voxels >70% of the maximum within the experiment was determined. The amplitudes of sensory-evoked responses were calculated as the change in the VSD signal (ΔF/F0) over a fixed time interval for each experiment. Finally, the time until peak response was determined as the time span between stimulation onset and peak amplitude within the ROI. Baseline was the 10 ms period immediately before the stimulus.
In VSD experiments, five experimental groups were examined: (1) effect of hyperlactemia (n = 4) on neuronal integrity, (2–4) evolution of the VSD signal after induction of severe hypoglycemia (insulin at 20 IU/kg, i.p.) and subsequent continuous infusion of saline (n = 5), glucose (n = 5), or lactate (n = 5), respectively, and (5) control experiments with saline infusion only (n = 3).
Radiotracer experiments
Radiotracer synthesis: production of 1-[11C]l-lactate.
Racemic 1-[11C]lactic acid was made by [11C]cyanohydrine synthesis starting from [11C]HCN (trapped as [11C]KCN) and acetaldehyde bisulfite adduct. The quantitatively formed 1-[11C]dl-lactonitrile was hydrolyzed by reflux in concentrated HCl, and the reaction mixture was introduced directly into a semipreparative polymeric HPLC column (250 × 10 mm, 0.03% H3PO4 as the mobile phase; Polymerx 10μ; Phenomenex) in which the product was isolated from the aggressive reaction matrix. Then the collected racemic 1-[11C]lactic acid was automatically introduced into a preparative chiral HPLC column coated with a penicillamine-derived chiral selector and eluted with 1 mm CuSO4 solution as the mobile phase in which both enantiomers were separated by ligand exchange chromatography. The fractions corresponding to [11C]d-lactic acid or [11C]l-lactic acid were passed through anion exchanger Sep-Pak cartridges (Accell Plus QMA, in CO32 form) in which the Cu2+ ions (as insoluble carbonate) and the [11C]lactate are retained. The latter was selectively washed out from the cartridges with 2 mmol/L sodium phosphate buffer, pH 7.4, to obtain injectable solutions of pure Na-[11C]d-lactate and l-lactate enantiomers. The quality control of the final product was done by chiral ligand exchange HPLC. The procedure led to a product of >99% chemical and enantiomeric purity. The specific activity at the end of synthesis was ∼400 GBq/μmol. FDG was obtained from the daily in-house production by the Radiochemistry Department of the University Hospital Zurich.
Beta-probe experiments using 1-[11C]l-lactate and FDG.
A recently developed two-channel system with surface probes was used for the measurement of the radioactivity concentration in brain cortices (Wyss et al., 2009). The used devices consist of scintillator crystals with a thickness of 0.2 mm and a diameter of 3 mm. The beta scintillators were made light-tight by applying a uniform coating of silver particles. The scintillations were measured using a photomultiplier tube and counting electronics (PerkinElmer Life and Analytical Sciences). The limited range of beta particles within biological tissues leads to a limited detection volume centered around the scintillating tip of the probe (Wyss et al., 2009). The sensitivities of the used scintillators were 0.31–0.60 cps/kBq/cc.
For beta-probe measurements, the arterial and venous lines were connected to form an arterio-venous (av) shunt. The av shunt was used for continuous monitoring of arterial blood pressure, injection of the radiotracers, continuous measurement of the total arterial blood radioactivity, and the collection of blood samples. For the recording of the total blood radioactivity, the shunt ran through a coincidence counter (GE Medical Systems) (for details, see Weber et al., 2002). Before the actual experimental measurements for 1-[11C]l-lactate, the kinetic properties and the buildup of metabolites of the novel radiotracer were characterized in 10 animals. In addition, in four of these animals, the first pass extraction fraction was determined with additional measurements of CBF (see below). All subsequent 1-[11C]l-lactate experiments consisted of two acquisitions. For each acquisition, 100–150 MBq of radiotracer was intravenously injected. First, the change of 1-[11C]l-lactate kinetics from baseline conditions to increased neuronal activity [electrical infraorbital nerve stimulation (ION): 2 mA, 2 Hz, 1 ms pulses applied during the whole acquisition period; n = 5] and the effect of monocarboxylic acid transporter blockade using α-cyano-4-hydroxy-cinnamate (CIN) (n = 5) was investigated. CIN was injected intraperitoneally (90 mg/kg) 30 min before tracer injection.
FDG was used to determine the local cerebral metabolic rate of glucose (LCMRglu). For each FDG acquisition, 50–80 MBq of tracer was injected. All FDG experiments consisted of two measurements. Accumulation of FDG was measured at baseline (SAL) and at artificially increased plasma lactate levels (LAC) (100 μmol · kg−1 · min−1 during the first 20 min; thereafter, 50 μmol · kg−1 · min−1, i.v.) during rest conditions (n = 4 for SAL; n = 13 for LAC) and during activation (n = 2 for SAL; n = 5 for LAC).
Characterization of 1-[11C]lactate.
The total radioactivity in arterial blood was continuously recorded over 40 min using a coincidence counter (GE Medical Systems). Whole blood activity was then corrected for (1) a different tracer concentration in whole blood and plasma and (2) the buildup of labeled metabolites. The ratio 11Cplasma/11Cwhole blood was determined in all animals at different time points (three to four blood samples per animal). The data of all animals were then pooled, and the time course of the ratio was approximated by fitting a quadratic polynomial to the data. This function was then used to convert counts in whole blood to counts in plasma.
Samples (about 400 μl) were collected at different time points after tracer injection, with a maximum of four blood samples per animal, to determine the time course of the ratio of the 11C activity in plasma to whole blood and for analysis of authentic tracer and metabolites. These samples were first centrifuged for 3 min at 2000 rpm. Proteins were then precipitated with 75 μl of acetonitrile in 50 μl of plasma. After centrifugation for 3 min at 2000 rpm, the composition of the 11C-derived radioactivity in the supernatant (80 μl) was analyzed by HPLC on a polymeric column (5 μm, 250 × 4.1 mm inner diameter; PRP-1; Hamilton) with 3 mmol/L phosphoric acid in water, pH 2.67, as the mobile phase (1 ml/min). The retention times of [11C]HCO3− (3.3 min) and lactic acid (5.1 min) were determined by using aqueous solution of NaHCO3 and dl-lactic acid as reference compounds, detected by UV absorption at 220 nm. The amount of authentic tracer was expressed as a fraction of total plasma 11C counts.
The fraction data of all animals was then pooled, and the time course of the fraction was approximated by a decaying biexponential function. This function was subsequently used to convert the total plasma activity to the time course of authentic [11C]lactate (input curve). At the end of these experiments, the rats were perfused with PBS, and brains were prepared for measurements with the HPLC system. Each brain was first homogenized before adding acetonitrile (150%). The subsequent procedure was the same as with the blood samples described above, except that the amount of supernatant injected into the HPLC system was 200 μl.
CBF measurements (used for estimating the first-pass extraction fraction of lactate).
The basis of the CBF calculation was the one-tissue compartment model, including a partition coefficient for H215O. The change of the radioactivity concentration in tissue Ctiss is then defined by the following differential equation:
where Ca is the arterial tracer concentration, and p is the tissue partition coefficient, e.g., the fraction of tissue that is H215O permeable. In this configuration, Ctiss is the concentration of H215O in 1 ml of brain, and it is assumed that H215O immediately reaches a homogeneous concentration in permeable space and no division into a vascular and a tissue compartment is necessary. The analytical solution of Equation 1 is as follows:
where ⊗ represents mathematical convolution.
Equation 2 was fitted to the data using least-squares fitting (Marquardt algorithm) implemented by the software PMOD (Mikolajczyk et al., 1998). Before data analysis, tissue time activity curves and arterial input curves were corrected for the physical decay of 15O.
Analysis of the radiotracer data
The calculations and parameter fitting were performed using the software PMOD (Mikolajczyk et al., 1998). Data were corrected for physical decay and multiplied by a calibration factor taking into account differences in sensitivities of the probe system and the coincidence counter. Before analysis, data were downsampled to obtain a 10 or 30 s resolution for 11C and 18F data, respectively. The investigated methods consisted of standard compartmental modeling using an arterial input function.
1-[11C]l-Lactate.
Tracer kinetic modeling was performed using the one-tissue compartment model. The parameters are as follows: K1 describes the uptake of tracer across the blood–brain barrier and is related to CBF and the first-pass extraction fraction EF (K1 = CBF * EF); and k2 represents the back-diffusion of label from the tissue to the vascular compartment.
Label exchange between the compartments is described by the following differential equation:
Because the total activity measured in a region is composed of counts from tissue and blood, all models contained a parameter (α) correcting for blood activity:
where CVOI is 11C activity measured by the tip of the beta scintillator, α is the percentage of intravascular space in the tissue, Ctiss is activity in the extravascular compartment, Cblood is total blood activity, and Ctiss was calculated by numerical integration of the differential equations. The vascular fraction α was included as a fit parameter that improved the least-squares fit.
The kinetic model was adjusted in two ways for the analysis of dual-injection data arising from use of the surface probe. (1) Two sets of rate constants (K1, k2) were used in the calculation of the operational equation: the first set for the time until the second injection, and the second set thereafter. The least-squares fit procedure resulted in estimates for all four rate constants (K1−1, k2−1 and K1−2, k2−2) and the vascular fraction. (2) The correction function to derive plasma activity from whole blood activity was adjusted to distinguish between the contributions from the two injections. To this end, the blood activity from the first injection was extrapolated using an exponential function, which was fitted to the blood activity 10 min before the second injection.
The interpretation of the 1-[11C]l-lactate experiments is based on the following assumptions, which are discussed in detail below: (1) parameter k2 is reflecting the activity of the enzyme lactate dehydrogenase (LDH), and (2) the total amount of lactate that is oxidized is the product of k2 and intracellular lactate concentration.
FDG.
FDG data were used for calculating LCMRglu. The procedure for the quantification of the probe studies followed the [14C]deoxyglucose method described by Sokoloff et al. (1977). A compartmental model with two tissue compartments and four kinetic rate constants (K1 to k4) was used. The operational equation, which was fitted to the probe time activity curve, additionally included a vascular fraction α as a fit parameter. This improved the least-squares fit compared with a fixed value of α = 5%. LCMRglu values were calculated from the fitted rate constants: LCMRglu = K1 * k3/(k2 + k3)Cplasma/LC, where Cplasma represents the plasma glucose concentration, and LC represents the lumped constant (LC of 0.437).
The kinetic model was adjusted in two ways for the dual-injection data analysis. (1) Two sets of four rate constants (K1 to k4) were used in the operational equation calculation: the first set for the time until the second injection, and the second set thereafter. The least-squares fit procedure resulted in estimates for all eight rate constants and the vascular fraction. (2) The correction function to derive plasma activity from whole blood activity was adjusted to distinguish between the contributions from the two injections. To this end, the blood activity from the first injection was extrapolated using an exponential function that was fitted to the blood activity 10 min before the second injection.
Statistics
Results are presented as mean ± SD if not otherwise stated. To test differences between different interventions for inter-individual comparisons, the nonparametric Wilcoxon's signed rank test was applied. A p value of 0.05 was taken as the significance limit.
Results
Lactate can sustain neuronal integrity as an alternative energy substrate
Severe glucose deprivation was induced by insulin injection (20 IU/kg, injection at time 0 min) while monitoring neural activity with VSD imaging. In three experimental groups, infusion of saline, glucose, or lactate was immediately initiated after the insulin injection. The total recording time for VSD imaging is limited because of dye washout and excitation light-related amplitude reductions of the signal (Lippert et al., 2007). Therefore, imaging commenced with delay, ∼100 min after the insulin injection. In animals infused with saline only, blood glucose levels reached 1.5 mmol/L already after 60 min. Plasma lactate levels varied on average between 1.0 and 1.5 mmol/L throughout the whole experimental period (Fig. 1F). Severe hypoglycemia reduced the amplitude of the VSD signal by >50% from the baseline after only 140 min after the application of insulin (Fig. 1A,B,E). Thereafter, the signal was further decreased as a result of severe glucose deprivation. In animals supplied continuously with glucose, plasma levels of glucose were not reduced by insulin action and remained at normoglycemic values (Fig. 1G). Not surprisingly, the neuronal signal did not show a significant decline throughout the period of data acquisition [270 min after the insulin injection; n = 5; not significant (n.s.) compared with baseline acquisition at 120 min (Fig. 1A,C,E)]. In the group in which sodium lactate was infused over the whole time period (plasma lactate levels between 5.5 and 9.7 mmol/L), plasma levels of glucose also exhibited a rapid decline to values ∼1 mmol/L within 60 min (Fig. 1H). Nevertheless, the amplitude of the VSD signal reflecting neuronal activity remained high (n = 5; n.s. compared with the 120 min time point) (Fig. 1A,D,E). The spread of activation remained constant in the glucose- and lactate-treated animals within the 240 min after the insulin injection, whereas the extent of activation in the animals receiving saline infusion only decreased considerably (data not shown). In the latter group, four of five animals showed no pixels surpassing 70% of the maximal amplitude after 150 min. In the fifth, the number decreased by >50%. The time course of the VSD signal revealed an accentuated delay of the signal in animals receiving lactate infusion compared with glucose-infused rats (Fig. 1C,D). In the animals receiving glucose infusion after insulin injection (n = 5), the times-to-peak were 21.8 ± 1.6, 21.6 ± 1.9, and 20.7 ± 1.5 ms after 120, 180, and 240 min, respectively. In the lactate group (n = 5), delays at the corresponding time points were 21.2 ± 0.9, 23.5 ± 4.4, and 36.2 ± 10.7 ms. The difference after 240 min was significant between the two groups (p < 0.05).
Figure 1.
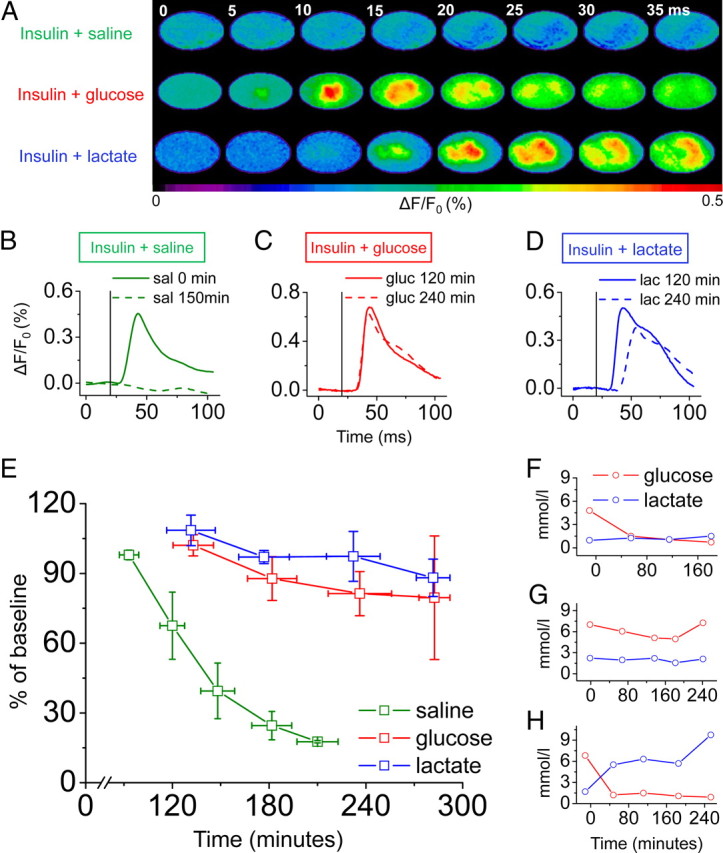
Lactate is able to sustain neuronal activity in the absence of glucose. A, A single electrical pulse applied to the rat's hindpaw evoked a transient increase in VSD fluorescence in the primary somatosensory cortex that was abolished after only 150 min in animals receiving saline after the insulin injection (top row) but remained stable in animals infused with glucose (middle row) or lactate (bottom row) after 240 min. B–D, The time courses of the VSD signal displayed a similar result in animals supplied with glucose (C; gluc) or lactate (D; lac) with sustained amplitude 240 min after insulin injection. In contrast, removal of the signal was observed in animals receiving saline infusion only (B; sal). In the group supplied with lactate, the signal onset was delayed (example shown in D). The vertical black line represents the time point of hindpaw stimulation. E, Comparison of amplitudes in all three groups injected with insulin at 0 min (mean ± SE; n = 5). F–H, Mean plasma levels of glucose and lactate plotted for saline (F), glucose (G), and lactate (H) animals.
Lactate is oxidized by the brain in an activity-dependent manner
To directly assess lactate use in the adult rodent brain in vivo, we synthesized radiolabeled 1-[11C]l-lactate. Characterization of 1-[11C]l-lactate revealed suitable properties for use in brain studies, allowing cerebral tracer kinetic measurements with high temporal resolution. An extraction fraction (EF = K1/CBF) of 30 ± 10% (n = 4) was calculated, which is sufficient to study physiological processes in the brain and is in good agreement with previous studies reporting the uptake of l-lactate to be 25–50% of that of glucose (Knudsen et al., 1991; Hassel and Bråthe, 2000). Kinetic modeling showed that a one-tissue compartment model was sufficient for the analysis of 1-[11C]l-lactate data, yielding K1 and k2 values measuring the uptake and the release of radiolabel, respectively. The time courses of the fraction of authentic tracer and metabolites in plasma are shown in Figure 2. As in brain, the main metabolite detected in blood was [11C]CO2. After 20 min, the fraction of true tracer fell to ∼40%. In the brain, the analysis revealed 58 ± 8% lactate and 42 ± 8% CO2 after 40 min (n = 7). Using for the first time 11C-radiolabeled lactate to measure cerebral lactate oxidation, we observed a noticeable increase of both rate constants K1 and k2 during increased neuronal activity (Fig. 3B, Table 1). Delivery of the tracer increased during electrostimulation of the infraorbital nerve as reflected by an increase of the CBF-dependent K1 from 0.09 ± 0.01 to 0.12 ± 0.03 ml · min−1 · ml−1 tissue (n = 5; change n.s.).
Figure 2.

1-[11C]l-lactate is a suitable tracer to study cerebral lactate oxidation. A, Measured radioactivity concentration in the brain (open circles), model fit (black line), and arterial input curve (gray line). The inset displays the residuals of the fitting to a one-tissue compartment model. The absence of any distribution bias supports the adequacy of the applied one-tissue compartment model. B, Fraction of native radiolabeled lactate over 40 min after intravenous injection of 1-[11C]l-lactate in blood. The filled circles represent data points from baseline experiments (n = 7) performed to characterize the radiotracer, and the solid line is the corresponding fit of a biexponential curve.
Figure 3.

The brain oxidizes lactate in an activity-dependent manner. A, Schematic of the biochemical pathways involved in the degradation of 1-[11C]l-lactate (11C-Lac) and the proposed interpretation of the rate constants K1 and k2 describing the kinetics of the radiotracer (for details, see Results). Briefly, K1 represents delivery of 1-[11C]l-lactate (orange arrow), and k2 reflects kinetically the loss of radiolabel after conversion of lactate to pyruvate and of pyruvate to acetyl-coenzyme A (purple arrows). During activation (B; S; n = 5), delivery (K1) and washout (k2) increased, whereas during MCT blockade (C; CIN; n = 5), the transfer of 1-[11C]l-lactate slowed down in both directions (Pyr, pyruvate; Ac CoA, acetyl coenzyme A; TCA cycle, tricarboxylic acid cycle). *p < 0.05, n.s., not significant.
Table 1.
1-[1 1C]l-lactate kinetics
| Animal number |
K1 (ml · min−1 · ml−1 tissue) |
k2 (min−1) |
||
|---|---|---|---|---|
| Baseline | Stimulation | Baseline | Stimulation | |
| 1 | 0.081 | 0.091 | 0.073 | 0.082 |
| 2 | 0.085 | 0.111 | 0.074 | 0.105 |
| 3 | 0.097 | 0.101 | 0.09 | 0.107 |
| 4 | 0.086 | 0.12 | 0.069 | 0.112 |
| 5 | 0.111 | 0.128 | 0.076 | 0.095 |
| Mean | 0.092 | 0.110 | 0.076 | 0.100 |
| SD | 0.01 | 0.01 | 0.01 | 0.01 |
| Baseline | CIN | Baseline | 4-CIN | |
|---|---|---|---|---|
| 6 | 0.138 | 0.076 | 0.116 | 0.052 |
| 7 | 0.139 | 0.083 | 0.115 | 0.071 |
| 8 | 0.132 | 0.107 | 0.102 | 0.082 |
| 9 | 0.128 | 0.091 | 0.109 | 0.076 |
| 10 | 0.115 | 0.08 | 0.098 | 0.081 |
| Mean | 0.130 | 0.087 | 0.108 | 0.072 |
| SD | 0.01 | 0.01 | 0.01 | 0.01 |
In parallel to the elevated delivery, radioactivity washout (k2) increased from 0.077 ± 0.009 to 0.105 ± 0.007 min−1 (n = 5; p < 0.05). In Discussion, we will argue that this is most likely related to increased LDH activity during stimulation.
There is strong evidence in our and other studies that the label leaves the tissue as CO2, which is cleaved off during the conversion of pyruvate to acetyl-CoA by pyruvate dehydrogenase (PDH) (Fig. 3A). In the present study, the metabolite analysis revealed only lactate and CO2 as labeled metabolites. This is in line with a study by van Hall et al. (2009). Using 1-[13C]lactate, they reported that 86 ± 15% of the label was released as CO2. The situation is completely different, if lactate is labeled at position 2 or 3. In that case, most of the label ends up in metabolites of the TCA cycle (van Hall, 1999; Bouzier et al., 2000; van Hall et al., 2009).
Increased uptake of blood-borne lactate and efficiency of lactate transport during activation
It is possible to calculate cerebral uptake of blood-borne lactate as the product of K1 and blood lactate concentration (in this group of experiments, plasma lactate levels ranged from 1.7 to 2.8 mmol/L) (Table 2). Based on these values, baseline mean cerebral lactate uptake was estimated to be 0.17 μmol · g−1 · min−1 and increased to 0.22 μmol · g−1 · min−1 during stimulation.
Table 2.
Plasma lactate levels and calculated cerebral lactate uptake
| Animal number | Plasma lactate level (mmol/L) |
Calculated cerebral lactate uptake (K1* lac; μmol · g−1 · min−1 |
||
|---|---|---|---|---|
| Baseline | Stimulation | Baseline | Stimulation | |
| 1 | 2 | 2.2 | 0.162 | 0.200 |
| 2 | 2 | 2.9 | 0.170 | 0.322 |
| 3 | 2 | 2.1 | 0.194 | 0.212 |
| 4 | 1.7 | 1.6 | 0.146 | 0.192 |
| 5 | 1.5 | 1.5 | 0.167 | 0.192 |
| Mean | 1.84 | 2.06 | 0.17 | 0.22 |
| SD | 0.2 | 0.6 | 0.02 | 0.06 |
lac, Plasma lactate level.
A detailed analysis of K1 revealed furthermore that the stimulation-induced increase cannot be completely explained by an increase in CBF only but must at least be partly attributable to an increased transport rate for lactate. We estimated this increase to be in the order of 30% using the following relationships:
where EF can be expressed according to Renkin (1959) and Crone (1963), and
Combining Equations 5 and 6 yields the following:
where PS is the permeability–surface area product. We performed a simulation assuming a constant PS from baseline to ION stimulation. In previous experiments, the applied electrostimulation of the ION led to a mean increase of CBF from 0.35 to 0.50 ml · min−1 · ml−1 tissue (Weber et al., 2003). In the present series of 1-[11C]l-lactate experiments, K1 increased from 0.09 ± 0.01 to 0.12 ± 0.03 ml · min−1 · ml−1 tissue during ION. Inserting these numbers into Equation 7 demonstrates a 32% increase of PS, from 0.104 to 0.137 ml · min−1 · ml−1. This suggests that monocarboxylate transporter (MCT) efficiency increases during increased demand.
Effect of MCT blockade on lactate uptake
Injection of CIN 30 min before [11C]lactate tracer kinetic measurements reduced K1 from 0.13 ± 0.01 to 0.09 ± 0.01 ml · min−1 · ml−1 tissue (n = 5; p < 0.05) and k2 from 0.11 ± 0.01 to 0.07 ± 0.01 min−1 (n = 5; p < 0.05) (Fig. 3C, Table 1). CIN has been shown to permeate the blood–brain barrier and to be effective 30 min after intraperitoneal injection (Schurr et al., 2001). The reduction in K1 after CIN administration is most likely attributable to the reduction of lactate transport by a partial blockade of MCTs. The concomitant reduction in k2 cannot be reliably interpreted without additional biochemical measurements. The fact that the relative decrease of k2 is of the same magnitude as the decrease of K1 (−33%) points to the possibility that k2 under CIN blockade represents back-diffusion of lactate into vascular space.
Increased blood lactate reduces the local metabolic rate of glucose
To address the question of how the brain processes blood-borne lactate compared with glucose, we artificially increased the supply of lactate to the brain by continuous intravenous infusion of sodium lactate (Fig. 4A). This led to a significant increase of plasma lactate levels (from 1.3 ± 0.6 to 5.8 ± 1.6 mmol/L; n = 10; p < 0.05). Plasma glucose levels were not affected by the continuous lactate infusions. In all examined animals, a clear reduction of LCMRglu was observed at resting conditions (decreases ranging from −25 to −49%; n = 13; p < 0.05) (Fig. 4B). The reduction of the cerebral glucose utilization was attributable to both a decreased delivery of glucose into the tissue (reflected by a change of K1 from 0.16 ± 0.05 to 0.12 ± 0.03 ml · min−1 · ml−1 tissue; n = 13; p < 0.05) and a reduced phosphorylation by hexokinase (k3 decreased from 0.04 ± 0.02 to 0.03 ± 0.01 min−1; n = 13; p < 0.05). The replacement of glucose by lactate as an energy substrate occurred in a dose-dependent manner (Fig. 4C).
Figure 4.

Lactate is preferred over glucose by the brain. A and D detail the protocol during baseline (A) and stimulation (D) conditions. Overall, hyperlactemia reduced LCMRglu by on average 38% in a dose-dependent manner (B, C) during baseline conditions. In activated cortex (S1contra), cerebral glucose utilization was further decreased. The effectiveness of stimulation was controlled autoradiographically (D). The ratio S1contra/S1ipsi decreased from 1.65 ± 0.47 to 1.26 ± 0.37 (E). 1, Acquisition 1; 2, acquisition 2; LAC, hyperlactemia; SAL, saline. *p < 0.05, n.s., not significant. n = 13 for LAC group, n = 4 for SAL group at baseline conditions, n = 5 for LAC group, and n = 2 for SAL at stimulation conditions.
Reduction in glucose utilization is balanced by the excess of lactate consumption
The observed mean reduction in glucose utilization was 21 μmol/100 g/min (Fig. 4B). Because glucose has a six-carbon skeleton, this corresponds to 42 μmol/100 g/min three-carbon substrates such as pyruvate. The following estimation demonstrates that the decrease in glucose utilization is quantitatively matched by an increase in lactate use. For this purpose, we made the following assumptions: (1) lactate transport from blood into the brain equals K1 * plasma lactate concentration ([Lac]), (2) K1 (= 0.09 min−1 at baseline) does not markedly change within the range of lactate concentrations found in our experiments, and (3) all the lactate, which is taken up, is ultimately oxidized by the brain.
Thus, lactate uptake during baseline conditions in the FDG experiments was 12 μmol/100 g/min (= 0.09 min−1 * 1.3 mmol/L; mean K1 determined in baseline 1-[11C]lactate experiments (Fig. 3B) and mean [Lac] from FDG experiments during saline infusion) and 52 μmol/100 g/min during hyperlactemia (= 0.09 min−1 * 5.8 mmol/L; mean [Lac] during lactate infusion). The difference of lactate uptake between baseline and hyperlactemia conditions is therefore 40 μmol/100 g/min (52 μmol/100 g/min − 12 μmol/100 g/min). This matches the above mentioned reduction of three-carbon constituents derived from glucose during hyperlactemia.
Reduction of glucose consumption during hyperlactemia is more pronounced during activation
Our results further demonstrate that the reduction of glucose utilization is not only dependent on blood lactate levels but also on the level of activation. As shown in Figure 4, D and E, the decrease of glucose utilization was more pronounced on the activated side, as reflected by the decrease of the ratio contralateral/ipsilateral in the primary somatosensory cortex (i.e., S1contra vs S1ipsi). During normolactemia, this ratio was 1.65 ± 0.47, but during hyperlactemia, it decreased to 1.26 ± 0.37 (n = 5; p < 0.05) (Fig. 4E). In two control animals, in which both acquisitions were performed under saline infusion, the ratio of LCMRglu between S1contra and S1ipsi remained the same during the first and the second acquisition (Fig. 4E). Plasma levels of glucose and lactate behaved in a similar manner as in the baseline group.
Neuronal activity is not reduced by lactate
To verify that the observed reduction in glucose utilization does not originate from a reduced requirement for energy of the brain attributable to impaired neural activity, we performed an additional series of VSD experiments during hyperlactemia. Under these conditions, increased lactate levels did not affect neuronal excitability (Fig. 5).
Figure 5.
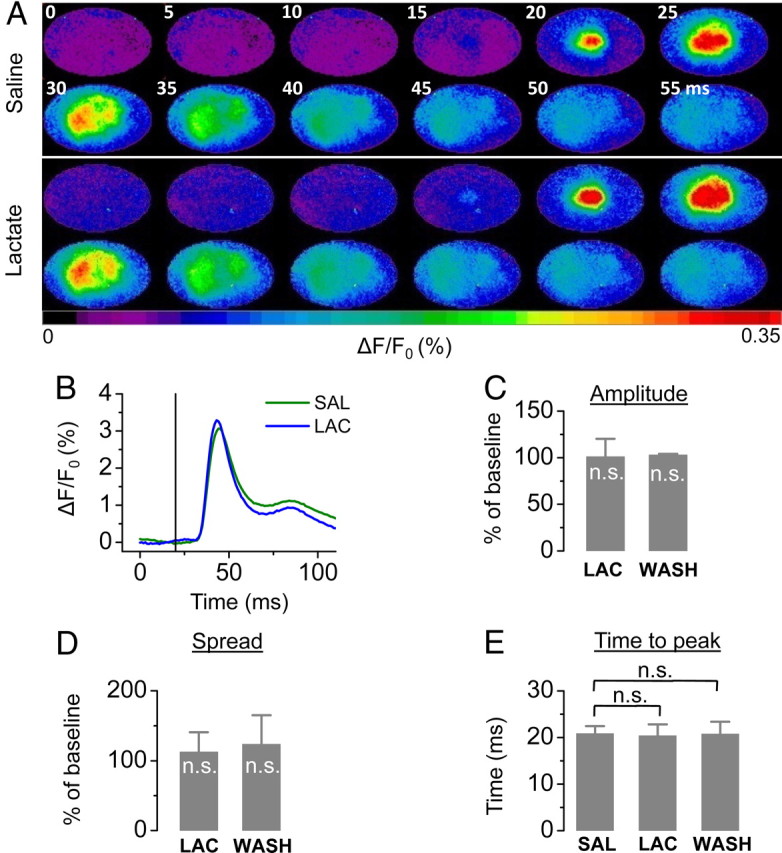
Cerebral integrity is not disrupted during hyperlactemia. A, B, A single example demonstrating the spread (A) and the amplitude over time (B) during saline (SAL) and lactate (LAC) infusion is shown. C–E, Mean results of changes in amplitude (C), spread (D), and time-to-peak (E) (n = 4; n.s., not significant compared with baseline).
Discussion
For a long time, no functional role has been assigned to cerebral lactate. Contrarily, the presence of lactate in the brain has been interpreted as a sign of cerebral harm and of hypoxia (Siesjö, 1981). Over the past few decades, this perspective has changed and it has been postulated that lactate may also possess physiological functions relevant to the CNS. Important roles have been attributed to lactate such as the modulation of CBF (Gordon et al., 2008), as a buffer to sustain neuronal energy supply (Kasischke et al., 2004) and most prominently as a pivotal element in the neuron–glia metabolic cooperation to regulate energy supply in an activity-dependent manner (for review, see Pellerin and Magistretti, 2003; Magistretti, 2006). Furthermore, it has been shown recently that lactate levels are sensed by a specific type of neuron (orexin neurons in the lateral hypothalamus) to coordinate the balancing of brain activity and energy supply (Lam et al., 2005; Shimizu et al., 2007; Erlichman et al., 2008; Parsons and Hirasawa, 2010). Nevertheless, the details of brain energy metabolism in general and of lactate as a cerebral energy substrate in particular are still highly debated.
Lactate serves as a metabolic source for neurons
It has been shown in vitro that lactate oxidation is able to sustain excitatory synaptic activity (Schurr et al., 1988; Rouach et al., 2008), and there have been reports about lactate protecting cerebral function during hypoglycemia (Maran et al., 1994; King et al., 1998). However, the latter in vivo studies did not use direct neuronal readouts to assess synaptic activity, and glucose levels were above 2.5 mmol/L. The present study is the first in vivo demonstration of the maintenance of neuronal excitability in the presence of lactate as the primary energy source. The amplitude of the stimulation-induced increase in neuronal activity was maintained when lactate was supplied during severe hypoglycemia (Fig. 1). However, the increased delay of the VSD signal may indicate that lactate alone may not quite suffice to completely sustain neuronal functioning. Indeed, it has been shown that glucose is used by neurons to maintain their antioxidant status via the pentose phosphate pathway (PPP), which cannot be fueled by lactate (Magistretti, 2008; Herrero-Mendez et al., 2009). Because of the low plasma glucose levels and subsequent small glucose concentration gradient, the transport into neurons is inadequate (Barros and Deitmer, 2010) and presumably not enough substrate is available for the neurons to efficiently stimulate their antioxidant PPP. Neurons, which are thought to be especially vulnerable against reactive oxygen and nitrogen species, might not be capable of completely avoiding oxidative damage, which may lead to the observed delay of the electrical signal in the absence of glucose. Furthermore, this finding supports the important role in functional neuroenergetics dedicated to glucose because glucose is required by the astrocyte to pump glutamate (Magistretti et al., 1999). Second, lactate is transported by MCTs in a cotransport with protons (Hertz and Dienel, 2005). Lactate elevations as performed in our study may lead to changes in lactate influx that result in alterations of intracellular and extracellular pH in nerve tissue. These changes of the proton gradient could disturb the conductance of action potentials along nerve axons and lead to a delayed response.
Physiological meaning of k2
Using 1-[11C]l-lactate, we demonstrated that the brain readily oxidizes lactate at normal plasma lactate levels in accordance with recent work using 1- and 3-[13C]l-lactate (Bouzier et al., 2000; van Hall et al., 2009; Boumezbeur et al., 2010). Our radiotracer approach enables the measurement of cerebral lactate oxidation without altering blood and tissue concentrations of lactate. In this respect, it is important to consider the physiological meaning of k2. By definition, it represents the loss of label from tissue (Fig. 3A). The model depicted in Figure 3A demonstrates that the label can be lost by back-diffusion of labeled lactate into vascular space and as labeled CO2. The latter pathway is more interesting because it is related to the oxidation of lactate, unless the diffusion of CO2 across the blood–brain barrier is rate limiting. This is unlikely because CO2 back-diffusion does not seem restricted (Paulson, 2002). Radiolabeled lactate is processed by LDH and PDH before the label is lost as CO2. Parameter k2 can therefore be related to either of them or to both, depending on whether one of these steps is rate limiting. A flux estimation suggests that the processing by LDH and not PDH is the rate-limiting step.
If k2 represented the conversion of pyruvate to acetyl-CoA, it would closely be related to the flux of pyruvate (FluxPDH), which in this model would be expressed as follows: FluxPDH = k2 * Cpyr, or Cpyr = FluxPDH/k2, where Cpyr is the intracellular pyruvate concentration. Inserting a measured FluxPDH value of 1.13 μmol · g−1 · min−1 (Oz et al., 2004) and the mean k2 from our [11C]l-lactate experiments (0.077 min−1) yields an unphysiological pyruvate concentration of ∼15 mmol/L. Measured pyruvate concentrations are in the range of 0.1–0.2 mmol/L (McIlwain, 1985).
In analogy, the conversion of lactate to pyruvate by LDH yields FluxLDH = k2 * Clac, where Clac is the intracellular lactate concentration. Using the numbers measured in humans by magnetic resonance spectroscopy [FluxLDH = 0.06 μmol · g−1 · min−1) (Boumezbeur et al., 2010)], the above equation yields an intracellular lactate concentration of 0.06/0.077 = 0.78 mmol/L. This is in the same range as the reported number [1 mmol/L at plasma lactate levels of 1.5 mmol/L (Boumezbeur et al., 2010)]. These estimations suggest that k2 is more related to the activity of LDH than PDH.
Evidence for increased lactate oxidation during stimulation
The increase of k2 can be seen as a facilitation of the flux of substrates through the oxidative chain. However, an effective increase of the lactate flux furthermore requires that there is no relevant stimulation-induced drop in another factor driving lactate oxidation, e.g., the intracellular lactate concentration. However, such a drop seems unlikely for two reasons. First, the extracellular lactate concentration has been shown to increase during stimulation (Hu and Wilson, 1997; Caesar et al., 2008). Second, there is evidence for a translocation of MCT2 to the membrane surface during stimulation (Pierre et al., 2009), which would augment lactate transport and lead to higher intracellular lactate concentration. Such a translocation could also explain the increased permeability surface product during stimulation observed in this study.
There is another line of evidence supporting an elevated neuronal lactate oxidation during increased neuronal activity. An increased conversion of lactate to pyruvate is facilitated by an increase of the lactate/pyruvate ratio. Besides the increase in the lactate concentration (see above), a decrease of the pyruvate concentration or both causes a drop in this ratio. A reduced pyruvate concentration is achieved by a reduced glycolytic flux. Indeed, there is evidence from experiments in cell cultures that glutamate inhibits neuronal glucose uptake (Porras et al., 2004; Castro et al., 2009) and that upregulation of glycolysis under stress conditions is prevented (Herrero-Mendez et al., 2009). Recently, it has been found in vivo that increased neuronal firing mediates inhibition of glucose transport in neurons while stimulating astrocytic glucose uptake (Chuquet et al., 2010).
Concluding remarks
The results presented demonstrate in vivo that lactate is metabolized by neurons. Indeed, VSD imaging enabled us to follow a neuronal readout, which is supported by the supplied lactate during the experimental period. Considering the facts that intracerebral glycogen stores are relatively limited and consumed in the absence of exogenous glucose within a few minutes (Brown and Ransom, 2007) and that gluconeogenic activity in the brain is negligible (Nelson et al., 1985), it is evident that the neurons must rely on lactate as an energy substrate under hypoglycemic conditions. In addition, hepatic gluconeogenesis is suppressed by the insulin action. Thus, it can be suggested that the neuroprotective effect of lactate observed in previous studies (Gjedde and Crone, 1975; Schurr et al., 2001; Mason et al., 2006; Berthet et al., 2009) is attributable to direct lactate oxidation in neurons. However, we cannot exclude astrocytic lactate uptake and oxidation.
It is important to note that the data reported here using radiolabeled lactate provide evidence for the ability of neurons to increase lactate use during stimulation. During increased activation, we observed an increased turnover of 1-[11C]lactate also under normoglycemia. Considering the fact that all the experiments were performed under anesthesia, thus reducing the overall metabolic rate by a factor of twofold to threefold (Hyder et al., 2006), it is reasonable to assume that an even larger contribution of lactate to brain metabolism may occur in the awake animal.
In summary, our multimodal experiments demonstrate that (1) lactate is capable of maintaining neuronal integrity to a large degree in the absence of glucose, (2) lactate is preferred over glucose if both substrates are available, (3) lactate is readily metabolized by the intact adult rodent brain, and (4) its metabolism is activity dependent.
Footnotes
M.T.W. and B.W. are supported by Swiss National Foundation Grants 31003A-124739/1 and PP0033-110751. R.J. is supported by grants from the Hartmann Müller Foundation, the Olga Mayenfisch Foundation, and the EMDO Foundation. We thank Florent Haiss for help in setting up the voltage-sensitive dye instrument and Felipe Barros for valuable discussions.
References
- Barros LF, Deitmer JW. Glucose and lactate supply to the synapse. Brain Res Rev. 2010;63:149–159. doi: 10.1016/j.brainresrev.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Berthet C, Lei H, Thevenet J, Gruetter R, Magistretti PJ, Hirt L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1780–1789. doi: 10.1038/jcbfm.2009.97. [DOI] [PubMed] [Google Scholar]
- Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2010;30:13983–13991. doi: 10.1523/JNEUROSCI.2040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier AK, Thiaudiere E, Biran M, Rouland R, Canioni P, Merle M. The metabolism of [3-(13)C]lactate in the rat brain is specific of a pyruvate carboxylase-deprived compartment. J Neurochem. 2000;75:480–486. doi: 10.1046/j.1471-4159.2000.0750480.x. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55:1263–1271. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- Caesar K, Hashemi P, Douhou A, Bonvento G, Boutelle MG, Walls AB, Lauritzen M. Glutamate receptor-dependent increments in lactate, glucose and oxygen metabolism evoked in rat cerebellum in vivo. J Physiol. 2008;586:1337–1349. doi: 10.1113/jphysiol.2007.144154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro MA, Beltrán FA, Brauchi S, Concha II. A metabolic switch in brain: glucose and lactate metabolism modulation by ascorbic acid. J Neurochem. 2009;110:423–440. doi: 10.1111/j.1471-4159.2009.06151.x. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, Buzsáki G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci. 2010;30:15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer JE. Substrate utilization and brain development. J Cereb Blood Flow Metab. 1982;2:394–407. doi: 10.1038/jcbfm.1982.45. [DOI] [PubMed] [Google Scholar]
- Crone C. The permeability of capillaries in various organs as determined by use of the indicator diffusion method. Acta Physiol Scand. 1963;58:292–305. doi: 10.1111/j.1748-1716.1963.tb02652.x. [DOI] [PubMed] [Google Scholar]
- Dombrowski GJ, Jr, Swiatek KR, Chao KL. Lactate, 3-hydroxybutyrate, and glucose as substrates for the early postnatal rat brain. Neurochem Res. 1989;14:667–675. doi: 10.1007/BF00964877. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Hewitt A, Damon TL, Hart M, Kurascz J, Li A, Leiter JC. Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: a test of the astrocyte-neuron lactate-shuttle hypothesis. J Neurosci. 2008;28:4888–4896. doi: 10.1523/JNEUROSCI.5430-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CN, Carpenter KL, Grice P, Howe DJ, Mason A, Timofeev I, Menon DK, Kirkpatrick PJ, Pickard JD, Sutherland GR, Hutchinson PJ. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain. 2009;132:2839–2849. doi: 10.1093/brain/awp202. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Crone C. Induction processes in blood-brain transfer of ketone bodies during starvation. Am J Physiol. 1975;229:1165–1169. doi: 10.1152/ajplegacy.1975.229.5.1165. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Bråthe A. Cerebral metabolism of lactate in vivo: evidence for neuronal pyruvate carboxylation. J Cereb Blood Flow Metab. 2000;20:327–336. doi: 10.1097/00004647-200002000-00014. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dienel GA. Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res. 2005;79:11–18. doi: 10.1002/jnr.20294. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem. 1997;69:1484–1490. doi: 10.1046/j.1471-4159.1997.69041484.x. [DOI] [PubMed] [Google Scholar]
- Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab. 2006;26:865–877. doi: 10.1038/sj.jcbfm.9600263. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- King P, Kong MF, Parkin H, MacDonald IA, Barber C, Tattersall RB. Intravenous lactate prevents cerebral dysfunction during hypoglycaemia in insulin-dependent diabetes mellitus. Clin Sci (Lond) 1998;94:157–163. doi: 10.1042/cs0940157. [DOI] [PubMed] [Google Scholar]
- Knudsen GM, Paulson OB, Hertz MM. Kinetic analysis of the human blood-brain barrier transport of lactate and its influence by hypercapnia. J Cereb Blood Flow Metab. 1991;11:581–586. doi: 10.1038/jcbfm.1991.107. [DOI] [PubMed] [Google Scholar]
- Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–947. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- Lippert MT, Takagaki K, Xu W, Huang X, Wu JY. Methods for voltage-sensitive dye imaging of rat cortical activity with high signal-to-noise ratio. J Neurophysiol. 2007;98:502–512. doi: 10.1152/jn.01169.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Brain energy metabolism. In: Squire L, Berg D, Bloom FE, du Lac S, Ghosh A, Spitzer N, editors. Fundamental neuroscience. San Diego: Academic; 2008. pp. 271–293. [Google Scholar]
- Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- Maran A, Cranston I, Lomas J, Macdonald I, Amiel SA. Protection by lactate of cerebral function during hypoglycaemia. Lancet. 1994;343:16–20. doi: 10.1016/s0140-6736(94)90876-1. [DOI] [PubMed] [Google Scholar]
- Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI. Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes. 2006;55:929–934. doi: 10.2337/diabetes.55.04.06.db05-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain H, Bachelard HS. Biochemistry and the central nervous system. Ed 5. New York: Churchill Livingstone; 1985. [Google Scholar]
- Mikolajczyk K, Szabatin M, Rudnicki P, Grodzki M, Burger C. A JAVA environment for medical image data analysis: initial application for brain PET quantitation. Med Inform (Lond) 1998;23:207–214. doi: 10.3109/14639239809001400. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Pereira de Vasconcelos A. Glucose and ketone body utilization by the brain of neonatal rats. Prog Neurobiol. 1993;40:163–221. doi: 10.1016/0301-0082(93)90022-k. [DOI] [PubMed] [Google Scholar]
- Nelson T, Lucignani G, Atlas S, Crane AM, Dienel GA, Sokoloff L. Reexamination of glucose-6-phosphatase activity in the brain in vivo: no evidence for a futile cycle. Science. 1985;229:60–62. doi: 10.1126/science.2990038. [DOI] [PubMed] [Google Scholar]
- Oz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Hirasawa M. ATP-sensitive potassium channel-mediated lactate effect on orexin neurons: implications for brain energetics during arousal. J Neurosci. 2010;30:8061–8070. doi: 10.1523/JNEUROSCI.5741-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson OB. Blood-brain barrier, brain metabolism and cerebral blood flow. Eur Neuropsychopharmacol. 2002;12:495–501. doi: 10.1016/s0924-977x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Food for thought: challenging the dogmas. J Cereb Blood Flow Metab. 2003;23:1282–1286. doi: 10.1097/01.WCB.0000096064.12129.3D. [DOI] [PubMed] [Google Scholar]
- Pierre K, Chatton JY, Parent A, Repond C, Gardoni F, Di Luca M, Pellerin L. Linking supply to demand: the neuronal monocarboxylate transporter MCT2 and the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid receptor GluR2/3 subunit are associated in a common trafficking process. Eur J Neurosci. 2009;29:1951–1963. doi: 10.1111/j.1460-9568.2009.06756.x. [DOI] [PubMed] [Google Scholar]
- Porras OH, Loaiza A, Barros LF. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci. 2004;24:9669–9673. doi: 10.1523/JNEUROSCI.1882-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkin EM. Transport of potassium-42 from blood to tissue in isolated mammalian skeletal muscles. Am J Physiol. 1959;197:1205–1210. doi: 10.1152/ajplegacy.1959.197.6.1205. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240:1326–1328. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Tseng MT, Rigor BM. Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 2001;895:268–272. doi: 10.1016/s0006-8993(01)02082-0. [DOI] [PubMed] [Google Scholar]
- Shackford SR, Schmoker JD, Zhuang J. The effect of hypertonic resuscitation on pial arteriolar tone after brain injury and shock. J Trauma. 1994;37:899–908. doi: 10.1097/00005373-199412000-00005. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Watanabe E, Hiyama TY, Nagakura A, Fujikawa A, Okado H, Yanagawa Y, Obata K, Noda M. Glial Nax channels control lactate signaling to neurons for brain [Na+] sensing. Neuron. 2007;54:59–72. doi: 10.1016/j.neuron.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Siesjö BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- van Hall G. Correction factors for 13C-labelled substrate oxidation at whole-body and muscle level. Proc Nutr Soc. 1999;58:979–986. doi: 10.1017/s0029665199001299. [DOI] [PubMed] [Google Scholar]
- van Hall G, Strømstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab. 2009;29:1121–1129. doi: 10.1038/jcbfm.2009.35. [DOI] [PubMed] [Google Scholar]
- Weber B, Burger C, Biro P, Buck A. A femoral arteriovenous shunt facilitates arterial whole blood sampling in animals. Eur J Nucl Med Mol Imaging. 2002;29:319–323. doi: 10.1007/s00259-001-0712-2. [DOI] [PubMed] [Google Scholar]
- Weber B, Späth N, Wyss M, Wild D, Burger C, Stanley R, Buck A. Quantitative cerebral blood flow measurements in the rat using a beta-probe and H2 15O. J Cereb Blood Flow Metab. 2003;23:1455–1460. doi: 10.1097/01.WCB.0000095799.98378.7D. [DOI] [PubMed] [Google Scholar]
- Wyss MT, Obrist NM, Haiss F, Eckert R, Stanley R, Burger C, Buck A, Weber B. A beta-scintillator for surface measurements of radiotracer kinetics in the intact rodent cortex. Neuroimage. 2009;48:339–347. doi: 10.1016/j.neuroimage.2009.06.077. [DOI] [PubMed] [Google Scholar]