

Abstract
Synthetic amyloid-β protein (Aβ) oligomers bind with high affinity to cellular prion protein (PrPC), but the role of this interaction in mediating the disruption of synaptic plasticity by such soluble Aβ in vitro is controversial. Here we report that intracerebroventricular injection of Aβ-containing aqueous extracts of Alzheimer's disease (AD) brain robustly inhibits long-term potentiation (LTP) without significantly affecting baseline excitatory synaptic transmission in the rat hippocampus in vivo. Moreover, the disruption of LTP was abrogated by immunodepletion of Aβ. Importantly, intracerebroventricular administration of antigen-binding antibody fragment D13, directed to a putative Aβ-binding site on PrPC, prevented the inhibition of LTP by AD brain-derived Aβ. In contrast, R1, a Fab directed to the C terminus of PrPC, a region not implicated in binding of Aβ, did not significantly affect the Aβ-mediated inhibition of LTP. These data support the pathophysiological significance of SDS-stable Aβ dimer and the role of PrPC in mediating synaptic plasticity disruption by soluble Aβ.
Introduction
The amyloid β-protein (Aβ) is strongly implicated in the cognitive decline of Alzheimer's disease (AD) (Selkoe, 2002), and great effort has been expended in characterizing the most pathogenically important Aβ assemblies (Shankar and Walsh, 2009). Water-soluble nonfibrillar assemblies of Aβ have been found to be highly toxic to synapses, providing a basis for the positive correlation between disease severity and postmortem concentration of soluble Aβ (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999). The concentration of water-soluble Aβ oligomers, in particular SDS-stable Aβ dimers, is elevated in the brains of AD patients (Mc Donald et al., 2010) and aqueous extracts of AD brain that contain such dimers powerfully inhibit long-term potentiation (LTP) in mouse hippocampal slices and impair avoidance learning in rats (Shankar et al., 2008). Several putative receptor sites have been proposed to mediate disruptive effects of Aβ on synaptic plasticity including proteoglycans (Snow et al., 1995); receptor for advanced glycation end products (Yan et al., 2009); LRP (Deane et al., 2004); integrins (Wright et al., 2007); and nicotinic (Wang et al., 2000), NMDA (Bi et al., 2002), insulin (Xie et al., 2002), and p75 neurotrophin (Yaar et al., 1997) receptors. The selectivity of these sites in mediating the disruptive effects as opposed to putative physiological roles of Aβ (Puzzo et al., 2008; Giuffrida et al., 2010) is unclear.
Recently, a broad range of different sized Aβ assemblies termed Aβ-derived diffusible ligands (ADDLs), but not Aβ monomers, were discovered to bind with high affinity to cellular prion protein (PrPc), specifically in a region that encompassed the amino acid sequence 95–105 (Laurén et al., 2009). Moreover, blocking PrPc with an antibody recognizing this region or a genetic knockout of PrPc prevented the inhibition of LTP by ADDLs in hippocampal slices. Although the potent and selective binding of PrPC by ADDLs has been confirmed (Balducci et al., 2010; Chen et al., 2010), whether or not such binding mediates their synaptic plasticity-disrupting actions is controversial. Recently, Kessels et al. (2010) failed to replicate the finding that the inhibition of LTP by ADDLs was absent in slices from Prnp−/− mice. Furthermore, an impairment of synaptic plasticity in hippocampal slices from APPPS1+ transgenic mice was not significantly affected when this strain of mice was crossed with Prnp−/− mice (Calella et al., 2010).
Given the potential clinical implications of a causative role for the binding of Aβ oligomers to PrPC in neuronal dysfunction (Kellett and Hooper, 2009) and the controversy over the mechanisms of the effects of synthetic and transgenic mouse-generated Aβ, we tested the involvement of PrPC in mediating in vivo synaptic plasticity disruption by the most disease-relevant preparation of Aβ, human brain-derived soluble Aβ (Shankar et al., 2008). Here we report that the inhibition of LTP by human brain extract containing SDS-stable dimers was prevented using an antibody fragment (Fab) directed to the 96–104, but not the 225–231, region of PrPC (Williamson et al., 1998) in the rat in vivo. These findings strongly support the potential value of immunotherapeutically targeting the binding of synaptotoxic Aβ assemblies to PrPC.
Materials and Methods
In vivo LTP.
Experiments were performed on urethane (1.5 g/kg, i.p.)-anesthetized male Wistar rats (250–300 g) under the approval of the ethical review committee of Trinity College Dublin and the Department of Health and Children, Ireland. The electrophysiological methods have been described previously (Klyubin et al., 2008). Briefly, twisted-wire, bipolar, Teflon-coated tungsten wires (62.5 μm inner core diameter, 75 μm external diameter) served as stimulation electrodes. Field EPSPs were recorded from the stratum radiatum in the CA1 area in response to stimulation of the Schaffer collateral–commissural pathway. Recording electrodes were located 3.4 mm posterior to bregma and 2.5 mm right of midline, and the stimulating electrode was located 4.2 mm posterior to bregma and 3.8 right of midline. The depth of the electrodes was optimized using electrophysiological criteria. Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity adjusted to elicit an EPSP amplitude of 50% of maximum. The high-frequency stimulation (HFS) protocol for inducing LTP consisted of 10 trains of 20 stimuli with an interstimulus interval of 5 ms (200 Hz) and an intertrain interval of 2 s, and the intensity was increased to evoke an EPSP of 75% of maximum amplitude. Samples were injected through a stainless steel guide cannula (22 gauge, 0.7 mm outer diameter, 13 mm length) that was implanted above the right lateral ventricle (1 mm lateral to the midline and 4 mm below the surface of the dura). Unless otherwise stated, two sequential intracerebroventricular injections were administered (5 μl of human brain extract 30 min after 10 μl of Fab in PBS or equivalent volumes of distilled water or PBS vehicle) via an internal cannula (28 gauge, 0.36 mm outer diameter).
Preparation and analysis of aqueous extracts of human brain.
Human brain tissue was obtained and used in accordance with the University College Dublin Human Research Ethics Committee guidelines (under Approval LS-E-10–10-Walsh). All three AD cases (a 75-year-old woman, an 80-year-old woman, and an 85-year-old man) were demented before death, and had fulminant amyloid and tangle pathology. A fourth brain was from a cognitively intact 90-year-old woman and lacked any significant AD pathology. Frozen samples of frontal or temporal cortex (1 g) were allowed to thaw on ice, chopped into small pieces with a razor blade, and then homogenized in 5 ml of ice-cold 20 mm Tris, pH 7.4, containing 150 mm NaCl (TBS) with 25 strokes of a Dounce homogenizer (Fisher). To separate water-soluble Aβ from membrane-bound or plaque Aβ, homogenates were centrifuged at 91,000 × g and 4°C in a TLA 55 rotor (Beckman-Coulter) for 78 min. To eliminate bioactive small molecules and drugs, the supernatant was exchanged into ammonium acetate using a 5 ml Hi-trap desalting column (GE Healthcare Bio-Sciences AB). Thereafter, the extract was divided into three parts: one aliquot was immunodepleted of Aβ by three rounds of 12 h incubation with the anti-Aβ antibody AW8 and protein A at 4°C. The second portion (the “mock immunodepletion”) was subjected to the same procedure as used with the first sample, but preimmune AW8 sera were used instead of AW8 antiserum. The third aliquot was not manipulated in any way. Aliquots of samples were stored at −80°C or were used to assess Aβ content using a sensitive immunoprecipitation/Western blotting procedure (Mc Donald et al., 2010). Briefly, samples were immunoprecipitated with AW8 at a dilution 1:80 and analyzed by Western blotting using a combination of the C-terminal monoclonal antibodies 2G3 and 21F12, each at a concentration of 1 μg/ml (kind gifts from Dr. Peter Seubert, Elan Pharmaceuticals). Immunoreactive bands were visualized using a fluorochrome coupled secondary antibody (Rockland) and were quantified by comparison to synthetic Aβ standards (Keck laboratory, Yale University, New Haven, CT) using a Licor Odyssey imaging system (Licor Biosciences).
Statistics.
LTP is expressed as the mean ± SEM percentage baseline field EPSP amplitude recorded over at least a 30 min baseline period. Statistical comparisons used ANOVA with post hoc Tukey test or paired and unpaired Student's t tests, as appropriate. Mann–Whitney U tests were used to analyze changes in baseline synaptic transmission.
Results
Human AD brain Aβ selectively inhibits LTP
The TBS-soluble extract of the cerebral cortex from AD patients used in the present studies contained readily detectible Aβ, which included SDS-stable dimer consistent with our previous findings (Mc Donald et al., 2010) (Fig. 1A).
Figure 1.
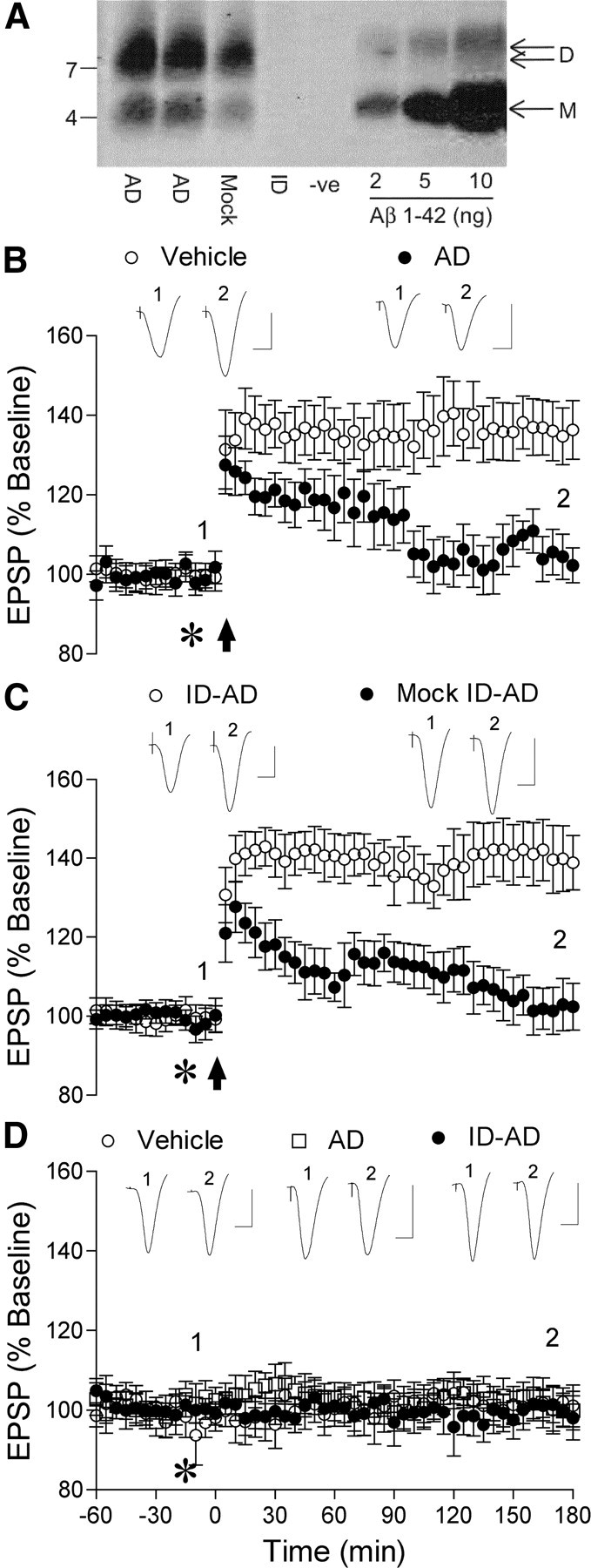
SDS-stable Aβ dimer-containing aqueous extract of human Alzheimer's disease brain inhibits LTP of synaptic transmission in the rat hippocampus in vivo. A, Unmanipulated TBS extracts from AD brain (AD), TBS (-ve), extract treated with preimmunine serum (Mock ID) or extract immunodepleted of Aβ (ID) were examined by immunoprecipitation/Western blotting as described in Materials and Methods. The Aβ content of each was estimated by reference to known amounts (2–10 ng) of synthetic Aβ1–42 loaded on the same gel. Molecular weight markers are on the left and the migration of Aβ monomer (M) and SDS-stable dimer (D) are indicated on the right. The blot was trimmed to the 13 kDa molecular weight standard. B, Application of high-frequency stimulation (arrow) induced robust LTP in animals that received vehicle (asterisk, 5 μl, n = 5), whereas acute injection of Aβ-containing AD brain extract (AD, 5 μl) completely inhibited LTP (n = 5). C, AD brain extract immunodepleted of Aβ (ID-AD, 5 μl) did not inhibit LTP (n = 4). In contrast, AD brain extract that had been processed in the same manner but with normal rabbit preimmune antiserum (mock ID-AD, 5 μl, n = 4) strongly inhibited LTP. D, Acute injection of Aβ-containing (asterisk, 5 μl, n = 4) or Aβ-immunodepleted (n = 4) AD brain extract did not affect baseline excitatory synaptic transmission, similar to vehicle-injected controls (5 μl, n = 4). Insets show representative traces at the times indicated. Calibration: 1.5 mV, 10 ms.
Acute intracerebroventricular injection of the soluble Aβ from AD brain (5 μl) 15 min before the application of high-frequency (200 Hz) conditioning stimulation strongly inhibited LTP. Thus, in control, vehicle-injected, rats LTP magnitude measured 135 ± 7% pre-HFS baseline at 3 h (n = 5, p < 0.05 compared with pre-HFS baseline), whereas in rats injected with the soluble Aβ-containing AD brain supernatant (69 pg of Aβ1–42 equivalent) LTP was completely inhibited (103 ± 5%; n = 5; p > 0.05 compared with pre-HFS baseline; p < 0.05 compared with vehicle) (Fig. 1B). Strong evidence that the Aβ was responsible for the inhibition of LTP was provided by the finding that TBS samples of the same brain extract treated with a polyclonal antibody, AW8, which immunodepleted Aβ (Fig. 1A), failed to inhibit LTP after intracerebroventricular (5 μl) injection (139 ± 8%; n = 4; p < 0.05 compared with pre-HFS baseline and nonimmunodepleted extract; p > 0.05 compared with vehicle) (Fig. 1C). Similar inhibition of LTP by Aβ-containing brain TBS extracts from two other AD patients (101 ± 6%; n = 5; p > 0.05 compared with pre-HFS baseline; p < 0.05 compared with vehicle) (Figs. 2, 3A), was also abrogated by immunodepletion of Aβ with AW8 [131 ± 6% (n = 5) and 139 ± 8% (n = 4), respectively; p < 0.05 compared with pre-HFS baseline and nonimmunodepleted extract; p > 0.05 compared with vehicle; data not shown]. Because the process of Aβ immunodepletion may remove other potentially important factors, we also tested TBS samples of AD brain extract that had been treated with preimmune serum. Such mock-immunodepleted extract still contained abundant SDS-stable Aβ dimer (38 pg of Aβ1–42 equivalent) (Fig. 1A) and strongly inhibited LTP (103 ± 6%, n = 4; p < 0.05 compared with vehicle-injected controls and animals injected with Aβ-immunodepleted samples) (Fig. 1C).
Figure 2.

Control human brain extract did not inhibit LTP. Application of HFS induced robust LTP after acute injection (asterisk, 5 μl) of control brain TBS extract (Non-AD) (n = 4) similar to vehicle-injected rats (n = 8). In contrast, injection of Aβ-containing TBS extract from the brain of another AD patient (AD) completely inhibited LTP at 3 h post-HFS (n = 5). Insets show representative traces at the times indicated. Calibration: 1.5 mV, 10 ms.
Figure 3.
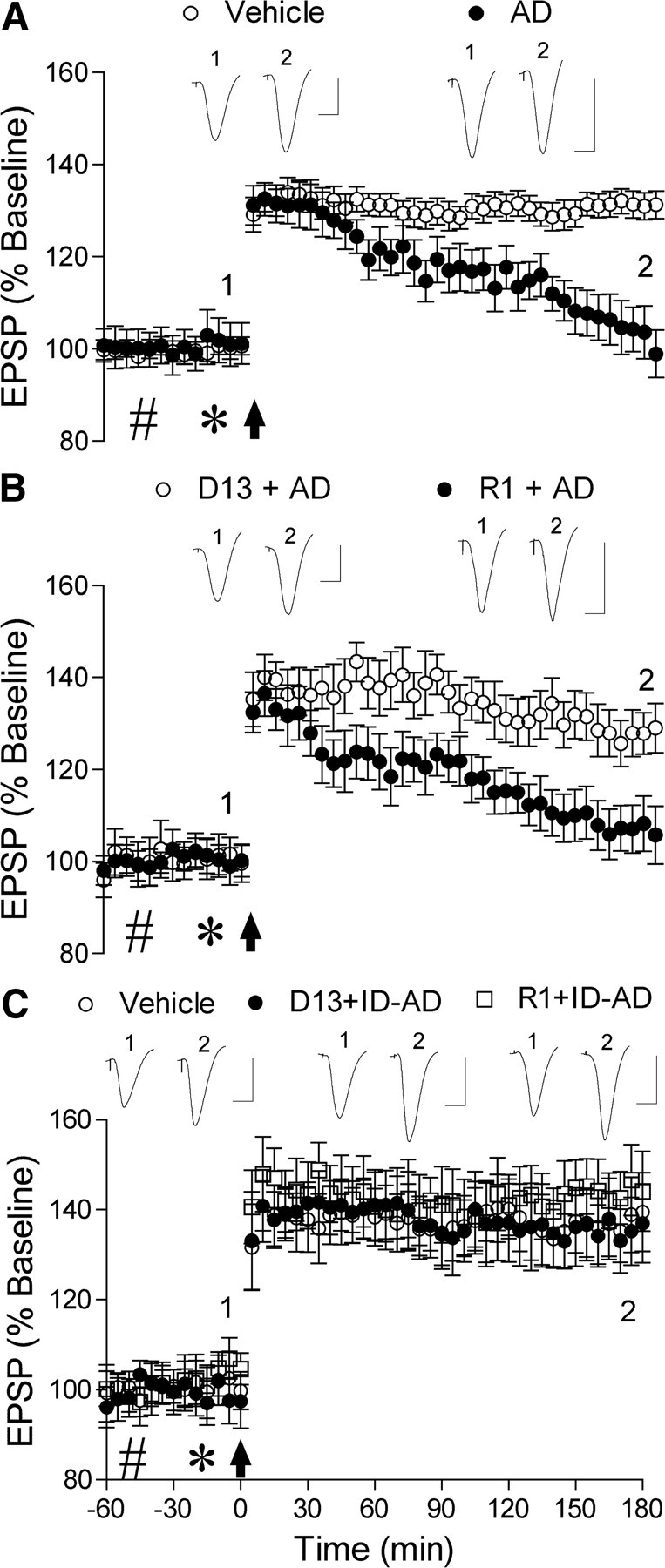
PrPC dependence of human AD brain Aβ-mediated inhibition of LTP. A, Acute injection of Aβ-containing AD brain extract (asterisk, 5 μl) completely inhibited LTP (n = 6) in contrast to animals that received two intracerebroventricular injections of vehicle (n = 5). B, Soluble AD brain Aβ-mediated inhibition of LTP was prevented by the antibody fragment D13 directed to PrPC96–104, but not the Fab R1 directed to PrPC225–231. Preinjection with D13 (11 μg in 10 μl, n = 5) prevented the inhibition of LTP by the brain extract. In contrast, the brain extract completely inhibited LTP in animals pretreated with R1 (11 μg, n = 5). C, Neither D13 nor R1 significantly affected control LTP. Preinjection with D13 (n = 5) or R1 (n = 4) before injection of Aβ-immunodepleted AD brain TBS extract did not significantly affect LTP compared with animals that received two injections of vehicle (n = 5). Insets show representative traces at the times indicated. Calibration: 1.5 mV, 10 ms.
The disruption of synaptic function by the Aβ-containing supernatant was selective for LTP over baseline synaptic transmission since injection of the dose of Aβ that completely inhibited LTP did not significantly affect baseline EPSPs (103 ± 2% at 3 h postinjection; n = 4; p > 0.05 compared with preinjection baseline or compared with 102 ± 2%, n = 4, in vehicle-injected rats) (Fig. 1D). Similarly immunodepleted aqueous brain extract did not significantly affect baseline synaptic transmission (102 ± 2%; n = 4; p > 0.05 compared with preinjection baseline or with vehicle-injected rats).
Importantly, samples of an equivalent TBS extract from the brain of a nondemented control subject, did not contain detectible amounts of soluble Aβ (data not shown) and failed to inhibit LTP (Fig. 2). Thus, HFS induced robust LTP in animals injected (5 μl, i.c.v.) with the control human brain TBS extract (136 ± 9%, n = 4, p < 0.05 compared with pre-HFS baseline; 133 ± 5% n = 8, p > 0.05 compared with vehicle).
PrPC dependence of human AD brain Aβ-mediated inhibition of LTP
Next we investigated the requirement for PrPC, in particular the putative ADDL binding sequence spanning residues 95–105 (Laurén et al., 2009; Chen et al., 2010), in the ability of Aβ-containing AD brain-soluble extract to inhibit LTP. We pretreated animals with the recombinant monovalent antigen-binding Fab fragment D13, which has a high affinity for an epitope 96–104 of PrPC (Williamson et al., 1998). We also studied the effects of the Fab fragment R1, which binds with high affinity to an epitope encompassing amino acids 225–231 (Williamson et al., 1998), which is located at the C-terminus region of PrPC and therefore distal to the putative binding sites for ADDLs (Laurén et al., 2009; Chen et al., 2010).
Injection of water-soluble, Aβ-containing AD brain extract (583 pg of Aβ1–42 equivalent in 5 μl) 15 min before HFS completely inhibited LTP measured at 3 h (101 ± 4%, n = 6, p > 0.05 compared with pre-HFS baseline; p < 0.05 compared with 131 ± 2%, n = 13, in animals that received two injections, 10 μl followed 15 min later with 5 μl, of vehicle) (Fig. 3A). Similarly, the Aβ-containing AD brain extract also completely inhibited LTP in animals preinjected with the PrPC225–231-binding Fab R1 (11 μg) (107 ± 6, n = 5; p > 0.05 compared with baseline; p < 0.05 compared with vehicle injections) (Fig. 3B). In marked contrast, preinjection of the PrPC96–104-binding Fab, D13 (11 μg in 10 μl) fully abrogated the inhibition of LTP by the human brain extract. Thus, HFS induced robust LTP (128 ± 2%, n = 5; p < 0.05 compared with baseline; p > 0.05 compared with vehicle-injected controls) in animals administered the Aβ-containing extract 30 min after intracerebroventricular injection of D13 (Fig. 3B). Importantly, neither D13 (11 μg) nor the control Fab R1 (11 μg) injected before immunodepleted AD brain extract (5 μl) significantly affected LTP (136 ± 7, n = 5, and 145 ± 9%, n = 4, respectively; p < 0.05 compared with baseline; p > 0.05 compared with 139 ± 8% in vehicle-injected controls n = 5) (Fig. 3C) at the dose used to investigate the role of PrPC in mediating the disruption of synaptic plasticity by Aβ-containing AD extract.
Discussion
The present findings strongly support and extend the proposal that the potent and selective disruption of synaptic plasticity by soluble Aβ assemblies is mediated through PrPC. Whereas previous research had focused on the effects of synthetic Aβ assemblies in the mouse in vitro, we provide convincing in vivo evidence in the rat that PrPC is necessary for the inhibition of LTP of hippocampal synaptic transmission by Aβ-containing AD brain extract. Moreover, consistent with a key role of a direct interaction of Aβ with the putative binding site in the 95–105 region of PrPC, we report that blocking PrPC96–104, but not the C terminus of PrPC was effective in preventing the inhibition of LTP.
Aqueous buffer-soluble extract of the cerebral cortex from AD patients that contained readily detectible SDS-stable dimer powerfully disrupted synaptic plasticity in vivo, consistent with our previous in vitro findings (Shankar et al., 2008). In these and the present studies, immunodepletion of Aβ effectively abrogated the inhibition of LTP, providing strong evidence for a critical role of Aβ. Here we found further support for this conclusion by the ability of mock-immunodepleted samples to fully inhibit LTP. Similar to our previous in vitro studies (Shankar et al., 2008), equivalent extract from the cortex of a non-AD control subject did not contain such soluble Aβ and did not inhibit LTP in vivo. The present findings are also consistent with our previous report that SDS-stable Aβ dimer-containing CSF from living human volunteers inhibited LTP in vivo, whereas samples that only contained detectible monomer were inactive (Klyubin et al., 2008).
The finding that PrPC is necessary for the inhibition of LTP in vivo by Aβ-containing AD brain TBS extract is consistent with the discovery that soluble synthetic Aβ assemblies bind with high affinity to PrPC to trigger disruption of synaptic plasticity in vitro (Laurén et al., 2009). However, Kessels et al. (2010) reported that synthetic Aβ oligomers strongly inhibited LTP in hippocampal slices from Prnp−/− mice. This conflict underlines a major difficulty with preparing equivalent preparations of synthetic Aβ oligomers. Synthetic Aβ preparations vary greatly in their composition even when great care is taken to follow rigid protocols to produce specific assemblies (Hepler et al., 2006). Unlike the synthetic Aβ oligomer preparation used by Kessels et al. (2010), which also caused a marked reduction in baseline excitatory synaptic transmission, the Aβ-containing AD brain extract selectively inhibited LTP in vivo. Indeed, the vast majority of previous reports found that considerably higher concentrations of synthetic Aβ are necessary to significantly reduce baseline excitatory synaptic transmission under the same conditions both in vivo and in vitro (Cullen et al., 1997; Shankar and Walsh, 2009), and it is likely that such higher levels act via additional sites, bypassing the need for an interaction with PrPC. Also in apparent conflict with the present findings and those of Laurén et al. (2009), in hippocampal slices from 4-month-old APPPS1+ mice crossed with Prnp−/− mice LTP was impaired to the same extent as in slices from age-matched pure APPPS1+ mice. However, at this age there is already extensive deposition of Aβ in plaques in the brain of this strain of mice (Grathwohl et al., 2009), and it is uncertain whether Aβ oligomers play a role in the observed impairment of synaptic plasticity seen in such mice.
That D13, an antibody fragment that binds PrPC96–104, prevented the inhibition of LTP by soluble AD brain extract, whereas R1, an antibody fragment that binds the C terminus of PrPC, failed to significantly alter the inhibition, points to the key role of the ADDL-binding sequence in PrPC (amino acids 95–105) identified by Laurén et al. (2009). Indeed, Laurén et al. (2009) found that a polyclonal antibody to the C-terminal part of PrPC failed to affect ADDL binding to cultured cells, whereas two antibodies to the region shared with D13 prevented binding. Chen et al. (2010) confirmed the importance of this region of PrPC but also found evidence that the N terminus of PrPC may act as an ancillary binding site for ADDLs.
Laurén et al. (2009) found that PrPC accounted for ∼50% of high-affinity binding sites on cultured hippocampal neuron cell membranes, which has been confirmed by Renner et al. (2010). Thus, in their open screen Laurén et al. (2009) reported that other sites included APLP1, and Renner et al. (2010) provided indirect evidence that PrPC binding by Aβ oligomers was colocalized with both mGlu5 (glutamate metabotropic subtype 5) and NMDA receptors. In the latter case, Aβ oligomer binding caused the accumulation of Aβ at synapses and aberrant changes in the membrane distribution of glutamate receptors. It was suggested that Aβ oligomers can act as an extracellular scaffold promoting the cross-linking of glutamate receptors and leading to disruption of synapses apparently by the formation of ectopic signaling platforms at the plasma membrane (Renner et al., 2010). Interestingly, PrPC is found in lipid raft-like domains of the plasma membranes, as are glutamate receptors (Francesconi et al., 2009; Kellett and Hooper, 2009; Delint-Ramirez et al., 2010), and aggregation of Aβ oligomers in such regions has been strongly implicated in mediating neurotoxicity (Zampagni et al., 2010).
How the present findings implicating PrPC in the synaptic plasticity-disrupting actions of Aβ in vivo relate to behavioral and cognitive changes remain to be determined. Intriguingly, crossing APP transgenic mice (APPPSde9) with Prnp−/−-null mice abrogated Aβ-associated spatial memory deficits (Gimbel et al., 2010), and systemic treatment with the anti- PrPC antibody 6D11 rapidly reversed impairment of radial maze learning in these mice (Chung et al., 2010). However, a rapid Aβ-mediated disruption of object recognition learning was not apparently reduced in Prnp−/− mice (Balducci et al., 2010), making it important to determine whether cognitive impairment caused by human brain-derived soluble Aβ is dependent on PrPC.
Footnotes
This work was supported by funding from Science Foundation Ireland (Grant 08/IN.1/B2033 to D.M.W., and Grants 06/IN.1/B88 and 10/IN.1/B3001 to M.J.R.), the Health Research Board (D.M.W. and M.J.R.), and the European Commission Seventh Framework Programme under Grant Agreements 200611 (D.M.W.) and 201159 (M.J.R.).
References
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Gall CM, Zhou J, Lynch G. Uptake and pathogenic effects of amyloid beta peptide 1-42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience. 2002;112:827–840. doi: 10.1016/s0306-4522(02)00132-x. [DOI] [PubMed] [Google Scholar]
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role of N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by β-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Deane R., Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Delint-Ramirez I, Fernández E, Bayés A, Kicsi E, Komiyama NH, Grant SG. In vivo composition of NMDA receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J Neurosci. 2010;30:8162–8170. doi: 10.1523/JNEUROSCI.1792-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi A, Kumari R, Zukin RS. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J Neurosci. 2009;29:3590–3602. doi: 10.1523/JNEUROSCI.5824-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, De Bona P, Pappalardo G, Nicoletti F, Rizzarelli E, Copani A. The monomer state of beta-amyloid: where the Alzheimer's disease protein meets physiology. Rev Neurosci. 2010;21:83–93. doi: 10.1515/revneuro.2010.21.2.83. [DOI] [PubMed] [Google Scholar]
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, Kinney G, Joyce JG. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- Kellett KA, Hooper NM. Prion protein and Alzheimer disease. Prion. 2009;3:190–194. doi: 10.4161/pri.3.4.9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–E4. doi: 10.1038/nature09217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of A beta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, Kimata K, Wight TN. Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the beta-amyloid protein of Alzheimer's disease. Arch Biochem Biophys. 1995;320:84–95. doi: 10.1006/abbi.1995.1345. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. Mapping the prion protein using recombinant antibodies. J Virol. 1998;72:9413–9418. doi: 10.1128/jvi.72.11.9413-9418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S, Malinin NL, Powell KA, Yednock T, Rydel RE, Griswold-Prenner I. Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiol Aging. 2007;28:226–237. doi: 10.1016/j.neurobiolaging.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22(RC221) doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Bierhaus A, Nawroth PP, Stern DM. RAGE and Alzheimer's disease: a progression factor for amyloid-beta-induced cellular perturbation? J Alzheimers Dis. 2009;16:833–843. doi: 10.3233/JAD-2009-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampagni M, Evangelisti E, Cascella R, Liguri G, Becatti M, Pensalfini A, Uberti D, Cenini G, Memo M, Bagnoli S, Nacmias B, Sorbi S, Cecchi C. Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J Mol Med. 2010;88:597–608. doi: 10.1007/s00109-010-0603-8. [DOI] [PubMed] [Google Scholar]