

Abstract
Neuronal precursors are continuously integrated into the adult olfactory bulb (OB). The vast majority of these precursor cells originates from the subventricular zone and migrates along the rostral migratory stream (RMS) en route to the OB. This process, called postnatal neurogenesis, results from intricate pathways depending both on cell-autonomous factors and extrinsic regulation provided by the local environment. Using electroporation in postnatal mice to label neuronal precursors with green fluorescent protein (GFP) and to reduce the expression levels of doublecortin (DCX) with short-hairpin (Sh) RNA, we investigated the consequences of impairing migration on the fate of postnatal-formed neurons. First, we showed that electroporation of Dcx ShRNA plasmid efficiently knocks down the expression of DCX and disrupts cells migration along the RMS. Second, we found misplaced anomalous migrating cells that displayed defects in polarity and directionality. Third, patch-clamp recordings performed at 5–7 days post-electroporation (dpe) revealed increased density of voltage-dependent Na+ channels and enhanced responsiveness to GABAA receptor agonist. At later time points (i.e., 12 and 30 dpe), most of the Dcx ShRNA+ cells developed in the core of the OB and displayed aberrant dendritic length and branching. Additional analysis revealed the formation of GABAergic and glutamatergic synaptic inputs on the mispositioned neurons. Finally, quantifying fate determination by numbering the proportion of GFP+/calretinin+ newborn neurons revealed that Dcx ShRNA+ cells acquire mature phenotype despite their immature location. We conclude that altering the pace of migration at early stages of postnatal neurogenesis profoundly modifies the tightly orchestrated steps of neuronal maturation, and unveils the influence of microenvironment on controlling neuronal development in the postnatal forebrain.
Introduction
The subventricular zone (SVZ), and to a lesser extent the rostral migratory stream (RMS) of the mammalian forebrain, contains neural progenitors that proliferate and migrate en route to the olfactory bulb (OB) throughout life (Altman, 1969). In the OB, postnatally generated neuroblasts differentiate into two types of local interneurons: granule cells (GCs) and periglomerular cells (Luskin, 1993; Lois and Alvarez-Buylla, 1994). The complex mechanism by which neural precursors become an OB interneuron is not fully deciphered. However, we know that a precisely orchestrated series of morpho-functional changes is requested to ensure the recruitment of neural precursors into their proper layers and, later, for developing correct connectivity with the host circuit (Petreanu and Alvarez-Buylla, 2002; Belluzzi et al., 2003; Carleton et al., 2003).
In the postnatal forebrain, the molecular signals that contribute to migration include polysialylated neural cell adhesion molecule (Chazal et al., 2000), ephrin-B (Conover et al., 2000), members of the Slits (Nguyen-Ba-Charvet et al., 2004; Kaneko et al., 2010) and integrin (Belvindrah et al., 2007) families, prokineticin 2 receptors (Ng et al., 2005), and ambient GABA (Lledo et al., 2006; Pathania et al., 2010). In addition, reelin and tenascin-R are two proteins that trigger the detachment of neuroblasts from the RMS (Hack et al., 2002; Saghatelyan et al., 2004). However, despite extensive investigation of the molecular signals regulating the migration of neuroblasts from the SVZ and controlling their arrival to the bulb, it remains poorly understood how much their innate developmental program impacts their ultimate functional identity.
During embryogenesis a widely accepted model proposes that external factors act in concert with cell-autonomous processes to ensure the correct neuronal maturation and connectivity of neuronal precursors (Ben-Ari and Spitzer, 2010). Extending this hypothesis to OB adult neurogenesis, our previous studies have shown that developing neurons receive synaptic inputs early, just after arrival into the bulbar circuit (Carleton et al., 2003; Panzanelli et al., 2009), thus implying developmental regulation by both cell-autonomous factors and synaptic activity (Ming and Song, 2005; Adam and Mizrahi, 2010).
Here, we investigated whether early alteration of neuroblast migration impacts morphological and functional maturation. Because newly formed neurons in the postnatal forebrain express neurotransmitter receptors, we hypothesized that initial changes in the local environment surrounding the migrating neuroblasts might impair the subsequent developmental stages leading to long-lasting alteration of cell fate. By reducing the expression levels of a protein that regulates the organization and stability of microtubules [e.g., doublecortin (DCX)], we found changes to cell targeting, dendritic development, and fate determination. Surprisingly, despite aberrant localization and cell morphology, DCX-deficient neuroblasts developing in ectopic positions receive GABAergic and glutamatergic synaptic inputs earlier when compared with their control counterparts in similar locations. Thus, variations in the microenvironment of developing postnatal-generated neurons have profound and long-lasting effects on their future function.
Materials and Methods
Animals.
C57BL/6J mice used in this study were obtained from Janvier. Animals were maintained in standard conditions (12 h light/dark cycle, ad libitum access to dry food and water). All the experimental procedures were accomplished according to the European Community Council Directives (86/609/EEC) and European Union General Guidelines. Those procedures were reviewed and approved by the animal welfare committee of the Pasteur Institute.
In vivo electroporations.
Postnatal day 3 (P3) to P4 mouse pups were first anesthetized on ice for 3 min. Then, they were maintained on a 4°C cold pad during the injection procedure. Mice were injected with the different plasmids directly into the lateral ventricle by transluminescence with an optical fiber (Leica). Approximately 2 μl of plasmid solutions at a concentration of 5 μg/μl (in endotoxin-free TE buffer; Qiagen) were injected with a 3.5 inch glass pulled pipette (Drummond) connected to a 15 μl Hamilton syringe. Then, the brain was placed between a 10 mm Tweezertrode, with the positive pole oriented on the injected side. Five, 50 ms square-wave pulses of 100 V, with a latency of 950 ms, were given with a BTX ECM 830 Electroporator (Harvard Apparatus). Reanimated mice were kept warm under a heating lamp and quickly returned to their mother after the procedure. Electroporated plasmids were as follows. Dcx-GFP, the minimal promoter for mouse Dcx gene (2 kb upstream genomic sequence of the Dcx gene) (Wang et al., 2007), was amplified by genomic PCR and cloned upstream to the GFP sequence from the EGFP-C1 plasmid (Clontech); pCig2-ires-GFP plasmid [CMV-enhancer/chicken β-actin promoter-GFP (CMV-GFP)] (kind gift from F. Polleux, The Scripps Research Institute); Dcx short-hairpin (Sh) RNA plasmid was produced from the SureSilencing ShRNA plasmid (SABiosciences Qiagen, Frederick, MD), where the following sequence, 5′-TCACCTGTCTCCATGATTTCT-3′ (targeting specifically dcx mRNA), was subcloned in the pGeneClip vector downstream to the U1 promoter to express the ShRNA. For control experiments related to migration studies, the same SureSilencing ShRNA plasmid was used with a scrambled noncoding sequence (5′-GGAATCTCATTCGATGCATAC-3′) instead of the dcx sequence, and no defect was observed. For long-term experiments (i.e., 30 dpe), the activity of Dcx promoter being repressed ∼15 d after neuronal precursor proliferation (Brown et al., 2003), pCig2-ires-GFP plasmid was used instead and mixed with Dcx ShRNA plasmid to a 1:1 ratio at a final concentration of 5 μg/μl for each vector.
Immunostaining and analysis.
For studies in mice at 5, 7, and 12 days post-electroporation (dpe), animals were anesthetized with isoflurane (Mundipharma) and rapidly decapitated, and brains were fixed in 4% paraformaldehyde overnight at 4°C. Then, dissected brains were embedded in 3.5% agarose (Invitrogen) and 8% sucrose. Sagittal sections (70 μm) were prepared using a VT1000S Vibratome (Leica), and immunostaining was performed on floating sections in 24-well plates. Sections were incubated with PBS supplemented with 10% bovine serum (Invitrogen) and 0.2% Triton X-100 (blocking buffer) for 1 h at room temperature, and incubated overnight at 4°C with the following antibodies in blocking buffer: rabbit polyclonal anti-DCX (Ab18723, Abcam), rabbit polyclonal anti-GFP (Invitrogen), chicken polyclonal anti-GFP (Millipore), rabbit polyclonal anti-calretinin (ab5054, Millipore), rabbit polyclonal anti-VGAT (anti-vesicular GABA transporter) (Millipore, AB5062P), and rabbit polyclonal anti-VGLUT1 (anti-vesicular glutamate transporter 1) (kind gift from S. El Mestikawy, INSERM, University Paris 6). Then, sections were rinsed three times with PBS during 10 min and incubated at room temperature during 2 h with secondary antibodies coupled to Alexa 488 or Alexa 568 (Invitrogen). For biocytin staining, an amplification with the Vectastain ABC kit PK 6100 (Vector Laboratories) was performed before incubation with Alexa 568-conjugated streptavidin (Invitrogen). Nuclei were counterstained for 10 min at room temperature with 4′,6′-diamidino-2-phenylindole dihydrochloride (1 μg/ml; Sigma-Aldrich). Sections were rinsed with PBS and mounted with ProLong Gold anti-fade mounting medium (Invitrogen). Stainings were analyzed with a confocal microscope (LSM 700, Zeiss) and, for synaptic markers (VGAT and VGLUT1), analyzed with Imaris software (Bitplane) for three-dimensional (3D) rendering and synaptic contact quantifications (all values are given as mean ± SEM, and a Student's t test was performed).
To quantify the neuronal polarity during migration, neuroblasts were categorized according to the number of processes: type I, single leading process; type II, bipolar neuroblast; or type III, multipolar cell. Countings were expressed as a percentage of total number of GFP+ cells in three different regions (RMS1, RMS2, and core of the OB). Three contiguous slices per animal were analyzed and six animals per condition were studied. All values are given as mean ± SEM. A Student's t test was used. To quantify the directionality of the migration, we estimated the angle between the longest leading processes of a migrating neuron along with the general direction of the tangential migration in the core OB (baseline) with Axiovision software (Zeiss). Then, cells were categorized according to the value of that angle: 0–45°, tangential; 45–90°, radial I; 90–135°, radial II; 135–180°, radial III. Values are expressed as a percentage of total analyzed GFP+ cells in the core of the OB. Three contiguous slices per animal were analyzed and five animals per condition were studied. All values are given as mean ± SEM. A Student's t test was performed. Because there was some variability between electroporation experiments, we quantified the neuronal differentiation as a ratio with the GFP+ cells, then taking into account the efficiency of electroporation. To count double-labeled GFP+/calretinin+ neurons, we used the QUIA Bio Image Analysis software (F. de Chaumont and J. C. Olivo-Marin, http://www.bioimageanalysis.org/index.html, Institut Pasteur, Paris France) based on a B3 wavelet-filtering unit. Briefly, after RMS and GC layer (GCL) external limit delineation, automatic counting was performed in concentric homogenous areas. Values are given as a percentage of GFP+ cells or double-labeled GFP+/calretinin+ cells. Three contiguous slices per animal were analyzed and six animals per condition were studied. All values are given as mean ± SEM, and a Student's t test was used. Finally, to quantify the length of the primary apical dendrite and the number of branchings per neuron, LSM Image Browser software (Zeiss) was used. Five animals per condition were studied and 50 cells in total for each condition were analyzed. All values are given as mean ± SEM, and a Student's t test was used.
Sholl analysis.
To quantify the complexity of the dendritic arborization at 12 and 30 dpe, we measured for each neuron the number of intersections of concentric circles (from the soma) with the dendrites. Confocal micrographs were analyzed using NIH ImageJ and Sholl analysis plug-in from the A. Ghosh laboratory (University of California, San Diego, La Jolla, CA). Values are expressed as mean ± SEM. Five animals per condition were studied and 50 cells in total for each condition were analyzed. A Student's t test was performed to compare control versus Dcx ShRNA+ populations.
Electrophysiological recordings.
For patch-clamp recordings, mice were deeply anesthetized with isoflurane (Mundipharma) and swiftly decapitated. The right hemispheres were rapidly dissected and placed in ice-cold artificial CSF (ACSF) (containing, in mm: 124 NaCl, 3 KCl, 1.3 MgSO4, 26 NaHCO3, 1.25 NaHPO4, 20 glucose, 2 CaCl; ∼310 mOsm, pH 7.3 when bubbled with a mixture of 95% O2 and 5% CO2; all chemicals from Sigma), before being placed, submerged in ice-cold ACSF, in the cutting chamber of a Leica Vibratome (VT 1200S). Sagittal slices (250 μm thick) of the OB were placed in bubbled ACSF in a warming bath at 35°C for 30 min and then at room temperature. For whole-cell recordings, individual slices were placed in a chamber mounted on a Zeiss Axioskop upright microscope, and continuously perfused (1.5 ml/min) with room temperature bubbled ACSF. Slices were visualized using a 40× water-immersion objective.
We obtained whole-cell patch-clamp recordings from visually targeted GFP+ neuroblasts or neurons. Patch pipettes, pulled from borosilicate glass (outer diameter 1.5 mm, inner diameter 1.17 mm, Harvard Apparatus; P-87 Flaming/Brown micropipette puller, Sutter Instrument), had resistances of 8–12 MΩ and were filled with a K-gluconate-based solution (in mm: 130 K-gluconate, 6 KCl, 2 NaCl, 10 Na-HEPES, 10 d-glucose, 0.2 Cs-EGTA, 0.3 GTP, 2 Mg-ATP, 0.2 cAMP, with 0.15% biocytin; 280–290 mOsm, pH 7.3). Rhodamine (0.1–0.4%) was included in the internal solution for GFP+ cell recordings. All voltages reported in the text are corrected for a liquid junction potential of +10 mV between our internal and external solutions. Recordings were obtained via an Axopatch 1D amplifier (Molecular Devices). Signals were Bessel filtered at 1–10 kHz, digitized, and sampled at intervals of 20–450 μs (2.2–20 kHz) according to the individual protocols.
Newly formed cells were identified either by double-labeling with both GFP and rhodamine during recording, or by the presence of GFP in the tip of the patch pipette after membrane rupture. GFP-targeted recordings that did not meet either of these criteria were discarded from our analyses. Cell membrane capacitance (Cm) and cell membrane resistance (Rm) were measured at −70 mV using a 5 mV voltage step. The Na+ and K+ currents were both measured under voltage-clamp conditions. Depolarizing pulses (100 ms) from −70 mV by incremental steps (5 mV), up to +10 mV, were given at a rate of 1 Hz. Na+ and K+ currents were measured after subtraction of scaled passive current responses to the appropriate voltage steps.
Before applying muscimol (25 μm) or kainic acid (50 μm), we recorded a 3 min stable baseline. We applied agonists until peak currents were reached (2–3 min) and quickly washed out. Spontaneous and evoked postsynaptic currents were analyzed with ELPHY software (Gérard Sadoc, Gif-sur-Yvette, France). To evoke postsynaptic currents, we used a small monopolar glass pipette (3–4 μm tip diameter, pulled from 1.5 mm outer diameter and 1.17 mm inner diameter borosilicate glass, and filled with ACSF), finely positioned using a micromanipulator. Stimulation intensities (100–200 μA, 100 μs) were set to obtain maximal responses. Amplitude and paired-pulse ratios (amp2nd/amp1st) were measured from averaged traces (10–30 traces). GABAergic postsynaptic currents were recorded at 0 mV and AMPAR-mediated EPSCs were recorded at −70 mV.
Results
The goal of this study was to investigate how early defects in neuroblast migration impacts cell fate. We used electroporation to deliver plasmid constructs encoding ShRNA (Yu et al., 2002) and GFP into neural progenitors located at the ventricular zone surface in 4-d-old mice. This method restricts, both in space and time, transfection to a subset of postnatal neuronal precursors. Because DCX is a key family member of the microtubule-associated proteins that pattern neuronal migration, both in the developing neocortex (Bai et al., 2003) and during adult neurogenesis (Kappeler et al., 2006; Koizumi et al., 2006), we chose a hairpin plasmid targeting specifically a region of the DCX coding sequence to alter neuronal migration.
Short-hairpin RNA targeting DCX impairs RMS neuroblast migration
Precursors cells in the SVZ give rise to neuroblasts that migrate rostrally to reach the OB, where they become mostly GABAergic interneurons (Lledo et al., 2006). Consistent with earlier findings (Brown et al., 2003), Figure 1A–C shows that postnatal-generated neuroblasts express DCX when migrating tangentially from the SVZ to the OB, or radially from the core of the OB to the distinct superficial layers. While a large number of labeled cells are distributed along the SVZ-RMS pathway at early time points (i.e., 5 dpe) (Fig. 1A), GFP+ cells also started populating the OB 2 d later (Fig. 1B), where they scattered throughout all layers at 12 dpe (Fig. 1C) while vacating the RMS.
Figure 1.

Electroporation model in postnatal mouse brain and validation of doublecortin knockdown. A–C, P4 electroporated mice with Dcx-GFP construct, analyzed with sagittal sections at 5, 7, and 12 dpe. Note the increasing number of radially migrating cells in the OB over time. D, E, Double-staining GFP (green)/dcx (red) in sagittal sections of 7 dpe mouse brain in RMS2 region after electroporation of control (D) or Dcx ShRNA (E) plasmid. F–H, High-magnification picture of double-staining GFP (green)/DCX (red) of 7 dpe mouse brain in RMS2 region after electroporation of control. The arrowhead indicates the double-labeled leading process. I, Quantification of double-labeled cells GFP+/DCX+ at three different levels of the rostrocaudal axis of the RMS (RMS1, RMS2, and the core OB) showing a strong reduction of doublecortin expression in GFP+ cells after Dcx ShRNA plasmid electroporation at 7 dpe. Note that the highest level of doublecortin knockdown is in the core OB (***p < 0.001). Scale bars: A, 210 μm, inset, 72 μm; D, 105 μm; F, 43 μm.
Because mutation in the DCX gene impairs neuroblast migration in the developing brain, leading to X-linked lissencephaly (Francis et al., 1999; Gleeson et al., 1999a), we hypothesized that knocking down DCX protein levels in RMS neuroblasts might impair the microtubule dynamics and thus neuroblast migration. Transfected cells with the control plasmid construct (Dcx-GFP) exhibit the previously described chain-like organization, with migrating RMS neuroblasts (Lois and Alvarez-Buylla, 1994) with elongated somas extending a single leading process pointing along the cell's direction of migration (Fig. 1D,F–H). In sharp contrast, when SVZ neural precursors were transfected with Dcx ShRNA, the organization of tangential migration was dramatically changed, with a strong reduction of cells migrating in parallel (Fig. 1E). To validate our approach, we processed 7 dpe Dcx ShRNA-electroporated brains for DCX immunocytochemistry (Fig. 1D–F). As expected, the percentage of transfected cells (i.e., GFP+) that were also immunoreactive for DCX protein decreased in Dcx ShRNA-transfected cells throughout the entire SVZ-OB pathway, with a stronger effect more rostrally (i.e., core OB in Fig. 1I) (RMS1, 93.9 ± 2.3% for Dcx-GFP+ and 49.5 ± 3.0% for Dcx ShRNA+; RMS2, 97.0 ± 1.2% for Dcx-GFP+ and 37.8 ± 1.8% for Dcx ShRNA+; core OB, 94.1 ± 0.8% for Dcx-GFP+ and 24.7 ± 2.6% for Dcx ShRNA+). This result validated the efficiency of our ShRNA approach at decreasing DCX protein expression levels in migrating postnatal-generated neuroblasts.
High-magnification examination of the forebrain (Fig. 2A,B) confirmed that in control transfections, cells migrated in parallel, both in the caudal (RMS1) and central (RMS2) part of the RMS (Fig. 2C,D), before detaching from the chains in the core of the OB (Fig. 2E). In contrast, while Dcx ShRNA+ migrating neuroblasts were still organized in parallel in the RMS1 (Fig. 2F), this organization was strongly disrupted both in the RMS2 (Fig. 2G) and in the core of the OB (Fig. 2H). Thus, we conclude that a normal DCX expression level is required to maintain migrating RMS neuroblasts in a parallel and compact organization.
Figure 2.

Altered neuronal migration of Dcx ShRNA+ cells at 7 dpe. A, B, Low-magnification picture of sagittal brain sections after electroporation with Dcx-GFP construct or Dcx ShRNA plasmid followed by anti-GFP immunostaining. C–H, Representative examples of GFP+ cells at three defined levels along the rostrocaudal axis of the RMS after control and Dcx ShRNA plasmid electroporation: RMS1 (C, F), RMS2 (D, G), core OB (E, H). Note that the migration is the most altered in the core OB. Scale bars: A, 210 μm; C, 115 μm.
Low levels of DCX impair cell polarity and directionality of neuronal migration
To further investigate the cellular consequences of reducing DCX expression, we analyzed the morphology of migrating neuroblasts in the forebrain at 7 dpe. Throughout the entire SVZ-OB system, control migrating Dcx-GFP+ neuroblasts were mostly monopolar with elongated soma extending a single leading process (Fig. 3A, type I). Rarely, we could see bipolar morphology (Fig. 3A, type II) and we almost never observed multipolar neuroblasts (Fig. 3A, type III), except a few in the core of the OB (Dcx-GFP7dpe+: RMS1, 94.2 ± 3.0% for type I; 5.7 ± 3.0% for type II; RMS2, 79.6 ± 1.4% for type I; 19.1 ± 2.5% for type II and 1.2 ± 0.2% for type III; Core OB, 64.8 ± 3.1% for type I; 33.5 ± 3.7% for type II; and 1.6 ± 0.3% for type III). In contrast, in all areas there was a significant decrease in Dcx ShRNA+ neuroblasts with monopolar morphology, and a significant increase in the proportion of neuroblasts with bipolar morphology (Dcx ShRNA7dpe+: RMS1, 71.2 ± 4.0% for type I; 28.7 ± 4.0% for type II; RMS2, 62.3 ± 3.9% for type I; 36.5 ± 3.5% for type II and 1.7 ± 0.3% for type III; core OB, 33.7 ± 2.4% for type I; 61.0 ± 2.4% for type II; and 5.1 ± 0.5% for type III). This result demonstrates that DCX expression is required for the morphology and polarity of neuroblasts during RMS migration in the postnatal forebrain.
Figure 3.

Impaired polarity and directionality during neuroblast migration after Dcx ShRNA plasmid electroporation at 7 dpe. A, Categorization of migrating neuroblasts according to their morphology, following the three predefined levels of the rostrocaudal axis of the RMS (RMS1, RMS2, and core OB): type I, single leading process, type II, bipolar shape, and type III, multipolar process neuroblasts. Note the increased proportion of type II migrating neuroblasts at the three levels (RMS1, RMS2, core OB) after electroporation with Dcx ShRNA plasmid. B, Quantification of directionality of the neuroblast migration after Dcx ShRNA plasmid electroporation in the core OB at 7 dpe. Migrating neuroblasts have been categorized (tangential, radial I, II, and III) according to the angle defined by the orientation of the longest neuronal leading process along with the tangential migration directionality. The yellow dashed line represents the baseline directionality defined by the tangential migration in the core OB. Note the increase of the radial I and II population (red arrows) vs tangential population (yellow arrows) after electroporation with Dcx ShRNA plasmid (*p < 0.05, **p < 0.01). Scale bar: B, 90 μm.
Next, we sought to investigate whether these morphological changes could alter the directionality of migration. To address this point, we quantified the direction of migrating neuroblasts by measuring the angle between the longest leading process and the central axis passing through the core of the OB (depicted by the dotted yellow lines in Fig. 3B). The angle values were used here to define the directionality of migration. As expected for the control condition, the vast majority of neuroblasts migrate in parallel to the central axis (89.5 ± 5.8%), while the rest (10.4 ± 2.5%) formed a 45–90° angle with the central axis (Fig. 3B, radial I). In Dcx ShRNA+ neuroblasts, the proportion of navigating cells in the radial I mode was dramatically increased at the expense of the number of tangentially migrating cells (55.1 ± 7.9% for tangential and 34.4 ± 5.7% for radial I). Some of the Dcx ShRNA+ neuroblasts were even located in the 90–135° sector (Fig. 3B, 10.3 ± 2.4% in radial II), where we never observed control neuroblasts. From these results, we conclude that migration directionality was significantly altered in Dcx ShRNA+ neuroblasts, at least when analyzed at 7 dpe, leading to mispositioned cells in the central portion of the RMS and the core of the OB.
Altered electrophysiological maturation of mutant neural cells
Because a large number of Dcx ShRNA+ neuroblasts were found scattered throughout the RMS-OB pathway (Fig. 3B), we wondered whether developing neuroblasts in ectopic locations would mature properly and would receive synaptic inputs. We investigated the precise developmental stages of the electrophysiological properties of RMS neuroblasts located in the three distinct regions of acute forebrain slices (Fig. 4A). Patch-clamp recordings, at 5–7 dpe, revealed that the membrane capacitance, membrane resistance, and maximum density of voltage-gated K+ currents all remained unchanged in Dcx ShRNA+ neuroblasts (Table 1; Fig. 4A). In contrast, the density of voltage-dependent Na+ channels was increased in Dcx ShRNA+ neuroblasts recorded in the core of the OB (Table 1; Fig. 4A). Furthermore, responsiveness to bath application of a GABAA receptor agonist, muscimol (25 μm), was increased twofold in Dcx ShRNA+ cells located in the RMS2 and the core of the OB (Table 1; Fig. 4B). This enhanced response to muscimol was specific to GABAA receptors, since the glutamate responsiveness remained constant (Table 1; Fig. 4C). These results show that altering neuronal migration by decreasing DCX expression levels influences the early functional maturation of neuroblasts.
Figure 4.

Early functional maturation is modified in Dcx ShRNA+ newborn cells (5–7 dpe). A, Electrophysiological properties of newly generated cells were assessed by whole-cell patch-clamp recordings in three different regions with respect to the rostrocaudal axis (RMS1, RMS2, core OB). Membrane capacitance (Cm), membrane resistance (Rm), and maximum density of voltage-dependent K+ channel-mediated currents (IK+) were measured. No significant differences were observed between control and Dcx ShRNA+ cells. In contrast, the maximum density of voltage-dependent Na+ channels (INa+) was significantly increased in Dcx ShRNA+ cells located in the core of the OB. B, GABA receptor (GABA-R)-mediated peak current amplitudes (IGABA) were measured after bath application of muscimol (25 μm) at 0 mV. Calibration: 20 s, 10 pA. C, Non-NMDA glutamate receptor (GluR)-mediated peak current amplitudes (IGLUT) were measured after bath application of kainic acid (50 μm) at −60 mV. Note that the GABA-R-mediated current is significantly increased in Dcx ShRNA+ cells in the RMS2 and in the core of the OB, although the amplitude of GluR-mediated currents remains constant. All cells were recorded between 5 and 7 dpe. Groups were compared 2 by 2 using a Mann–Whitney test (**p < 0.01, ***p < 0.001). Calibration: 2 min, 10 pA.
Table 1.
Functional properties of Dcx-GFP+ or Dcx ShRNA+ cells along the RMS (5–7 dpe)
| RMS1 |
RMS2 |
Core OB |
||||
|---|---|---|---|---|---|---|
| Dcx-GFP+ | Dcx ShRNA+ | Dcx-GFP+ | Dcx ShRNA+ | Dcx-GFP+ | Dcx ShRNA+ | |
| Cm (pF) | 3.6 ± 0.3, n = 7 | 3.9 ± 0.5, n = 5 | 3.4 ± 0.2, n = 10 | 3.0 ± 0.3, n = 6 | 4.1 ± 0.9, n = 21 | 3.5 ± 0.2 n = 18 |
| Rm (GΩ) | 3.2 ± 0.5, n = 6 | 4.0 ± 0.9, n = 5 | 4.1 ± 0.9, n = 8 | 3.9 ± 0.6, n = 8 | 4.2 ± 1.0, n = 12 | 4.5 ± 0.9, n = 13 |
| IK+ (pA/pF) | 18.9 ± 5.2, n = 5 | 20.8 ± 2.5, n = 4 | 30.6 ± 4.3, n = 10 | 33.1 ± 3.3, n = 7 | 41.2 ± 3.9, n = 19 | 46.1 ± 7.8, n = 16 |
| INa+ (pA/pF) | 11.1 ± 1.4, n = 5 | 14.4 ± 1.3, n = 2 | 11.7 ± 2.6, n = 9 | 19.7 ± 4.8, n = 7 | 10.4 ± 1.2, n = 18 | 19.0 ± 2.5, n = 11** |
| IGABA (pA/pF) | 1.0 ± 0.2, n = 9 | 1.5 ± 0.4, n = 5 | 0.9 ± 0.1, n = 11 | 2.5 ± 0.5, n = 8** | 2.3 ± 0.3, n = 18 | 4.9 ± 0.4, n = 17*** |
| IGLUT (pA/pF) | 0.9 ± 0.3, n = 6 | 1.4 ± 0.5, n = 5 | 1.1 ± 0.3, n = 6 | 1.4 ± 0.3, n = 6 | 3.1 ± 0.9, n = 9 | 2.4 ± 0.7, n = 10 |
The three different regions were defined with respect to the rostrocaudal axis (RMS1, RMS2, core OB). Cm, Rm, and IK+ were not different between control and Dcx ShRNA+ cells. In contrast, INa+ was significantly increased in Dcx ShRNA+ cells located in the core of the OB. The GABA receptor-mediated current is significantly increased in Dcx ShRNA+ cells in the RMS2 and in the core of the OB, although the amplitude of glutamate receptor-mediated currents remains constant. Groups were compared 2 by 2 using a Mann–Whitney test (**p < 0.01, ***p < 0.001).
Dcx ShRNA+ neurons showed altered dendritic arbor architecture and earlier fate determination
Because previous works have established a clear link between early GABAergic signaling and neuroblast maturation during adult neurogenesis (for review, see Ge et al., 2007; Platel et al., 2010), we sought to investigate whether higher GABAergic responses seen at 5 dpe in Dcx ShRNA+ cells would lead to morphological changes a few days later. In cells located in the core of the OB at 12 dpe, the overall morphology of mutant developing neurons appeared quite different from that of control cells (Fig. 5A,B), and this difference was accentuated at 30 dpe (Fig. 5C). A thorough quantification of dendritic arbor complexity by Sholl analysis revealed control neurons form unbranched dendrites, whereas Dcx ShRNA+ neurons exhibited intricate arborization at 12 dpe that became even more complex at 30 dpe (Fig. 5D). We quantified the dendritic arbor architecture by measuring the length of the primary apical dendrite and the average number of branchings per neuron. Already at 12 dpe, we observed a twofold increase in the number of branchings in mutant cells, and by 30 dpe, the longest dendritic length had doubled (length of apical dendrite: Dcx-GFP12dpe+, 50.0 ± 3.4 μm; Dcx ShRNA12dpe+, 49.9 ± 3.1 μm; Dcx ShRNA30dpe+, 102.8 ± 17.8 μm; average branching: Dcx-GFP12dpe+, 1.71 ± 0.1 μm; Dcx ShRNA12dpe+, 3.5 ± 0.3 μm; Dcx ShRNA30dpe+, 4.0 ± 0.2 μm; Fig. 5E).
Figure 5.

Impaired morphological maturation after doublecortin knockdown at 12 and 30 dpe. A–C, Morphological changes of GFP+ cells between control Dcx-GFP+ cells (A) and Dcx ShRNA+ cells at 12 dpe (B) and Dcx ShRNA+ cells blocked in the core OB at 30 dpe (C). D, Sholl analysis of control and mutant neurons at 12 and 30 dpe. Note the increased complexity of cellular morphology of Dcx ShRNA+ cells. E, Quantification of the length of the primary apical dendrite and the average number of branchings per neuron (*p < 0.05, ***p < 0.001). Scale bars: A–C, 18 μm.
These morphological changes led us to ask whether the dendritic alteration seen in mispositioned migrating cells was accompanied by changes in their fate. We concentrated our analysis on the appearance of calretinin expression (Fig. 6A,B). We specifically used calretinin since it is the most predominant marker for GCs generated during the postnatal period (Kosaka and Kosaka, 2007; Whitman and Greer, 2007; Batista-Brito et al., 2008). Nevertheless, we cannot rule out the possibility that the mutant cells switch to other subtypes since we lack additional markers to confirm this. When we quantified the laminar distribution by counting the number of GFP+ cells found in concentric sectors starting from the RMS toward the more superficial portion of the GCL, the proportion of GFP+ neuroblasts accumulated in the core of the RMS was increased after electroporation with Dcx ShRNA plasmid (Fig. 6C; control CMV-GFP30dpe+, 17.6 ± 3.0%; Dcx ShRNA30dpe+, 41.9 ± 12.9%). Remarkably, the percentage of double-labeled GFP+/calretinin+ cells accumulated in the core of the OB was also significantly increased in Dcx ShRNA+ neuroblasts (Fig. 6D; control CMV-GFP30dpe+, 19.0 ± 3.0%; Dcx ShRNA30dpe+, 39.5 ± 12.9%), indicating that migrating cells trapped in the core of the OB nevertheless differentiate into calretinin+ neurons.
Figure 6.

Abnormally differentiated Dcx ShRNA+ neurons in the core OB are calretinin+ at 30 dpe. A, C, Double-staining calretinin (red)/GFP (green), 30 dpe after either control CMV-GFP or Dcx ShRNA plasmid electroporation. Note the presence of accumulated GFP+ cells in the core of the OB. Inset in C, Example of double-labeled GFP+/calretinin+ cell (arrowhead). B, D, Quantification of percentage of GFP+ cells (B) and percentage of double-labeled GFP+/calretinin+ cells (D) according to concentric sectors from the RMS toward the more superficial portion of the GCL. Inset in B, Schematic representation of concentric sectors of the OB for quantification of GFP+ and calretinin+ cells (D, dorsal; V, ventral; A, anterior; P, posterior). Note the increased percentage of double-labeled GFP+/calretinin+ cells in the core OB (RMS) after electroporation with Dcx ShRNA plasmid (*p < 0.05, **p < 0.01). Scale bars: A, 210 μm; C inset, 37 μm.
Synaptic integration of mispositioned developing neurons
Next, we asked whether the dendritic alteration and change in cell fate determination were accompanied by physiological changes. For this purpose, patch-clamp recordings were performed in the core of the OB at two time points following electroporation: 12 and 30 dpe (Fig. 7A). Intrinsic membrane properties indicated that mutant cells had higher membrane capacitance (Cm = 3.7 ± 0.2 pF, n = 8 for control Dcx-GFP12dpe+; Cm = 10.1 ± 1.4 pF, n = 12 for Dcx ShRNA12dpe+; Cm = 14.5 ± 1.2 pF, n = 9 for ShRNA30dpe+) but lower input membrane resistance (Rm = 3.3 ± 1.0 GΩ, n = 5 for Dcx-GFP12dpe+; 1.1 ± 0.3 GΩ, n = 6 for Dcx ShRNA12dpe+; 0.4 ± 0.1 GΩ, n = 9 for Dcx ShRNA30dpe+) and lower density of voltage-dependent K+ currents (IK+ = 52.2 ± 8.2 pA/pF, n = 6 for Dcx-GFP12dpe+; 20.8 ± 3.2 pA/pF, n = 10 for Dcx ShRNA12dpe+; 26.2 ± 4.8 pA/pF, n = 7 for Dcx ShRNA30dpe+) (Fig. 7B), consistent with Dcx ShRNA+ cells having advanced maturation. This assumption was also supported by the increase of voltage-gated Na+ channel density (INa+ = 28.85 ± 7.55 pA/pF, n = 3 for Dcx-GFP12dpe+; 44.1 ± 7.7, n = 7 for Dcx ShRNA12dpe+; 118.7 ± 7.5, n = 8 for Dcx ShRNA30dpe+; Fig. 7B). A thorough detailed investigation of the synaptic activity reveals similar functional maturation. Whereas Dcx-GFP12dpe+ neurons located in the core of the OB had not yet received synaptic inputs, both GABAA receptor-mediated postsynaptic currents (IPSCs) and glutamatergic receptor-mediated postsynaptic currents (EPSCs) were recorded in mutant neurons at 12 and 30 dpe. The frequency of spontaneous IPSCs (0.03 ± 0.01 Hz, n = 6 at 12 dpe; 0.12 ± 0.03 Hz, n = 7 at 30 dpe) (Fig. 7C) and the amplitude of IPSCs evoked by electrical stimulations delivered in the GCL (74.0 ± 15.5 pA, n = 5 at 12 dpe; 236.0 ± 36.0 pA, n = 7 at 30 dpe) (Fig. 7D) increased between 12 and 30 dpe, suggesting that the number of GABAergic synapses impinging onto Dcx ShRNA+ neurons was enhanced over time. We found no change in the amplitude of spontaneous IPSCs (22.9 ± 3.1 pA, n = 6 at 12 dpe; 25.3 ± 3.2 pA, n = 7 at 30 dpe; Fig. 7C) or in the paired-pulse ratio of evoked responses [amp2nd/amp1st; 0.8 ± 0.1, n = 5 at 12 dpe, 0.7 ± 0.1, n = 7 at 30 dpe with interstimulus interval (ISI) = 150 ms; Fig. 7D], indicating no major presynaptic or postsynaptic modification of these synapses between 12 and 30 dpe.
Figure 7.

Synaptic integration of Dcx ShRNA+ cells in the core of the OB. A, Example of Dcx ShRNA+ cells recorded at 12 and 30 dpe in the core of the OB. The presence of biocytin in the pipette solution confirmed the presence of GFP in the recorded cell and allowed visualization of cell position and morphology. Scale bar, 37 μm. B, Intrinsic membrane properties of control cells recorded at 12 dpe and Dcx ShRNA+ cells at 12 and 30 dpe in the core of the OB. Membrane capacitance (Cm), membrane resistance (Rm), and maximum density of voltage-dependent K+ channel (IK+)- and Na+ channel (INa+)-mediated currents were measured. Note that although the Dcx ShRNA+ cells are blocked in the core of the OB, the changes of their intrinsic membrane properties confirm that they are undergoing maturation. C, Properties of spontaneous IPSCs at 0 mV received by Dcx ShRNA+ cells in the core of the OB at 12 and 30 dpe. Note that there is an increase of Dcx ShRNA+ cells' spontaneous IPSC frequency, but not amplitude, over the maturation time. Calibration: 3 ms, 10 pA. D, Properties of IPSCs evoked by paired electrical stimulation in the GCL (voltage clamp = 0 mV; ISI = 150 ms) onto Dcx ShRNA+ cells in the core of the OB at 12 and 30 dpe. Note the increase of IPSC amplitude without modification of the paired-pulse ratio. Representative traces are averages of >10 IPSCs. Calibration: 50 ms, 50 pA. E, Properties of EPSCs evoked by paired electrical stimulation in the GCL (voltage clamp = −70 mV; ISI = 50 ms) onto Dcx ShRNA+ cells in the core of the OB at 12 and 30 dpe. Representative traces are averages of >10 IPSCs (calibration: 10 ms, 25 pA). Note the increase of IPSC amplitude and decrease of the paired-pulse ratio. Then, despite their ectopic location, Dcx ShRNA+ cells undergo synaptic integration. Groups were compared 2 by 2 using a Mann–Whitney test (*p < 0.05, **p < 0.01, ***p < 0.001).
Concerning the glutamatergic inputs, we only analyzed EPSCs evoked by electrical stimulations delivered in the GCL because of the very low occurrence of spontaneous events at 12 dpe (data not shown). The amplitude of EPSCs was dramatically increased (39.80 ± 8.31 pA, n = 5 at 12 dpe; 153.9 ± 51.42 pA, n = 7 at 30 dpe) and the paired-pulse ratio was decreased (ISI = 50 ms; amp2nd/amp1st, 1.81 ± 0.16, n = 5 at 12 dpe; 1.13 ± 0.07, n = 7 at 30 dpe) (Fig. 7E), suggesting an increase in the probability of glutamate release over time (Zucker and Regehr, 2002).
The rapid synaptic integration of ectopically maturing Dcx ShRNA+ neurons, followed by an increase in the frequency of synaptic events between 12 and 30 dpe, suggests that the density of input nerve terminals might increase during the same time frame. Analyzing the presence of presynaptic staining apposed to GFP+ neurons confirmed this hypothesis (Fig. 8). At 12 dpe, a double staining of GFP with VGAT (Fig. 8A,B) revealed GABAergic contacts impinging onto Dcx ShRNA+ neurons. Closer views at the proximal portion of the dendrites indicated that GABAergic terminals were apposed, and sometimes occurred on dendritic spines along the dendrites of maturing Dcx ShRNA+ neurons (Fig. 8C). Quantifications of those contacts indicated an increase of VGAT+ punctae between 12 and 30 dpe (Fig. 8H). Similarly, the double staining of GFP with VGLUT1 (Fig. 8D,E) indicated fewer densities of glutamatergic apposition along dendrites of Dcx ShRNA+ neurons when counted at 12 dpe and increased dramatically at 30 dpe, with glutamatergic terminals being found both in apposition along GFP+ dendrites and contacting GFP+ spines (Fig. 8F–H). Collectively, these data demonstrate that Dcx ShRNA+ neurons located deep inside the OB precociously assumed features of Dcx-expressing interneurons, but also exhibited novel features never seen in Dcx-expressing interneurons at early stages of postnatal neurogenesis.
Figure 8.
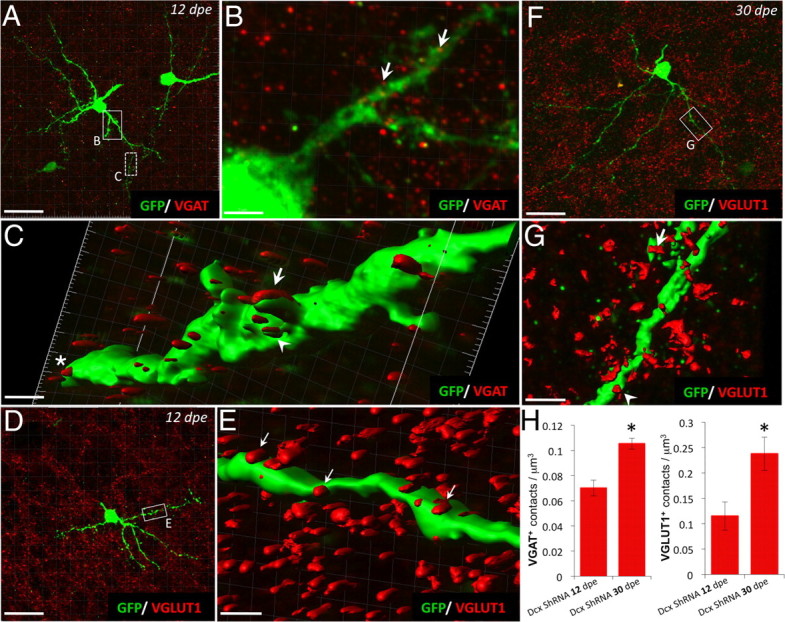
Abnormally differentiated Dcx ShRNA+ neurons receive GABAergic and glutamatergic inputs at different time points of maturation after electroporation. A–C, Double-staining GFP (green)/VGAT (red) (A) at 12 dpe in the core OB after electroporation with Dcx ShRNA plasmid (box with plain line is enlarged in B and dashed line box is represented in C as a 3D rendering of that region). B, Red dots (VGAT staining) are closely apposed along the neuronal process (arrows). C, 3D rendering of the GFP+ dendrite of Dcx ShRNA+ neuron at 12 dpe. Note that VGAT staining (red) is present along the dendrite as appositions (star), developing spine (arrowhead), or spine (arrow). D–G, Double-staining GFP (green)/VGLUT1 (red) on differentiated Dcx ShRNA+ neurons at 12 and 30 dpe in the core OB. Box in D is enlarged in E (with 3D rendering); box in F is enlarged in G. Arrows in E and G indicate the presence of glutamatergic apposition (red) along the GFP+ dendrite (green). Note the presence of VGLUT1+ appositions on GFP+ spine at 30 dpe (G). H, Quantifications of VGAT+ and VGLUT1+ contacts per μm3. Note the increase of VGAT+ and VGLUT1+ contact densities between 12 and 30 dpe. Scale bars: A, D, F, 26 μm; B, 3.46 μm; C, 0.72 μm; E, 1.17 μm; G, 2.34 μm.
Discussion
Neuronal migration in the postnatal RMS supplies the OB with immature neurons fated to replace preexisting neurons located in the GCL and the glomerular layer. Many genetic models affecting neuronal migration, such as neural cell adhesion molecule(NCAM) (Chazal et al., 2000), integrins (Belvindrah et al., 2007), ephrins (Conover et al., 2000), prokineticin (Ng et al., 2005), Slits (Nguyen-Ba-Charvet et al., 2004; Kaneko et al., 2010), DCX (Koizumi et al., 2006; Kappeler et al., 2007), and cyclin-dependent kinase 5 (CDK5) (Hirota et al., 2007), have highlighted the importance of the migratory pathway to convey the neuronal precursors at the right place and at the right time. Here, our study provides for the first time insights on how the determination of neuronal fate is dependent on cellular migration in the postnatal RMS. Specifically, our data reveal unexpected features of newly formed neurons when their journey in the RMS is artificially prolonged during early postnatal life. Reducing the expression levels of DCX in migrating neuroblasts altered their migration and placed them in atypical locations. Surprisingly, developing neurons in aberrant positions continue to mature and develop a multibranching dendritic arborization that receives anomalous synaptic contacts as early as 12 dpe, and that are maintained for several weeks. Thus, early defects in the migration generated during the first postnatal days had profound and long-lasting modifications in the subsequent developmental stages of postnatal-generated neurons, including their morphology, their fate determination, and their cellular physiology. Together, these findings highlight the surprisingly high degree of cellular plasticity of maturing neurons.
DCX downregulation modifies the temporal sequence of neuroblast maturation
Correct neuronal targeting is essential during postnatal neurogenesis for normal bulbar functions (Belvindrah and Lledo, 2011). RMS tangential migration is a dynamic process involving both cell-autonomous and environmental factors. One of the key players involved in cell motility is DCX, which binds to and stabilizes microtubules (Gleeson et al., 1999b; Schaar et al., 2004). Our Dcx ShRNA approach decreased DCX protein expression levels in migrating postnatal-generated neuroblasts. During normal maturation, newly generated neurons begin to express mature neuronal markers such as the calcium-binding protein calretinin upon reaching mature bulbar circuits. At the same time, DCX immunoreactivity decreases sharply and remains undetectable thereafter. Because DCX naturally turns off (Brown et al., 2003), our knockdown strategy forces this normal change to occur at an abnormally early period of neural maturation. Future studies will be conducted to address the possibility that DCX acts as a tonic inhibitor preventing neural cells to achieve an advanced program of maturation by maintaining them in an immature state. Our study shows that early alteration in tangential migration has profound effects not only on the ultimate neuronal positioning, but also on the cell's morphology and wiring with host circuits. To confirm that this defect in temporal maturation of neuroblasts is a general consequence of alteration of tangential migration, it would be interesting to revisit the phenotype of different genetic models of neuronal migration perturbations such as NCAM, β1-integrins, prokineticin, or CDK5 mutant mice with the same toolbox used in the present study (Chazal et al., 2000; Ng et al., 2005; Belvindrah et al., 2007; Hirota et al., 2007).
At a temporal level, when the precise expression pattern of DCX is altered, most of the mutant neurons show an atypical morphology and extend prematurely exuberant multipolar processes, suggesting impairment in growth of radial neurites. Nevertheless, some cells attained bipolar morphologies and migrated properly into the superficial GCL. We do not know yet whether these neurons represent an insensitive population of cells to DCX knockdown or, more likely, whether these neurons express normal levels of DCX because they have been transfected with lower copy numbers of Dcx ShRNA plasmids.
Our study also revealed that the migratory behavior of multipolar mutant cells is quite distinct from that of bipolar control neuroblasts characterized by a leading and a trailing process. We found that the mutant neuroblasts had no preferential migratory direction. As a consequence, the overall migration rate was much slower, thus leading to longer residence time in the RMS. We still do not know whether the premature expression of neuronal marker (e.g., calretinin) originates from changes in the pattern of cell migration or from modification of their cellular morphology. According to the former hypothesis, the apparent increase in time spent during their journey in the RMS would lead to differential exposure to signaling or cellular interactions. Alternatively, it is possible that the dramatic morphological changes could result from direct effects of DCX on the cellular physiology since DCX can form a complex with cytoplasmic dynein (Tanaka et al., 2004), a retrograde molecular motor involved in nucleosomal translocation coupling and intracellular transport. In this context, DCX is required directly in migrating cells for the appropriate cytoskeletal dynamic and could also regulate the intracellular signaling and trafficking required to sense external signals that direct tangential migration. Among others, BDNF is a recent candidate that has been demonstrated to regulate bidirectional RMS flow via different receptors such as TrkB (Bagley and Belluscio, 2010) and p75NTR (Snapyan et al., 2009).
Developing postnatal-generated neurons in ectopic areas
At a spatial level, the downregulation of the DCX expression generates neurons that develop in ectopic areas, a unique situation never seen in control neuroblasts (Belvindrah et al., 2009; Platel et al., 2010). The aberrant positions include neurons found in the RMS of the OB and in the deep portion of the GCL, in sharp contrast to their control counterparts already integrated in the more superficial portion of the GCL. The concomitant modification of neuronal morphology and physiology with the mispositioning of the neurons highlights the importance of fate determinants provided by the local environment and/or the host circuits. This is demonstrated by the fact that mutant neuroblasts in the RMS showed an aberrant phenotype even before reaching the host circuit. Our results suggest that the mean residence time in the RMS could be an important factor in determining the cell fate. Moreover, the instructive function fulfilled by the migratory path represents a unique process specific to adult neurogenesis, where surrounding astrocytes and blood vessels actively participate in modulating migration (Snapyan et al., 2009; Kaneko et al., 2010). For instance, the GABA release from neuroblasts suppresses proliferation in the SVZ but also reduces the pace of neuroblast migration in the RMS (Bolteus and Bordey, 2004; Mejia-Gervacio et al., 2011). In contrast, during embryogenesis, the migrating neuroblasts receive fewer messengers (i.e., GABA, glutamate, trophic factors, etc.) sent by immature surrounding tissues. Future transplantation experiments will address the precise impact of the host circuit in shaping the fate of developing neurons and how the context of the microenvironment could potentiate fate programming and neuronal commitment versus a potential direct implication of DCX in dendritic development.
Ectopic neurons exhibit unique features
The misplaced developing Dcx ShRNA+ neurons extended a complex multibranched dendritic arbor in the RMS of the OB that was clearly distinguishable from the morphology of control developing neurons of similar ages in the GCL. Control neurons migrated out of the RMS toward superficial layers of the GCL and projected a long apical dendrite into the external plexiform layer, where they formed dendro-dendritic reciprocal synapses with mitral/tufted cells (Shepherd et al., 2004). Then, because of the absence of an appropriate spatiotemporal control neuronal population, we deliberately concentrated our efforts on a temporal analysis of mutant neurons that continue to develop in ectopic areas. This provided a unique opportunity to evaluate the functional properties of developing neurons located in inappropriate positions using a genetic model of a human neuronal developmental disorder. In contrast to the results gained from cortical DCX-deficient neurons, which exhibited delayed maturation rate (Ackman et al., 2009), we found that ectopic mutant neurons display anticipated maturation of their functional properties, at least for some parameters. For instance, at 7 dpe in the RMS, these developing neurons expressed a higher density of voltage-dependent Na+ channels and a greater responsiveness to bath application of muscimol. This effect was specific to GABAA receptors, since the glutamate responsiveness remained unchanged. The presence of synaptic events further indicates that despite their erroneous location, mutant neurons have integrated into an ectopic network. This peculiarity raises the question of the identity of the ectopic presynaptic terminals impinging onto the newly formed neurons.
Remarkably, the maturing Dcx ShRNA+ neurons located deep inside the bulb developed some features of mature bulbar interneurons such as early GABAergic synaptic inputs (Panzanelli et al., 2009) and expression of calretinin. We still do not know whether the synaptic inputs trigger early maturation, and what function the multibranching GABAergic ectopic neurons might fulfill in these aberrant locations.
Despite possessing some hallmarks of postnatal-born developing OB interneurons, other electrophysiological properties were unique to the mutant neurons. Most puzzling was the early increase of Na+ currents together with the downregulation of K+ currents that produced precocious spiking activity. This observation supports previous findings obtained with animal models that have revealed increased neuronal excitability, and seizure risk, when neuronal migration is disrupted in the neocortex (Roper et al., 1995; Lee et al., 1997; Nosten-Bertrand et al., 2008). Similarly, in the hippocampus, despite a strong defect in morphological development after reduction of DISC-1 (disrupted in schizophrenia 1 protein) expression, aberrant neurons display an accelerated maturation (Duan et al., 2007). It could also explain to some degree why most of the neuronal migration disorders in humans are risk factors for seizure (Guerrini et al., 1999; Sheen and Walsh, 2003). In the future, it will be important to determine which specific molecular mechanisms are responsible for the developmental restriction of Na+ current in developing postnatal control neurons.
The increased expression of GABAA receptors together with voltage-gated Na+ channels in mutant neurons, well before synapses appeared, demonstrates that the development of these neurons is not a fully automated unidirectional process that remains insensitive to the local environment. In contrast, this result supports the claim that activation of transmitter signaling provides a phenotypic checkpoint (Ben-Ari and Spitzer, 2010). Nevertheless, those fate switches and plasticity properties are different according to excitatory versus inhibitory neurogenesis and/or embryogenesis versus postnatal development. For instance, DCX mutant excitatory precursors generated from the ventricular zone during cortical development are in a “freezing” state during which the fate of these neurons could be rescued by reexpression of DCX, but not changed to another cell type (Manent et al., 2009). On the other hand, the inhibitory GC precursors, once misrouted from the OB, demonstrated their ability to very rapidly acquire a mature phenotype distinct from their original fate. This finding reveals the high level of plasticity of postnatal precursors, and the existence of a phenotype “critical period,” which varies according to the timing or the nature of neurogenesis (embryonic vs adult).
Implication for disorders of neuronal migration and for cellular brain therapies
Human mutations in the X-linked gene Dcx are one of the most common genetic defaults of double-cortex syndrome and of lissencephaly (Dobyns and Truwit, 1995). Our study suggests that human developmental disorders that originate from disrupting neuroblast migration do not only produce neurons that fail to reach their target circuits, but that also imperfectly recapitulate the original program of maturation. As a result, these neurons synaptically integrate into inappropriate neural networks.
Previous studies have shown that endogenous or transplanted neuronal progenitors in the CNS can migrate to sites of injury (Imitola et al., 2004). If rerouting neuroblasts could be therapeutically useful, our study indicates that such approaches would need to take into account the impact of modifying neuronal migration on cell fate determination and on cellular physiology, particularly when neuronal precursors are guided outside of the olfactory system. For instance, in some experimental demyelinating models intended to replicate the symptoms of multiple sclerosis, it has been shown that SVZ precursors have the ability to generate oligodendrocytes, whereas most progenitors are fated to generate interneurons (Nait-Oumesmar et al., 1999). In the case of cortical lesions, neuroblasts can emigrate toward the site of injury, likely through an epidermal growth factor pathway (Sundholm-Peters et al., 2005). This emphasizes how important it is to characterize what endogenous factors modulate the migration of neuronal precursors and, thus, control cell fate.
To conclude, we have demonstrated that alteration of neuronal migration by knocking down DCX generates permanent changes to the fate and the physiology of neuronal precursors. Consequently, therapeutic strategies aiming at directing neuroblasts toward lesion sites should take into account where the neuroblasts have to migrate during their journey to provide the damaged circuit with the desired neuronal phenotype.
Footnotes
The “Aprionis” group, the Fondation pour la Recherche Médicale (FRM team), the Ecole des Neurosciences de Paris, and “ERA-Net Neuron” of the FP7 program by the European Commission supported this study. R.B. obtained a fellowship from the Association pour la Recherche sur le Cancer. We thank Fabrice de Chaumont and Jean-Christophe Olivo-Marin (Institut Pasteur) for helping us with quantitative image analysis.
References
- Ackman JB, Aniksztejn L, Crépel V, Becq H, Pellegrino C, Cardoso C, Ben-Ari Y, Represa A. Abnormal network activity in a targeted genetic model of human double cortex. J Neurosci. 2009;29:313–327. doi: 10.1523/JNEUROSCI.4093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam Y, Mizrahi A. Circuit formation and maintenance—perspectives from the mammalian olfactory bulb. Curr Opin Neurobiol. 2010;20:134–140. doi: 10.1016/j.conb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Altman J. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol. 1969;137:433–457. doi: 10.1002/cne.901370404. [DOI] [PubMed] [Google Scholar]
- Bagley JA, Belluscio L. Dynamic imaging reveals that brain-derived neurotrophic factor can independently regulate motility and direction of neuroblasts within the rostral migratory stream. Neuroscience. 2010;169:1449–1461. doi: 10.1016/j.neuroscience.2010.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Batista-Brito R, Close J, Machold R, Fishell G. The distinct temporal origins of olfactory bulb interneuron subtypes. J Neurosci. 2008;28:3966–3975. doi: 10.1523/JNEUROSCI.5625-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluzzi O, Benedusi M, Ackman J, LoTurco JJ. Electrophysiological differentiation of new neurons in the olfactory bulb. J Neurosci. 2003;23:10411–10418. doi: 10.1523/JNEUROSCI.23-32-10411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvindrah R, Lledo P. Postnatal neurogenesis of the forebrain. Comp Dev Neurosci. 2011 in press. [Google Scholar]
- Belvindrah R, Hankel S, Walker J, Patton BL, Müller U. Beta1 integrins control the formation of cell chains in the adult rostral migratory stream. J Neurosci. 2007;27:2704–2717. doi: 10.1523/JNEUROSCI.2991-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvindrah R, Lazarini F, Lledo PM. Postnatal neurogenesis: from neuroblast migration to neuronal integration. Rev Neurosci. 2009;20:331–346. doi: 10.1515/revneuro.2009.20.5-6.331. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Spitzer NC. Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 2010;33:485–492. doi: 10.1016/j.tins.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolteus AJ, Bordey A. GABA release and uptake regulate neuronal precursor migration in the postnatal subventricular zone. J Neurosci. 2004;24:7623–7631. doi: 10.1523/JNEUROSCI.1999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo PM. Becoming a new neuron in the adult olfactory bulb. Nat Neurosci. 2003;6:507–518. doi: 10.1038/nn1048. [DOI] [PubMed] [Google Scholar]
- Chazal G, Durbec P, Jankovski A, Rougon G, Cremer H. Consequences of neural cell adhesion molecule deficiency on cell migration in the rostral migratory stream of the mouse. J Neurosci. 2000;20:1446–1457. doi: 10.1523/JNEUROSCI.20-04-01446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–1097. doi: 10.1038/80606. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Truwit CL. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132–147. doi: 10.1055/s-2007-979744. [DOI] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, Liu CY, Ganesan S, Cheng HJ, Ming GL, Lu B, Song H. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Ge S, Pradhan DA, Ming GL, Song H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends Neurosci. 2007;30:1–8. doi: 10.1016/j.tins.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Minnerath SR, Fox JW, Allen KM, Luo RF, Hong SE, Berg MJ, Kuzniecky R, Reitnauer PJ, Borgatti R, Mira AP, Guerrini R, Holmes GL, Rooney CM, Berkovic S, Scheffer I, Cooper EC, Ricci S, Cusmai R, Crawford TO, et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999a;45:146–153. doi: 10.1002/1531-8249(199902)45:2<146::aid-ana3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999b;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Canapicchi R, Dobyns WB. Epilepsy and malformations of the cerebral cortex. Neurologia. 1999;14(Suppl 3):32–47. [PubMed] [Google Scholar]
- Hack I, Bancila M, Loulier K, Carroll P, Cremer H. Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat Neurosci. 2002;5:939–945. doi: 10.1038/nn923. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Ohshima T, Kaneko N, Ikeda M, Iwasato T, Kulkarni AB, Mikoshiba K, Okano H, Sawamoto K. Cyclin-dependent kinase 5 is required for control of neuroblast migration in the postnatal subventricular zone. J Neurosci. 2007;27:12829–12838. doi: 10.1523/JNEUROSCI.1014-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko N, Marín O, Koike M, Hirota Y, Uchiyama Y, Wu JY, Lu Q, Tessier-Lavigne M, Alvarez-Buylla A, Okano H, Rubenstein JL, Sawamoto K. New neurons clear the path of astrocytic processes for their rapid migration in the adult brain. Neuron. 2010;67:213–223. doi: 10.1016/j.neuron.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler C, Saillour Y, Baudoin JP, Tuy FP, Alvarez C, Houbron C, Gaspar P, Hamard G, Chelly J, Métin C, Francis F. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet. 2006;15:1387–1400. doi: 10.1093/hmg/ddl062. [DOI] [PubMed] [Google Scholar]
- Kappeler C, Dhenain M, Phan Dinh Tuy F, Saillour Y, Marty S, Fallet-Bianco C, Souville I, Souil E, Pinard JM, Meyer G, Encha-Razavi F, Volk A, Beldjord C, Chelly J, Francis F. Magnetic resonance imaging and histological studies of corpus callosal and hippocampal abnormalities linked to doublecortin deficiency. J Comp Neurol. 2007;500:239–254. doi: 10.1002/cne.21170. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci. 2006;9:779–786. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- Kosaka T, Kosaka K. Heterogeneity of nitric oxide synthase-containing neurons in the mouse main olfactory bulb. Neurosci Res. 2007;57:165–178. doi: 10.1016/j.neures.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Lee KS, Schottler F, Collins JL, Lanzino G, Couture D, Rao A, Hiramatsu K, Goto Y, Hong SC, Caner H, Yamamoto H, Chen ZF, Bertram E, Berr S, Omary R, Scrable H, Jackson T, Goble J, Eisenman L. A genetic animal model of human neocortical heterotopia associated with seizures. J Neurosci. 1997;17:6236–6242. doi: 10.1523/JNEUROSCI.17-16-06236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–1148. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- Manent JB, Wang Y, Chang Y, Paramasivam M, LoTurco JJ. Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat Med. 2009;15:84–90. doi: 10.1038/nm.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia-Gervacio S, Murray K, Lledo PM. NKCC1 controls GABAergic signaling and neuroblast migration in the postnatal forebrain. Neural Dev. 2011;6:4. doi: 10.1186/1749-8104-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Van Evercooren AB. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science. 2005;308:1923–1927. doi: 10.1126/science.1112103. [DOI] [PubMed] [Google Scholar]
- Nguyen-Ba-Charvet KT, Picard-Riera N, Tessier-Lavigne M, Baron-Van Evercooren A, Sotelo C, Chédotal A. Multiple roles for slits in the control of cell migration in the rostral migratory stream. J Neurosci. 2004;24:1497–1506. doi: 10.1523/JNEUROSCI.4729-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosten-Bertrand M, Kappeler C, Dinocourt C, Denis C, Germain J, Phan Dinh Tuy F, Verstraeten S, Alvarez C, Métin C, Chelly J, Giros B, Miles R, Depaulis A, Francis F. Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS One. 2008;3:e2473. doi: 10.1371/journal.pone.0002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzanelli P, Bardy C, Nissant A, Pallotto M, Sassoè-Pognetto M, Lledo PM, Fritschy JM. Early synapse formation in developing interneurons of the adult olfactory bulb. J Neurosci. 2009;29:15039–15052. doi: 10.1523/JNEUROSCI.3034-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, Yan LD, Bordey A. A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology. 2010;58:865–876. doi: 10.1016/j.neuropharm.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Alvarez-Buylla A. Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J Neurosci. 2002;22:6106–6113. doi: 10.1523/JNEUROSCI.22-14-06106.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel JC, Stamboulian S, Nguyen I, Bordey A. Neurotransmitter signaling in postnatal neurogenesis: the first leg. Brain Res Rev. 2010;63:60–71. doi: 10.1016/j.brainresrev.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper SN, Gilmore RL, Houser CR. Experimentally induced disorders of neuronal migration produce an increased propensity for electrographic seizures in rats. Epilepsy Res. 1995;21:205–219. doi: 10.1016/0920-1211(95)00027-8. [DOI] [PubMed] [Google Scholar]
- Saghatelyan A, de Chevigny A, Schachner M, Lledo PM. Tenascin-R mediates activity-dependent recruitment of neuroblasts in the adult mouse forebrain. Nat Neurosci. 2004;7:347–356. doi: 10.1038/nn1211. [DOI] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell SK. Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron. 2004;41:203–213. doi: 10.1016/s0896-6273(03)00843-2. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Walsh CA. Developmental genetic malformations of the cerebral cortex. Curr Neurol Neurosci Rep. 2003;3:433–441. doi: 10.1007/s11910-003-0027-8. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Chen WR, Greer CA. Olfactory bulb. In: Shepherd GM, editor. The synaptic organization of the brain. Ed 5. New York: Oxford UP; 2004. [Google Scholar]
- Snapyan M, Lemasson M, Brill MS, Blais M, Massouh M, Ninkovic J, Gravel C, Berthod F, Götz M, Barker PA, Parent A, Saghatelyan A. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 2009;29:4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundholm-Peters NL, Yang HK, Goings GE, Walker AS, Szele FG. Subventricular zone neuroblasts emigrate toward cortical lesions. J Neuropathol Exp Neurol. 2005;64:1089–1100. doi: 10.1097/01.jnen.0000190066.13312.8f. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Qiu R, Tsark W, Lu Q. Rapid promoter analysis in developing mouse brain and genetic labeling of young neurons by doublecortin-DsRed-express. J Neurosci Res. 2007;85:3567–3573. doi: 10.1002/jnr.21440. [DOI] [PubMed] [Google Scholar]
- Whitman MC, Greer CA. Synaptic integration of adult-generated olfactory bulb granule cells: basal axodendritic centrifugal input precedes apical dendrodendritic local circuits. J Neurosci. 2007;27:9951–9961. doi: 10.1523/JNEUROSCI.1633-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]