

Abstract
Activation of Group I metabotropic glutamate receptors (mGluRs) in rat hippocampus induces a form of long-term depression (LTD) that is dependent on protein synthesis. However, the intracellular mechanisms leading to the initiation of protein synthesis and expression of LTD after mGluR activation are only partially understood. We investigated the role of several pathways linked to mGluR activation, translation initiation, and induction of LTD. We found that Group I mGluR-dependent protein synthesis and associated LTD, as induced by the agonist (RS)-3,5-dihydrophenylglycine (DHPG) or paired-pulse synaptic stimulation, was dependent on activation of calcium/calmodulin-dependent protein kinase IIα (CaMKII). DHPG induced a transient increase in the level of phospho-CaMKII (phospho-CaMKIIT286) in synaptoneurosomes prepared from whole hippocampus and in CA1 minislices. In synaptoneurosomes, DHPG also induced an increase in phosphorylation of eIF4E, and an increase in protein synthesis that was abolished by translation inhibitors and the CaMKII inhibitors 1-[N,O-bis(5-isoquinolinesulphonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine (KN62) and 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chloro-cinnamyl)-N-methylbenzylamine (KN93). In field recordings from CA1, both the translation inhibitor cycloheximide and KN62 significantly reduced DHPG-induced LTD. Combined application did not further reduce the LTD, suggesting a common mechanism. In whole-cell recordings, a third CaMKII inhibitor, AIP (autocamtide-2-related inhibitory peptide), significantly reduced the DHPG-induced LTD of synaptic currents. Inhibition of the classical pathway mediating many Group I mGluR effects by blocking PKC (protein kinase C) or PLC (phospholipase C) did not impair DHPG-induced protein synthesis or LTD. Collectively, these findings demonstrate an important role for CaMKII in mediating the initiation of protein synthesis that then supports the postsynaptic expression of DHPG-induced LTD.
Introduction
Activation of Group I metabotropic glutamate receptors (mGluRs) leads to induction of long-term depression (LTD) (mGluR-LTD) in the hippocampus (Palmer et al., 1997; Naie et al., 2007), which is ultimately expressed through internalization of postsynaptic AMPA receptors (AMPARs) (Snyder et al., 2001) and is typically dependent on both rapid dendritic de novo protein synthesis (Huber et al., 2000) and posttranslational changes occurring within the postsynaptic terminal. The few proteins known to be upregulated by Group I mGluRs are also involved in AMPAR cycling (Waung and Huber, 2009).
The mechanisms by which mGluRs initiate the synthesis of new proteins, and how these proteins are involved in mGluR-LTD, are not yet fully understood. Group I mGluRs can couple directly to pathways involved in the regulation of translation, but not those regulating transcription, to control LTD (Huber et al., 2000). There are at least two signaling pathways that link mGluR activation to the initiation of translation and LTD: activation of extracellular signal-regulated kinase (ERK) by mitogen-activated protein kinase kinase (MEK), and activation of mammalian target of rapamycin (mTOR) through phosphoinositide-3-kinase (PI3K) and Akt. Thus, (RS)-3,5-dihydrophenylglycine (DHPG) increases levels of phosphorylated ERK and mTOR (Choe and Wang, 2001a,b; Gallagher et al., 2004; Banko et al., 2006; Antion et al., 2008) and initiates translation through the phosphorylation and activation of eIF4E (Banko et al., 2006). Furthermore, mGluR activation increases other regulators of protein synthesis such as mRNA polyadenylation (Shin et al., 2004) and p70S6 kinase activity (Antion et al., 2008). In contrast, the classical mGluR pathway involving the G-protein-mediated activation of phospholipase C (PLC), production of diacylglycerol and inositol triphosphate (IP3), and subsequent activation of protein kinase C (PKC) and release of calcium from intracellular stores, does not seem to be involved in at least one form of mGluR-LTD (Schnabel et al., 1999b).
One additional kinase that can regulate translation, but that has received relatively little attention regarding its role in DHPG-induced protein synthesis and LTD, is calcium/calmodulin-dependent protein kinase IIα (CaMKII). Initially, mGluR-mediated hippocampal LTD was reported to be potentiated by inhibition of CaMKII activity (Schnabel et al., 1999b), but a number of biochemical studies have since linked the activity of CaMKII to mGluR- (Choe and Wang, 2001b) and glutamate-stimulated activation of ERK (Vanhoutte et al., 1999) and mTOR (Lenz and Avruch, 2005). Furthermore, CaMKII activation can activate translation factors (Atkins et al., 2004, 2005) and promote protein synthesis (Atkins et al., 2004) in the hippocampus. Collectively, these studies suggest that CaMKII may mediate mGluR activation of translational processes, and thus possibly LTD.
The present study investigated pathways involved in the regulation of DHPG-induced protein synthesis and LTD with particular emphasis on the role of CaMKII. We found that DHPG induced phosphorylation of CaMKII and that CaMKII inhibitors completely blocked DHPG-induced protein synthesis in hippocampal synaptoneurosomes. Furthermore, the protein synthesis-dependent component of DHPG-induced LTD was blocked by CaMKII inhibitors in hippocampal slices. These findings indicate that CaMKII plays a significant role in the regulation of mGluR-induced protein synthesis and LTD.
Materials and Methods
Animals.
In all biochemical and electrophysiological experiments, tissue was prepared from young adult male Sprague Dawley rats (42–70 d), as described previously (Mockett et al., 2004, 2007). All experimental procedures were performed in accordance with regulations laid down by the Animal Ethics Committee of the University of Otago.
Drugs and reagents.
For the biochemical analyses, chloramphenicol, orthovanadate, emetine, rapamycin, and cycloheximide were purchased from Sigma-Aldrich. The kinase inhibitors 1-[N,O-bis(5-isoquinolinesulphonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine (KN62), 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chloro-cinnamyl)-N-methylbenzylamine (KN93), chelerythrine, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059), 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole (SB203580), and lavendustin were purchased from Tocris Bioscience, whereas N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl (H89) was supplied by BIOMOL. The Group I mGluR antagonist 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495) was purchased from Tocris Bioscience. Complete protease inhibitor was purchased from Roche Diagnostics. [35S]Methionine and biodegradable counting scintillant were supplied by GE Healthcare.
For the electrophysiological experiments, all salts were supplied by BDH Chemicals. DHPG, CNQX, 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122), and APV were purchased from Tocris Bioscience and dissolved in double-distilled water as stock solutions, whereas picrotoxin, also from Tocris Bioscience, was dissolved in ethanol. QX-314 (Tocris Bioscience) was dissolved directly into the electrode solution. Calbiochem supplied 1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidine-dione (U73343), the inactive analog of U73122. K-methanesulfonate, Na2ATP, NaGTP, phosphocreatine, and EGTA-4Na were purchased from Sigma-Aldrich. All stock drug solutions were diluted 1:1000 with artificial CSF (aCSF) for the final working concentration.
Synaptic protein synthesis assays.
Protein synthesis was measured in synaptoneurosomes (a synapse-enriched fraction) (Hollingsworth et al., 1985; Williams et al., 2003, 2007), prepared from either whole hippocampus or isolated dentate gyrus and area CA1, by monitoring the incorporation of [35S]methionine into insoluble material after precipitation with trichloroacetic acid (TCA) (Claasen et al., 2009). Synaptoneurosomes (100 μg) were preincubated (15 min; 37°C) with 0 or 20 μm of the Group I mGluR agonist DHPG in HEPES buffer, pH 7.4 [50 mm HEPES, 124 mm NaCl, 3.2 mm KCl, 1.06 mm KH2PO4, 2.5 mm CaCl2, 1.3 mm MgCl2, 26 mm NaHCO3, 10 mm glucose, Complete protease inhibitor (Roche), 0.7 mm chloramphenicol]. Newly synthesized proteins were then labeled by the addition of [35S]methionine (10 μCi; in vivo labeling quality) in the continued presence of DHPG. After 30 min, cold methionine (200 μg) was added (10 min; 37°C), synaptoneurosomes were collected by centrifugation, and proteins were precipitated by addition of 0.5 ml of 10% TCA/0.1% methionine followed by centrifugation (13,000 × g; 5 min; 4°C). Protein pellets were washed three times in 5% TCA/0.1% methionine and collected by centrifugation between each wash. The pellets were solubilized in 1 m NaOH (37°C; overnight), neutralized with 1 m HCl, and [35S]methionine incorporation was measured by addition of biodegradable counting scintillant using a liquid scintillation counter (Wallac; MicroBeta Trilux). In some experiments, eukaryotic protein synthesis inhibitors (cycloheximide, 60 μm; emetine, 30 μm; or rapamycin, 20 nm), protein kinase inhibitors [CaMKII: KN62, 10 μm; KN93, 10 μm; protein kinase C: chelerythrine, 1 μm; p42/44 MAPK: PD98059, 50 μm; p38 MAPK: SB203580, 1 μm; tyrosine kinase: lavendustin, 10 μm; protein kinase A: H89, 10 μm], tyrosine phosphatase inhibitor (orthovanadate, 1 mm) or mGluR antagonist (LY341495, 100 μm), were added to the 15 min preincubation. [35S]Methionine incorporation was expressed as a ratio relative to matched no-drug controls.
Slice preparation and incubation for biochemical analysis.
Tissue was dissected and slices cut in a manner similar to that described for the electrophysiological experiments, except that for some experiments slices were further dissected manually to contain predominantly area CA1. These slices were placed in tissue culture dishes (35 mm; four to six slices/dish) containing 1 ml of aCSF and transferred to the upper compartment of a static incubation chamber. This was the same chamber used to equilibrate slices before electrophysiological experiments and has been described in detail previously (Mockett et al., 2007). DHPG (0, 100 μm) and other drug treatments were bath applied in fresh oxygenated and warmed (32°C) aCSF. At the conclusion of the experiment, minislices were transferred to 1.5 ml Eppendorf tubes and immediately snap-frozen on dry ice. All slices were stored at −80°C until analyzed by quantitative Western blot analysis.
Western blot analysis.
To determine whether DHPG altered the total levels or phosphorylation status of CaMKII or translation factors, and to determine the time course for any changes, hippocampal synaptoneurosomes were treated as per the protein synthesis assay but in the absence of isotope. To investigate rapid changes in phosphorylation status, synaptoneurosomes were treated with DHPG (0, 20 μm) with or without KN62 (10 μm), and reactions were terminated at 0 (nominal), 5, 10, or 20 min. All incubations were terminated by rapid cooling on ice, precipitation by centrifugation, and lysis in SDS-PAGE loading buffer before separation through 9 or 12% SDS-PAGE and transfer to nitrocellulose membrane (Schleicher & Schuell). Whole-cell extracts were prepared from four to five CA1 minislices as described previously (Williams et al., 1998; Mockett et al., 2007) by homogenization and sonication in ice-cold extraction buffer, and then separated by SDS-PAGE before being transferred to nitrocellulose membrane. Western blot analysis was performed using the following antibodies: CaMKIIA (Sigma-Aldrich; C6974), phospho-CaMKIIAT286 (Sigma-Aldrich; P-247), eEF2 (Abcam; ab33208), phospho-eEF2Thr56 (Cell Signaling Technology; 2331), eIF4E (Cell Signaling Technology; 9742), and phospho-eIF4ESer209 (Abcam; ab76256). For chemiluminescence detection of antibody binding, incubations were performed essentially as described previously (Williams et al., 1998), and antibody binding was detected using Supersignal West Pico (Pierce). X-ray films were scanned using a Bio-Rad imaging densitometer and quantified using Molecular Analyst software. For fluorescence detection of antibody binding, membranes were blocked in LI-COR blocking buffer (LI-COR Biosciences) and antibody binding was detected using IgG-IRDye 680/800-conjugated secondary antibodies (Millennium Science; 926-32221, 926-32210) in LI-COR blocking buffer and visualized using the LI-COR Odyssey Infrared Fluorescence Imaging system. Fluorescent images were quantified using the LI-COR Odyssey software, and background levels were subtracted from the relative intensity of each band. For all Western blot experiments, all values derived from phosphospecific antibodies were normalized to their respective pan-antibodies, and data for the DHPG-treated samples were expressed relative to the matched no-drug samples for each time point. For all experiments, group means were compared statistically by two-tailed independent Student's t tests at the p < 0.05 significance level.
Hippocampal field potential electrophysiology.
Transverse hippocampal slices (400 μm) were prepared as described previously (Mockett et al., 2004, 2007). In brief, rats were anesthetized with ketamine (100 mg/kg, i.p.) and decapitated, and 400 μm slices were prepared. Slices were then transferred to an oxygenated and humidified static incubation chamber where they were placed on Millicell culture plate inserts (Millipore) in interface contact with aCSF and held at 32°C for at least 2 h to equilibrate. The aCSF consisted of the following (in mm): 124 NaCl, 3.2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 10 d-glucose, and was equilibrated with carbogen (95% O2–5% CO2). Slices were then submerged in a recording chamber through which aCSF equilibrated with carbogen was superperfused continuously at a rate of 2 ml/min and maintained at a temperature of 32.5°C. Baseline field EPSPs (fEPSPs) were elicited in area CA1 by stimulation of the Schaffer collateral–commissural pathway at 0.017 Hz (diphasic pulses, 0.1 ms half-wave duration) using a Teflon-coated 50 μm tungsten wire monopolar electrode (A-M Systems). Pulses were delivered in pairs separated by 50 ms to enable the measurement of paired-pulse facilitation (PPF). Evoked responses were recorded with a glass microelectrode filled with 2 m NaCl (1–2 MΩ) and placed in stratum radiatum of area CA1. During periods of baseline recording, the stimulation intensity was adjusted to elicit a fEPSP with an amplitude of 1.0 mV. Baseline stability was visually assessed immediately before bath perfusion of the drugs and by linear regression at the conclusion of each experiment (Raymond et al., 2000). Group averages included only those slices whose baseline slope, as determined by linear regression, changed by <10% over the 30 min baseline. Chemically mediated Group I mGluR-LTD was induced by bath applying DHPG (100 μm) for 10 min, whereas synaptically mediated mGluR-LTD was induced by paired-pulse stimulation (1200 pairs) delivered at 1 Hz with a 50 ms interpulse interval. Stimulus intensity was increased during this time to evoke a 2 mV EPSP. APV (50 μm) was bath perfused for the duration of the experiment.
Patch-clamp electrophysiology.
Animals were deeply anesthetized with ketamine (100 mg/kg, i.p.), and the brains were removed and chilled in ice-cold oxygenated aCSF for which sucrose was substituted for NaCl (composition in mm: 210 sucrose, 20 glucose, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 3 MgCl2, 2 ascorbic acid, and 3 Na-pyruvate, pH 7.4, when gassed with 95% O2–5% CO2). Transverse hippocampal slices (400 μm) were cut on a Vibroslicer (Campden Instruments) and transferred to a static incubation chamber containing oxygenated aCSF (composition in mm: 124 NaCl, 20 glucose, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 2 ascorbic acid, and 3 Na-pyruvate, pH 7.4, when gassed with 95% O2–5% CO2) at 34°C where they were placed on a Millicell culture plate insert in interface conditions for 30 min (Ireland and Abraham, 2009). The incubation chamber was then allowed to cool to room temperature and the slices held for an additional 90 min. Slices were then individually transferred to a recording chamber and superfused at 2 ml/min with oxygenated aCSF (lacking ascorbic acid and pyruvate) at 32°C.
Standard whole-cell patch-clamp techniques were used to record AMPAR-mediated EPSCs from the cell body of visualized CA1 pyramidal neurons. Patch electrodes were formed from soft borosilicate glass (WPI) and filled with a K+-based electrode solution (composition in mm: 145 K-methanesulfonate, 10 HEPES, 4 Na2ATP, 0.4 NaGTP, 4 MgCl2, 10 Na2 phosphocreatine, 0.2 EGTA-4Na, and 1 QX-314, pH 7.4, when adjusted with CsOH, 310 mOsm when adjusted with water) to give resistances of 2.5–3 MΩ. For experiments in which CaMKII inhibition was required, autocamtide-2-related inhibitory peptide (AIP) (50 μm) was dissolved in the electrode solution. Once whole-cell recording was achieved, at least 40–60 min were allowed for the electrode solution to equilibrate with the cytosol, and for AIP to diffuse to the dendrites, before recordings were commenced. LTD was induced by bath application of DHPG (100 μm, 10 min). Access resistance (typically 10–20 MΩ) was assessed at the beginning and end of each experiment, and experiments that differed by >30% of baseline values were discarded. AMPAR-mediated EPSCs were isolated by bath application of APV (50 μm) and picrotoxin (20 μm) and evoked once every 30 s by electrical stimulation of the Schaffer collaterals via a wide-bore patch electrode filled with aCSF. Single AMPA EPSCs (150–300 pA) were recorded in voltage-clamp mode at a holding potential of −65 mV using an Axopatch 1D amplifier and pCLAMP 9 software (Molecular Devices). Responses were sampled at 10 kHz and filtered at 2 kHz.
Data analysis.
The initial slopes of the fEPSPs were measured and expressed as a percentage change from the baseline level, calculated as the average of the last 15 min of the baseline recording period (Mockett et al., 2002). EPSC peak amplitudes were measured relative to the prestimulus baseline and expressed as a percentage change from the baseline level, calculated from an average of the last 10 min of the baseline recording period (Ireland and Abraham, 2009). The degree of LTD for each experiment was measured as the average of the last 5 min of the post-DHPG recording period, whereas the group means were expressed as the percentage change ± SEM. PPF was calculated as the ratio of the initial slope of the first and second evoked paired-pulse responses and expressed as a percentage change from the average of the last 15 min of the baseline recording period. For all experiments, group means were compared statistically by two-tailed independent Student's t tests and one-way ANOVAs with Dunnett's post hoc tests at the p < 0.05 significance level.
Results
Group I mGluR-activated protein synthesis is CaMKII dependent
To test our central hypothesis that CaMKII mediates an increase in protein synthesis after Group I mGluR activation by DHPG, we measured [35S]methionine incorporation into TCA-precipitated protein in synaptoneurosomes prepared from whole hippocampi and assessed its sensitivity to the global mGluR blocker LY341495. The isolated synaptoneurosome preparation was developed previously for its enrichment of synaptic proteins and its suitability for posttreatment biochemical analysis (Hollingsworth et al., 1985). These features of synaptoneurosomes are particularly pertinent to the focus of the present work as studies have established that synaptodendritic protein synthesis is an integral step in the LTD induced by mGluR activation (Huber et al., 2000). In response to DHPG treatment (20 μm), protein synthesis increased nearly twofold over basal levels (1.92 ± 0.12; n = 14; p = 0.008) (Fig. 1A). This effect was completely abolished by LY341495 (100 μm, 1.0 ± 0.06; n = 4; p < 0.001), as well as concurrent addition of cycloheximide (60 μm, 0.53 ± 0.06; n = 4; p < 0.001 relative to DHPG alone), emetine (30 μm, 0.56 ± 0.04; n = 6; p < 0.001), or rapamycin (20 nm, 0.76 ± 0.06; n = 4; p < 0.001) (Fig. 1A). In the absence of DHPG, the translation inhibitors cycloheximide and emetine, and the mTOR inhibitor rapamycin, reduced basal protein synthesis to approximately one-half of basal “no-drug” levels (cycloheximide, 0.55 ± 0.04, n = 7; emetine, 0.45 ± 0.03, n = 5; rapamycin, 0.52 ± 0.06, n = 4) (Fig. 1B). Notably, DHPG failed to initiate a significant increase in protein synthesis compared with that recorded in the presence of cycloheximide or emetine alone. A small but significant increase in protein synthesis was induced by DHPG (p < 0.05) in the presence of rapamycin, compared with rapamycin alone, although this failed to even reach “no-drug” basal levels. These data are in agreement with previous studies showing that Group I mGluR activation increases synaptic translation (Weiler et al., 1996; Huber et al., 2000) and suggest that DHPG-induced protein synthesis is dependent on both a rapamycin-sensitive mTOR pathway and an alternative pathway (Gallagher et al., 2004; Page et al., 2006).
Figure 1.
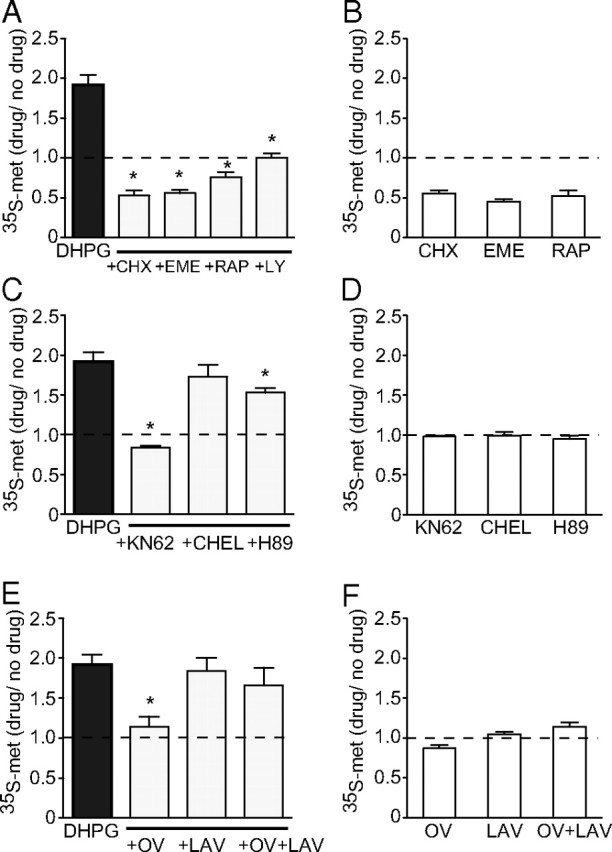
Identification of intracellular pathways involved in the induction and expression of Group I mGluR-dependent protein synthesis. A, DHPG-induced protein synthesis (20 μm) was significantly reduced in synaptoneurosomes by the translation inhibitors cycloheximide (CHX) (60 μm), emetine (EME) (30 μm), and rapamycin (RAP) (20 nm). The presence of the global mGluR blocker LY341495 abolished the DHPG response (LY, 100 μm). B, All three translation inhibitors by themselves reduced basal protein synthesis. C, Inhibition of CaMKII with KN62 (10 μm) caused a significant reduction in induced protein synthesis, whereas inhibition of PKA had a more modest effect (H89, 10 μm). Inhibition of PKC with chelerythrine (CHEL) (1 μm) had no significant effect. D, Neither KN62, CHEL, nor H89 alone had any effect on basal protein synthesis. E, Dephosphorylation of a tyrosine residue is required for the expression of DHPG-induced protein synthesis. Application of the PTP inhibitor orthovanadate (OV) (1 mm) completely prevented the increase in protein synthesis, whereas application of the PTK inhibitor lavendustin (LAV) (10 μm) alone had no significant effect by itself. Importantly, lavendustin blocked the effect of orthovanadate when applied with it (OV+LAV). F, Neither OV, LAV, nor OV+LAV alone had any effect on basal protein synthesis. *Significantly different from no-drug controls (p < 0.05; n = 4–7). Error bars indicate SEM.
Group I mGluRs are known to be linked to a number of intracellular pathways capable of initiating translation. To test our hypothesis that CaMKII is linked to at least one of these pathways, we applied the CaMKII inhibitor KN62 (10 μm) before and during DHPG incubation in synaptoneurosomes. Under these conditions, a complete block of stimulated protein synthesis was observed (0.84 ± 0.02; n = 4; p < 0.001) (Fig. 1C). Applied alone, KN62 had no effect on basal levels of protein synthesis (0.98 ± 0.01; n = 4; p = 0.326) (Fig. 1D).
Group I mGluRs are also known to activate PKC and release calcium from intracellular stores leading to activation of calcium-sensitive adenylate cyclases. To investigate the role of these pathways in mediating DHPG-induced facilitation of protein synthesis, we applied inhibitors of PKC and protein kinase A (PKA) together with DHPG and measured [35S]-methionine incorporation. Inhibition of PKC with chelerythrine chloride (1 μm) had no significant effect on DHPG-induced protein synthesis (1.73 ± 0.15; n = 4; p = 0.257 compared with DHPG alone), whereas inhibition of PKA with H89 (10 μm) induced a modest but significant reduction (1.53 ± 0.06; n = 5; p = 0.008) (Fig. 1C). Neither chelerythrine nor H89 had any effect on basal levels of protein synthesis when applied alone (Fig. 1D). These findings suggest that, of these kinases tested, CaMKII plays a particularly strong role in the facilitation of protein synthesis by DHPG.
In recent studies, an additional possible mechanism mediating mGluR-LTD has emerged. Moult et al. (2002, 2006) have shown that activation of a protein tyrosine phosphatase (PTP) is involved in the expression of mGluR-LTD, possibly through dephosphorylation of the GluA2 AMPAR subunit. We investigated whether this mechanism might also be involved in regulating DHPG-induced protein synthesis. Application of the PTP inhibitor orthovanadate (1 mm) completely abolished the DHPG-induced increase in protein synthesis (1.14 ± 0.13; n = 6; p < 0.001 compared with DHPG alone), whereas the protein tyrosine kinase (PTK) inhibitor lavendustin (10 μm) had no significant effect (1.84 ± 0.16; n = 6; p = 0.53) (Fig. 1E). However, coapplication of lavendustin with orthovanadate prevented the effect of orthovanadate on protein synthesis (1.67 ± 0.22; n = 6; p = 0.278), suggesting that PTP plays a permissive rather than direct role in the regulation of DHPG-induced protein synthesis (Fig. 1E). Neither drug alone, nor in combination, significantly affected basal levels of protein synthesis (Fig. 1F).
Regional regulation of DHPG-induced protein synthesis
Previous studies have revealed regional variations within the hippocampus in the mechanisms mediating DHPG-LTD (Gallagher et al., 2004; Wang et al., 2007). We investigated whether this was also reflected in the facilitation of protein synthesis and the role of CaMKII. We prepared synaptoneurosomes from microdissected dentate gyrus (DG) and CA1 regions and examined the effect of DHPG and selected inhibitors on protein synthesis. In both DG and CA1 synaptoneurosomes, DHPG (20 μm; 10 min) caused a robust increase in protein synthesis similar to that observed in whole hippocampus preparations (DG, 1.92 ± 0.05, n = 4, p < 0.001; CA1, 1.51 ± 0.12, n = 4, p = 0.026) (Fig. 2). Coapplication of a second CaMKII inhibitor (KN93, 10 μm) caused a complete block of DHPG-induced protein synthesis in both the DG (0.80 ± 0.01; n = 4; p < 0.001) and CA1 (0.99 ± 0.03; n = 4; p = 0.026) compared with their respective control groups, thus highlighting the involvement of CaMKII in DHPG-stimulated protein synthesis in both subregions of the hippocampus.
Figure 2.

Comparison of mechanisms regulating DHPG-induced protein synthesis between hippocampal subregions. In synaptoneurosomes prepared from the DG and CA1, DHPG (20 μm) caused a large increase in protein synthesis. This was completely blocked in both regions by the CaMKII inhibitor KN93 (10 μm). Inhibition of the MEK/MAPK pathway with the ERK1/2 inhibitor PD98059 (50 μm) significantly reduced the level of protein synthesis in both DG and CA1, whereas the p38 inhibitor SB203580 (1 μm) was effective only in the DG. Inhibition of PLC with U73122 (10 μm) had no significant effect in either region. *Significantly different from no-drug controls (p < 0.05; n = 4–5). Error bars indicate SEM.
We next examined the contribution of MEK/ERK and p38 MAPK to protein synthesis in CA1 and dentate gyrus. These pathways are activated by Group I mGluRs and have previously been shown to be involved in the initiation of protein synthesis in the hippocampus (Gallagher et al., 2004; Banko et al., 2006; Antion et al., 2008). In both the DG and CA1, the MEK inhibitor PD98059 (50 μm) significantly reduced DHPG-induced protein synthesis (DG, 1.17 ± 0.05, n = 4, p < 0.001; CA1, 1.03 ± 0.04, n = 5, p = 0.022) (Fig. 2). Inhibition of p38 MAPK with SB203580 (1 μm) also caused a significant, but incomplete, reduction in the DG (1.35 ± 0.07; n = 5; p < 0.001), but no significant effect was observed in CA1 (1.30 ± 0.08; n = 5; p = 0.213) (Fig. 2). These findings suggest that, under our conditions, ERK1/2 in particular plays a significant role in DHPG-induced protein synthesis in both the DG and CA1, whereas the p38 MAPK plays a more minor role. These findings are in agreement with some (Rush et al., 2002; Gallagher et al., 2004), but not all previous studies (Huang et al., 2004; Wang et al., 2007) of ERK and p38MAPK contributions to mGluR-LTD, suggesting that experimental factors such as age and induction protocol may affect the pathways activated.
The present findings showing a lack of effect of PKC inhibition, as reported previously (Schnabel et al., 1999a; Rush et al., 2002), suggests that DHPG-LTD is not mediated by the classically described mGluR/PLC/PKC intracellular pathway. To confirm this, we tested whether PLC inhibition blocks DHPG-stimulated protein synthesis. Inhibition of PLC with 10 μm U73122, a dose previously shown to block other actions of DHPG (Cohen et al., 1998), had no significant effect on DHPG-induced protein synthesis in either the DG or CA1, although slight decreases were observed (DG, 1.54 ± 0.16, n = 5, p = 0.074; CA1, 1.36 ± 0.09, n = 5, p = 0.353) (Fig. 2). This classical mGluR-linked pathway, therefore, does not play a major role in mediating the stimulation of protein synthesis by Group I mGluR activation.
Group I mGluR activation increases CaMKII phosphorylation
CaMKII is activated by autophosphorylation at Thr286, leading to an upregulation of activity through rendering the kinase activity calcium independent (Lisman et al., 2002). We used Western blot analysis to determine whether a relative change in phosphorylated CaMKII at Thr286 (phospho-CaMKII) could be detected after exposure of synaptoneurosomes to DHPG. We found that synaptoneurosomes treated with DHPG (20 μm) showed a reduction in phospho-CaMKII levels (0.73 ± 0.08; n = 6; p = 0.02) at the nominal 0 min time point, which was rapidly followed by a biphasic increase in phospho-CaMKII peaking at 5 and 20 min after commencement of DHPG administration. Total levels of CaMKII did not change significantly in response to any period of DHPG treatment (Fig. 3A). Interestingly, the increase in phospho-CaMKII at 5 min (1.25 ± 0.10; n = 6; p = 0.05) was only partially sensitive to coincubation with KN62 (1.14 ± 0.09; n = 5), a value intermediate between the DHPG group and no-drug controls, and not significantly different from either. Together, these data are indicative of an early activation of protein phosphatase activity that is then rapidly inhibited to permit increased CaMKII autophosphorylation, likely driven by a combination of newly activated CaMKII (KN62 sensitive) and existing synaptic pools of basally autophosphorylated CaMKII.
Figure 3.
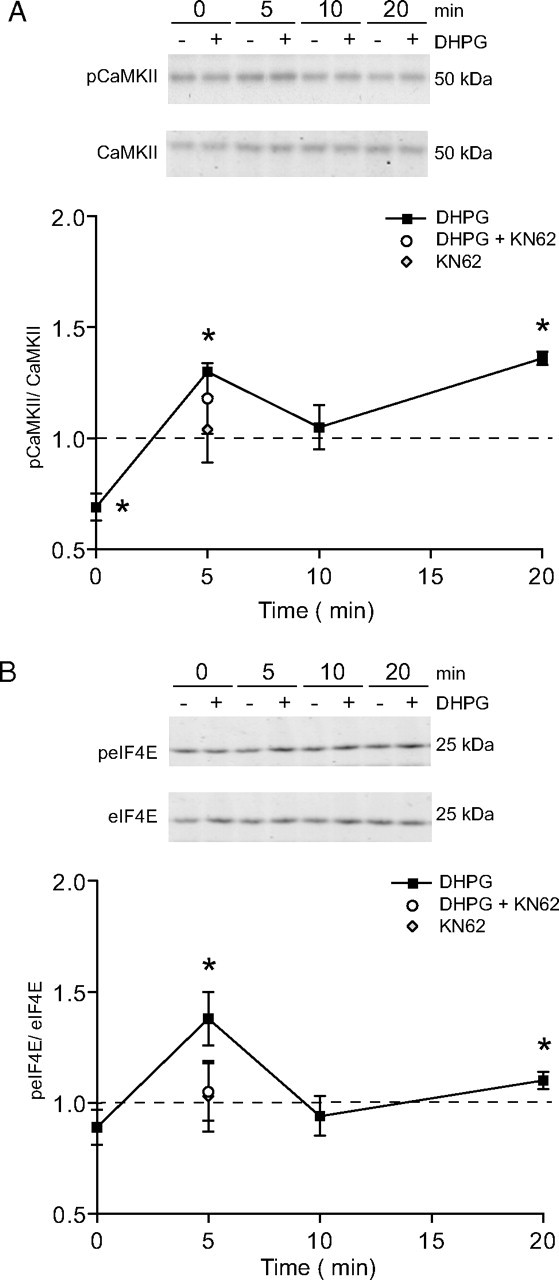
Group I mGluR activation causes an increase in phosphorylated CaMKII and enhances eIF4E phosphorylation in a CaMKII-dependent manner. Representative Western blots and summary data showing a biphasic response in phospho-CaMKII normalized to total CaMKII (A) and phospho-eIF4E normalized to total eIF4E (B) in response to DHPG (20 μm) applied to isolated synaptoneurosomes prepared from whole hippocampi. Total CaMKII and eIF4E levels were not altered by this treatment. KN62 also abolished the DHPG-induced increase in phospho-eIF4E levels (5 min). *Significantly different from no-drug controls (p < 0.05; n = 6). Error bars indicate SEM.
To ensure that the increased CaMKII phosphorylation was not specific to the synaptoneurosome preparation, we tested the effect of DHPG treatment (0, 100 μm; 10 min) on phospho-CaMKII levels in CA1 minislices. In this preparation, we found that DHPG markedly increased the relative level of phospho-CaMKII (2.03 ± 0.29; n = 5; p = 0.023 compared with control slices). This pronounced effect was also attenuated by the CaMKII inhibitor KN62 (10 μm, 1.15 ± 0.14; n = 5; p = 0.336 relative to no drug controls) (data not shown). These results show that Group I mGluR activation of both whole hippocampal synaptoneurosomes as well as isolated CA1 slices leads to a rapid, although transient, increase in CaMKII autophosphorylation.
CaMKII regulation of DHPG effects on protein synthesis factors
The initiation and elongation phases are key control points of protein synthesis, and indeed DHPG is known to rapidly increase phosphorylation of the initiation factor eIF4E within 5 min (Banko et al., 2006). In contrast, dephosphorylation of the elongation control factor eEF2 promotes protein synthesis, but it is not clear whether DHPG affects eEF2 phosphorylation status. Here, we assessed DHPG effects on eIF4E and eEF2 phosphorylation status in synaptoneurosomes, and the role of CaMKII in mediating any effects. We found that neither the phosphorylation status of eEF2 nor its total levels were affected by DHPG treatment (20 μm) (data not shown). Although mGluR-mediated changes in phospho-eEF2 have been described in other preparations (Park et al., 2008), the present data indicate that eEF2 is not modulated by DHPG treatment in rat hippocampal synaptoneurosomes. In contrast, DHPG increased the levels of phospho-eIF4E in a biphasic manner that closely paralleled that of phospho-CaMKII, with the exception of the nominal 0 min time point. Enhanced eIF4E phosphorylation relative to total eIF4E was evident after 5 min of DHPG treatment (1.38 ± 0.12; n = 6; p = 0.02) (Fig. 3B), no longer detectable at 10 min, but modestly elevated again at 20 min (1.10 ± 0.4; n = 6; p = 0.045). Furthermore, we found that the rapid increase in phospho-eIF4E levels was completely blocked by coincubation with KN62 (1.05 ± 0.13; n = 5) (Fig. 3B). KN62 treatment alone had no effect on basal phospho-eIF4E levels (1.04 ± 0.06; n = 6) (Fig. 3B). These data suggest a possible role of the eIF4E phosphorylation pathway in mediating CaMKII-dependent protein synthesis.
Expression of Group I mGluR-mediated LTD is protein synthesis and CaMKII dependent
Having shown that CaMKII is critically involved in the stimulation of protein synthesis, we predicted that CaMKII inhibitors would also block protein synthesis-dependent LTD, as induced by DHPG. To test this, we first monitored the LTD in fEPSPs induced by DHPG (100 μm, 10 min) in stratum radiatum of CA1. DHPG at 50–100 μm has previously been shown to induce a robust LTD that is rapid in onset, specific to Group I mGluR activation, and NMDAR independent (Huber et al., 2001; Schnabel et al., 2001; Huang and Hsu, 2006). In this experiment, we routinely recorded the responses to paired-pulse stimuli (50 ms interstimulus interval) throughout each experiment to look for presynaptic changes in transmitter release probability. We also included APV (50 μm) in the bathing medium throughout the recording period to prevent any NMDAR-dependent effects.
DHPG caused a rapid reduction in synaptic transmission that partially recovered on washout to form a persistent and stable LTD lasting the duration of the 60 min post-DHPG recording period (−36 ± 3.%; n = 7) (Fig. 4). To assess whether or not protein synthesis is required for the expression of this LTD, we perfused the slices with cycloheximide (60 μm) throughout the experiment. This treatment caused a significant, but partial, reduction in the magnitude of both the initial induction and the later expression of the DHPG-induced LTD (−20 ± 6%; n = 5; p = 0.023 compared with DHPG alone). This suggests that Group I mGluR-dependent LTD is only partially dependent on protein synthesis under our conditions and in our age animals (Fig. 4). Analysis of the effect of DHPG on PPF revealed an acute, but transient, increase in the level of facilitation during perfusion, and a gradual decline during washout to a small but stably increased level 60 min after DHPG (9 ± 4%; n = 7; p = 0.007 compared with baseline). PPF in the presence of DHPG plus cycloheximide was not significantly different from DHPG alone (8 ± 4%; n = 5; p = 0.777) (Fig. 4). The fact that the small and presumed presynaptically mediated PPF change caused by DHPG was not affected by cycloheximide indicates that this change is protein synthesis independent. Conversely, the protein synthesis-dependent component of the LTD is probably postsynaptically mediated, supporting previous findings (Nosyreva and Huber, 2005; Huang and Hsu, 2006).
Figure 4.

Inhibition of protein synthesis causes a partial reduction in the magnitude of Group I mGluR-dependent LTD. DHPG-induced LTD (100 μm) was partially reduced by the translation inhibitor cycloheximide (CHX) (60 μm), suggesting that new protein synthesis is required for the full expression of this LTD. No effect on PPF was observed. Inset, Sample fEPSP recordings are the average of 10 traces taken at the times indicated by the corresponding numbers on the average response plot. Calibration: 1 mV, 10 ms.
We then tested whether CaMKII activity was important for the protein synthesis component of the DHPG-induced LTD. In initial experiments, continuous KN62 (10 μm) applied alone induced a slowly developing and nonstabilizing depression in baseline fEPSPs. This effect was not attributable to a general deterioration of the slices as washout of the drug was accompanied by a recovery of transmission back to near baseline levels (−6.8 ± 6.8%; n = 4) (data not shown). This depression had the potential to obscure any influence exerted by KN62 on LTD induced by DHPG. To remove this artifact from later experiments, the average effect of KN62 applied alone was subtracted from each experiment in which DHPG was applied in the presence of KN62. Using this normalization procedure, KN62 caused a significant but incomplete reduction in both the magnitude of the initial depression and in the LTD measured after 60 min of DHPG washout (−14 ± 5%; n = 11; p = 0.006) (Fig. 5A). Notably, this effect was similar in pattern to that observed for inhibition of protein synthesis by cycloheximide (Fig. 1). We predicted that if KN62 blocks the same transduction pathway as the protein synthesis inhibitor, then application of both drugs would give no more reduction of LTD than either one alone. Indeed, combined drug treatment again caused only a partial reduction in the magnitude of the DHPG-induced LTD, no greater than either drug alone (DHPG control, −42 ± 3%, n = 18; KN62 plus cycloheximide, −26 ± 5%, n = 5; p = 0.026) (Fig. 5B). Thus, the influence of CaMKII on DHPG-induced LTD is likely to be exerted through the regulation of protein synthesis rather than through posttranslational modification of existing synaptic proteins. In both experimental protocols, KN62, like cycloheximide, had no effect on the pattern of PPF changes, supporting the view that the protein synthesis-dependent LTD is independent of changes in the probability of transmitter release (Fig. 5A,B).
Figure 5.
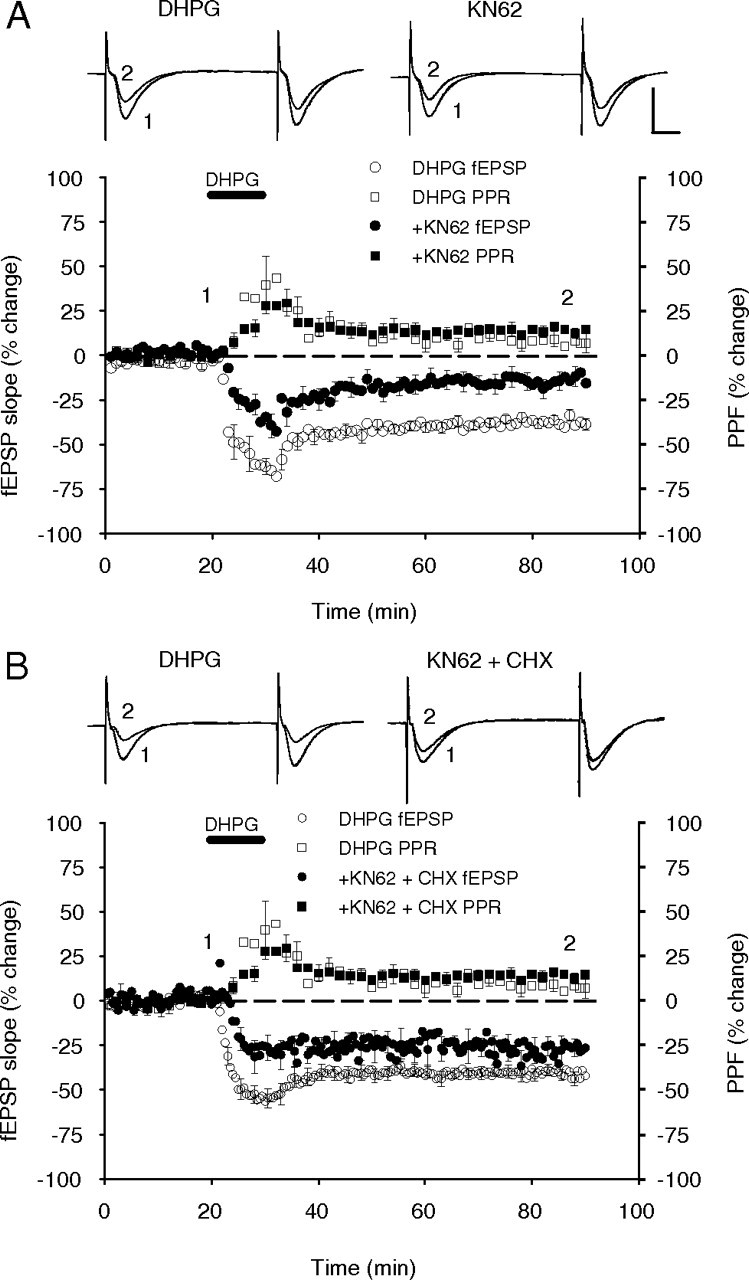
Group I mGluR-dependent LTD is partially dependent on CaMKII activation. A, Inhibition of CaMKII with KN62 (10 μm) caused a significant but incomplete reduction in the magnitude of DHPG-induced (100 μm) LTD. No effect on the PPF was observed. B, Combined inhibition of CaMKII activity with KN62 and protein synthesis with cycloheximide (CHX) (60 μm) did not induce a cumulative reduction of DHPG-induced LTD, suggesting these drugs targeted the same pathway. No significant effect on the PPF was observed. Inset, Sample fEPSP recordings are the average of 10 traces taken at the times indicated by the corresponding numbers on the average response plot. Calibration: 1 mV, 10 ms.
Recent reports have suggested that chemically and synaptically induced mGluR-LTD are expressed through similar, but not identical, mechanisms (Naie et al., 2007; Moult et al., 2008). We therefore considered the possibility that the more physiologically relevant synaptically induced mGluR-LTD may have different requirements for CaMKII activity. To examine this question, we induced mGluR-LTD using a synaptic stimulation protocol consisting of paired pulses delivered at 1 Hz [paired-pulse low-frequency stimulation (PP-LFS)] in the presence of APV (Kemp and Bashir, 1999; Moult et al., 2008). In these experiments, paired-pulse synaptic stimulation invoked an acute depression of basal transmission that partially recovered to form a persistent and stable LTD measured 75 min after PP-LFS (−22 ± 4%; n = 11) (Fig. 6). Coadministration of the global mGluR blocker LY341495 (100 μm) blocked LTD induction demonstrating that the depression was a mGluR-dependent LTD (13 ± 7%; n = 4) (data not shown). Notably, synaptically induced mGluR-LTD was significantly reduced when induced in the presence of KN62 (10 μm, −8 ± 3%; n = 7; p = 0.015) (Fig. 6). Collectively, these results indicate that induction of mGluR-LTD in CA1 by either protocol involves a common signal transduction mechanism that requires the activation of CaMKII.
Figure 6.

mGluR-LTD induced with PP-LFS is partially dependent on CaMKII activation. Synaptic stimulation using paired-pulse stimulation (1200 pairs, 50 ms interstimulus interval) delivered at 1 Hz in the presence of APV induced an LTD that was mGluR dependent, as it was completed blocked by the application of LY341495 (data not shown). Stimulation delivered in the presence of KN62 partially blocked LTD expression indicating that, like DHPG-induced LTD, synaptically induced mGluR-LTD is dependent on CaMKII activation.
To establish whether these effects were specifically the result of CaMKII activity and not other CaMKs affected by the KN drug family, we applied the specific CaMKII inhibitor AIP (50 μm) via the electrode solution in whole-cell patch-clamp experiments. In these experiments, DHPG induced an LTD of AMPAR-mediated EPSCs (−50 ± 5%; n = 10) that was significantly reduced by AIP (−26 ± 5%; n = 10; p = 0.003) (Fig. 7C). The partial effect on LTD was consistent with the partial effect observed in the field potential experiments. Application of the AMPAR blocker CNQX (10 μm) after DHPG washout confirmed these were AMPAR-mediated currents (Fig. 7A,B). Together, these experiments strongly support a role for CaMKII in mediating DHPG-induced LTD.
Figure 7.
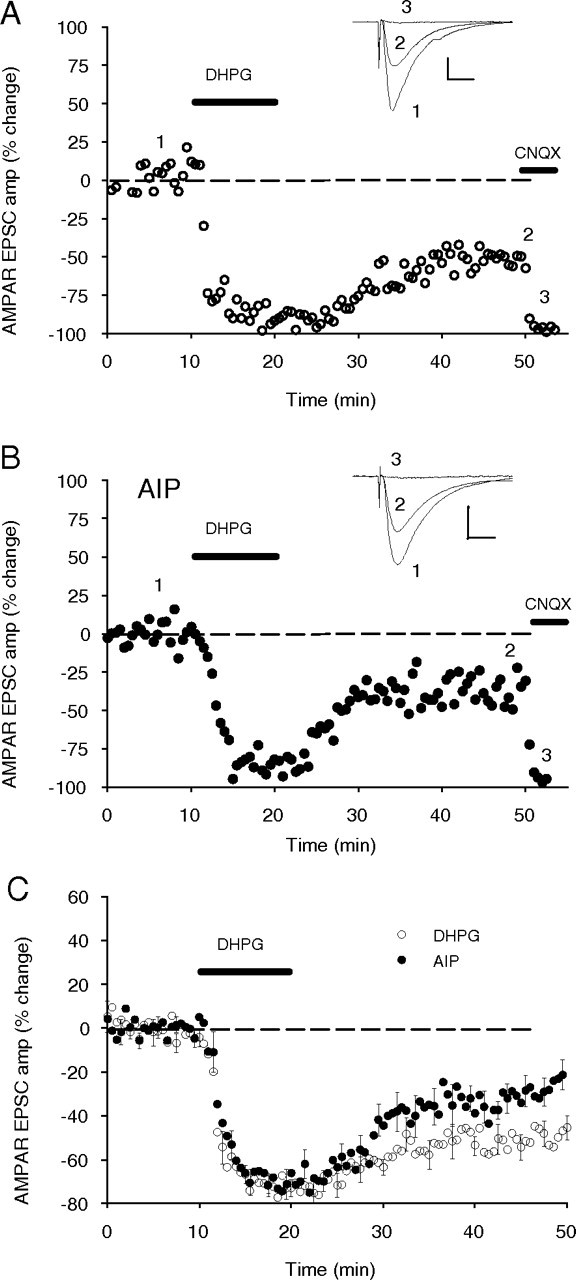
Inhibition of CaMKII with intracellularly applied AIP reduces the magnitude of the DHPG-induced LTD. A, Single experiment showing DHPG (100 μm), in the presence of bath-applied picrotoxin (100 μm) and APV (50 μm), induced an acute depression of the EPSCAMPA, which stabilized to a persistent depression as measured after 30 min of washout (−51%). Subsequent application of CNQX (10 μm) almost completely abolished this current (−97% of post-DHPG baseline) showing it was primarily AMPAR mediated. Inset, Sample EPSP recordings are the average of 10 traces taken at the times indicated by the corresponding numbers. Calibration: 10 ms, 150 pA. B, Single experiment as in A, except that AIP was included in the patch pipette internal solution (50 μm) and allowed to diffuse into the cell for 60 min before the application of DHPG. LTD of the EPSCAMPA was reduced to −39%. CNQX reduced the current to −95% of the post-DHPG baseline. Calibration: 10 ms, 100 pA. C, AIP (50 μm) significantly reduced the depression induced by DHPG (100 μm).
Classical Group I mGluR-activated second messengers do not mediate DHPG-induced LTD
In the final series of experiments, we tested the hypothesis that inhibitors of PKC and PLC, which did not affect the DHPG facilitation of protein synthesis, would also not affect DHPG-induced LTD. The PKC inhibitor chelerythrine, applied 60 min before the DHPG, induced a slowly developing depression in baseline transmission. This had stabilized by the time of DHPG delivery and, in inhibitor alone experiments, the depression reversed back to baseline after washout (data not shown), suggesting it was a nontoxic effect on synaptic function. In support of our previous biochemical data, chelerythrine (10 μm) did not significantly alter the initial induction or the maintenance of DHPG-induced LTD (−37 ± 4%; n = 5; p = 0.82) (Fig. 8A). Surprisingly, inhibition of PLC with U73122 (10 μm) significantly facilitated DHPG-induced LTD (−51 ± 3%; n = 4; p = 0.007). This effect appears to have resulted from an earlier stabilization of the LTD after DHPG washout rather than a greater level of LTD induction (Fig. 8B). Perfusion with the inactive analog U73343 (10 μm) had no effect on DHPG-induced LTD (−43 ± 6%; n = 7; p = 0.86) (Fig. 8C). PPF after DHPG perfusion was not altered by chelerythrine treatment compared with controls, but was significantly elevated in the U73122 perfused group, consistent with the increased LTD (control, 9 ± 4%, n = 7; U73122, 34 ± 9%, n = 4; p = 0.009) (Fig. 8B). Figure 9 summarizes the effects of DHPG, alone and in combination with enzyme inhibitors, on fEPSP LTD (Fig. 9A) and PPF (Fig. 9B).
Figure 8.
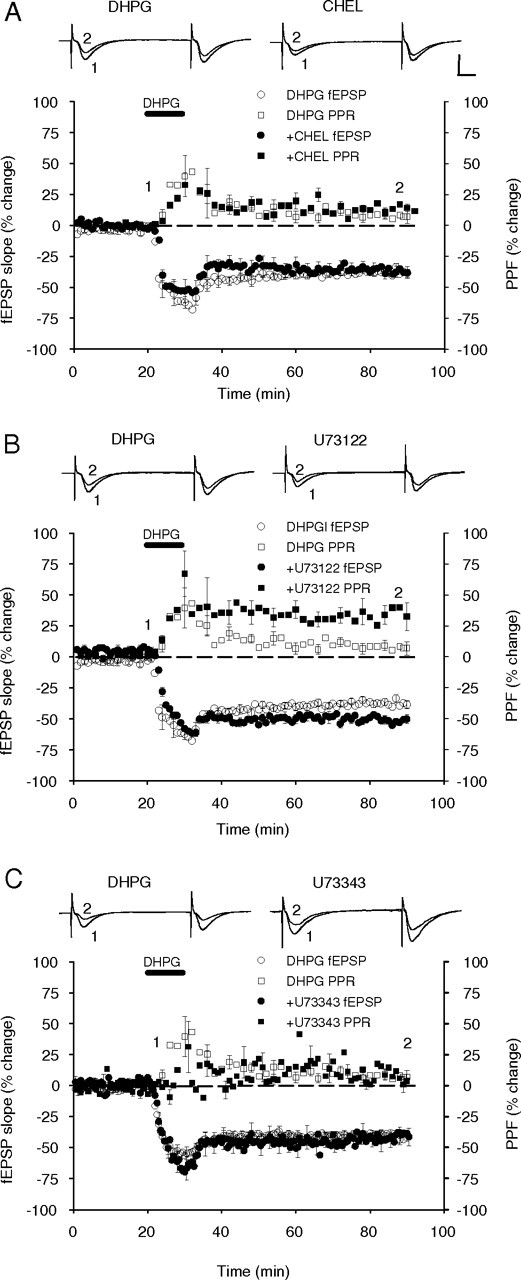
Inhibition of intracellular enzymes activated by Group I mGluR activation has differential effects on DHPG-induced LTD. A, Inhibition of PKC with chelerythrine (CHEL, 10 μm) had no significant effect on DHPG-induced LTD (100 μm) or on the PPF. B, Inhibition of PLC with U73122 (10 μm) induced a significant increase in DHPG-induced LTD and the PPF. C, U73343 (10 μm), the inactive analog of U73122, had no significant effect on either parameter. Inset, Sample fEPSP recordings are the average of 10 traces taken at the times indicated by the corresponding numbers on the average response plot. Calibration: 1 mV, 10 ms.
Figure 9.
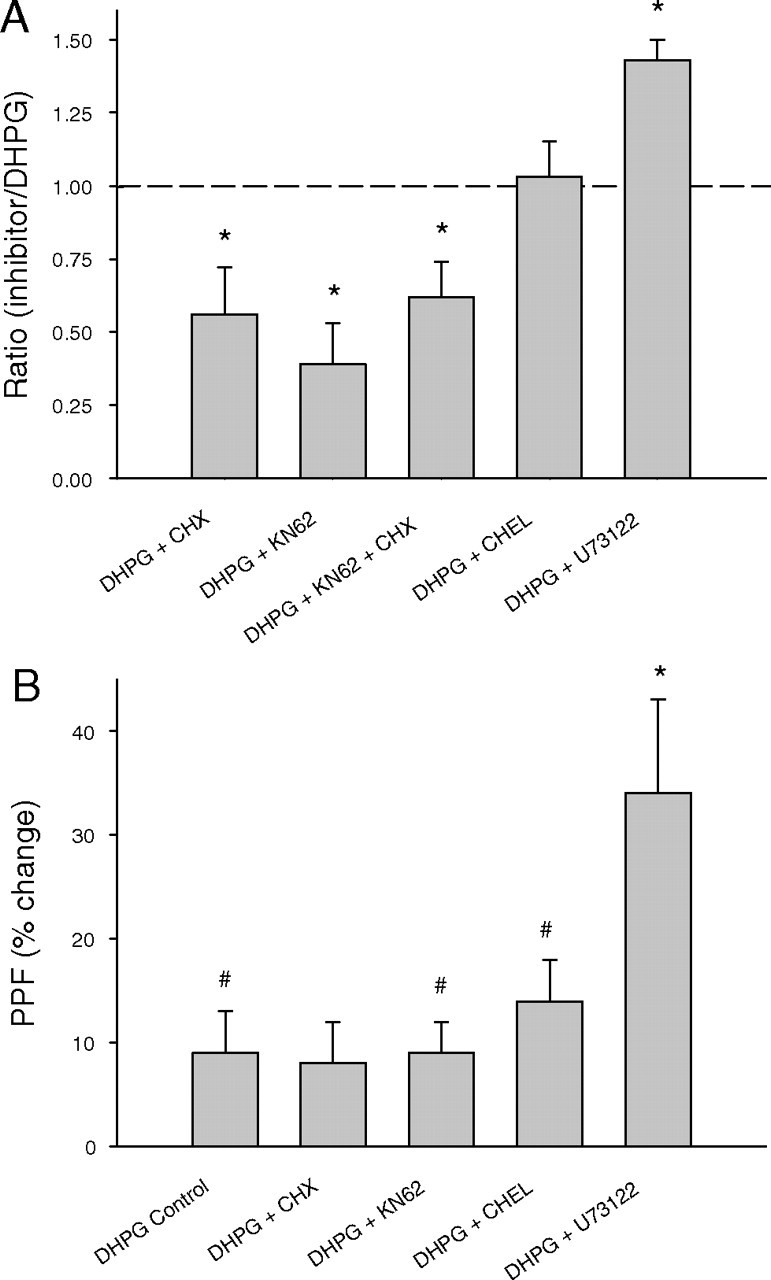
Summary histograms showing the effect of intracellular pathway inhibitors on DHPG-induced LTD and PPF. A, Inhibition of PLC increased the magnitude of the LTD, whereas both CaMKII activity and mRNA translation are necessary for its full expression. Inhibition of PKC had no significant effect. Data are mean ratio of inhibitor LTD relative to LTD induced by DHPG alone. B, PLC inhibition, but not PKC, CaMKII, or translation inhibition, significantly altered the PPF, suggesting that PLC facilitation of LTD is mediated presynaptically. *Significantly different from DHPG control; #significantly different from pre-DHPG baseline (p < 0.05). Error bars indicate SEM.
Discussion
The present work was designed to examine a potential role for CaMKII in mediating the effects of Group I mGluR activation on biochemical and electrophysiological parameters of neuronal function of hippocampal pyramidal cells, given the conflicting findings that have surrounded the issue of Group I mGluR–CaMKII coupling (Schnabel et al., 1999b; Choe and Wang, 2001b). We thus determined whether DHPG could elicit an increase in phosphorylation of CaMKII, a step critical to its activation (Lisman et al., 2002), and whether inhibition of this and other enzymes linked to Group I mGluR signal transduction could reduce the stimulation of protein synthesis (Huber et al., 2000; Raymond et al., 2000) and induction of LTD that is triggered by DHPG (Huber et al., 2001). We found that DHPG induced a rapid reduction in phosphorylated CaMKII levels, which was followed by a biphasic rise in phospho-CaMKII with increased phospho-CaMKII being first evident at 5 min, followed by a second phase of increase beginning 20 min after drug delivery. This shows that pharmacological activation of Group I mGluRs in the hippocampus rapidly regulates CaMKII activity.
To test for functional effects of activated CaMKII, we examined its contribution to DHPG-stimulated protein synthesis. We confirmed (Huber et al., 2000) that activation of Group I mGluRs with DHPG in the hippocampus can induce a rapid rise in protein synthesis in both acute slices and in synaptoneurosomes. That the elevated protein synthesis could be blocked by the translation inhibitors emetine and cycloheximide, and with the mTOR inhibitor rapamycin, as well as the general mGluR blocker LY341495, is in agreement with previous reports demonstrating activation of somatodendritic protein synthesis machinery by Group I mGluR activation (Weiler et al., 1996; Raymond et al., 2000) and that at least one pathway leading to translation initiation involves activation of mTOR (Hou and Klann, 2004; Gong et al., 2006). Importantly, two inhibitors of CaMKII, KN62 and KN93, which had no effect on basal protein synthesis, completely abolished the effects of DHPG. Moreover, KN62, and a third CaMKII inhibitor, AIP, significantly reduced DHPG-induced LTD. The use of three CaMKII inhibitors in this study was designed to resolve uncertainties regarding their CaMKII specificity (Sihra and Pearson, 1995; Baraldi et al., 2000; Gao et al., 2006). AIP in particular has very high specificity for CaMKII (Ishida and Fujisawa, 1995; Ishida et al., 1995) and the present findings with this inhibitor strongly support those of KN62 and KN93. Collectively, these findings suggest that CaMKII is an essential mediator of DHPG-induced translation initiation leading to de novo protein synthesis and associated LTD.
Our findings of a coupling between Group I mGluRs and CaMKII activation are in accord with the observation that in vivo infusion of DHPG into rat striatum increased phosphorylated CaMKII immunoreactivity (Choe and Wang, 2001b). This effect was significantly attenuated by simultaneous infusion of KN62, as was the increased phosphorylation of the translation regulator ERK, as well as Elk-1 and CREB. Although this appears to be the only other study demonstrating a link between Group I mGluR activation and CaMKII phosphorylation, more recent studies add support to the proposed regulatory role of CaMKII in protein synthesis. Thus, both translation regulators ERK1/2 and mTOR are phosphorylated after Group I mGluR activation in striatum and cortex (Choe and Wang, 2001a; Gong et al., 2006; Volk et al., 2006; Antion et al., 2008), and CaMKII also activates these regulators (Vanhoutte et al., 1999), although the role of Group I mGluRs was not addressed in this latter study. Together with the present study, these studies indicate that Group I mGluR activation of protein synthesis via the MEK–ERK1/2–Mnk1 and PI3K–Akt–mTOR signaling cascades involves activation of CaMKII.
The mechanism by which CaMKII contributes to protein synthesis remains unclear. As phosphorylation of eIF4E closely paralleled that of the early-phase CaMKII phosphorylation, and was dependent on CaMKII activity, this implies that CaMKII may contribute to the MAPK cascade. A potential mechanism by which this occurs may be via CaMKII-dependent phosphorylation of PEA15, a protein that functions to sequester ERK and that has recently been implicated in spatial learning (Ramos et al., 2009). As PEA15 binding to ERK is inhibited by phosphorylation mediated by either CaMKII or PKC (Kubes et al., 1998), phosphorylation of PEA15 represents a potential pathway linking activation of CaMKII to ERK and therefore the regulation of protein translation. Alternatively, CaMKII may increase protein synthesis through activation of the polyadenylation factor CPEB (cytoplasmic polyadenylation element-binding protein) (Atkins et al., 2004, 2005; Shin et al., 2004).
Mechanism of CaMKII activation
The mechanism of CaMKII activation appears to involve a combination of calcium/calmodulin-dependent and -independent pathways since it was only partially blocked by KN62. The pattern of immediate dephosphorylation followed by increased phosphorylation bears a strong inverse correspondence to the pattern of PP2A activation by DHPG (Narayanan et al., 2007), suggesting that the CaMKII activation is under strong control by this phosphatase, known to regulate specifically soluble CaMKII (Strack et al., 1997), and possibly mGluR-LTD (Schnabel et al., 2001). As KN62 and KN93 completely blocked the DHPG-induced increase in protein synthesis and LTD, it appears that newly activated kinase is critical for these outcomes. This conclusion is supported by previous studies showing that mGluR-LTD is blocked by postsynaptic calcium chelators (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997) (but see Fitzjohn et al., 2001). The source of the calcium driving CaMKII activation, however, remains unclear. The classical pathway for mGluR-mediated rises in calcium is via PLC activation and release of calcium from intracellular stores via IP3. However, in the present study, an inhibitor of PLC only slightly reduced DHPG-stimulated protein synthesis and had no blocking effect on the LTD, consistent with the non-PLC-mediated contribution of MAP kinase to mGluR-stimulated protein synthesis (Klann and Dever, 2004) and LTD (Gallagher et al., 2004). A potential alternative source of calcium entry is via G-protein-independent activation of calcium-permeable transient receptor potential channels (Gee et al., 2003).
Role of tyrosine phosphorylation in regulating DHPG-induced protein synthesis
We have shown previously that DHPG blocks the slow afterhyperpolarization in CA1 pyramidal neurons by a tyrosine phosphorylation gating mechanism, such that although the DHPG blocking effect is reduced by a tyrosine phosphatase inhibitor, the effect is restored if a tyrosine kinase inhibitor is coapplied with the phosphatase inhibitor (Ireland et al., 2004). A similar pattern of results was reported for DHPG-induced LTD (Moult et al., 2008). In the present experiments, we likewise observed a block of DHPG-induced protein synthesis by orthovanadate that was prevented by coadministration of the kinase inhibitor lavendustin. Collectively, these findings suggest that the tyrosine phosphorylation state of an unknown protein (enhanced by orthovanadate through disinhibition of tyrosine kinases) gates both DHPG-induced protein synthesis and LTD.
CaMKII and the presynaptic versus postsynaptic expression of DHPG-LTD
In addition to blocking the DHPG-induced protein synthesis, CaMKII inhibitors also blocked approximately one-half of the DHPG-induced and synaptically induced LTD. This contrasts with a previous report (Schnabel et al., 1999b) in which KN62 facilitated DHPG-LTD. The reasons for these discrepant results are not clear. However, the present finding is consistent with our other observations that CaMKII mediates DHPG-induced protein synthesis, that a protein synthesis inhibitor also only blocked ∼50% of the LTD, and that the KN62 and cycloheximide effects on LTD were not additive.
The locus of the expression mechanism of DHPG-LTD in CA1 has been the source of considerable debate. Recent reports indicate that differences in findings may be attributable to developmental age differences, as a shift occurs from a presynaptic locus during the first few postnatal weeks to a protein synthesis-dependent postsynaptic locus in adolescent and adult rats,tk;1 (Nosyreva and Huber, 2005; Kumar and Foster, 2007). Under our conditions and using young adult rats 6–10 weeks of age, we seemed to observe an LTD containing both presynaptic and postsynaptic components. For example, the DHPG-LTD was accompanied by a rise in PPF of ∼10% above baseline, suggesting that presynaptic expression mechanisms were engaged. However, protein synthesis and postsynaptic CaMKII inhibitors partially blocked the LTD without affecting the PPF enhancement, thus revealing a postsynaptic and protein synthesis-dependent component to the LTD, as previously shown by Huber et al. (2000, 2001). In contrast, we made the new observation that PLC inhibition significantly increased DHPG-induced LTD and PPF changes, indicating a facilitation of the presynaptic form of mGluR-LTD. Modulation of neurotransmitter release induction by PLC has previously been reported (Diamant et al., 1990).
Summary
Emerging evidence indicates that CaMKII may play an important role in mediating the initiation of DHPG-induced protein synthesis. Our data provide direct support for this view by showing that DHPG induces CaMKII phosphorylation and that CaMKII inhibition blocks DHPG-induced protein synthesis and LTD. Other studies have shown that Group I mGluR activation increases ERK1/2 and mTOR phosphorylation, possibly through an increase in CaMKII activity, and that CaMKII may regulate the activity of translation factors involved in the expression of DHPG-LTP-related proteins. Additional studies are required, however, to establish which specific substrates are targets for CaMKII and what their roles are in the ultimate expression of DHPG-LTD.
Footnotes
This work was supported by grants from the New Zealand Health Research Council and the New Zealand Marsden Fund.
References
- Antion MD, Hou L, Wong H, Hoeffer CA, Klann E. mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol Cell Biol. 2008;28:2996–3007. doi: 10.1128/MCB.00201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Nozaki N, Shigeri Y, Soderling TR. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2004;24:5193–5201. doi: 10.1523/JNEUROSCI.0854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Davare MA, Oh MC, Derkach V, Soderling TR. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1 during hippocampal long-term potentiation. J Neurosci. 2005;25:5604–5610. doi: 10.1523/JNEUROSCI.5051-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi PG, Romagnoli R, Tabrizi MA, Falzoni S, di Virgilio F. Synthesis of conformationally constrained analogues of KN62, a potent antagonist of the P2X7-receptor. Bioorg Med Chem Lett. 2000;10:681–684. doi: 10.1016/s0960-894x(00)00083-4. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ. Group I metabotropic glutamate receptor activation increases phosphorylation of cAMP response element-binding protein, Elk-1, and extracellular signal-regulated kinases in rat dorsal striatum. Brain Res Mol Brain Res. 2001a;94:75–84. doi: 10.1016/s0169-328x(01)00217-0. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ. Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci Lett. 2001b;313:129–132. doi: 10.1016/s0304-3940(01)02258-3. [DOI] [PubMed] [Google Scholar]
- Claasen AM, Guévremont D, Mason-Parker SE, Bourne K, Tate WP, Abraham WC, Williams JM. Secreted amyloid precursor protein-alpha upregulates synaptic protein synthesis by a protein kinase G-dependent mechanism. Neurosci Lett. 2009;460:92–96. doi: 10.1016/j.neulet.2009.05.040. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Raymond CR, Abraham WC. Priming of long-term potentiation induced by activation of metabotropic glutamate receptors coupled to phospholipase C. Hippocampus. 1998;8:160–170. doi: 10.1002/(SICI)1098-1063(1998)8:2<160::AID-HIPO8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Diamant S, Schwartz L, Atlas D. Potentiation of neurotransmitter release coincides with potentiation of phosphatidyl inositol turnover. A possible in vitro model for long term potentiation. Neurosci Lett. 1990;109:140–145. doi: 10.1016/0304-3940(90)90552-k. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U. Group I metabotropic glutamate receptors activate a calcium-sensitive TRP-like conductance in rat hippocampus. J Physiol. 2003;546:655–664. doi: 10.1113/jphysiol.2002.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem. 2006;96:179–194. doi: 10.1111/j.1471-4159.2005.03527.x. [DOI] [PubMed] [Google Scholar]
- Huang CC, You JL, Wu MY, Hsu KS. Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI. Rab5 complex. Potential role in (S)-3,5-dihydroxyphenylglycene-induced long-term depression. J Biol Chem. 2004;279:12286–12292. doi: 10.1074/jbc.M312868200. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC. Mechanisms of group I mGluR-dependent long-term depression of NMDA receptor-mediated transmission at Schaffer collateral-CA1 synapses. J Neurophysiol. 2009;101:1375–1385. doi: 10.1152/jn.90643.2008. [DOI] [PubMed] [Google Scholar]
- Ireland DR, Guevremont D, Williams JM, Abraham WC. Metabotropic glutamate receptor-mediated depression of the slow afterhyperpolarization is gated by tyrosine phosphatases in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2004;92:2811–2819. doi: 10.1152/jn.01236.2003. [DOI] [PubMed] [Google Scholar]
- Ishida A, Fujisawa H. Stabilization of calmodulin-dependent protein kinase II through the autoinhibitory domain. J Biol Chem. 1995;270:2163–2170. doi: 10.1074/jbc.270.5.2163. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- Kubes M, Cordier J, Glowinski J, Girault JA, Chneiweiss H. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes: identification of Ser104 and Ser116 phosphorylated, respectively, by protein kinase C and calcium/calmodulin kinase II in vitro. J Neurochem. 1998;71:1307–1314. doi: 10.1046/j.1471-4159.1998.71031307.x. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Shift in induction mechanisms underlies an age-dependent increase in DHPG-induced synaptic depression at CA3 CA1 synapses. J Neurophysiol. 2007;98:2729–2736. doi: 10.1152/jn.00514.2007. [DOI] [PubMed] [Google Scholar]
- Lenz G, Avruch J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem. 2005;280:38121–38124. doi: 10.1074/jbc.C500363200. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Mockett B, Coussens C, Abraham WC. NMDA receptor-mediated metaplasticity during the induction of long-term depression by low-frequency stimulation. Eur J Neurosci. 2002;15:1819–1826. doi: 10.1046/j.1460-9568.2002.02008.x. [DOI] [PubMed] [Google Scholar]
- Mockett BG, Brooks WM, Tate WP, Abraham WC. Dopamine D1/D5 receptor activation fails to initiate an activity-independent late-phase LTP in rat hippocampus. Brain Res. 2004;1021:92–100. doi: 10.1016/j.brainres.2004.06.039. [DOI] [PubMed] [Google Scholar]
- Mockett BG, Guévremont D, Williams JM, Abraham WC. Dopamine D1/D5 receptor activation reverses NMDA receptor-dependent long-term depression in rat hippocampus. J Neurosci. 2007;27:2918–2926. doi: 10.1523/JNEUROSCI.0838-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Schnabel R, Kilpatrick IC, Bashir ZI, Collingridge GL. Tyrosine dephosphorylation underlies DHPG-induced LTD. Neuropharmacology. 2002;43:175–180. doi: 10.1016/s0028-3908(02)00110-7. [DOI] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Corrêa SA, Collingridge GL, Fitzjohn SM, Bashir ZI. Co-activation of p38 mitogen-activated protein kinase and protein tyrosine phosphatase underlies metabotropic glutamate receptor-dependent long-term depression. J Physiol. 2008;586:2499–2510. doi: 10.1113/jphysiol.2008.153122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naie K, Tsanov M, Manahan-Vaughan D. Group I metabotropic glutamate receptors enable two distinct forms of long-term depression in the rat dentate gyrus in vivo. Eur J Neurosci. 2007;25:3264–3275. doi: 10.1111/j.1460-9568.2007.05583.x. [DOI] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group 1 mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Page G, Khidir FA, Pain S, Barrier L, Fauconneau B, Guillard O, Piriou A, Hugon J. Group I metabotropic glutamate receptors activate the p70S6 kinase via both mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK 1/2) signaling pathways in rat striatal and hippocampal synaptoneurosomes. Neurochem Int. 2006;49:413–421. doi: 10.1016/j.neuint.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos JW, Townsend DA, Piarulli D, Kolata S, Light K, Hale G, Matzel LD. Deletion of PEA-15 in mice is associated with specific impairments of spatial learning abilities. BMC Neurosci. 2009;10:134. doi: 10.1186/1471-2202-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Thompson VL, Tate WP, Abraham WC. Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci. 2000;20:969–976. doi: 10.1523/JNEUROSCI.20-03-00969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci. 2002;22:6121–6128. doi: 10.1523/JNEUROSCI.22-14-06121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Palmer MJ, Kilpatrick IC, Collingridge GL. A CaMKII inhibitor, KN-62, facilitates DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999a;38:605–608. doi: 10.1016/s0028-3908(98)00229-9. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999b;38:1585–1596. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–1101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CY, Kundel M, Wells DG. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J Neurosci. 2004;24:9425–9433. doi: 10.1523/JNEUROSCI.2457-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihra TS, Pearson HA. Ca/calmodulin-dependent kinase II inhibitor KN62 attenuates glutamate release by inhibiting voltage-dependent Ca2+-channels. Neuropharmacology. 1995;34:731–741. doi: 10.1016/0028-3908(95)00051-7. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pagès C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chang L, Rowan MJ, Anwyl R. Developmental dependence, the role of the kinases p38 MAPK and PKC, and the involvement of tumor necrosis factor-R1 in the induction of mGlu-5 LTD in the dentate gyrus. Neuroscience. 2007;144:110–118. doi: 10.1016/j.neuroscience.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Waung MW, Huber KM. Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr Opin Neurobiol. 2009;19:319–326. doi: 10.1016/j.conb.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Childers WS, Greenough WT. Calcium ion impedes translation initiation at the synapse. J Neurochem. 1996;66:197–202. doi: 10.1046/j.1471-4159.1996.66010197.x. [DOI] [PubMed] [Google Scholar]
- Williams JM, Mason-Parker SE, Abraham WC, Tate WP. Biphasic changes in the levels of N-methyl-d-aspartate receptor-2 subunits correlate with the induction and persistence of long-term potentiation. Brain Res Mol Brain Res. 1998;60:21–27. doi: 10.1016/s0169-328x(98)00154-5. [DOI] [PubMed] [Google Scholar]
- Williams JM, Guévremont D, Kennard JT, Mason-Parker SE, Tate WP, Abraham WC. Long-term regulation of N-methyl-d-aspartate receptor subunits and associated synaptic proteins following hippocampal synaptic plasticity. Neuroscience. 2003;118:1003–1013. doi: 10.1016/s0306-4522(03)00028-9. [DOI] [PubMed] [Google Scholar]
- Williams JM, Guévremont D, Mason-Parker SE, Luxmanan C, Tate WP, Abraham WC. Differential trafficking of AMPA and NMDA receptors during long-term potentiation in awake adult animals. J Neurosci. 2007;27:14171–14178. doi: 10.1523/JNEUROSCI.2348-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]