

Abstract
Brain-derived neurotrophic factor (BDNF), secreted from target tissues, binds and activates TrkB receptors, located on axonal terminals of the innervating neurons, and thereby initiates retrograde signaling. Long-range anterograde transport of TrkB in axons and dendrites requires kinesin-mediated transport. However, it remains unknown whether anterograde TrkB transport mechanisms are the same in axons versus in dendrites. Here, we show that c-Jun NH2-terminal kinase-interacting protein 3 (JIP3) binds directly to TrkB, via a minimal 12 aa domain in the TrkB juxtamembrane region, and links TrkB to kinesin-1. The JIP3/TrkB interaction selectively drives TrkB anterograde transport in axons but not in dendrites of rat hippocampal neurons. Moreover, we find that TrkB axonal transport mediated by JIP3 could regulate BDNF-induced Erk activation and axonal filopodia formation. Our findings demonstrate a role for JIP3-mediated TrkB anterograde axonal transport in recruiting more TrkB into distal axons and facilitating BDNF-induced retrograde signaling and synapse modulation, which provides a novel mechanism of how the TrkB anterograde transport can be coupled to BDNF signaling in distal axons.
Introduction
Neurons develop axons and dendrites to communicate with distant cells. The precise targeting and localization of proteins within these domains are critical to every aspect of neuronal function (Hirokawa, 1998; Burack et al., 2000). Long-range anterograde transport of molecules in axons and dendrites is mainly mediated by microtubule-dependent motors, kinesins (Guzik and Goldstein, 2004). In neurons, conventional kinesin (kinesin-1), which consists of two heavy chains (KHC) and two light chains (KLC) (Vale et al., 1985; Hirokawa, 1998), is a multifunctional transporter of both axonal cargo such as synapsin and GAP43 (Ferreira et al., 1992) and dendritic cargo such as mRNA (Severt et al., 1999) and the AMPA receptor (Kim and Lisman, 2001). Several binding partners for kinesin-1 have been identified that are soluble adaptor proteins and could mediate the attachment of membrane-bound cargoes to kinesin-1 (Hammond et al., 2008; Verhey and Hammond, 2009). However, in neurons, the cargo proteins transported by kinesin-1 remain primarily unknown.
Neurotrophins have been considered to play important roles in the differentiation, neurite outgrowth, and survival of a variety of neurons, as well as in modulating the function of neural circuits (Bibel and Barde, 2000; Huang and Reichardt, 2001; Poo, 2001; Chao, 2003). In particular, brain-derived neurotrophic factor (BDNF) plays an essential role in activity-dependent changes in synaptic function (Lu, 2003; Nagappan and Lu, 2005). BDNF, released from target tissues, binds and activates TrkB receptors located on axonal terminals of the innervating neurons and thereby initiates retrograde signaling (Segal, 2003). Therefore, BDNF signaling in neurons depends on proper cellular processing, transport, and localization of its TrkB receptor (Huang and Reichardt, 2003). Delivery of TrkB in the anterograde direction along axons or dendrites (from soma to nerve terminals) requires kinesin-1-mediated transport (Butowt and von Bartheld, 2007). It is not known whether the mechanisms responsible for TrkB anterograde transport in neurons are different in axons versus dendrites. Recently, it was reported that TrkB transport can be mediated by an adaptor complex comprising Slp1, Rab27B, and CRMP-2 to kinesin-1 and can be anterogradely transported (Arimura et al., 2009). However, knockdown of any of these adaptors only reduces axonal targeting by 30–50%, suggesting the existence of additional transport mechanisms.
c-Jun NH2-terminal kinase (JNK)-interacting protein 3 (JIP3) is exclusively expressed in the brain and distributed in dendrites, perikarya, and axons of neurons (Miura et al., 2006). JIP3 was originally identified as a JNK-binding protein and functions as a scaffold protein to trigger specific JNK signaling modules (Ito et al., 1999; Kelkar et al., 2000). Subsequent genetic studies revealed that JIP3 is an ortholog of both Drosophila Sunday driver (syd) and the Caenorhabditis elegans UNC-16, which are implicated as adaptor proteins in kinesin-dependent cargo transport to axons (Bowman et al., 2000; Byrd et al., 2001). It has been shown that JNK3 can bind to JIP3 and be trafficked in the anterograde axonal transport pathways (Cavalli et al., 2005). However, it remains to be determined whether JIP3 can directly bind to other cargoes and mediate their anterograde transport. In this study, we show that JIP3 directly mediates TrkB interaction with kinesin-1 light chain (KLC1) and drives TrkB axonal but not dendritic anterograde transport, which is essential for subsequent BDNF-triggered signaling and filopodia formation.
Materials and Methods
Reagents and antibodies.
Human recombinant BDNF was obtained from PeproTech. Antibodies were purchased as follows: rabbit anti-TrkB antibodies from Millipore (catalog #07-225); mouse anti-Flag (M2), rabbit anti-HA, mouse IgG Sepharose, and mouse anti-tubulin antibodies from Sigma-Aldrich; rabbit anti-Flag from Thermo Fisher Scientific; mouse or rabbit anti-JIP3, mouse anti c-Myc, goat anti-KLC1, goat anti-Rab27B, and mouse anti-p-Tyr (pY99) antibodies from Santa Cruz Biotechnology; rabbit anti c-Myc antibodies from Bethyl Laboratories; mouse anti-HA (HA.11) antibodies from Covance Research Products; rabbit anti-p44/42 MAP kinase; mouse anti-phospho-p44/42 (Erk1/2; Thr202/Tyr204), rabbit anti-phospho-TrkA (pY490), and rabbit anti-His-Tag antibodies from Cell Signaling Technology; Alexa Fluor 488- or 594-conjugated goat anti-mouse or rabbit IgG heavy and light chains (H+L) from Invitrogen; Alexa Fluor 488-phalloidin from Invitrogen; Cy5-conjugated goat anti-mouse IgG (H+L) from Jackson ImmunoResearch Laboratories; horseradish peroxidase-conjugated goat anti-mouse or rabbit IgG antibodies from Calbiochem. The restriction enzymes were purchased from Fermentas. Vectashield mounting medium was obtained from Vector Laboratories. The other reagents were from Sigma-Aldrich.
Plasmid constructs.
Rat TrkB-FL, TrkB.T1, Flag-rTrkB-FL-GFP, and TrkA constructs were prepared as described previously (Chen et al., 2005b; Huang et al., 2009; Zhao et al., 2009). HA-tagged mouse JIP3 and its mutants (1–625, 1–424, ΔCC1, ΔCC2, ΔLZ), Flag-tagged human interleukin-2 receptor (IL2R), Flag-tagged rat TrkC, Flag-rTrkB-FL-mRFP, HAJIP3-RFP, and Myc-tagged human KLC1 were subcloned into pCDNA3.1 expression plasmid (Invitrogen). All the IL2R-JM chimeric constructs and the TrkA-BJM1 construct were generated by means of a two-step PCR. All the constructs were confirmed by DNA sequencing to exclude potential PCR-introduced mutations. The pSuper-RFP or pSuper-EGFP vectors (OligoEngine) were used to transcribe functional small interfering RNA (siRNA). In the vectors, oligonucleotides targeting different genes were inserted into the downstream of H1 promoter, with their veracity confirmed by double digestion and sequencing. The target sequences for each of the genes were as follows: human JIP3, CAGGCGGAGGAGAAATTCA; rat JIP3, CAGGCCGAGGAGAAATTCA; human and rat KLC1, GAATTGCTTAGTGATGAAT; human Rab27B, CAAATTCATCACTACAGTA; rat Rab27B, CAGAGTTTCTTGAATGTCA. The expression levels of corresponding proteins in HEK293 or PC12 cells transfected with the resulting siRNA or the scramble siRNA were analyzed by immunoblotting with individual antibodies.
Neuronal cultures and transfection.
Cultures of hippocampal neurons from timed-pregnant Sprague Dawley rats were prepared as described previously (Zhao et al., 2009). Briefly, embryos were removed from the rats at embryonic day 18 and then placed into HBSS without Ca2+ and Mg2+. Hippocampi were dissected from the embryos, digested with 0.05% trypsin–EDTA for 5 min at 37°C, and dissociated by mechanical trituration with a sterile, fire-polished glass Pasteur pipette in a 1:1 mixture of DMEM/F-12 (Invitrogen) plus 10% fetal bovine serum. Neurons were cultured on coverslips coated with 0.1 mg/ml poly-d-lysine (Sigma-Aldrich) in six-well plates in Neurobasal medium, which was supplemented with 2% B27 and 0.5 mm glutamine. An incubator with saturated humidity, 5% CO2, and invariant temperature at 37°C was used for the culture. Neurons cultured for 3 d in vitro (DIV) were transfected using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Invitrogen) according to the manufacturer's instructions.
Immunofluorescence analysis.
Hippocampal neurons cultured for 5 d were fixed with 4% paraformaldehyde in PBS for 10 min. Cultures were washed with PBS three times and then permeabilized with 0.4% Triton X-100 in PBS for 10 min. After being washed in PBS again, the cells were incubated with a blocking solution (PBS containing 10% normal goat serum) for 1 h at room temperature. After incubation with the primary antibodies at 4°C overnight, neurons were washed with PBS and incubated with fluorescent secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Cy5 for 1 h at room temperature. All the immunostained cells were observed with an LSM710 confocal microscope (Carl Zeiss), and quantification of labeling intensities was performed using MetaMorph software (Molecular Devices).
For analysis of the sciatic nerve, fresh-frozen sections were incubated with blocking solution and then incubated at 4°C overnight with a combination of rabbit anti-TrkB and mouse anti-JIP3 antibodies in dilution buffer (PBS containing 1% goat serum and 0.03% Triton X-100). Primary antibodies were visualized using fluorescent secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 594. Images were collected on a specimen using the LSM710 confocal microscope (Carl Zeiss).
For analysis of the density of filopodia, neurons at 5 DIV, transfected with the indicated constructs, were treated with 100 ng/ml BDNF for 20 min and then fixed with prewarmed (37°C) 4% paraformaldehyde in PBS at a 1:1 ratio with cell culture medium for 10 min to protect filopodia from collapsing. After fixation, neurons were washed for 5 min with PBS three times and permeabilized with 0.4% Triton X-100 in PBS for 10 min. After being washed in PBS again, the neurons were incubated with blocking solution for 1 h at room temperature. Then neurons were incubated with Alexa 488-phalloidin (which specifically labels F-actin) in PBS for 20 min at room temperature. After being washed two times with PBS, the neurons were incubated with diluted mouse anti-HA antibodies overnight at 4°C. Then the neurons were washed with PBS and incubated with fluorescent secondary antibodies conjugated to Alexa 594 for 1 h at room temperature. Neurons were mounted after being washed three times with PBS. Acquired images were analyzed using MetaMorph software.
To visualize the plasma membrane level of Flag-TrkB-FL-GFP in distal axons of hippocampal neurons transfected with siRNA-RFP or JIP3-RFP constructs, cells were fixed with 4% paraformaldehyde in PBS for 5 min without permeabilization. Cells were then stained with M2 anti-Flag monoclonal antibody, followed by Cy5-conjugated goat anti-mouse secondary antibody (1:1000; Jackson Immuno Research). The GFP fluorescence represented total TrkB amount. Acquired images were analyzed using MetaMorph software.
Coimmunoprecipitation assay.
Forty-eight hours after being electroporated (Amaxa Biosystems) with the indicated constructs, HEK293 cells were extracted by TNE buffer (10 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol with protease inhibitors). Lysates were clarified by centrifugation at 14,000 × g for 15 min at 4°C. After centrifugation, the soluble supernatants were incubated with the indicated antibodies for >2 h at 4°C. The immunocomplex was then precipitated with protein A- or G-Sepharose (Sigma) overnight at 4°C. The beads were then washed three times with TNE buffer, eluted by boiling in sample buffer for SDS-PAGE, and, at last, subjected to immunoblotting to analyze with the indicated antibodies.
For endogenous interaction, rat brain was homogenized and lysed in TNE buffer (8 ml of TNE/g brain lysate) with protease inhibitors and clarified by centrifugation at 14,000 × g for 15 min at 4°C. After centrifugation, the proteins were quantified using bicinchoninic acid protein assay. Approximately 10 mg samples of rat brain lysates were incubated with mouse anti-JIP3 antibodies, and the same amount of lysates was incubated with mouse IgG-Sepharose as a negative control. The immunocomplex was precipitated using protein G-Sepharose beads. The bound proteins were eluted and analyzed by immunoblotting with anti-JIP3, anti-TrkB, or anti-KLC1 antibodies. To confirm the result, rat brain lysates were also incubated with goat anti-KLC1 antibodies and precipitated using protein G-Sepharose beads. Immunoblotting was done with the above antibodies to detect the bound proteins.
Preparation of glutathione S-transferase fusion proteins and in vitro binding assay.
The various glutathione S-transferase (GST)–TrkB juxtamembrane (JM) mutant constructs are as follows: pGEX-TrkB-JM1 containing the TrkB JM domain encoding amino acids 454–465; pGEX-TrkB-JM2 containing the TrkB JM domain encoding amino acids 454–485; pGEX-TrkB-JM3 containing the TrkB JM domain encoding amino acids 454–508; pGEX-TrkB-JM containing the TrkB JM domain encoding amino acids 454–537. These cDNA fragments were generated by PCR and subcloned into the pGEX-4T-1 vector (GE Healthcare), respectively, using the BamHI and NotI sites. All the fusion proteins were expressed in BL21 Escherichia coli (DE3 strain; Novagen) and immobilized on glutathione-Sepharose 4B beads (GE Healthcare). The JIP3-CC1 (amino acids 451–520) fragment was subcloned into pET28a (Novagen) to yield His-CC1. His-tagged CC1 was purified using Ni2+ affinity purification (Qiagen). The purified His-CC1 proteins were incubated with GST-fusion protein-bound beads in pull-down buffer (20 mm Tris, 1% BSA, 0.5% Triton X-100) at 4°C for 1–2 h. After an extensive wash with PBS buffer, the bound proteins were analyzed by SDS-PAGE followed by staining with Coomassie Blue (Sigma) or immunoblotting with anti-His antibodies.
Erk1/2 activation assays.
Hippocampal neurons were electroporated with siJIP3 or HAJIP3 constructs, respectively. Five days later, neurons were serum starved for 12 h and stimulated with BDNF (50 ng/ml) for different time periods (0 min, 15 min). The neurons were incubated with rabbit anti-Erk1/2 and mouse anti-pERK1/2 antibodies, followed by immunostaining with corresponding Alexa Fluor 594-conjugated goat anti-rabbit antibodies and Cy5-conjugated goat anti-mouse antibodies. For quantitation of the phosphorylation level of Erk1/2, the total Erk1/2 and phosphorylated Erk1/2 fluorescence intensity of individual neurons was analyzed with MetaMorph software. PC12 cells (stably expressing Flag-TrkB-FL) were electroporated with empty vector, HAJIP3, or siJIP3 and induced to differentiation for 2 d with 50 ng/ml NGF. Then cells were serum starved for 8 h and stimulated with BDNF (50 ng/ml) or NGF (50 ng/ml) for different time periods (0 min, 15 min). The phosphorylation levels of Erk1/2 and TrkA were analyzed by immunoblotting. Phosphorylation of TrkB was analyzed by immunoprecipitation with rabbit anti-Flag antibodies and immunoblotting with anti-phosphoTyr antibodies. For densitometric analysis, immunoreactive bands were scanned and quantitated.
Sciatic nerve ligation.
Male rats were anesthetized with 5% chloral hydrate, and the sciatic nerve on one side was ligated using a 5-0 polypropylene monofilament. One day after ligation, the animals were killed, and the sciatic nerves were frozen in liquid nitrogen immediately. For immunohistochemistry staining of sciatic nerve, fresh longitudinal frozen sections were prepared (20 μm). The contralateral sciatic nerve sections were prepared in parallel as a control. For immunoblotting analysis of sciatic nerve, ligated and contralateral unligated sciatic nerves (3 mm apart from the ligature) were dissected, and extracts were analyzed with the indicated antibodies. Tubulin is used as a loading control. Then immunoblotting panels were analyzed by NIH ImageJ (Scion).
Microscopic quantitative analysis.
To quantitatively analyze the amount of endogenous TrkB in distal axons and dendrites of hippocampal neurons at 5 DIV, TrkB staining levels in the distal 30 μm of the axons or 10 μm of the dendrites were measured by the MetaMorph software. We used several methods to distinguish axons from dendrites (Dotti et al., 1988). For example, dendrites have irregular contours, taper gradually, and branch at relatively acute angles. Axons, in contrast, display a smoother contour, have a relatively even diameter along their course, and extend much farther away from the cell body. Among these traits, particularly valuable in our study is the distinction that all of the long processes in the hippocampal culture represent axons (Dotti et al., 1988). In every experiment, a consistent set of acquisition parameters was used for each set of images. At least three independent experiments were conducted for each manipulation, and >50 cells were examined in each experiment. Each error bar represents the mean ± SEM, and the data were analyzed by one-way ANOVA.
Filopodia were defined as any protrusion under 10 μm in length. The number of filopodia and the length of the axonal or dendritic shaft were then computed to obtain the filopodia density (number of filopodia per 10 μm). For each neuron, an axon or dendrite length ranging between 30 and 80 μm was analyzed. Each experimental condition was repeated at least three times. In each experiment, >35 cells were examined at random, the results of more than three independent experiments were compiled, and the mean ± SEM was calculated.
To quantitatively analyze the ratio of surface/total TrkB in distal parts of axons, the intensities of Cy5 (anti-Flag staining) or GFP for each ROI by selecting the region of distal axons in every images were measured by the MetaMorph software. The Cy5 staining image represented the surface TrkB fluorescence of each ROI, which is shown with blue pseudocolor; the total TrkB fluorescence of each ROI was the signal measured in the GFP image, and the RFP fluorescence expressed by siRNA or JIP3 construct is shown with red color. Then the signal ratio of Cy5 channel/green channel was calculated to get the surface/total ratio in distal axons.
Live cell imaging.
For the hippocampal neurons used for live cell imaging, the transfection of constructs was performed by the Nucleofector device (Amaxa Biosystems) according to the manufacturer's instructions, before plating. Five days after being seeded in the glass-bottomed 35 mm dishes (Willco Wells) coated with 0.1 mg/ml poly-d-lysine, the transfected neurons were observed by an Eclipse TE 2000-U inverted fluorescence microscope (Nikon) equipped with a motorized Z drive using a 40× oil-immersion objective lens (1.0 NA). The cells selected for imaging had healthy morphologies, and images were acquired every 1 s continuously for 1 min with a HQ2 cooled CCD camera. The positive vesicle was tracked and processed by MetaMorph software. A kymograph was generated using NIH ImageJ. The relative frequency of immobile, directional, and bidirectional movements was calculated and presented as the average of some neurites. For RNAi experiments, 80–120 vesicles of 20 axons or 40 dendrites were counted in each condition and shown the relative frequency. These results were performed at least in triplicate, and the data were analyzed by one-way ANOVA using SPSS 13.0.
Statistical analysis.
Statistical significance was assessed using the Student's t test or ANOVA, followed by post hoc tests. Data were presented as mean ± SEM, and p < 0.05 was considered significant.
Results
Both TrkB-FL and TrkB.T1 receptors are anterogradely transported in sciatic nerve
Sciatic nerve ligation analysis is a well established in vivo tool used to identify transported molecules in axons biochemically and immunohistochemically. Western immunoblotting analyses of anterogradely and retrogradely transported molecules in 1 d ligated rat sciatic nerve confirmed previous findings (Yano et al., 2001) that TrkB-FL protein levels are increased in extracts from both proximal (anterograde) and distal (retrograde) sides of the ligation (Fig. 1A,B). Interestingly, we found that the truncated TrkB isoform, TrkB.T1 receptor, which lacks the tyrosine kinase domain, is also anterogradely transported (Fig. 1A,B). A previous study has indicated that TrkB receptors undergo anterograde transport via the interaction between the TrkB tyrosine kinase domain and the Slp1/Rab27B/CRMP-2/kinesin-1 complex (Arimura et al., 2009). However, the anterograde transport of truncated TrkB.T1 receptor suggests that TrkB tyrosine kinase domain-independent anterograde transport mechanisms exist. It is known that TrkB.T1 and TrkB-FL receptors share a small part of JM domain containing 12 amino acids (K454–G465) (Fig. 1C), which might be a key domain involved in TrkB anterograde transport. We screened a human brain cDNA library using the 12 aa as bait by yeast two-hybrid assay. Among the 35 positive clones, three clones, which encoded JIP3, were isolated. JIP3 has been implicated as an adaptor protein in kinesin-1-dependent anterograde transport (Bowman et al., 2000; Byrd et al., 2001; Cavalli et al., 2005). Therefore, we investigated whether JIP3 could mediate TrkB anterograde transport.
Figure 1.

Both TrkB-FL and TrkB.T1 receptors undergo anterograde transport. A, Sciatic nerves were ligated unilaterally at the midpoint for 1 d and processed for immunoblotting. Ligated and contralateral unligated sciatic nerves were dissected, and the extracts were analyzed with the indicated antibodies. Tubulin is used as a loading control. B, Quantification of the proximal and distal pool fraction. Data were shown as the mean ± SEM from five independent experiments (*p < 0.05, the ligated pool vs the control unligated pool; one-way ANOVA). Values were normalized to the level of the unligated pool. C, Schematic representation of the structure of TrkB-FL and TrkB.T1 receptors. The shared identical intracellular region (454–465 aa) was marked with black color.
JIP3 colocalizes and interacts with TrkB in vitro and in vivo
We first compared the subcellular distribution of JIP3 and TrkB in cultured hippocampal neurons (5 DIV) by immunocytochemistry staining. We used TrkB and JIP3 antibodies to examine the localization of endogenous TrkB and JIP3. The results indicated that TrkB-containing vesicles were present in the perinuclear region, axons, and dendrites, partially colocalized with JIP3 (Fig. 2A). To test the hypothesis that anterograde transport of TrkB is associated with JIP3 in vivo, we performed sciatic nerve ligation to follow the localization of TrkB and JIP3. The sciatic nerve was ligated for 1 d and then removed, sectioned longitudinally, and processed for immunohistochemical staining. A remarkable accumulation of TrkB receptor was observed in the proximal portion of the sciatic nerve after ligation (Fig. 2B). When the expression of JIP3 was examined in the same sections, a pattern similar to the TrkB staining was observed, and JIP3 and TrkB showed higher colocalization in the proximal sides of the ligation (Fig. 2C,D), which is consistent with their in vivo interaction.
Figure 2.

Colocalization of JIP3 and TrkB in vitro and in vivo. A, Subcellular colocalization of endogenous TrkB and JIP3 in hippocampal neurons visualized by confocal microscopy. Staining was performed with rabbit anti-TrkB (green) and mouse anti-JIP3 (red) antibodies. Colocalization of both proteins was shown in yellow. Bottom, Enlarged images of the framed regions, with the white arrows indicating the colocalization of TrkB and JIP3. Scale bar, 20 μm. B, The sciatic nerve from a male rat was ligated for 24 h, and longitudinal sections of ligated (left sciatic nerve) and the control unligated sections (right sciatic nerve) were immunostained with anti-TrkB antibodies. The bilateral part around the ligation in the white rectangle was enlarged in C. Scale bar, 100 μm. C, Colocalization of TrkB and JIP3 in the ligated sciatic nerve. Longitudinal sections from a ligated sciatic nerve were double immunostained with anti-TrkB (green) and anti-JIP3 (red) antibodies; the proximal portion is at the top, whereas the distal end of the ligation is at the bottom. The proximal part near the ligation in the white rectangle was enlarged in D. Scale bar, 50 μm. Colocalization of TrkB and JIP3 is indicated in yellow.
Next, we examined whether JIP3 could form a complex with TrkB. First, we transfected FlagTrkB-FL or FlagTrkB.T1 with N-terminal HAJIP3 in HEK293 cells. After immunoprecipitation of JIP3 with HA antibodies, an association of TrkB-FL or TrkB.T1 with JIP3 was observed as assessed by immunoblotting analysis using Flag antibodies, although TrkB.T1 showed weaker association (Fig. 3A). The complex between the FlagTrkB-FL/FlagTrkB.T1 and HAJIP3 was also detected after immunoprecipitation of Flag and immunoblotting with anti-HA antibodies (Fig. 3A). To examine whether this interaction occurs under physiological conditions, we performed endogenous JIP3/TrkB coimmunoprecipitation assays from brain lysates. Lysates from rat brain were immunoprecipitated with anti-JIP3 antibodies, and we found TrkB coimmunoprecipitated with JIP3 under the endogenous level (Fig. 3B). As expected, KLC1 also associated with the TrkB/JIP3 complex, which was further confirmed by the immunoprecipitation with anti-KLC1 antibodies from brain lysate (Fig. 3B). Thus, we determined that TrkB/JIP3/KLC1 could form a complex both in vitro and in vivo.
Figure 3.

Ternary complex formation of JIP3, TrkB, and KLC1 in vitro and in vivo. A, Coimmunoprecipitation of JIP3 with TrkB-FL/TrkB.T1. Lysates from HEK293 cells transfected with HAJIP3 and Flag-tagged TrkB-FL/TrkB.T1 constructs were immunoprecipitated with anti-HA antibodies or anti-Flag antibodies. Then, immunoblotting analysis was performed to detect immunoprecipitated proteins. B, Endogenous JIP3 associates with TrkB and KLC1. Rat brain lysates were subjected to immunoprecipitation with mouse anti-JIP3 or goat anti-KLC1 antibodies, and the protein complex was eluted; the TrkB, KLC1, and JIP3 were detected by immunoblotting. C, JIP3-dependent interaction between TrkB and KLC1. Lysates from HEK293 cells cotransfected with MycKLC1 and FlagTrkB-FL/FlagTrkB.T1 constructs were immunoprecipitated with anti-Myc antibodies. Then, immunoblotting analysis was performed to detect immunoprecipitated proteins. For FlagTrkB-FL: TrkB-FL wild-type group, cotransfected with FlagTrkB-FL and MycKLC1, with endogenous JIP3 expression; JIP3 overexpressed group, cotransfected with FlagTrkB-FL, MycKLC1, and HAJIP3; siJIP3 group, cotransfected with FlagTrkB-FL, MycKLC1, and siJIP3. For FlagTrkB.T1: TrkB.T1 wild-type group, cotransfected with FlagTrkB.T1 and MycKLC1, with endogenous JIP3 expression; siJIP3 group, cotransfected with FlagTrkB.T1, MycKLC1, and siJIP3. Quantitation of immunoblotting in the top panel of Western blots in C is shown as a percentage of TrkB coimmunoprecipitation by KLC1 in different groups compared with TrkB-FL wild-type group. Data are shown as the mean ± SEM from three independent experiments [n = 3; *p < 0.05, vs FlagTrkB-FL wild-type (WT) group; ##p < 0.01, vs FlagTrkB.T1 WT group, one-way ANOVA]. D, The KLC1-independent interaction between TrkB-FL and JIP3. Lysates from HEK293 cells transfected with HAJIP3 and FlagTrkB-FL constructs were immunoprecipitated with anti-HA antibodies. Then, immunoblotting analysis was performed to detect immunoprecipitated proteins. Wild-type group, Cotransfected with FlagTrkB-FL and HAJIP3, with endogenous KLC1 expression; KLC1 overexpressed group, cotransfected with FlagTrkB-FL, MycKLC1, and HAJIP3; siKLC1 group, cotransfected with FlagTrkB-FL, HAJIP3, and siKLC1. E, Simultaneous knockdown of JIP3 and Rab27B further decreases the association between TrkB-FL and KLC1. Lysates from HEK293 cells cotransfected with MycKLC1 and TrkB-FL constructs were immunoprecipitated with anti-Myc antibodies. Wild-type group, Cotransfected with FlagTrkB-FL and MycKLC1, with endogenous JIP3 and Rab27B expression; siJIP3 group, cotransfected with FlagTrkB-FL, MycKLC1, and siJIP3; siRab27B group, cotransfected with FlagTrkB-FL, MycKLC1, and siRab27B; siJIP3+siRab27B group, cotransfected with FlagTrkB-FL, MycKLC, siJIP3, and siRab27B. Quantitation of immunoblotting in the top panel of Western blots in E is shown as a percentage of TrkB coimmunoprecipitation by KLC1 in different groups compared with wild type. Data are shown as the mean ± SEM from three independent experiments (n = 3; *p < 0.05; **p < 0.01, vs the FlagTrkB-FL WT group; #p < 0.05, vs siJIP3+siRab27B group; one-way ANOVA). IP, Immunoprecipitation; CO-IP, coimmunoprecipitation.
Given TrkB/JIP3/KLC1 could form a complex; the next question was which molecule is the intermediate bridge between TrkB and KLC1. To address this question, we examined whether JIP3 could contribute to the TrkB and KLC1 interaction. We found overexpression or knockdown of JIP3 significantly increased or decreased the association between TrkB-FL and KLC1 by three molecule coimmunoprecipitation studies (Fig. 3C). Conversely, overexpression or siRNA knockdown of KLC1 had no effect on the association between TrkB-FL and JIP3 (Fig. 3D). These results indicate that JIP3 is an intermediate interacting protein that links TrkB-FL binding with KLC1.
Interestingly, we found the elimination of TrkB-FL binding with KLC1 by knockdown of JIP3 was not complete (Fig. 3C). It has been previously reported that Slp1/Rab27/CRMP-2 could also mediate the association between TrkB-FL and KLC1 via the TrkB tyrosine kinase domain (Arimura et al., 2009). We found double knockdown of JIP3 and Rab27B could further decrease the association between TrkB-FL and KLC1, compared with knockdown of JIP3 or Rab27B alone (Fig. 3E). This result suggests that TrkB-FL could synchronously form a complex with KLC1 through JIP3 and Slp1/Rab27/CRMP-2, respectively, and these two systems appear to function in a subadditive manner. Since TrkB.T1 does not have the tyrosine kinase domain and could not interact with the Slp1/Rab27/CRMP-2 complex, we found binding of TrkB.T1 to KLC1 was more substantially reduced with JIP3 knockdown than that of TrkB-FL (Fig. 3C). This result reveals greater dependence of TrKB.T1–kinesin association on JIP3, since it does not have the alternative binding option.
Mapping the TrkB/JIP3 interaction domain
TrkB-FL and TrkB.T1 both interact with JIP3, and they share a consensus 12 amino acids in the intracellular JM domain. To address whether these 12 amino acids play an important role in this interaction, various mutants of TrkB receptor were constructed: TrkB-JM1 retaining only the 12 aa in the TrkB intracellular domain, TrkB-JM0 with the entire TrkB intracellular domain deletion, and TrkBΔJM1 with the 12 aa deletion in the TrkB intracellular domain (Fig. 4A). The coimmunoprecipitation assay indicated that the TrkB mutant with only the 12 aa in juxtamembrane domain (TrkB-JM1) could still interact with JIP3; however, the JM1 deletion (12 aa, K454–G465) abolished the TrkB/JIP3 interaction. This result suggests that the JM1 domain is necessary for TrkB/JIP3 interaction. To further investigate whether the JM1 domain is sufficient for TrkB/JIP3 interaction, we divided the 80 amino acid JM domain into five parts (see Fig. 4B for details) based on published information (Yano et al., 2001) and, respectively, transplanted them to the C terminal of IL2R, which has a very short cytoplasmic tail. We found IL2R itself could not interact with JIP3 and the TrkBJM1 transplantation could confer its binding ability with JIP3 (Fig. 4B), which suggests that the JM1 domain in TrkB is not only necessary but also sufficient for TrkB/JIP3 interaction. The TrkB/JIP3 association could be enhanced by the amino acids between JM2 and JM3 (Fig. 4B), which explained the weaker association of JIP3 with TrkB.T1 compared with the TrkB-FL. It is well known that there are three most common types of Trk receptors: TrkA, TrkB, and TrkC. We want to know whether JIP3 could form complex with all three Trk receptors. Interestingly, we found that JIP3 could also bind with TrkC but not with TrkA (Fig. 4C). Amino acid sequence alignment of the JM1 domain among Trk receptors revealed that TrkB and TrkC showed higher alignment similarity compared with TrkA (Fig. 4C). Swapping the TrkB JM1 domain (BJM1) into TrkA conferred TrkA the binding ability with JIP3 (Fig. 4D). This result indicates that JIP3 could specifically mediate TrkB/TrkC but not TrkA binding to kinesin-1, and this binding depends on their JM1 domain.
Figure 4.

The JM1 domain of TrkB interacts directly with JIP3. A, Schematic representation of the TrkB mutants generated. HEK293 cells were transiently cotransfected with Flag-tagged TrkB-FL or TrkB mutants and HA-tagged JIP3. Immunoprecipitation was performed using rabbit anti-HA antibodies, and immunoblotting was performed with mouse anti-HA or mouse anti-Flag antibodies. B, Schematic representation of the TrkB juxtamembrane domain, IL2R, and chimeric IL2R–BJM mutants. HEK293 cells were transiently transfected with the indicated constructs. Immunoprecipitation was performed using mouse anti-Flag antibodies, and immunoblotting was performed with rabbit anti-HA or rabbit anti-Flag antibodies. C, Coimmunoprecipitation of JIP3 with TrkB-FL, TrkA-FL, and TrkC-FL. HEK293 cells were cotransfected with Flag-tagged Trks and HAJIP3; immunoprecipitation was performed using mouse anti-Flag antibodies, followed by immunoblotting with rabbit anti-Flag or rabbit anti-HA antibodies. Multiple sequence alignment on the JM1 domain of three Trk receptors was performed by ClustalW2. D, TrkB-JM1 confers TrkA the binding ability with JIP3. A schematic representation of TrkA and chimeric TrkA-BJM1 mutant is shown. Immunoprecipitation was performed using mouse anti-Flag antibodies, and immunoblotting was performed with rabbit anti-HA or rabbit anti-Flag antibodies.
To verify which domain in JIP3 is involved in the JIP3/TrkB interaction, we constructed a series of deletion mutants of JIP3 and tested their ability to bind to TrkB-FL by coimmunoprecipitation studies (Fig. 5A,C). The results indicate that the 424–625 amino acid region of JIP3 is critical for TrkB-FL and JIP3 interaction. The 424–625 amino acid region in JIP3 contains three conserved domains named leucine Zipper-like domain (LZ), coiled-coil 1 (CC1), and coiled-coil 2 (CC2) (Hammond et al., 2008). To define more precisely the region required for its binding with TrkB, three JIP3 mutants with respective LZ, CC1, or CC2 domain deletions were constructed, and their ability to bind to TrkB-FL was analyzed by coimmunoprecipitation studies (Fig. 5A,C). The results indicate that the CC1 deletion could abolish the JIP3 and TrkB-FL interaction (Fig. 5A). We also examined the ability of these JIP3 mutants to bind to KLC1 and found that the LZ deletion mutant lost its ability to bind to KLC1 (Fig. 5B). Thus, our data suggests that JIP3 associates with TrkB-FL and KLC1 via its CC1 domain and LZ domain, respectively. We next examined whether the CC1 deletion mutant of JIP3 (JIP3ΔCC1) could compete with wild-type JIP3 to bind with KLC1 and thus reduce the TrkB-FL and KLC1 association. As shown in Figure 5D, JIP3ΔCC1 overexpression significantly decreased the TrkB-FL and KLC1 interaction, which suggests that JIP3ΔCC1 could be used as a dominant-negative (DN) construct to evaluate the role of JIP3 in regulating TrkB-FL transport and function.
Figure 5.
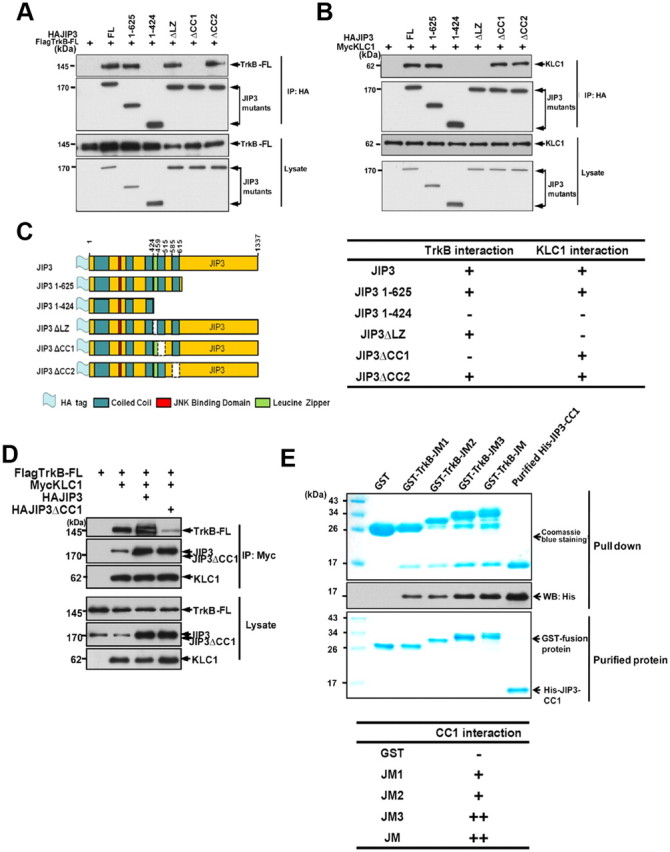
Mapping the JIP3 binding domain with TrkB. A, Coimmunoprecipitation of TrkB-FL with JIP3 mutants. HEK293 cells were cotransfected with FlagTrkB-FL and HAJIP3 mutants, and immunoprecipitation was performed using rabbit anti-HA antibodies, immunoblotted with mouse anti-Flag or mouse anti-HA antibodies. B, Coimmunoprecipitation of KLC1 with JIP3 mutants. HEK293 cells were cotransfected with MycKLC1 and HAJIP3 mutants, and immunoprecipitation was performed using rabbit anti-HA antibodies, immunoblotted with mouse anti-Myc or mouse anti-HA antibodies. C, Schematic representation of the HAJIP3 mutants. Amino acid numbers correspond to appropriate domains within the JIP3 deletion constructs. A summary of TrkB-FL/KLC1 interactions with the JIP3 mutants is provided. D, Interaction of TrkB with KLC1 was reduced by HAJIP3ΔCC1. HEK293 cells were cotransfected with MycKLC1, FlagTrkB-FL, and HAJIP3/JIP3ΔCC1 constructs, and immunoprecipitation was performed using rabbit anti-Myc antibodies, followed by immunoblotting with mouse anti-Myc, anti-Flag, or anti-HA antibodies. E, Direct interaction of the TrkB-JM1 domain with the JIP3-CC1 domain in vitro. GST-JM1, GST-JM2, GST-JM3, and GST-JM fusion proteins were incubated with the purified His-CC1. The precipitate was subjected to Coomassie Brilliant Blue staining and immunoblotting analysis using anti-His antibodies. A summary of JM mutants' interactions with CC1 is provided.
All the results above indicated that JIP3 may play an important role in the interaction between TrkB and KLC1. To confirm that JIP3 does interact directly with TrkB, we performed a GST pull-down in vitro interaction study between JIP3 and TrkB. Bacterial-expressed 6×His-tagged JIP3 CC1 domain (451–520 amino acids) was purified and tested for its direct binding to bacterial-expressed GST-fusion proteins (GST-JM1, GST-JM2, GST-JM3, GST-JM, or GST). The in vitro binding assay reveals that JIP3-CC1 could directly bind with the TrkB JM1 domain and the amino acids between the JM2 and JM3 could enhance this interaction (Fig. 5E), which is consistent with our previous coimmunoprecipitation experiments.
JIP3 regulates axonal but not dendritic TrkB anterograde transport
It has been reported that Slp1/Rab27B/CRMP-2 mediated anterograde TrkB-FL transport by kinesin-1. Since we showed that TrkB/JIP3/KLC1 could form a complex, it is conceivable that JIP3 may also mediate TrkB transport by kinesin-1. To address this question, we first examined the effect of overexpression/knockdown of JIP3 on the subcellular localization of TrkB-FL-GFP in differentiated PC12 cells (Fig. 6A). In a control group (transfected with scrambled siRNA), TrkB-FL-GFP accumulated at the tips of neurites. Depletion of JIP3 by siRNA or transfection of JIP3 DN construct JIP3ΔCC1 could significantly decrease this distal localization of TrkB-FL-GFP (Fig. 6A,B). On the contrary, overexpression of JIP3 led to a significant increase in the amount of TrkB-FL-GFP at the tips of neurites. These results suggest that JIP3 facilitates anterograde transport of TrkB-FL-GFP in PC12 cells. We repeated the experiments in cultured hippocampal neurons to investigate whether JIP3 could enhance TrkB anterograde transport under endogenous conditions in neurons. TrkB staining levels in the distal 30 μm of the axons was significantly enhanced by JIP3 overexpression (Fig. 6C,D). In contrast, either overexpression of a dominant-negative form of JIP3 (JIP3ΔCC1), which could form a complex with KLC1 but cannot interact with TrkB, or JIP3 knockdown via siJIP3 transfection decreased TrkB localization at the distal part of axon (Fig. 6C,D). Interestingly, the involvement of JIP3 in TrkB anterograde transport is specific for axons, as overexpression/knockdown of JIP3 had no effect on distal TrkB dendritic distribution (Fig. 6D). Simultaneously knocking down JIP3 and Rab27B could further decrease the endogenous TrkB distal axonal localization compared with their respective knockdown (Fig. 6D), which supports the idea that JIP3/TrkB and Slp1/Rab27B/CRMP-2/TrkB complex functions in an additive fashion to facilitate TrkB anterograde transport.
Figure 6.
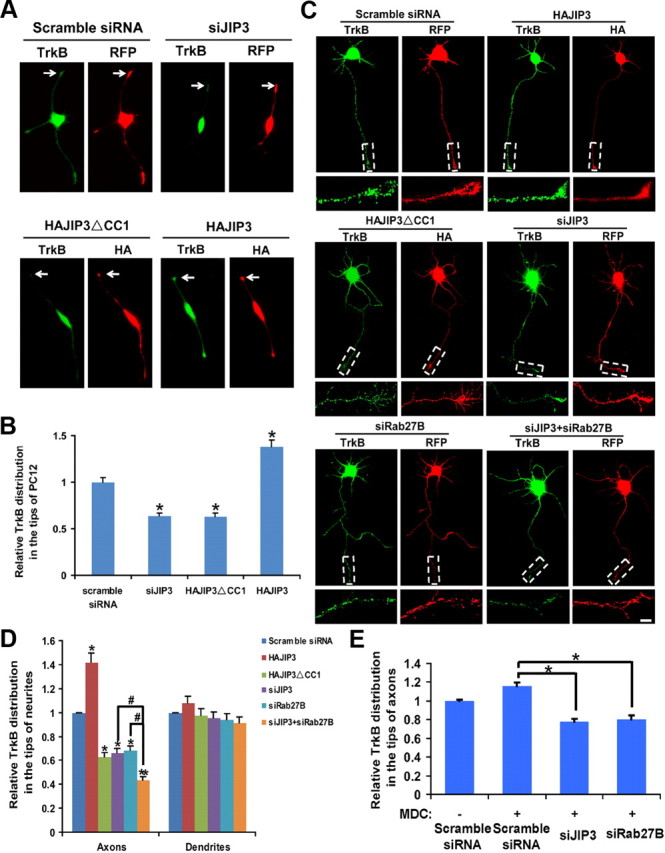
JIP3 mediates anterograde TrkB transport. A, JIP3 influences the localization of TrkB-FL-GFP in PC12 cells. Distribution of TrkB-FL-GFP in the tips of differentiated PC12 cells, which were cotransfected with TrkB-FL-GFP and distinctive constructs (scramble siRNA, siJIP3, HAJIP3, or HAJIP3ΔCC1) and induced to differentiation for 2 d with 50 ng/ml NGF. The cells were fixed, and the relative TrkB-FL-GFP distribution in the tips of cells was measured. Arrows indicate the tips of PC12. The transfected siRNA is shown in red. The expression of JIP3 or JIP3ΔCC1 was detected by immunostaining with anti-HA antibodies (red); the localization of TrkB-FL-GFP is shown in green. B, Quantitative analysis of the localization of TrkB-FL-GFP in the tips of PC12 cells in A. The values expressed as mean ± SEM from three independent experiments (n = 3; *p < 0.05, vs the scramble siRNA group; one-way ANOVA). C, JIP3 influences the localization of endogenous TrkB at the distal axon. Cultured hippocampal neurons transfected with HAJIP3 or the dominant-negative form of JIP3 (HAJIP3ΔCC1), siJIP3, siRab27B, or siJIP3 + siRab27B at 3 DIV were stained for the endogenous TrkB at 5 DIV. The transfected siRNA is shown in red. The expression of HAJIP3 or HAJIP3ΔCC1 was detected by immunostaining with anti-HA antibodies (red), and the endogenous TrkB was detected by immunostaining with anti-TrkB antibodies (green). Scale bar, 10 μm. D, Quantitative analysis of the localization of endogenous TrkB at a distal axon or dendrites in C. The values shown are the mean ± SEM from three independent experiments (n = 3; *p < 0.05; **p < 0.01, vs scramble siRNA group; #p < 0.05, vs siJIP3 + siRab27B group; one-way ANOVA). E, Quantitative analysis for the localization of endogenous TrkB at a distal axon in the presence of MDC. Data shown are the mean ± SEM of three independent experiments (n = 3; *p < 0.05, vs the scramble siRNA + MDC group; one-way ANOVA).
It was reported recently that TrkA in the plasma membrane of cell bodies could undergo internalization and targeting to the axon terminus in response to NGF stimulation (Ascano et al., 2009). Thus, we wanted to determine whether the TrkB transported via JIP3 was coming from the membrane of cell bodies or was a newly synthesized protein derived from the Golgi network. To address this question, we used MDC (50 μm, 12 h) to block TrkB internalization (Rougier et al., 2011) and found knockdown of JIP3 or Rab27B could still decrease TrkB anterograde transport in the presence of MDC (Fig. 6E), which suggests the TrkB transported by JIP3 or Rab27B is mainly from the biosynthetic pathway.
To further examine the function of JIP3 in TrkB transport under live cell conditions, we transfected neurons with TrkB-FL-mRFP and monitored TrkB-FL-mRFP-containing vesicle movement in neurons at 5 DIV by using time-lapse fluorescence microscopy. TrkB-FL-mRFP-containing vesicles were found to be highly mobile, in axons, visibly moving anterogradely, retrogradely, or bidirectionally (Fig. 7A–C). Compared with axons, a weaker dynamic movement of TrkB-FL-mRFP vesicles was observed in dendrites (Fig. 7C). To quantify the TrkB-FL-mRFP-containing vesicle movements, we subdivided the vesicles into four categories: (1) the anterograde group (vesicles that moved only in the anterograde direction); (2) the retrograde group (vesicles that moved only in the retrograde directions); (3) the bidirectional group (vesicles that moved in both directions); and (4) the immobile group (vesicles that were immobile). Axonal and dendritic TrkB-FL-mRFP-containing vesicles had similar proportions of the four groups (Fig. 7D). Compared with the control group, neurons cotransfected with TrkB-FL-mRFP and siJIP3 had a significant decreased percentage of the anterogradely transported vesicles and the corresponding increased percentage of the immobile vesicles in axons (Fig. 7D). JIP3 knockdown had no effect on the percentage of TrkB-containing vesicle categories in dendrites (Fig. 7D). These results indicate that JIP3 has important functions on anterograde TrkB transport preferentially in axons.
Figure 7.

Depletion of JIP3 results in the inhibition of anterograde TrkB vesicle transport. A, A 3 DIV hippocampal neuron cotransfected with EGFP and TrkB-FL-mRFP; the white box indicates the area shown in B. Scale bar, 20 μm. B, Movement of TrkB-FL-mRFP. TrkB-FL-mRFP vesicles can be transported retrogradely (down arrows), anterogradely (arrowheads), bidirectionally (up arrows). Immobile TrkB-FL-mRFP- vesicles (bold arrows) are also observed. The numbers indicate seconds. C, Kymograph of some neurites indicated in A. The kymograph of the axon and three of the dendrites for the neuron in A is shown. Images were captured every 1 s for 60 s in hippocampal neurons. Scale bar, 20 μm. D, Quantitative analysis of relative frequency of anterograde (Antero), retrograde (Retro), bidirectional (Bidir), or immobile vesicles of TrkB-FL-mRFP in siJIP3-transfected neurons The numbers of cells analyzed for each group are >60. Data shown are the mean ± SEM of three independent experiments (n = 3; *p < 0.05, vs the scramble siRNA group; one-way ANOVA).
JIP3-dependent TrkB anterograde transport is required for BDNF-induced signaling
To assess the functional consequences of JIP3-dependent TrkB anterograde transport, next we investigated whether JIP3 could affect BDNF-induced signaling. The addition of BDNF (50 ng/ml) to hippocampal neurons for 15 min induced activation of Erk1/2, which is a well established downstream target of TrkB activation. Because of the low transfection efficiency in neurons, the phosphorylation of Erk1/2 induced by BDNF was analyzed by immunocytochemical staining. Phosphorylated Erk1/2 (pErk1/2) was attenuated by ∼35% in JIP3 knockdown neurons despite identical levels of total Erk1/2, whereas pErk1/2 was increased by about 40% in JIP3-overexpressed neurons. The effect of JIP3 on BDNF-induced Erk1/2 phosphorylation was more obvious in the axonal shaft and terminus (Fig. 8A,B). This result underscores the importance of JIP3 for efficient TrkB-mediated signaling. JIP3 was originally identified as a JNK-binding protein and functions as a scaffold protein to modulate JNK signaling (Matsuura et al., 2002; Takino et al., 2005). Several studies indicated that JIP3 cannot affect Erk kinase activity (Kelkar et al., 2000; Matsuura et al., 2002); however, in the JIP3 knock-out mouse, the activation but not the distribution of Erk in axons of the developing brain was reduced (Ha et al., 2005). We speculated that JIP3 might indirectly modulate Erk1/2 activation via regulating TrkB transport. To address this question, we analyzed the BDNF-induced Erk1/2 phosphorylation in the undifferentiated and differentiated PC12 cells that stably express Flag-TrkB-FL. We found overexpression or knockdown of JIP3 could enhance or decrease BDNF-induced Erk1/2 activation only in differentiated, but not in undifferentiated, PC12-TrkB-FL cells (Fig. 8C,D). Next we found overexpression or knockdown of JIP3 also changed the TrkB phosphorylation level accordingly in differentiated PC12-TrkB-FL cells (Fig. 8C,D), which is more specific than Erk1/2 activation. This result suggests that efficient BDNF-induced signaling depends on JIP3-mediated TrkB transport. To further exclude the possibility that JIP3 may alter Erk1/2 activation in a manner that does not involve changes in TrkB, we investigated the effect of JIP3 on Erk1/2 activation in response to NGF in differentiated PC12 cells. We found that overexpression or knockdown of JIP3 had no effect on NGF-induced TrkA phosphorylation and Erk1/2 activation, which suggests that JIP3 modulates Erk1/2 activation via its role in TrkB anterograde transport regulation. In all, these results support the model that the localization of TrkB mediated by JIP3/kinesin-1 to the axon terminus is critical for the signal transmission induced by BDNF.
Figure 8.

JIP3 is involved in BDNF-induced Erk1/2 phosphorylation in neurons. A, BDNF-induced Erk1/2 phosphorylation in siJIP3- or HAJIP3-transfected neurons. Immunostaining of Erk1/2 and pErk1/2 was performed in cultured neurons. The hippocampal neurons were treated for 15 min with BDNF (50 ng/ml), and levels of total Erk1/2 and pErk1/2 were examined using anti-Erk1/2 and anti-pErk1/2 antibodies. GFP indicates the transfected neurons. Arrowheads indicate the untransfected neurons, and arrows indicate the transfected neurons. Scale bar, 50 μm. Bottom panels show enlarged images of the framed regions indicating immunostaining in axon terminals. B, Quantitative analysis of the mean fluorescence intensity of phosphorylation of Erk1/2 in HAJIP3- or siJIP3-transfected neurons compared with the control condition (scramble siRNA group). Data shown are the mean ± SEM from three independent experiments (n = 3; *p < 0.05; **p < 0.01, vs the scramble siRNA group; one-way ANOVA). C, Immunoblotting using anti-Erk1/2 and anti-pErk1/2 antibodies to assess activation of Erk1/2 in PC12 cells stably expressed TrkB (differentiated or undifferentiated) with JIP3 overexpression or knockdown when treated with BDNF. PhosphoTrkB was also analyzed by immunoprecipitation with rabbit anti-Flag antibodies and immunoblotting with anti-phosphoTyr antibodies. D, Histogram shows pErk1/2 and pTrkB levels in each group in C. Data were shown as the mean ± SEM from three independent experiments (n = 3; *p < 0.05, vs the vector control group; one-way ANOVA). E, Immunoblotting using anti-Erk1/2 and anti-pErk1/2 antibodies to assess activation of Erk1/2 in differentiated PC12 cells (with endogenous expression of TrkA) with JIP3 overexpression or knockdown when treated with NGF (50 ng/ml). PhosphoTrkA was also analyzed by immunoblotting. F, Histogram shows pErk1/2 and pTrkA levels in each group in E. Data were shown as the mean ± SEM from three independent experiments.
Since JIP3 mediates axonal TrkB anterograde transport and modulates BDNF-induced TrkB signaling, we next investigated whether JIP3 could facilitate TrkB-FL cell-surface insertion. According to our previous report (Zhao et al., 2009), we used ratiometric fluorescence assay to measure the TrkB cell-surface level. We found that overexpression or knockdown of JIP3 could increase or decrease total and cell-surface TrkB-FL levels in distal axons, respectively. However, the ratio of surface TrkB-FL versus total TrkB-FL remained unchanged (Fig. 9). Knockdown of Rab27B also had no effect on the surface/total TrkB-FL ratio (Fig. 9). These results suggest that anterogradely transported TrkB-FL is successively inserted into plasma membrane and JIP3 might not play a significant role in TrkB plasma membrane insertion.
Figure 9.

JIP3 does not facilitate TrkB plasma membrane insertion. A, Hippocampal neurons expressing FlagTrkB-FL-GFP were stained with M2 anti-Flag antibody under nonpermeabilized conditions to label surface TrkB-FL. Total TrkB-FL was represented by GFP fluorescence. The neurons were transfected with HAJIP3-RFP, siJIP3, or siRab27B, respectively. Immunofluorescence images of the tips of axons are from a representative cell for each condition. B, Quantification of surface TrkB-FL-GFP levels in the tips of axons by ratiometric fluorescence assay of immunofluorescence images shown in A. C, Quantification of total TrkB-FL-GFP distribution in the tips of axons by ratiometric fluorescence assay of immunofluorescence images shown in A. D, Quantification of TrkB surface/total ratio in the tips of axons by ratiometric fluorescence assay of immunofluorescence images shown in A. The data were normalized to the scramble siRNA group. Data were shown as the mean ± SEM from three independent experiments (n = 3; *p < 0.05; **p < 0.01, vs the scramble siRNA group; one-way ANOVA).
JIP3 regulates axonal filopodia formation in response to BDNF
BDNF/TrkB signaling could drive activity-dependent synaptic morphogenesis (Poo, 2001; Cohen-Cory, 2002). To investigate the role of JIP3-dependent TrkB transport in BDNF-triggered synaptogenesis, we examined whether JIP3 could regulate BDNF-induced filopodia formation, a process involved in synaptogenesis and neuronal development. Filopodia are thin, actin-rich plasma membrane protrusions that extend beyond the leading edge of migrating cells and growth cones, often from the edges of lamellipodia. After staining with Alexa 488-phalloidin, filopodia were defined as any protrusion under 10 μm in length. Hippocampal neurons were transfected, respectively, with scramble siRNA, siJIP3, JIP3ΔCC1 (JIP3 DN construct), or JIP3, and the formation of filopodia was examined after a 20 min administration of BDNF. Consistent with the previous report (Gehler et al., 2004), treatment of hippocampal neurons with BDNF induced the filopodia formation in both axons and dendrites (Fig. 10A,B). Compared with the control group, siJIP3 and JIP3 DN transfection abolished the BDNF-induced axonal filopodia formation, and overexpression of JIP3 could augment the axonal filopodia formation elicited by BDNF stimulation (Fig. 10A,B). On the contrary, JIP3 had no effect on the augmentation of dendritic filopodia density in response to BDNF treatment (Fig. 10A,B). A previous report has shown that axonal filopodia, which are known to originate presynaptic specializations, are regulated in response to BDNF, and this response is dependent on MAPK kinase (Menna et al., 2009). Together, these results suggest that JIP3 could selectively influence BDNF-induced axonal filopodia formation, which might be mediated by the role of JIP3 in BDNF signaling.
Figure 10.
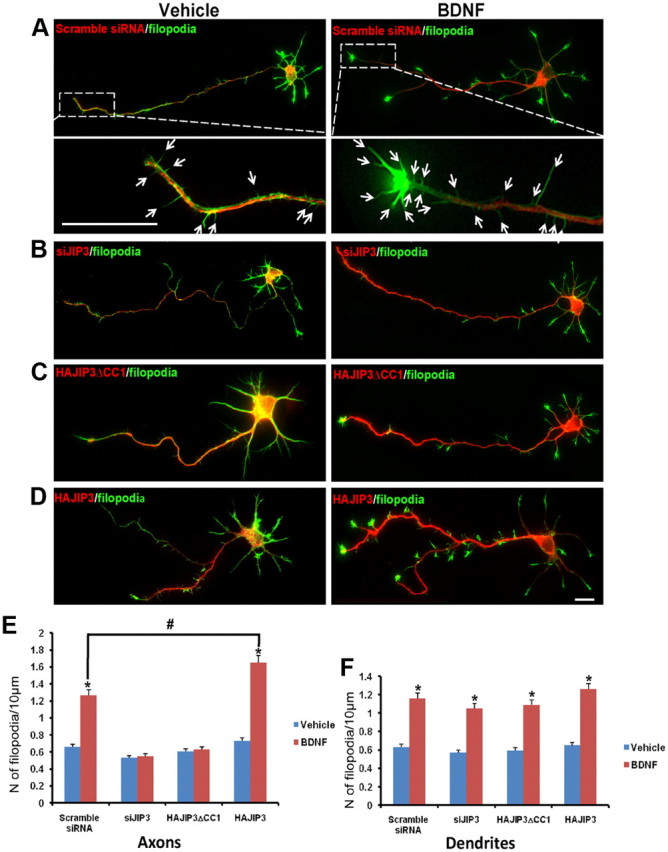
JIP3 regulates axonal filopodia formation in response to BDNF in hippocampal neurons. After staining with Alexa 488-phalloidin, filopodia were defined as any protrusion under 10 μm in length. The number of filopodia and the length of axonal or dendritic shaft were then computed to obtain the filopodia density (number of filopodia per 10 μm). A–D, Hippocampal neurons (5 DIV) transfected with scramble siRNA (A), siJIP3 (B), HAJIP3ΔCC1(C), or HAJIP3 (D) were treated with BDNF (100 ng/ml) for 20 min, with the unstimulated group as vehicle control. Existence of transfected siRNA could be detected by RFP fluorescence; the expression of HAJIP3 or HAJIP3ΔCC1 was detected by immunoblotting with HA antibodies (red). Bottom panels in A showed enlarged images of the framed regions with arrows highlighting the filopodia. E, F, The number of filopodia in axons (E) or dendrites (F) per 10 μm were calculated. For each neuron, the length of axon or dendrite length ranging between 30 and 80 μm was analyzed. The results of more than three independent experiments were compiled. Results are presented as the mean ± SEM (*p < 0.05, vs the vehicle control; #p < 0.05, vs scramble siRNA group stimulated with BDNF; one-way ANOVA). Scale bars: A, D, 10 μm.
Discussion
In neurons, the BDNF receptor TrkB has been shown to be present in dendritic spines, axon initial segments, axon terminals, dendritic shafts, and cell bodies (Yan et al., 1997; Drake et al., 1999). TrkB in specific neuronal subdomains has been suggested to be important for selective functions. For example, a number of studies have indicated that presynaptic TrkB may mediate enhancement of glutamate release and retrograde survival signaling from target-derived BDNF, whereas postsynaptic TrkB may enhance NMDA receptor function in central neurons (Lessmann, 1998; Poo, 2001; Lu, 2003). Thus, the targeting and localization of TrkB receptors must be precisely regulated to exert appropriate BDNF responses (Yano and Chao, 2004). Recently, it was reported that an adaptor complex comprising Slp1, Rab27B, and CRMP-2 directly links the cytoplasmic tail of Trk to kinesin-1 (Arimura et al., 2009). Yet, knockdown of one or more of these adaptors reduces, but does not completely abrogate, anterograde Trk transport and distal axon localization, suggesting that alternative adaptor complexes may work in concert with Rab27B to traffic Trk receptors to the synapse. From our studies, we conclude that JIP3 could directly bind with TrkB via a minimal 12 aa domain in the TrkB juxtamembrane region and link TrkB to kinesin-1. Moreover, the JIP3/TrkB interaction could selectively drive TrkB anterograde transport in axons but not in dendrites. Finally, we showed that JIP3-mediated TrkB axonal transport could regulate BDNF-induced Erk activation and axonal filopodia formation.
Our studies provide three new insights into the regulation of TrkB axonal anterograde transport. To our knowledge, this is the first evidence that JIP3, a KLC1 adaptor protein, directly binds to TrkB and links TrkB to the motor protein kinesin-1. We find that endogenous JIP3 and TrkB interact when coimmunoprecipitated from brain lysates, as well as colocalize in sciatic nerve and hippocampal neurons (Figs. 2, 3). Overexpression or knockdown of JIP3 could, respectively, enhance or reduce the interaction between TrkB and KLC1, suggesting that JIP3 bridges the binding of TrkB with KLC1. Simultaneous knockdown of both JIP3 and Rab27B has the largest effect in reducing TrkB/KLC1 interaction, which suggests that JIP3 and Rab27B mediate TrkB binding with KLC1 and that they may have a cooperative effect in TrkB anterograde transport (Fig. 3). We have mapped a minimal 12 aa domain in the TrkB juxtamembrane domain that is necessary and sufficient for TrkB binding with JIP3. This 12 aa domain is different from the TrkB/Slp1/Rab27b interaction domain that located in the TrkB tyrosine kinase region. The 12 aa TrkB/JIP3 interacting domain also exists in the truncated TrkB isoform, TrkB.T1, suggesting that JIP3 is also involved in regulating TrkB.T1 anterograde transport. It has been reported that TrkB.T1 may not only serve as a dominant-negative regulator of TrkB-FL (Haapasalo et al., 2001) but is also capable of regulating cell morphology and activating Rho GTPase in a BDNF-dependent manner (Baxter et al., 1997; Rose et al., 2003). Thus, the JIP3-mediated axonal transport of TrkB.T1 may help to regulate its physiological function in distal axons.
Interestingly, JIP3 does not interact with TrkA. To our knowledge, JIP3 is the first adaptor protein that has been found to specifically link TrkB (or TrkC), but not its homolog TrkA, to motor proteins, which suggests that different Trk members might use distinct long-distance transport machineries (Fig. 4C).
Second, we found that JIP3 selectively mediates TrkB anterograde transport in axons but not in dendrites (Fig. 6). Neurons develop axons and dendrites, which have different constituents and functions. Whether the anterograde transport machinery for TrkB is the same in axons versus in dendrites remains unclear. It has been shown that kinesin-1 exists in axons, soma, and dendrites under normal conditions (Niclas et al., 1994; Kanai et al., 2000). JIP3 binding with kinesin-1 could steer kinesin predominantly to axons (Setou et al., 2002), which demonstrated that a specific adaptor protein can direct a motor and its cargo to distinct neuronal subdomains. Drosophila JIP3 ortholog SYD mutants have axonal accumulations that contain numerous synaptic vesicle proteins (Bowman et al., 2000). These proteins are present in axonal accumulations in kinesin-I complex mutants as well (Martin et al., 1999). These data suggest that JIP3 may transport synaptic vesicle in neurons; however, only a few cargos have been identified that could bind to JIP3 and be anterogradely transported. JIP3 has been known to bind to JNK3 and mediate JNK3 anterograde axonal transport. To our knowledge, TrkB is the second direct cargo that could be anterogradely transported in axon via JIP3. Postsynaptic targeting of TrkB receptor plays an important role in long-term potentiation (Poo, 2001; Lu, 2003; Nagappan and Lu, 2005); however, the mechanism underlying TrkB anterograde transport to dendrites still remains to be characterized. Dendritic TrkB anterograde transport may be mediated by a unique dendritic plus-end-directed motors such as KIF21B (Marszalek et al., 1999) or via another adaptor such as GRIP1, which enables kinesin to transport cargos in dendrite (Setou et al., 2002). It has been reported recently that dynein could drive cargo anterograde transport into dendrites because of the mixed microtubule array in dendrites (Kapitein et al., 2010). Thus, dynein may also mediate TrkB dendritic anterograde transport since TrkB binds dynein directly (Yano et al., 2001).
Third, we showed that JIP3-mediated TrkB anterograde axonal transport regulates BDNF actions (Fig. 8). Localized application of BDNF to the tip of an immature neurite consistently induces its axon differentiation and elongation, suggesting distal TrkB has important roles in axon formation (Shelly et al., 2007). In the present study, we found that depletion of JIP3 reduced the amount of TrkB in the axon and decreased axonal Erk1/2 signaling after BDNF stimulation, which suggests the importance of JIP3-mediated recruitment of TrkB into the distal axon for efficient BDNF/TrkB signaling. BDNF has been shown to augment the number and density of axonal and dendritic filopodial formation (Luikart et al., 2008; Menna et al., 2009), a process with crucial impacts on synapse formation. We found depletion of JIP3 selectively inhibits BDNF-induced axonal filopodia formation, which is in accordance with our previous finding that JIP3 knockdown reduces TrkB distal distribution in axons but not in dendrites and suggests that the JIP3/TrkB transport may play a role in presynaptic specializations. These data suggest that JIP3-mediated TrkB anterograde axonal transport could recruit more TrkB into distal axons and facilitate BDNF-induced retrograde signaling, which provides a mechanism of how the TrkB anterograde transport is coupled to BDNF signaling.
A recent report showed that sortilin could interact with Trk receptors and facilitate their anterograde axonal transport (Vaegter et al., 2011). In contrast to the JIP3 and TrkB/TrkC interaction, sortilin could bind with all three Trk receptors (TrkA, TrkB, and TrkC), and the extracellular domains of Trk receptors are responsible for their association. Since the majority of sortilin resides in the Golgi apparatus and sortilin has been shown to facilitate intracellular sorting of BDNF into the regulated secretion pathway (Nielsen et al., 2001; Chen et al., 2005a), we speculate sortilin could act upstream of the conventional anterograde transport machinery to ensure translocation of Trk proteins from the trans-Golgi network into kinesin-dependent transport vesicles.
In all, our studies have identified an adaptor molecule, JIP3, which could directly bridge TrkB with kinesin-1 and specifically regulate TrkB anterograde axonal transport. Furthermore, we demonstrated that JIP3-mediated TrkB axonal anterograde transport is essential in BDNF-induced signaling and filopodia formation. These findings provide insights into the mechanistic link between anterograde TrkB axonal transport and BDNF retrograde signaling and synapses modulation. Since depletion of both JIP3 and Rab27B could not completely abolish the TrkB interaction with kinsesin-1, future studies will try to identify additional adaptor proteins that mediate TrkB anterograde transport.
Footnotes
This work was supported by the National Natural Science Foundation of China (Grants 30725020, 31071254, and 30900717), the National 973 Basic Research Program of China (Grants 2010CB912004 and 2009CB941403), the State Program of National Natural Science Foundation of China for Innovative Research Group (Grant 81021001), the Foundation for Excellent Young Scientist of Shandong Province (Grant BS2010SW022), the research fund for the Doctoral Program of Higher Education of China (Grant 200804221070), and the Independent Innovation Foundation of Shandong University.
References
- Arimura N, Kimura T, Nakamuta S, Taya S, Funahashi Y, Hattori A, Shimada A, Menager C, Kawabata S, Fujii K, Iwamatsu A, Segal RA, Fukuda M, Kaibuchi K. Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell. 2009;16:675–686. doi: 10.1016/j.devcel.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Ascano M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Kinesin-dependent axonal transport is mediated by the Sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Burack MA, Silverman MA, Banker G. The role of selective transport in neuronal protein sorting. Neuron. 2000;26:465–472. doi: 10.1016/s0896-6273(00)81178-2. [DOI] [PubMed] [Google Scholar]
- Butowt R, von Bartheld CS. Conventional kinesin-I motors participate in the anterograde axonal transport of neurotrophins in the visual system. J Neurosci Res. 2007;85:2546–2556. doi: 10.1002/jnr.21165. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG, Nykjaer A, Hempstead BL, Lee FS. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005a;25:6156–6166. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Tanowitz M, Lee FS. A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol Biol Cell. 2005b;16:5761–5772. doi: 10.1091/mbc.E05-07-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002;298:770–776. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. 1999;19:8009–8026. doi: 10.1523/JNEUROSCI.19-18-08009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Niclas J, Vale RD, Banker G, Kosik KS. Suppression of kinesin expression in cultured hippocampal neurons using antisense oligonucleotides. J Cell Biol. 1992;117:595–606. doi: 10.1083/jcb.117.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC. Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci. 2004;24:10741–10749. doi: 10.1523/JNEUROSCI.2836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol. 2004;16:443–450. doi: 10.1016/j.ceb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Ha HY, Cho IH, Lee KW, Lee KW, Song JY, Kim KS, Yu YM, Lee JK, Song JS, Yang SD, Shin HS, Han PL. The axon guidance defect of the telencephalic commissures of the JSAP1-deficient brain was partially rescued by the transgenic expression of JIP1. Dev Biol. 2005;277:184–199. doi: 10.1016/j.ydbio.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Koponen E, Hoppe E, Wong G, Castren E. Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem Biophys Res Commun. 2001;280:1352–1358. doi: 10.1006/bbrc.2001.4296. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Griffin K, Jih GT, Stuckey J, Verhey KJ. Co-operative versus independent transport of different cargoes by Kinesin-1. Traffic. 2008;9:725–741. doi: 10.1111/j.1600-0854.2008.00722.x. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang SH, Zhao L, Sun ZP, Li XZ, Geng Z, Zhang KD, Chao MV, Chen ZY. Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB.T1. J Biol Chem. 2009;284:15126–15136. doi: 10.1074/jbc.M809763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Kelkar N, Gupta S, Dickens M, Davis RJ. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol Cell Biol. 2000;20:1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A labile component of AMPA receptor-mediated synaptic transmission is dependent on microtubule motors, actin, and N-ethylmaleimide-sensitive factor. J Neurosci. 2001;21:4188–4194. doi: 10.1523/JNEUROSCI.21-12-04188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V. Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. Gen Pharmacol. 1998;31:667–674. doi: 10.1016/s0306-3623(98)00190-6. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Zhang W, Wayman GA, Kwon CH, Westbrook GL, Parada LF. Neurotrophin-dependent dendritic filopodial motility: a convergence on PI3K signaling. J Neurosci. 2008;28:7006–7012. doi: 10.1523/JNEUROSCI.0195-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Weiner JA, Farlow SJ, Chun J, Goldstein LS. Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J Cell Biol. 1999;145:469–479. doi: 10.1083/jcb.145.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura H, Nishitoh H, Takeda K, Matsuzawa A, Amagasa T, Ito M, Yoshioka K, Ichijo H. Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J Biol Chem. 2002;277:40703–40709. doi: 10.1074/jbc.M202004200. [DOI] [PubMed] [Google Scholar]
- Menna E, Disanza A, Cagnoli C, Schenk U, Gelsomino G, Frittoli E, Hertzog M, Offenhauser N, Sawallisch C, Kreienkamp HJ, Gertler FB, Di Fiore PP, Scita G, Matteoli M. Eps8 regulates axonal filopodia in hippocampal neurons in response to brain-derived neurotrophic factor (BDNF) PLoS Biol. 2009;7:e1000138. doi: 10.1371/journal.pbio.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Fukaya M, Sato T, Sugihara K, Asano M, Yoshioka K, Watanabe M. Expression and distribution of JNK/SAPK-associated scaffold protein JSAP1 in developing and adult mouse brain. J Neurochem. 2006;97:1431–1446. doi: 10.1111/j.1471-4159.2006.03835.x. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Niclas J, Navone F, Hom-Booher N, Vale RD. Cloning and localization of a conventional kinesin motor expressed exclusively in neurons. Neuron. 1994;12:1059–1072. doi: 10.1016/0896-6273(94)90314-x. [DOI] [PubMed] [Google Scholar]
- Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, Pohlmann R, Petersen CM. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20:2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature. 2003;426:74–78. doi: 10.1038/nature01983. [DOI] [PubMed] [Google Scholar]
- Rougier JS, Albesa M, Abriel H Viard P Neuronal Precursor. Cell-expressed developmentally down-regulated 4–1 (NEDD4–1) controls the sorting of newly synthesized CaV1.2 calcium channels. J Biol Chem. 2011;286:8829–8838. doi: 10.1074/jbc.M110.166520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Severt WL, Biber TU, Wu X, Hecht NB, DeLorenzo RJ, Jakoi ER. The suppression of testis-brain RNA binding protein and kinesin heavy chain disrupts mRNA sorting in dendrites. J Cell Sci. 1999;112:3691–3702. doi: 10.1242/jcs.112.21.3691. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Takino T, Nakada M, Miyamori H, Watanabe Y, Sato T, Gantulga D, Yoshioka K, Yamada KM, Sato H. JSAP1/JIP3 cooperates with focal adhesion kinase to regulate c-Jun N-terminal kinase and cell migration. J Biol Chem. 2005;280:37772–37781. doi: 10.1074/jbc.M505241200. [DOI] [PubMed] [Google Scholar]
- Vaegter CB, Jansen P, Fjorback AW, Glerup S, Skeldal S, Kjolby M, Richner M, Erdmann B, Nyengaard JR, Tessarollo L, Lewin GR, Willnow TE, Chao MV, Nykjaer A. Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat Neurosci. 2011;14:54–61. doi: 10.1038/nn.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yano H, Chao MV. Mechanisms of neurotrophin receptor vesicular transport. J Neurobiol. 2004;58:244–257. doi: 10.1002/neu.10321. [DOI] [PubMed] [Google Scholar]
- Yano H, Lee FS, Kong H, Chuang J, Arevalo J, Perez P, Sung C, Chao MV. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci. 2001;21(RC125):1–7. doi: 10.1523/JNEUROSCI.21-03-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Sheng AL, Huang SH, Yin YX, Chen B, Li XZ, Zhang Y, Chen ZY. Mechanism underlying activity-dependent insertion of TrkB into the neuronal surface. J Cell Sci. 2009;122:3123–3136. doi: 10.1242/jcs.047712. [DOI] [PubMed] [Google Scholar]