

Abstract
In vivo and in vitro motoneuron survival depends on the support of neurotrophic factors. These factors activate signaling pathways related to cell survival or inactivate proteins involved in neuronal death. In the present work, we analyzed the involvement of the nuclear factor-κB (NF-κB) pathway in mediating mouse spinal cord motoneuron survival promoted by neurotrophic factors. This pathway comprises ubiquitously expressed transcription factors that could be activated by two different routes: the canonical pathway, associated with IKKα/IKKβ kinase phosphorylation and nuclear translocation RelA (p65)/p50 transcription factors; and the noncanonical pathway, related to IKKα kinase homodimer phosphorylation and RelB/p52 transcription factor activation. In our system, we show that neurotrophic factors treatment induced IKKα and IKKβ phosphorylation and RelA nuclear translocation, suggesting NF-κB pathway activation. Protein levels of different members of the canonical or noncanonical pathways were reduced in a primary culture of isolated embryonic motoneurons using an interference RNA approach. Even in the presence of neurotrophic factors, selective reduction of IKKα, IKKβ, or RelA proteins induced cell death. In contrast, RelB protein reduction did not have a negative effect on motoneuron survival. Together these results demonstrated that the canonical NF-κB pathway mediates motoneuron survival induced by neurotrophic factors, and the noncanonical pathway is not related to this survival effect. Canonical NF-κB blockade induced an increase of Bim protein level and apoptotic cell death. Bcl-xL overexpression or Bax reduction counteracted this apoptotic effect. Finally, RelA knockdown causes changes of CREB and Smn protein levels.
Introduction
Neuronal survival and apoptosis play a critical role in the development of the nervous system by eliminating superfluous neurons and ensuring proper connections. One family of molecules that plays a role in the maintenance of neuronal survival, blocking apoptosis, is the transcription factor nuclear factor-κB (NF-κB) (Bhakar et al., 2002). Inhibition of NF-κB activity causes neuronal death in a variety of neurodegenerative models (Koulich et al., 2001); reduced NF-κB activity has been involved in disorders such as Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis (Mattson et al., 2000).
NF-κB is a ubiquitously expressed transcription factor system that consists of homodimers and heterodimers of five structurally related proteins: RelA/p65, RelB, c-Rel, p50, and p52, of which the p50/RelA heterodimer is the most abundant and widely expressed. RelA, RelB, and c-Rel contain domains that activate transcription without the help of other NF-κB subunits (Kaltschmidt et al., 2005). NF-κB is held in an inactive form in the cytosol by interaction with the inhibitor protein IκB (Hayden and Ghosh, 2004). There are distinct NF-κB activation pathways. The most frequently observed is the canonical pathway that consists of the transcription factor dimer RelA/p65, and is typified by the phosphorylation of IκBα on serine 32/36 residues and proteasome degradation. Liberated RelA/p65 translocates to the nucleus, resulting in gene induction or gene repression. Canonical pathway activity depends on the catalytic subunits IKKα and IKKβ of the IκB kinase complex (IKK) (Perkins, 2007).
In the nervous system, NF-κB is activated by a variety of neurotrophic factors (NTFs). NF-κB promotes neuroprotection or neurodegeneration, but also regulates neuronal functions such as neurite growth (Kaltschmidt et al., 2005; Gallagher et al., 2007; Gutierrez et al., 2008). In the present work, we investigated whether NF-κB signaling mediates the survival effect induced by NTFs on the spinal cord motoneurons (MNs). The large variety of neurotrophic molecules that can support MN survival in vivo and in vitro indicates that developing and postnatal MNs depend on a cooperation of these molecules that is so far not fully understood (Oppenheim, 1996; Holtmann et al., 2005). NTFs promoting MN survival in vivo and in vitro belong to different gene families and activate several signaling pathways (Soler et al., 1999; Dolcet et al., 2001). To maintain MNs alive in culture, we (Gou-Fabregas et al., 2009) and others (Arce et al., 1999) use a cocktail of NTFs to achieve the maximal cell survival rates. We analyze the intracellular mechanisms involved in MN survival induced by the NTF cocktail. Using a primary culture of embryonic mouse MNs and reducing the protein level of several NF-κB family members, we demonstrate that the NF-κB canonical pathway is involved in the survival process. We show an apoptotic cell death effect on MNs when the canonical pathway is blocked; cell death is abolished when the antiapoptotic Bcl-xL protein is overexpressed or when proapoptotic Bax protein is reduced. Finally, we show that RelA knockdown causes CREB and Smn protein reduction, suggesting the involvement of NF-κB signaling in the regulation of these proteins.
Materials and Methods
Mouse MN isolation and culture.
MN cultures were prepared from embryonic day 12.5 (E12.5) CD1 male and female mouse spinal cords, essentially as described previously (Arce et al., 1999; Gou-Fabregas et al., 2009), with modifications. Briefly, mouse embryo spinal cords were dissected and the dorsal half removed. Ventral cords were chopped and incubated in GHEBS buffer (137 mm NaCl, 2.7 mm KCl, 22.2 mm glucose, 25 mm HEPES buffer, pH 7.4, and 20 IU/ml penicillin plus 20 mg/ml streptomycin) containing trypsin (Sigma; final concentration 0.025%) for 10 min at 37°C, followed by mechanical dissociation, then collected under a 4% bovine serum albumin (BSA) cushion. The largest cells were isolated by centrifugation (10 min at 520 × g) on an Iodixanol (OptiPrep, Axis-Shield) density gradient. Iodixanol solution (11.5%) was freshly prepared in GHEBS. At the end of this procedure, cells were again centrifuged through a BSA cushion. The collected cells were pooled in a tube containing culture medium and plated at various densities (see MN culture and survival evaluation). Cultured MNs enriched by Iodixanol were clearly identified by morphological criteria (Gou-Fabregas et al., 2009). All of the procedures were in accordance with the Spanish Council on Animal Care and approved by the University of Lleida Advisory Committee on Animal Services.
Isolated MNs were plated in four-well tissue culture dishes (Nunc, Thermo Fisher Scientific) for survival experiments (15,000 neurons per well), or for Western blot analysis (70,000 cells per well). Wells were coated with polyornithine/laminin as described previously (Soler et al., 1998). Culture medium was Neurobasal (Invitrogen) supplemented with B27 (Invitrogen), horse serum (2% v/v), l-glutamine (0.5 mm), and 2-mercaptoethanol (25 μm).
Measures of MN survival and apoptotic cell death.
MN survival evaluation was performed as described previously (Arce et al., 1999), with modifications. Briefly, cells were plated in complete medium containing a cocktail of recombinant NTFs (1 ng/ml BDNF, 10 ng/ml GDNF, and 10 ng/ml CNTF; PeproTech). Three hours after plating, medium was replaced and complete medium containing lentiviral particles (see below) was added. Twenty hours later, medium was washed and fresh medium containing NTFs was added. Photomicrographs of different microscopic areas from each dish were performed (four central areas per well, three wells for each condition), and we counted the number of large phase-bright neurons with long neurite processes present in the photomicrographs. The number of cells present in each dish was considered our initial 100%. Photomicrographs and counts were performed at indicated times in the same microscopic areas as the initial count. Survival was expressed as the percentage of cells counted with respect to the initial value (100%). Survival experiments performed in nontransduced cultures were performed as described in text and figure legends.
For apoptotic nuclear morphology assessment, cells were fixed in 4% paraformaldehyde and 100% methanol, and stained with 0.05 μg/ml Hoechst 33258 for 30 min at room temperature. Uniformly stained nuclei were scored as healthy, viable cells, whereas condensed or fragmented nuclei were considered as dead cells. For survival and cell death quantification experiments, we used an Olympus IX71 microscope equipped with epifluorescence optics.
Immunofluorescence of RelA nuclear translocation.
To determine RelA translocation to the nucleus, cells were incubated for 24 h in the presence or absence of the NTF cocktail. The cultures were fixed in 4% paraformaldehyde and 100% methanol, incubated overnight at 4°C with a polyclonal anti-RelA antibody (1:100; Santa Cruz Biotechnology), and further incubated with an anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (Invitrogen) for 1 h at room temperature protected from light. Nuclear staining was obtained with Hoechst 33258, as described above. Micrographs were obtained using a FluoView 500 Olympus confocal microscope.
Western blot analysis.
Western blot analysis was performed as described previously (Pérez-García et al., 2004). Cells were rinsed in ice-cold PBS, pH 7.2, after stimulation or lentiviral transduction. Total cell lysates were collected and resolved in SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride Immobilon-P transfer membrane filters (Millipore), using a GE Healthcare semidry Trans-Blot according to the manufacturer's instructions. The membranes were blotted with the specific antibodies: anti-Bax (1:1000); anti-Cleaved Caspase-3 (1:1000); anti-RelA (1:1000); anti-phospho-IKK (1:1000); anti-Ser 32/36 IκBα (1:500); anti-phospho-serine 473-Akt (1:1000); anti-CREB (1:1000); or anti-phospho-CREB (1:1000) from Cell Signaling Technology; anti-Bcl-xL (1:3000) or anti-Smn (1:5000) from BD Transduction Laboratories; anti-IKKβ (1:1000) or anti-IKKα (1:1000) from Calbiochem; anti-RelB (1:1000) (C-19), anti-IκBα (1:1000), or anti-Akt (1:1000) from Santa Cruz Biotechnology; or anti-Bim (1:500) from Stressgen, following the instructions of the providers. Unless stated otherwise, to control the specific protein content per lane, membranes were reprobed with a monoclonal anti-α-Tubulin antibody (1:50000, Sigma), as described by the provider. Blots were developed using the Super Signal chemiluminescent substrate (Pierce) or the ECL Advance Western Blotting detection kit (GE Healthcare).
NF-κB inhibitor SN50, the inactive control peptide SN50M, and the proteasome inhibitor MG132 were purchased from Calbiochem. The PI 3-kinase inhibitor LY294002 was from Tocris Bioscience.
Plasmids and production of lentiviral particles.
Lentiviral-based vectors for RNA-interference-mediated gene silencing (FSVi) were performed as described previously (Encinas et al., 2008). FSVi consisted of a U6 promoter for expression of short-hairpin RNAs (shRNAs) and the Venus variant of green fluorescent protein (GFP) under the control of an SV40 promoter for monitoring transduction efficiency. Lentiviruses were propagated in HEK293T cells using the polyethyleneimine (PEI, Sigma) cell transfection method. Twenty micrograms of the above plasmids containing the shRNAs or the empty vector, 13 μg of pSPAX2, and 7 μg of pM2G were transfected to HEK293T cultures. Cells were allowed to produce lentivirus for 4 d. Then the medium was centrifuged at 1200 × g for 5 min, and the supernatant was filtered using a 45 μm filter. The medium containing the lentiviruses was stored at 4°C. Biological titers of the viral preparations, expressed as the number of transducing units per milliliter (TU/ml), were determined by transducing HEK293T cells in limiting dilutions. After 48 h, the percentage of GFP-positive cells was measured, and viruses at 4 × 105 to 1 × 106 TU/ml were used for the experiments. For lentiviral transduction, MNs were plated in four-well dishes and 2 h later the medium containing lentivirus (2 TU/cell) was added. The medium was changed 20 h later and infection efficiency was monitored in each experiment by directly counting GFP-positive cells. The frequency of infection rose 99%. RNA interference efficiency was monitored by Western blot analysis using specific antibodies.
The following sequences were used for generation of shRNAs: Bax: 5′-GCAGCTGACATGTTTGCTGAT-3′; IKKα: 5′-CAGGCTCTTTCAGGGCAT-3′; IKKβ1: 5′-GCTGCACATTTGAATCTGTAT-3′; IKKβ2: 5′-GCTCTTAGATACCTTCATGAA-3′; scrambled IKK control: 5′-GCTCTCTTGAAGGTTGATGTA-3′; RelA: 5′-AGATCTTGAGCTCGGCAGTG-3′; RelB: 5′-GCATGCGCTTCCGCTACGAGT-3′. shRNAs of IKKs and RelA have been described by Dolcet et al. (2006).
Statistical analysis.
All experiments were performed at least three times. Values were expressed as mean ± SEM. The data obtained from the independent experiments were used for statistical analysis. We used one-way ANOVA to assess survival differences between groups for variable treatment. If the ANOVA test was statistically significant, we performed post hoc pairwise comparisons using the Bonferroni test; p values lower than 0.05 were considered significant.
Results
Neurotrophic factor stimulation induces NF-κB pathway activation on spinal cord MNs
Addition of NTFs to culture medium induces neuronal survival through the activation of intracellular pathways. One of the well known pathways activated after NTFs stimulation is the PI 3-kinase/Akt [for review, see Airaksinen and Saarma (2002) and Chao (2003)]. However, NTFs can also activate other pathways that are crucial for the neuronal physiology. In some neuronal populations, the NF-κB pathway is activated by cytokines and is responsible for cell survival and/or neurite outgrowth (Maggirwar et al., 1998; Gallagher et al., 2007). Because no information exists about the involvement of the NF-κB pathway mediating MN survival, we decided to analyze this subject in our MN model. Spinal cord MNs respond to a variety of NTFs; requirements for these factors vary depending on the MN subpopulations and the developmental period (Holtmann et al., 2005). Our previous work (our unpublished results and Gou-Fabregas et al., 2009) and that of others (Arce et al., 1999) showed that using a cocktail of NTFs enhanced MN survival compared to the survival rate obtained by each NTF separately. Thus, we decided to analyze the role of the NF-κB pathway when optimal survival conditions were present in the culture medium. The maximal survival level obtained was when we added to the medium the following cocktail: 1 ng/ml BDNF, 10 ng/ml CNTF, and 10 ng/ml GDNF (our unpublished results and Fig. 1).
Figure 1.
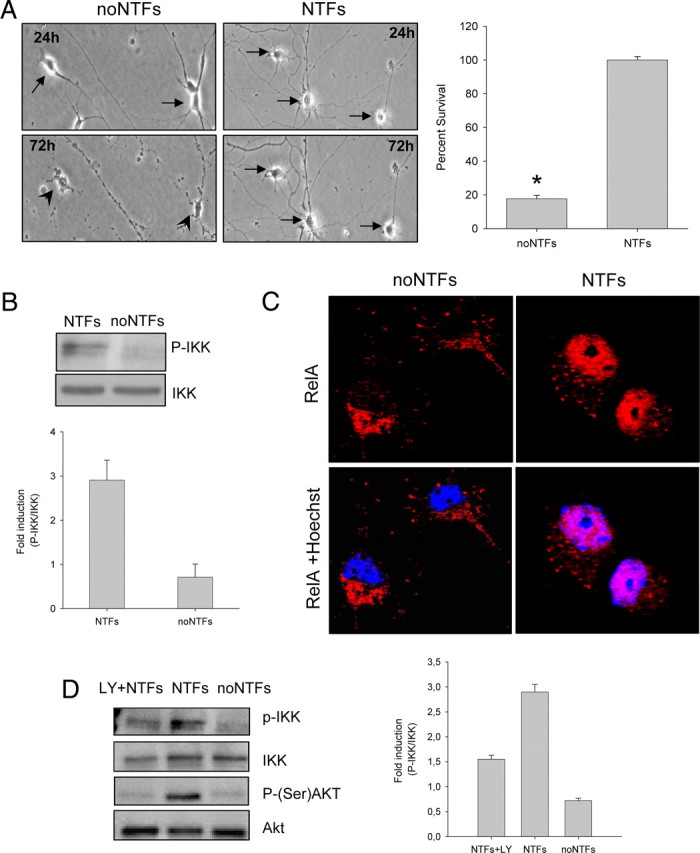
Effect of NTFs on IKK phosphorylation and p65 nuclear translocation. A, Representative microscopy images of 24 and 72 h cultured spinal cord MNs in the presence (right panels) or the absence (left panels) of NTFs (10 ng/ml CNTF, GDNF, HGF, BDNF). Arrows indicate surviving cells. Arrowheads indicate dying cells. The percentage of cell survival was measured as described in Materials and Methods. Values of the graph are the mean of the percentage of cell survival for each condition of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.01). B, Protein extracts of NTF-stimulated (10 min) or nonstimulated cultures were probed with an anti-phospho-IKK (P-IKK) antibody by Western blot analysis. Membranes were stripped and reprobed with an antibody against IKK, used as a loading control. C, Cells were cultured in the presence or the absence of the NTF cocktail. Twenty-four hours later, cultures were fixed and immunofluorescence was performed using an anti-RelA antibody. Representative confocal images of p65 (red) and Hoechst nuclear staining (blue). D, MN cultures were stimulated with NTFs in the presence or the absence of 25 μm LY294002, or left untreated, and protein extracts were probed with P-IKK and anti-phospho-Ser473 Akt antibodies. LY294002 was pretreated for 45 min before NTF stimulation. Membranes were stripped and reprobed with an antibody against IKK or Akt, respectively. Graph values in B and D represent measures of phospho-IKK versus total IKK from three independent experiments ± SEM.
After 72 h in the presence of NTFs, ∼100% of MNs were alive; only ∼20% of MNs survived in the absence of NTFs (Fig. 1). To analyze the ability of NTFs to activate the NF-κB pathway, we tested both IKK phosphorylation and RelA nuclear translocation. The IKK protein complex becomes phosphorylated and activated in response to the stimuli that induce NF-κB pathway activation (Delhase et al., 1999). Using an anti-phospho-IKK antibody (P-IKK), we evaluated IKK phosphorylation after 10 min of NTFs stimulation. We observed that NTF stimulation increases IKK phosphorylation level as compared to nonstimulated cultures, indicating that NTFs were able to induce IKK phosphorylation (Fig. 1B).
After IKK and IκB phosphorylation, NF-κB complex is released, relocates to the nucleus, and binds to the κB DNA elements, resulting in gene induction or gene repression (Perkins, 2007). To analyze the effect of NTF treatment on RelA translocation to the nucleus, MNs were cultured for 24 h in the presence or absence of NTFs. Using an anti-RelA antibody, immunofluorescent confocal images showed that NTF treatment induced nuclear relocation of RelA (Fig. 1C). However, in the absence of NTFs, RelA is maintained primarily in the cytoplasm. The percentage of MNs with RelA localized in the nucleus of NTF-treated cultures was 85 ± 6.6%, whereas in the absence of NTFs, the percentage of cells with RelA localized in the nucleus was only 17.3 ± 3.3%. Together, these results indicate that NTF treatment induces NF-κB pathway activation in MNs. We mentioned above that one of the main intracellular pathways related to NTF-induced neuronal survival is the PI 3-kinase/Akt. To examine whether this pathway has a role in NF-κB activation in MNs, we used the PI 3-kinase inhibitor LY294002 and analyzed IKK phosphorylation by Western blot. When 25 μm LY294002 was present in the culture medium, the level of IKK phosphorylation was 50% reduced when compared to the NTF-treated controls (Fig. 1D). This result shows that PI 3-kinase blockade results in IKK phosphorylation decrease, suggesting the involvement of this pathway in NF-κB activation after NTF treatment.
NTFs induce canonical NF-κB pathway activation
At least two pathways to activate NF-κB exist: canonical and noncanonical. The canonical pathway depends upon the phosphorylation status of the two kinases IKKα and IKKβ, which together with the regulatory subunit NEMO/IKKγ constitute the catalytic part of the IKK complex; activation of the noncanonical pathway depends on the phosphorylation of an IKKα homodimer. The most frequently observed is the canonical pathway, which is induced in response to various stimuli. This pathway is typified by the rapid phosphorylation of the IKK complex, IκB degradation, the release of the RelA/p50 complex, and its translocation to the nucleus (Li et al., 1999; Perkins, 2007). To analyze the activation of the canonical pathway in our experimental model, we first examined whether NTFs induced IκB endogenous protein reduction. Western blots showed a clear decrease in total IκB protein after 30–45 min followed by a characteristic resynthesis of IκBα that was evident after 60 min of NTF treatment (Fig. 2A).
Figure 2.

Effect of NTFs on IκB and p65 phosphorylation. A, Representative Western blots for total IkBα (left) and phospho-Ser 32/36 IkBα (right) in protein extracts of MNs treated with NTFs in the presence or the absence of 5 μm MG132. Time 0 indicates the basal level of total or phospho-IkBα. B, Representative Western blot for phospho-Ser536 RelA and total RelA in protein extracts of cultures stimulated (20 min) or nonstimulated with NTFs. Graph indicates the increase of RelA phosphorylation compared with the total RelA protein. C, MNs were cultured in the presence of NTFs and the NFκB inhibitor SN50 (20 μm) or its inactive control SN50M (20 μm). Cell survival was evaluated 3 d after treatment. Graph values are the mean of the percentage of cell survival for each condition of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.001).
In the canonical pathway, NF-κB is activated by phosphorylation of IκBα on serine residues 32 and 36 (Hayden and Ghosh, 2004). To ascertain whether NTFs increases IκBα phosphorylation in these serine residues, protein extracts from MNs treated with NTFs for times ranging from 30 to 60 min were analyzed by Western blot and probed with an anti-IκB phosphoserine 32/36 antibody. To observe better IκB phosphorylation, we avoid the proteasome-mediated degradation of IκB by adding the proteasome inhibitor MG132 in the culture medium. As we show in Figure 2A, NTFs promoted a marked increase in phosphoserine IκBα within 30 min, confirming the activation of the canonical NF-κB signaling pathway.
Finally, we also analyzed RelA serine phosphorylation using an anti-phosphoserine 536 RelA antibody. This serine phosphorylation in RelA is mediated by IKK complex (Perkins, 2007). Twenty minutes after NTF stimulation, we observed a clear increase of phosphor-serine 536-RelA compared to the nonstimulated controls (Fig. 2B). All these findings together strongly suggest the activation by NTFs of the canonical NF-κB signaling pathway in MNs.
Effect of IKKβ or IKKα inhibition on the NTF-induced MN survival
To assess the requirement for NF-κB signaling in the NTF-induced MN survival, we inhibited several different steps in the NF-κB signaling network. First, we pharmacologically inhibited the NF-κB pathway using the SN50 inhibitor peptide, a cell-permeable peptide that blocks the nuclear localization sequence of p50 (Lin et al., 1995). To this end, cultures were treated with NTFs and 20 μm SN50 or its inactive control peptide SN50M, and 3 d later MN survival was evaluated. The presence of NF-κB inhibitor in the culture medium significantly decreased MN survival induced by NTF treatment when compared with its inactive control (Fig. 2C).
To further assess the relevance of the NF-κB pathway to mediating NTF-induced MN survival, we performed lentiviral-based knockdown of IKKβ and IKKα. First, we generated two shRNA sequences targeting two specific sites of mouse IKKβ (shIKKβ1 and shIKKβ2) and generated lentiviral particles containing the shRNAs. MNs were isolated from E12.5 mouse embryos and maintained in the presence of the NTFs. Two hours after plating, culture medium was changed and medium containing NTFs and lentivirus of shIKKβ1 or shIKKβ2 or shRNA empty vector control was added. Twenty hours later, the medium was washed and replaced by fresh medium supplemented with NTFs. As shown in Figure 3A, the frequency of transduction rose to 99% of the cells present in the culture dish (GFP-positive cells monitored by fluorescence microscope). After 5 d, Western blot analysis of cultures transduced with lentivirus carrying shIKKβ1 or shIKKβ2 exhibited a strong reduction of IKKβ protein compared with the control cultures transduced with the empty vector (Ctrl) (Fig. 3B). The low levels of IKKβ were maintained throughout the experiment (data not shown). MNs transduced with these lentiviral constructions were used for a time course survival analysis of the effect of endogenous IKKβ knockdown in NTF-supplemented cultures. To assess cell survival, we calculated the percentage of cells present in the tissue culture dish at various days after transduction compared to the number of cells counted in the same microscopic fields at the beginning of the experiment. Cultures transduced with the empty vector control showed a level of cell survival ∼100% from day 1 to day 6 (end of the experiment) (Fig. 3B). Control survival experiments using a scrambled RNA control vector for the shIKK knockdown were also performed. When cells were transduced with the scrambled shIKK, cell survival was similar to the empty vector controls (3 d after transduction: 86.3 ± 4.8% and 93.3 ± 2.8%, respectively; 5 d after transduction: 82.2 ± 0.5% and 80.8 ± 3.8%, respectively). Time course survival experiments demonstrated that when IKKβ protein level was reduced, cell survival dramatically decreased from day 4 to day 6 (80.6 ± 5.5% and 6.9 ± 1.7%, respectively) (Fig. 3B). We also generated a shRNA sequence targeting specific sites of mouse IKKα (shIKKα). After 5 d of lentiviral transduction, IKKα protein level was reduced in shIKKα cultures compared to empty vector control cells (Fig. 3A), and this reduction was maintained through the end of day 6 (data not shown). We also performed survival experiments after IKKα protein knockdown, following the same cell-counting protocol described above and in Materials and Methods. Results showed that MN survival was compromised when endogen IKKα was reduced. The percentage of survival of shIKKα cultures decreased gradually from day 1 to day 6 (Fig. 3B). Western blots of IKK protein reduction revealed that IKKβ dropped abruptly at day 5 after transduction and IKKα decreased gradually from the day 2. Together, all these results demonstrated that both IKKβ and IKKα contribute to NTF-induced MN survival.
Figure 3.

Effect of IKKβ or IKKα knockdown on MN survival in the presence of NTFs. Mouse MNs were transduced with lentivirus containing the shIKKβ1, shIKKβ2, shIKKα, or empty vector (Ctrl) constructs. A, Representative microscopy images of 3 d shIKKβ-, shIKKα-, or Ctrl-transduced cultures: phase contrast (left) and GFP (right). GFP indicates green fluorescent protein-expressing cells in the same microscopic field. B, MNs were transduced with shIKKβ1, shIKKβ2, shIKKα, or Ctrl for 5 d. Total cell lysates were probed by Western blot with an anti-IKKβ or an anti-IKKα antibody. For the loading control, membranes were reprobed using an antibody against α-Tubulin. Graph values are the mean of the percentage of IKKβ or IKKα protein level in MNs transduced with shIKKβ1, shIKKβ2, or shIKKα compared to the Ctrl from three independent experiments. C, Cells were transduced with shIKKβ1, shIKKβ2, shIKKα, or Ctrl constructs and cultured in the presence of NTFs. A time course cell survival evaluation was performed, and the percentage of surviving MNs was measured as described in Materials and Methods. Graph values are the mean of the percentage of cell survival for each condition of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.001). Bottom, Representative Western blots of IKKα or IKKβ protein decrease at different time points after shIKKα or shIKKβ lentiviral transduction.
RelA, but not RelB, inhibition causes cell death in NTF-maintained MNs
Active binding to specific DNA sequences is performed by heterodimers or homodimers of NF-κB subunits. The most prominent and extensively studied dimer is that of RelA (also called p65) and p50, which is activated in the canonical pathway; RelB and p52 form a dimer that participates in the noncanonical scheme (Perkins, 2007). To determine which of these pathways is involved in MN survival, we analyzed the effect of RelA or RelB knockdown. We generated shRNA targeting specific sites of mouse RelA (shRelA) or RelB (shRelB) sequences. As shown in Figures 4 and 5, the frequency of transduction was near 100% of the cells present in the culture (GFP-positive cells monitored by fluorescence microscopy). When analyzed by Western blot using specific antibodies, the level of RelA or RelB proteins in shRelA- or shRelB-transduced cultures, respectively, was clearly reduced in comparison to the empty vector control cells. Figure 4A shows a time course analysis of RelA protein reduction; and Figure 5C shows RelB protein reduction (day 5 after transduction). We performed survival experiments following the same protocol described above. Reduction of endogen RelA protein caused MN cell death (Fig. 4B); nevertheless, RelB protein knockdown had no effect on MN survival (Fig. 5C). After 6 d of transduction, no surviving cells were found in shRelA-transduced cultures, whereas in shRelB at the same time period the percentage of cell survival was similar to empty vector control cultures (∼90%). These results strongly suggest that NTFs induce MN survival through activation of the NF-κB canonical pathway, but not by the noncanonical pathway. The activation of the noncanonical pathway was shown by Western blot using an antibody against the p100 and the cleaved form p52. When cells were stimulated by NTFs, we observed the presence of a band corresponding to p52, whereas in non-NTF-stimulated protein extracts, p52 was absent in three independent experiments (Fig. 5A). Thus we demonstrated that even though the noncanonical pathway is activated by NTFs, its activation is not involved in the survival-promoting effect of these factors on MNs.
Figure 4.

RelA knockdown reduced NTF-promoted MN survival. A, Representative microscopy images of 3 d shRelA-transduced MNs: phase contrast (left) and GFP (right) of the same microscopic field. Western blot analysis of RelA protein levels after 0, 3, 4, or 5 d of shRelA transduction. Protein extracts were collected at the different time points and analyzed using an antibody against RelA protein. Membranes were reprobed with an antibody against α-Tubulin, used as a loading control. B, Representative microscopy images of the same microscopic field of 0, 3, or 5 d shRelA-transduced cultures of MNs. Arrows indicate surviving cells. Arrowheads indicate dying or dead cells. The graph in B represents the percentage of surviving MNs at several time points after shRelA or empty vector transduction, as described in Materials and Methods. Graph values are the mean of the percentage of cell survival for each condition of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.001).
Figure 5.

RelB protein is not implicated in mouse MNs survival. A, Representative Western blot for p100/p52 in protein extracts of cultures stimulated (20 min) or nonstimulated with NTFs. The same membrane was reprobed with an antibody against α-Tubulin, used as a loading control. B, Microscopic images of 5 d shRelB-transduced MNs: phase contrast and GFP (left and right, respectively). Total cell lysates of transduced cells (shRelB or empty vector control, Ctrl) were collected 5 d after transduction and probed with an anti-RelB antibody by Western blot; the membrane was reprobed with an antibody against α-Tubulin, used as a loading control. C, Cell survival was evaluated at 3 or 6 d after shRelB or control transduction. Graph represents the mean of the percentage of cell survival for each condition of nine wells for three independent experiments ± SEM (error bars).
Inhibition of the NF-κB pathway causes apoptotic cell death
Trophic factor deprivation induces apoptotic cell death in MNs (Soler et al., 1999; Dolcet et al., 2001). It is also known that in some neuronal models, inhibition of NF-κB activation induces apoptosis (Piccioli et al., 2001); and NF-κB regulates antiapoptotic gene transcription (Tian et al., 2005). To determine whether NF-κB pathway inactivation induces apoptotic cell death in our cultured cells, we analyzed both the presence of apoptotic nuclei and the activity of Caspase-3 as indicators of classical apoptosis. To this end, MNs were transduced using the following lentiviral constructs: shIKKβ, shIKKα, shRelA, or the empty vector control; 24 h later, medium containing lentiviral particles was washed and replaced by fresh medium containing NTFs. Five days later, we measured Caspase-3 activity by Western blot using a specific antibody (Fig. 6A). As expected, shIKKβ-, shIKKα-, and shRelA-transduced cultures showed an increase of the activation-specific Caspase-3 fragment (p17). Control cultures also displayed activation of Caspase-3 when deprived of NTFs (data not shown).
Figure 6.

IKKα, IKKβ, or RelA protein knockdown causes apoptotic cell death. A, MNs were transduced with shIKKα, shIKKβ, shRelA, or empty vector (Ctrl) constructs and cultured in the presence of NTFs. After 5 d, total cell lysates were analyzed by Western blot using an antibody against Cleaved Caspase-3. Membranes were reprobed with an antibody against α-Tubulin, as a loading control; or antibodies against IKKα, IKKβ, or RelA as protein reduction control. B, Five days after transduction, cultures were fixed and stained using Hoechst nuclear dye. Representative microscopy images of Hoechst staining of apoptotic (b) or nonapoptotic (a) nuclear morphology. The graph in B represents the percentage of apoptotic nuclei present in shIKKα-, shIKKβ-, shRelA-, or Ctrl-transduced cultures; values are the mean of six wells for each condition from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.05; **p < 0.001), compared to the empty vector control.
To further analyze the apoptotic origin of the cell death observed, we counted the number of nuclei showing apoptotic morphology, using Hoechst 33258 staining (Fig. 6B). After 5 d of transduction, the percentage of apoptotic cells was increased in shIKKβ, shIKKα, and shRelA compared to the empty vector control treatment. Around 50% of the cells that received shIKKβ showed this particular morphology, which was the highest proportion of apoptotic nuclei. shIKKα or shRelA treatment also induced a significant increase of apoptotic nuclei (13 ± 3.9% and 25.7 ± 3.9%, respectively). Thus, when the NF-κB pathway was inactivated, the dying cells displayed apoptotic nuclear morphology, characterized by chromatin condensation and shrinkage of the nucleus even in the presence of NTFs. Control experiments of nontransduced cultures also showed an increase of the percentage of apoptotic nuclei when deprived (NTF-supplemented cultures: 7 ± 1.2%; NTF-deprived cultures: 55 ± 3%), confirming that in our system NTF deprivation induces apoptotic cell death. Together these results demonstrated that inhibition of the NF-κB pathway induced apoptotic cell death in NTF-maintained MNs.
Bcl-xL overexpression or Bax knockdown protects MN cell death after IKKβ, IKKα, or RelA inhibition
We show here that NTFs induce MN survival through the NF-κB pathway. When this pathway is blocked, the consequence is an apoptotic cell death even in the presence of NTFs. The same type of cell death is observed when MNs are deprived of NTFs (see above and our unpublished results). In other neuronal models, this apoptotic cell death could be blocked by overexpression or inhibition of antiapoptotic or proapoptotic proteins (respectively). Because Bcl-xL is one of the strongest antiapoptotic proteins present in the nervous system (González-García et al., 1995), we decided to overexpress human Bcl-xL (hBcl-xL; kindly provided by Dr. J. X. Comella, Institut de Recerca Hospital Vall d'Hebron, Barcelona, Spain) in our cell model. Thus, shIKKβ, shIKKα, shRelA, or the empty vector control MN cultures were cotransduced with a lentiviral vector carrying an expression construct containing the human Bcl-xL gene. Twenty hours after cotransduction, medium containing lentiviral particles was washed and replaced by fresh medium containing NTFs. By Western blot analysis, we observed that Bcl-xL was overexpressed after 5 d of lentiviral transduction (Fig. 7). To determine the effect of Bcl-xL overexpression in these cultures, survival experiments were performed after 3 and 5 d as described above. As observed in Figure 7A, Bcl-xL significantly rescued cells from the death effect caused by IKKβ, IKKα, or RelA protein level reduction.
Figure 7.

Bcl-xL overexpression protects MNs from apoptotic cell death caused by IKKα, IKKβ, or RelA interference. Mouse MNs were cotransduced with lentivirus containing the hBcl-xL construct plus shIKKα, shIKKβ, shRelA, or the empty vector (Ctrl) or transduced with shIKKα, shIKKβ, shRelA, or Ctrl lentivectors, and survival was measured as indicated in the diagram. A, Graphs represent the mean of the percentage of cell survival for each condition mentioned above of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.001). B, Total cell lysates from different time points (0, 24, 48, and 72 h) of nontransduced cultures treated with or without NTFs were analyzed by Western blot using an antibody against Bcl-xL. The same membrane was reprobed with an antibody against α-Tubulin as a loading control. C, Protein extracts of 5 d transduced (shIKKα, shIKKβ, shRelA, or Ctrl) cultures were probed with an anti-Bcl-xL antibody by Western blot. Membrane was reprobed with an antibody against α-Tubulin, as a loading control. D, Total cell lysates of 5 d cotransduced cultures with hBcl-xL plus shIKKα or shIKKβ or cultures transduced with shIKKα, shIKKβ, or Ctrl were probed with antibodies against IKKβ or IKKα by Western blot. The same membranes were reprobed with an antibody against α-Tubulin as a loading control.
To establish whether NTF cocktail increased Bcl-xL protein levels in our experimental paradigm, we analyzed changes of this protein in the presence or the absence of NTFs. MNs were isolated and cultured for different time periods. Western blot analysis of protein extracts obtained from cultures with or without NTFs was performed using a specific antibody against Bcl-xL. Results showed that the level of Bcl-xL protein was clearly reduced in NTF-deprived cultures compared to NTF-treated cells (Fig. 7B). Thus, we proceeded to evaluate whether NF-κB pathway inhibition induces changes in Bcl-xL protein level. Western blot analysis of protein extracts from 5 d cultures transduced with shIKKβ, shIKKα, shRelA, or empty vector control did not show significant differences in Bcl-xL protein, indicating that in our system NF-κB inhibition neither increased nor reduced Bcl-xL (Fig. 7C). Finally, we checked whether Bcl-xL overexpression affected IKKβ, IKKα, or RelA protein expression in shRNA-transduced cultures. Protein extracts were analyzed by Western blot and results revealed that Bcl-xL overexpression did not affect IKKβ or IKKα (Fig. 7D) or RelA (data not shown) protein level.
To further confirm that apoptosis was controlling the cell death process caused by NF-κB pathway blockade and that increasing the cellular antiapoptotic mechanisms could rescue MNs from death caused by NF-κB inhibition, we reduced the levels of the proapoptotic member of the Bcl-2 family, Bax. Bax has been related to mitochondrial dysfunction and apoptosis (Wei et al., 2001). Thus, after plating, we transduced MNs with a shRNA targeting specific sites of mouse Bax sequence (shBax) or left them untransduced; after 3 d, cultures were cotransduced or transduced (respectively) with shIKKβ, shIKKα, shRelA, or the empty vector control (considered as time 0 of the experiment; see diagram in Fig. 8). Using a specific antibody, we found that Bax protein level was significantly reduced after 3 d of shBax transduction. After 3 and 5 d of shIKKβ, shIKKα, or shRelA cotransduction, cell survival was evaluated as described in Materials and Methods (Fig. 8). Results showed that in cultures cotransduced with shBax and shIKKβ or shRelA (Fig. 8A) or shIKKα (data not shown), survival percentage was significantly increased compared with cultures with no Bax cotransduction. As described above for Bcl-xL protein, IKKβ or RelA reduction did not change Bax protein level (Fig. 8B).
Figure 8.

Bax protein knockdown rescues MNs from cell death caused by IKKβ or RelA interference. MNs were transduced with shBax or nontransduced; 3 d later, they were cotransduced or transduced with shIKKβ, shRelA, or empty vector (Ctrl). Bax protein reduction was analyzed by Western blot using a specific antibody (see diagram). A, Cell survival was measured as described in Materials and Methods. Graphs represent for each condition the mean percentage of survival of nine wells from three independent experiments ± SEM (error bars). Asterisk indicates significant differences between data from the experiments using one-way ANOVA test and Bonferroni's post hoc multiple comparisons (*p < 0.001). B, Protein extracts were collected from 5 d transduced cultures (shIKKβ, shRelA, or Ctrl) and analyzed by Western blot using an anti-Bax antibody. Membranes were reprobed with an anti-α-Tubulin antibody as a loading control.
Effect of NF-κB pathway inhibition on Bim protein level
It has been demonstrated that during apoptosis, Bim, the BH3-only member of Bcl-2 family, can directly or indirectly activate the proapoptotic Bax and the subsequent mitochondrial apoptotic pathway. Moreover, Bim displaces Bcl-xL in the mitochondria and promotes Bax translocation during TNFα-induced apoptosis (Zhang et al., 2008). In this context, we decided to analyze whether Bim protein levels changed when the canonical NF-κB pathway was knocked down. Thus, MNs were transduced with shRelA, and protein extracts were collected 5 d after transduction. By Western blot analysis using an antibody against BimEL, we observed that Bim protein level increased in shRelA cultures compared with the empty vector control condition (Fig. 9A). These results strongly suggest that after canonical NF-κB signaling inhibition, Bim increase can induce the apoptotic cell death observed.
Figure 9.

RelA interference causes Bim protein increase and Smn and CREB protein reduction. A, Protein extracts of MNs transduced with shIKKβ, shRelA, or empty vector (Ctrl) constructs and cultured in the presence of NTFs were probed with an anti-BimEL antibody, the same membrane was reprobed with an antibody against α-Tubulin, used as a loading control. B, Representative Western blots for Smn, RelA, phospho-CREB, and total CREB in protein extracts of 5 d shRelA- or control empty vector-transduced cultures. The same membranes were reprobed with an antibody against α-Tubulin as a loading control. C, Schematic model showing the proposed intracellular events of NTF-induced NF-κB activation in MNs. The blockade of the canonical NF-κB pathway induces an increase of Bim protein and apoptotic cell death and a decrease of CREB protein levels, leading to the reduction of Smn protein level.
RelA knockdown causes Smn protein decrease
Smn (survival motor neuron) protein is a ubiquitously expressed protein with nuclear and cytoplasm functions. Its deficiency causes the neurodegenerative disease spinal muscular atrophy. This disease is characterized by the specific loss of ventral horn spinal cord MNs (Sumner, 2006). Because the NF-κB pathway is involved in MN survival, we wanted to know whether the NF-κB signaling pathway might be affecting the Smn protein level. MNs were transduced with shRelA, and cell extracts were collected 4 d after transduction and analyzed by Western blot using a specific antibody against Smn. Results obtained showed that after RelA knockdown, Smn protein levels were reduced compared with the empty vector control cultures (38 ± 6% and 100%, respectively). It has been previously described that the transcription factor CREB (cAMP-response element-binding) regulates Smn protein expression (Majumder et al., 2004). In this context, we decided to evaluate changes in CREB phosphorylation and protein level. MNs were transduced with shRelA, and 4 d later, cell extracts were collected. Western blot analysis using specific antibodies against CREB and phospho-CREB demonstrated that the endogenous CREB protein level was reduced (Fig. 9B). All these results together suggest that CREB and Smn protein expression is dependent on NF-κB signaling pathway activity.
Discussion
In the present work, we demonstrate that the canonical NF-κB pathway mediates cell survival in a primary culture of isolated mouse embryonic MNs. Reducing protein level of several members of the NF-κB signaling pathway revealed that these proteins are essential to the ability of NTFs to promote MN survival. Inhibition of IκB kinases or the DNA-binding subunit RelA caused a very substantial reduction of MN survival and induced apoptotic cell death. However, reduction of RelB protein did not affect MN survival. The NF-κB pathway could be activated by two different routes: the canonical or classical and the noncanonical. The canonical pathway is characterized by the activation and the translocation of p50/RelA heterodimers to the nucleus, whereas the noncanonical pathway results from p52/RelB heterodimers activation and translocation (Perkins, 2007). It is known that p52/RelB have affinity for κB elements distinct from p50/RelA, and might therefore regulate a distinct subset of NF-κB target genes. It is also believed that the subunit composition of the complexes may vary depending on factors such as the developmental state of the neurons and their location within the nervous system. In our culture system, we reduced RelA or RelB proteins to analyze the involvement of the canonical or the noncanonical NF-κB pathway on MN survival. Our results clearly demonstrated that RelA reduction caused cell death, whereas RelB did not, indicating that only the canonical pathway is involved in NTF-induced MN survival, even though both pathways are activated in MNs after NTF treatment. The noncanonical signaling is characterized by IKKα-dependent processing of p100 to p52 (Xiao et al., 2004), whereas both IKKα and IKKβ are associated with the activation of the canonical pathway (Perkins, 2007). The phosphorylation of these two kinases induces RelA release and translocation to the nucleus. In the present work, we show that NTF stimulation induces IKK phosphorylation, IκBα serine phosphorylation and degradation, and RelA translocation to the nucleus, indicating the activation of the canonical pathway. We also show p100 processing, demonstrating that NTFs also activate the noncanonical signaling. After IKKα or IKKβ knockdown, we observed reduction of cell survival and increase of apoptotic cell death. However, when RelB protein level was reduced, MN survival was not affected. This result reinforced the initial hypothesis that NF-κB canonical pathway mediates MN survival and the cell death effect observed by IKKα knockdown is due to its role in the canonical signaling activation.
The NF-κB canonical pathway has been related to cell survival during development. In fact, IKKβ (Li et al., 1999) or RelA (Beg et al., 1995) knock-out mice show embryonic lethality accompanied with a massive degeneration of the liver by apoptosis. The role of the canonical pathway during the development of the nervous system has been revealed by IKKα and IKKβ double-deficient mice. The double knock-out demonstrated that both kinases are essential for NF-κB activation and have an important role in protecting neurons against excessive apoptosis during development (Li et al., 2000). These authors also postulated that both IKKs might have a genetic redundancy during nervous system development.
NF-κB has been studied more extensively in the immune system; however, it is known that in some neuronal populations NF-κB activity induces cell survival. Early indications that NF-κB activation promotes neuronal survival came from studies of embryonic hippocampal cells under stressing conditions (Cheng et al., 1994; Mattson et al., 1997), but there are other examples: in cerebellar granule cells, trophic factor withdrawal induces NF-κB inactivation (Kovács et al., 2004) and NF-κB activity contributes to promote survival in sympathetic NGF-dependent neurons (Maggirwar et al., 1998). Thus, strong evidence suggests that the NF-κB pathway regulates cell survival of the central and peripheral nervous systems. Here we show for the first time that NF-κB is one of the central survival pathways of embryonic spinal cord MNs. It is also known that NF-κB activity is related to other neuronal functions such as neurite outgrowth (Gallagher et al., 2007; Gutierrez et al., 2008), suggesting an important role of this pathway in neuronal development and probably in some neuronal diseases (Mattson and Meffert, 2006). For example, genes regulating NF-κB pathway activity have been related to a novel form of lower motor neuron disease (Maystadt et al., 2007), indicating that indirect NF-κB deregulation might be involved in neuronal diseases. The loss of NF-κB activation could remove protection of neurons against apoptosis and thereby compromise survival. The recent description that IKKβ regulates pathogenesis of Huntington's disease by inducing cleavage of the mutated huntingtin (Khoshnan et al., 2009) suggests a direct involvement of NF-κB in neurodegenerative disorders. In our hands, when we knock down RelA, Smn protein level decreases. Smn protein is important during MN embryonic development (Schrank et al., 1997), and Smn reduction causes the lower MN neurodegenerative disorder spinal muscular atrophy (Sumner, 2006). The disease is characterized by the specific degeneration of MNs when Smn is reduced. The molecular mechanisms involved in this process are still unknown, and several hypotheses have been proposed. Here we show for the first time that the NF-κB pathway could regulate Smn protein level. In shRelA cultures, we also observed a decrease of CREB protein. It is also known that CREB protein increases SMN promoter activity (Majumder et al., 2004), suggesting the role of CREB in the molecular regulation of Smn level.
The relevance of the NF-κB pathway to cell survival is related to the induction of target genes whose products inhibit some aspects of the apoptotic machinery. In some cell lines and primary cultures, NGF or TNF induced an increase of Bcl-xL protein level through the activation of the NF-κB pathway (Bui et al., 2001). In our system, the NTF cocktail also increased the levels of Bcl-xL. Bcl-xL protein increases when MNs are maintained in the presence of NTFs, but not when NTFs are absent in the culture medium. However, we did not observe differences in Bcl-xL levels when NF-κB was blocked by shIKKβ, shRelA, or shIKKα, indicating that the NF-κB pathway is not directly regulating Bcl-xL protein level. However, when Bcl-xL protein was increased by overexpression, the apoptosis caused by NF-κB pathway blockade, was completely abolished. In the same line of evidence, we show that Bax protein reduction is able to rescue cells from apoptosis when NF-κB is reduced. Thus, from these results we can conclude that the NF-κB pathway could be regulating at least a Bcl-xL upstream mechanism associated with NF-κB activity. Our work demonstrates that the NF-κB family of proteins is necessary for MN survival; knockdown of these proteins probably deregulates gene expression of proteins that exert their function upstream of the mitochondria. It has been recently demonstrated that the BH3-only protein, Bim, displaces Bcl-xL in the mitochondria and promotes Bax translocation during TNFα-induced apoptosis in PC12 cells (Zhang et al., 2008). In our model, we observed in shRelA-treated cultures an increase of Bim protein level, suggesting that this protein could be regulating the apoptotic cell death process caused by the blockade of NF-κB signaling.
In summary, our results demonstrate that NF-κB can mediate NTF-induced cell survival in developing MNs and regulate the expression of proteins involved in MN neurodegenerative disorders. We describe in Figure 9C a schematic model showing the proposed mechanism of the effect of NTF treatment on the NF-κB pathway through the activation of PI 3-kinase and the later expression of Smn, CREB, and Bim proteins.
Footnotes
This work was supported by grants to R.M.S. from Instituto de Salud Carlos III–Fondo de Investigaciones Sanitarias (PI080267), the Ministerio de Ciencia e Innovación [Consolider-Ingenio 2010 (CSD2007-00020)], and the Generalitat de Catalunya (Suport Grups de Recerca Emergents) (2009SGR740); and to M.E. from the Ministerio de Ciencia e Innovación (BFU2007-67619). S.M. holds a fellowship from Comissionat per a Universitats i Recerca del Departament d'Innovació, Universitats i Empresa de la Generalitat de Catalunya i del Fons Social Europeu; A.G. holds a postdoctoral contract from GENOMA España (GENAME project); M.G.-F. holds a fellowship from Universitat de Lleida. We especially thank Prof. Joan X. Comella for providing the Bcl-xL plasmid construct, Dr. Montserrat Rue for statistics support, and Berta Daussa for technical assistance. We also thank Dr. Elaine Lilly (Writer's First Aid) for English language revision of the manuscript.
References
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Arce V, Garces A, de Bovis B, Filippi P, Henderson C, Pettmann B, deLapeyrière O. Cardiotrophin-1 requires LIFRbeta to promote survival of mouse motoneurons purified by a novel technique. J Neurosci Res. 1999;55:119–126. doi: 10.1002/(SICI)1097-4547(19990101)55:1<119::AID-JNR13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-κB activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor kappaB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IkappaBalpha. J Cell Biol. 2001;152:753–764. doi: 10.1083/jcb.152.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- Dolcet X, Soler RM, Gould TW, Egea J, Oppenheim RW, Comella JX. Cytokines promote motoneuron survival through the Janus kinase-dependent activation of the phosphatidylinositol 3-kinase pathway. Mol Cell Neurosci. 2001;18:619–631. doi: 10.1006/mcne.2001.1058. [DOI] [PubMed] [Google Scholar]
- Dolcet X, Llobet D, Encinas M, Pallares J, Cabero A, Schoenenberger JA, Comella JX, Matias-Guiu X. Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281:22118–22130. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- Encinas M, Rozen EJ, Dolcet X, Jain S, Comella JX, Milbrandt J, Johnson EM., Jr Analysis of Ret knockin mice reveals a critical role for IKKs, but not PI 3-K, in neurotrophic factor-induced survival of sympathetic neurons. Cell Death Differ. 2008;15:1510–1521. doi: 10.1038/cdd.2008.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D, Gutierrez H, Gavalda N, O'Keeffe G, Hay R, Davies AM. Nuclear factor-κB activation via tyrosine phosphorylation of inhibitor κB-α is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J Neurosci. 2007;27:9664–9669. doi: 10.1523/JNEUROSCI.0608-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-García M, García I, Ding L, O'Shea S, Boise LH, Thompson CB, Núñez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci U S A. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou-Fabregas M, Garcera A, Mincheva S, Perez-Garcia MJ, Comella JX, Soler RM. Specific vulnerability of mouse spinal cord motoneurons to membrane depolarization. J Neurochem. 2009;110:1842–1854. doi: 10.1111/j.1471-4159.2009.06278.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, O'Keeffe GW, Gavaldà N, Gallagher D, Davies AM. Nuclear factor κB signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J Neurosci. 2008;28:8246–8256. doi: 10.1523/JNEUROSCI.1941-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Holtmann B, Wiese S, Samsam M, Grohmann K, Pennica D, Martini R, Sendtner M. Triple knock-out of CNTF, LIF, and CT-1 defines cooperative and distinct roles of these neurotrophic factors for motoneuron maintenance and function. J Neurosci. 2005;25:1778–1787. doi: 10.1523/JNEUROSCI.4249-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–299. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Tescu S, Brundin P, Patterson PH. IKKalpha and IKKbeta regulation of DNA damage-induced cleavage of huntingtin. PLoS One. 2009;4:e5768. doi: 10.1371/journal.pone.0005768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulich E, Nguyen T, Johnson K, Giardina C, D'Mello S. NF-kappaB is involved in the survival of cerebellar granule neurons: association of IkappaBbeta [correction of Ikappabeta] phosphorylation with cell survival. J Neurochem. 2001;76:1188–1198. doi: 10.1046/j.1471-4159.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- Kovács AD, Chakraborty-Sett S, Ramirez SH, Sniderhan LF, Williamson AL, Maggirwar SB. Mechanism of NF-kappaB inactivation induced by survival signal withdrawal in cerebellar granule neurons. Eur J Neurosci. 2004;20:345–352. doi: 10.1111/j.1460-9568.2004.03493.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev. 2000;14:1729–1733. [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Varadharaj S, Ghoshal K, Monani U, Burghes AH, Jacob ST. Identification of a novel cyclic AMP-response element (CRE-II) and the role of CREB-1 in the cAMP-induced expression of the survival motor neuron (SMN) gene. J Biol Chem. 2004;279:14803–14811. doi: 10.1074/jbc.M308225200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- Maystadt I, Rezsöhazy R, Barkats M, Duque S, Vannuffel P, Remacle S, Lambert B, Najimi M, Sokal E, Munnich A, Viollet L, Verellen-Dumoulin C. The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am J Hum Genet. 2007;81:67–76. doi: 10.1086/518900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron. 1996;17:195–197. doi: 10.1016/s0896-6273(00)80151-8. [DOI] [PubMed] [Google Scholar]
- Pérez-García MJ, Ceña V, de Pablo Y, Llovera M, Comella JX, Soler RM. Glial cell line-derived neurotrophic factor increases intracellular calcium concentration. Role of calcium/calmodulin in the activation of the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2004;279:6132–6142. doi: 10.1074/jbc.M308367200. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Piccioli P, Porcile C, Stanzione S, Bisaglia M, Bajetto A, Bonavia R, Florio T, Schettini G. Inhibition of nuclear factor-kappaB activation induces apoptosis in cerebellar granule cells. J Neurosci Res. 2001;66:1064–1073. doi: 10.1002/jnr.1251. [DOI] [PubMed] [Google Scholar]
- Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler RM, Egea J, Mintenig GM, Sanz-Rodriguez C, Iglesias M, Comella JX. Calmodulin is involved in membrane depolarization-mediated survival of motoneurons by phosphatidylinositol-3 kinase- and MAPK-independent pathways. J Neurosci. 1998;18:1230–1239. doi: 10.1523/JNEUROSCI.18-04-01230.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler RM, Dolcet X, Encinas M, Egea J, Bayascas JR, Comella JX. Receptors of the glial cell line-derived neurotrophic factor family of neurotrophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci. 1999;19:9160–9169. doi: 10.1523/JNEUROSCI.19-21-09160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner CJ. Therapeutics development for spinal muscular atrophy. NeuroRx. 2006;3:235–245. doi: 10.1016/j.nurx.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280:17435–17448. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xing D, Chen M. Bim(L) displacing Bcl-x(L) promotes Bax translocation during TNFalpha-induced apoptosis. Apoptosis. 2008;13:950–958. doi: 10.1007/s10495-008-0226-5. [DOI] [PubMed] [Google Scholar]