

Abstract
Amyloid-β peptide (Aβ) plays an important role in the pathogenesis of Alzheimer's disease (AD). Aβ is generated by proteolysis of β-amyloid precursor protein (APP) and is cleared by enzyme-mediated degradation and phagocytosis by microglia and astrocytes. Some cytokines, such as TGF-β1, stimulate this phagocytosis. In contrast, cellular upregulation of HSP70 expression provides cytoprotection against Aβ. HSP70 activity in relation to inhibition of Aβ oligomerization and stimulation of Aβ phagocytosis has also been reported. Although these in vitro results suggest that stimulating the expression of HSP70 could prove effective in the treatment of AD, there is a lack of in vivo evidence supporting this notion. In this study, we address this issue, using transgenic mice expressing HSP70 and/or a mutant form of APP (APPsw). Transgenic mice expressing APPsw showed less of an apparent cognitive deficit when they were crossed with transgenic mice expressing HSP70. Transgenic mice expressing HSP70 also displayed lower levels of Aβ, Aβ plaque deposition, and neuronal and synaptic loss than control mice. Immunoblotting experiments and direct measurement of β- and γ-secretase activity suggested that overexpression of HSP70 does not affect the production Aβ. In contrast, HSP70 overexpression did lead to upregulation of the expression of Aβ-degrading enzyme and TGF-β1 both in vivo and in vitro. These results suggest that overexpression of HSP70 in mice suppresses not only the pathological but also the functional phenotypes of AD. This study provides the first in vivo evidence confirming the potential therapeutic benefit of HSP70 for the prevention or treatment of AD.
Introduction
Alzheimer's disease (AD) is characterized pathologically by the accumulation of neurofibrillary tangles and senile plaques, the latter of which are composed of amyloid-β peptide (Aβ), such as Aβ40 and Aβ42 (Hardy and Selkoe, 2002; Mattson, 2004). To generate Aβ, β-amyloid precursor protein (APP) is first cleaved by β-secretase and then by γ-secretase (Hardy and Selkoe, 2002; Mattson, 2004). Aβ can be cleared from the brain via three main pathways: degradation by enzymes, such as neprilysin, insulin-degrading enzyme (IDE), and endothelin-converting enzyme 2 (ECE-2), phagocytosis by microglia and astrocytes, and transport into the blood and lymph nodes (Miners et al., 2008; Zlokovic, 2008; Rodríguez et al., 2009). However, monomeric Aβ easily self-assembles to form oligomers, protofibrils, and fibrils, and it is now believed that less aggregated forms of Aβ, such as oligomers and protofibrils, are more important than the highly aggregated forms (Haass and Selkoe, 2007). Therefore, cellular factors that affect the production and clearance of Aβ and/or oligomerization of Aβ may be good targets for the development of drugs to prevent or treat AD.
When cells are exposed to stressors, heat shock proteins (HSPs) are induced, and the cellular upregulation of their expression, especially that of HSP70, provides resistance as the HSPs refold or degrade denatured proteins produced by the stressors (Morimoto and Santoro, 1998; Muchowski and Wacker, 2005). Not only AD but also other neurodegenerative diseases display aggregation of proteins, and overexpression of HSP70 (polyglutamine diseases) or HSP104 (Parkinson's disease) in animal models suppresses the aggregation of each pathogenic protein and ameliorates the corresponding disease symptoms (Adachi et al., 2003; Katsuno et al., 2005; Muchowski and Wacker, 2005; Lo Bianco et al., 2008). An increased level of expression of HSPs, such as small HSPs and HSP70 in the brain of AD patients, has been reported in a number of studies (Perez et al., 1991; Muchowski and Wacker, 2005), with in vitro experiments suggesting that expression of HSPs, in particular HSP70, could suppress the progression of AD (Magrané et al., 2004; Muchowski and Wacker, 2005; Evans et al., 2006; Kumar et al., 2007; Yoshiike et al., 2008). In terms of in vivo studies, the effect of overexpression of HSPs on the pathogenesis of AD has not been examined in vertebrate models. In this study, we investigated the effect of overexpression of HSP70 on AD-related phenotypes, using transgenic mice expressing HSP70 and/or a mutant (Swedish) type of APP (APPsw). Our results demonstrate that overexpression of HSP70 suppresses not only the pathological phenotypes of AD but also the resultant cognitive deficits, possibly through its effects on antiaggregation, neuroprotection, and stimulation of Aβ clearance.
Materials and Methods
Materials.
Eagle's minimal essential medium (EMEM) was obtained from Nissui Pharmaceutical. A fluorescent substrate for β-secretase [H2N-Arg-Glu-(EDANS)-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys-(DABCYL)-Arg-O] was purchased from Calbiochem and that for γ-secretase [Nma-Gly-Gly-Val-Val-Ile-Ala-Thr-Val-Lys(Dnp)-d-Arg-d-Arg-d-Arg-NH2] and synthetic Aβ were obtained from Peptide Institute. Alexa Fluor 488 goat anti-mouse IgG, Neurobasal medium, B27, and an antibody to Aβ oligomer (A11) were purchased from Invitrogen. Sandwich ELISA (sELISA) kit for Aβ oligomers was obtained from Immunobiological Laboratories. An antibody to actin was obtained from Santa Cruz. Fetal bovine serum (FBS), thioflavin-S, and antibodies to the C-terminal fragment (CTF) of APP and synaptophysin were purchased from Sigma-Aldrich. An antibody to Aβ (6E10) came from Covance Research Products. An antibody to HSP70 was from Assay Designs, and those against neuronal nuclei (NeuN) and presenilin 1 (PS1) were from Millipore Bioscience Research Reagents. The RNeasy kit was obtained from QIAGEN. PrimeScript first-strand cDNA Synthesis kit was from Takara Bio, and iQ SYBR Green Supermix was purchased from Bio-Rad. ELISA kits for tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 were from Pierce. An ELISA kit for TGF-β1 was obtained from R&D Systems. Sandwich ELISA kits for Aβ40 and Aβ42 and ara-C (cytarabine) were from Wako. Mounting medium for immunohistochemical analysis (VECTASHIELD) was from Vector Laboratories. Mayer's hematoxylin and mounting medium for histological examination (malinol) were purchased from Muto Pure Chemicals. The Envision kit was from Dako.
Animals.
Transgenic mice that express APPsw (APP23, C57BL/6) were a gift from Dr. M. Staufenbiel (Novartis Institutes for BioMedical Research, Basel, Switzerland) (Sturchler-Pierrat et al., 1997). Transgenic mice expressing HSP70 were gifts from Drs. C. E. Angelidis and G. N. Pagoulatos (University of Ioannina, Ioannina, Greece) and crossed with C57BL/6 wild-type mice (WT/WT) 10 times to generate WT/HSP70 mice (Tanaka et al., 2007). APP23 male mice were crossed with WT/HSP70 female mice to generate APPsw/HSP70 mice. Parallel crosses were made between APP23 mice and WT/WT mice to generate APPsw/WT animals. All experiments in this study were done using female mice.
The experiments and procedures described here were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health and were approved by the Animal Care Committee of Kumamoto University.
Morris water maze test.
The Morris water maze test was conducted in a circular 90-cm-diameter pool filled with water at a temperature of 22.0 ± 1°C, as described previously (Kobayashi et al., 2000; Huang et al., 2006), with minor modifications. In the hidden platform test, a circular platform (10 cm in diameter) was submerged 0.5 cm below water level. Swimming paths were tracked for 60 s with a camera and stored in a computer (Video Tracking System CompACT VAS/DV; Muromachi kikai). The mice were given four trials (one block) per day for 7 consecutive days, during which the platform was left in the same position. The time taken to reach the platform (escape latency) was measured, and the average of four trials was determined.
Twenty-four hours after the last trial of the hidden platform test, the mice were subjected to a transfer test in which the platform was removed, and their swimming path was recorded for 60 s. Percentage search time for each quadrant and crossing time in the area where the platform had been located were determined.
sELISA for Aβ or Aβ oligomer and ELISA for cytokines.
Cells were cultured for 48 h, and the conditioned medium was subjected to sELISA for Aβ, as described previously (Tomita et al., 1998b; Hoshino et al., 2007a).
Aβ40, Aβ42, and cytokine levels in the brain were determined as described previously (Iwata et al., 2004). Briefly, the brain hemispheres were homogenized in 50 mm Tris/HCl buffer, pH 7.6, containing 150 mm NaCl, and then centrifuged. Guanidine/HCl (0.5 m, final concentration) was added to the supernatants (soluble fractions). The precipitates were solubilized by sonication in 6 m guanidine/HCl, after which the solubilized pellet was centrifuged and the resulting supernatant diluted (insoluble fractions). The amount of Aβ40 and Aβ42 in each fraction was determined by sELISA. An ELISA assay for Aβ oligomers or cytokines was performed on the soluble fractions (but without guanidine/HCl), according to the manufacturer's instructions.
Thioflavin-S staining and immunohistochemical analyses.
The brain hemispheres were fixed in 4% buffered paraformaldehyde and embedded in paraffin before being cut into 4-μm-thick sections, which were then deparaffinized and washed in PBS.
For thioflavin-S staining, sections were stained with 1% thioflavin-S solution. Samples were mounted with malinol and inspected using a BX51 microscope (Olympus). Fluorescence microscopic images at region (1.0 mm2) in the hippocampus and cerebral cortex were used to calculate the area stained with thioflavin-S using the LuminaVision (Mitani). By determining the threshold optical density, we divided into thioflavin-S-positive and -negative area and the percentage of thioflavin-S-positive area to total area was determined. We prepared three sections per mouse and calculated the average of values in three sections.
For immunohistochemical analysis to detect NeuN and HSP70, sections were incubated with 0.3% hydrogen peroxide in methanol for removal of endogenous peroxidase. They were then blocked with 2.5% goat serum for 10 min, and incubated for 12 h with antibody to NeuN (1:1000 dilution) or HSP70 (1:100 dilution) in the presence of 2.5% bovine serum albumin (BSA), followed by incubation for 1 h with peroxidase-labeled polymer conjugated to goat anti-mouse (for NeuN) or anti-rabbit (for HSP70) Ig. 3,3′-Diaminobenzidine was applied to the sections, which were then incubated with Mayer's hematoxylin. Samples were mounted with malinol and inspected using a BX51 microscope. NeuN-positive cells in the pyramidal cell layer of the hippocampal CA3 region (within 500 μm from the edge of the dentate gyrus) were counted. We prepared three sections per mouse and calculated the average of values in three sections.
For immunohistochemical analysis to detect synaptophysin, sections were blocked with 2.5% goat serum (v/v) for 10 min, incubated for 12 h with antibody to synaptophysin (1:200 dilution) in the presence of 2.5% BSA, and then incubated with Alexa Fluor 488 goat anti-mouse IgG. Samples were mounted with VECTASHIELD and inspected with the aid of a BX51 fluorescence microscope. Fluorescence intensity in area (100 × 150 μm) of the hippocampal CA3 region was determined using LuminaVision and shown relative to fluorescence intensity in wild-type mice. We prepared three sections per mouse and calculated the average of values in three sections.
Immunoblotting analysis.
Whole-cell extracts were prepared as described previously (Hoshino et al., 2003). The protein concentration of each sample was determined by the Bradford method (Bradford, 1976). Samples were applied to SDS polyacrylamide gels (Tris/tricine gel for detection of Aβ, CTFα, and CTFβ or Tris/glycine gel for detection of other proteins) and subjected to electrophoresis, after which proteins were immunoblotted with each antibody.
A dot blotting assay for Aβ oligomer was performed on the soluble fractions (but without guanidine/HCl), as described previously (Wang et al., 2007). Proteins (4 μg) of soluble fractions were applied to nitrocellulose membrane, after which proteins were detected with antibody against Aβ oligomer (A11).
β- and γ-secretase-mediated peptide cleavage assay.
β- and γ-secretase activity was monitored as previously reported (Fukumoto et al., 2002; Farmery et al., 2003). Solubilized membranes were incubated for 1 h at 37°C in 200 μl of 50 mm acetate buffer, pH 4.1, containing 100 mm sodium chloride, 0.025% BSA, and 10 μm β-secretase fluorescent substrate or for 4 h at 37°C in 200 μl of 50 mm Tris/HCl buffer, pH 6.8, containing 2 mm EDTA, 0.25% CHAPSO (3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate), and 10 μm γ-secretase fluorescent substrate. Fluorescence was measured using a plate reader (Fluostar Galaxy; BMG Labtech) with an excitation wavelength of 355 nm and an emission wavelength of 510 nm (for β-secretase) or 440 nm (for γ-secretase).
Real-time reverse transcription-PCR analysis.
Real-time reverse transcription (RT)-PCR was performed as previously described (Mima et al., 2005) with some modifications. Total RNA was extracted from the brain or cultured cells using an RNeasy kit according to the manufacturer's protocol. Samples (1 μg of RNA) were reverse-transcribed using a first-strand cDNA synthesis kit. Synthesized cDNA was used in real-time RT-PCR (Chromo 4 instrument; Bio-Rad) experiments using iQ SYBR GREEN Supermix, and then analyzed with Opticon Monitor Software. Specificity was confirmed by electrophoretic analysis of the reaction products and by inclusion of template- or reverse transcriptase-free controls. To normalize the amount of total RNA present in each reaction, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA was used as an internal standard.
Primers were designed using the Primer3 website. The primers used were as follows (name: forward primer, reverse primer): gapdh: 5′-aactttggcattgtggaagg-3′, 5′-acacattgggggtaggaaca-3′; neprilysin: 5′-gcagcctcagccgaaactac-3′, 5′-caccgtctccatgttgcagt-3′; ide: 5′-accaggaaatgttggctgtc-3′, 5′-tctgagaggggaactctcca-3′; ece-2: 5′-gctatgcccatgtacccagt-3′, 5′-tggcatccagagtacccttc-3′; il-1β: 5′-gatcccaagcaatacccaaa-3′, 5′-ggggaactctgcagactcaa-3′; il-6: 5′-ctggagtcacagaaggagtgg-3′, 5′-ggtttgccgagtagatctcaa-3′; tnf-α: 5′-cgtcagccgatttgctatct-3′, 5′-cggactccgcaaagtctaag-3′; tgf-β1: 5′-tgacgtcactggagtacgg-3′, 5′-ggttcatgtcatggatggtgc-3′.
Cell culture.
The primary culture of cortical neurons was done as described previously (Saito et al., 2008). The cortex of embryonic day 15.5 mice was dissected. Neurons were spread in a buffer containing papain and cultured at 5 × 104 cells cm−2 in Neurobasal medium containing B27 and antibiotics on poly-d-lysine-coated dishes.
Primary cultures of microglia and astrocytes were prepared as described previously (Kauppinen and Swanson, 2005). The cortex of 1-d-old mice was dissected. Cells were spread in a buffer containing papain and DNase and cultured at 1 × 106 cells cm−2 in EMEM containing 10 mm HEPES/KOH and 10% FBS for 2 weeks. Microglial cells were obtained by mildly shaking and collecting the floating cells. For preparation of astrocytes, cells were further incubated in EMEM containing 10 mm HEPES/KOH, 10% FBS, and 20 μm ara-C for 3 d to induce microglial cell death.
Statistical analysis.
All values are expressed as the mean ± SEM or SD. Two-way ANOVA followed by Tukey's test was used to evaluate differences between more than three groups. Student's t test for unpaired results was used for the evaluation of differences between two groups. Differences were considered to be significant for values of p < 0.05.
Results
Effect of overexpression of HSP70 on cognitive function in transgenic mice expressing APPsw
To examine the effect of overexpression of HSP70 on the pathogenesis of AD, we used transgenic mice expressing APPsw (APP23, AD model mice) crossed to transgenic mice that express human HSP70 under the control of the human β-actin promoter (Plumier et al., 1995). The overexpression of HSP70 in the mice has been demonstrated in various organs, including the brain (Plumier et al., 1997). We first confirmed the expression of HSP70 in the brain by immunohistochemical analysis. As shown in Figure 1A, a higher level of HSP70 expression was observed in various brain regions, including the hippocampus and cerebral cortex, in the HSP70 transgenic mice (APPsw/HSP70) at the age of 18 months when compared with the control animals (WT/WT and APPsw/WT). We also compared the level of HSP70 in various parts of the brain by immunoblotting. Higher levels of HSP70 were observed in all regions of the brain tested (hippocampus, cerebral cortex, temporal cortex, diencephala, and cerebellar cortex) in the APPsw/HSP70 mice at the age of 18 months than in the APPsw/WT mice (Fig. 1B). Similar results were observed APPsw/HSP70 mice at the age of 12 months (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Expression of HSP70 in the brain. A, B, Brains were removed from WT/WT, APPsw/WT, and APPsw/HSP70 mice at the age of 18 months. A, Sections were prepared and subjected to immunohistochemical analysis with an antibody to HSP70. Scale bar, 500 μm. B, Brains were divided into fractions containing the hippocampus, cerebral cortex, temporal cortex, diencephala, and cerebellar cortex. Whole-cell extracts were then prepared from each fraction and subjected to immunoblotting with an antibody to HSP70 or actin. C, The band intensity of HSP70 was determined, corrected with that of actin, and expressed relative to the control (n = 3). **p < 0.01. Error bars indicate SEM.
We then used a Morris water maze to examine the effect of overexpression of HSP70 on spatial learning and memory. Four strains of mice (WT/WT, WT/HSP70, APPsw/WT, and APPsw/HSP70) were trained to learn the location of a hidden platform four times per day for 7 d, and the time required to reach the platform (escape latency) was measured. As shown in Figure 2A, APPsw/WT mice took a significantly longer time than WT/WT mice to reach the platform, a result that is consistent with previous reports (Van Dam et al., 2003) and suggests a deficiency in spatial learning and memory in the former group. This impaired ability of APPsw/WT mice to reach the hidden platform did not reflect reduced swimming ability, as swimming speed and ability to locate a visible platform were indistinguishable between the four strains of mice (data not shown). APPsw/HSP70 mice took a significantly shorter time to reach the platform than APPsw/WT mice (Fig. 2A). Furthermore, there was no significant difference in the escape latency between APPsw/HSP70 and WT/WT mice or between WT/HSP70 and WT/WT mice (Fig. 2A). These results suggest that expression of APPsw leads to disturbances in spatial learning and memory, an effect that can be ameliorated by overexpression of HSP70.
Figure 2.
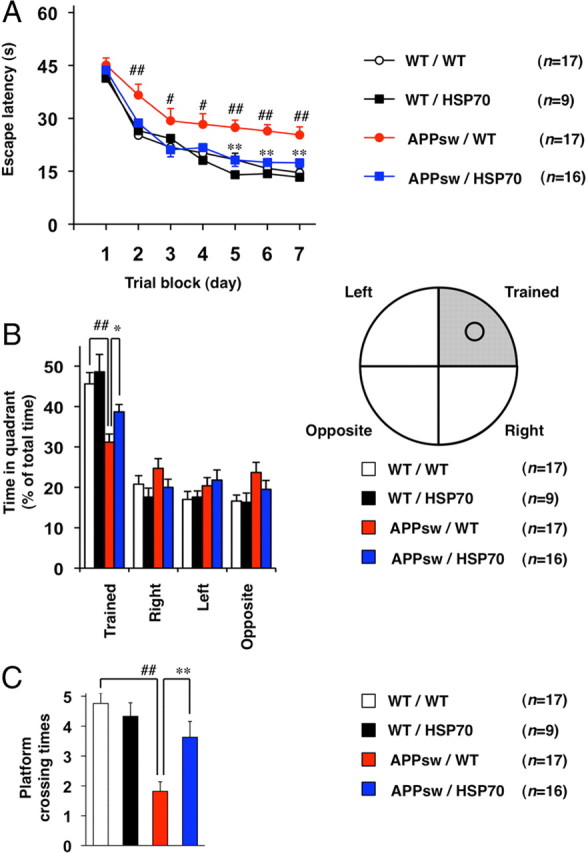
Effects of overexpression of HSP70 on spatial learning and memory in transgenic mice expressing APPsw. Cognitive behavioral tests were performed, using the Morris water maze, on 12-month-old WT/WT (n = 17), WT/HSP70 (n = 9), APPsw/WT (n = 17), and APPsw/HSP70 mice (n = 16) as described in Materials and Methods. The average (4 tests) escape latency in each trial block was measured for 7 d (A), after which the mice were subjected to a transfer test in which the platform was removed. B, C, The spatial memory for a platform location was estimated by percentage search time for each quadrant (the platform had been located in the “trained” quadrant) (B) or platform crossing times (C). Values are given as mean ± SEM. **p < 0.01, *p < 0.05, versus APPsw/WT mice; ##p < 0.01, #p < 0.05, versus WT/WT mice.
We next performed a transfer test to estimate the spatial memory of platform location. After a 7 d training period (see above), each mouse was subjected to a Morris water maze test in which the platform was removed and the percentage search time for each quadrant was measured. As shown in Figure 2B, the ratio of time spent in the trained quadrant was lower for the APPsw/WT group than for either the WT/WT or the APPsw/HSP70 mice. Furthermore, the crossing time of the area where the platform had been located, another indicator of spatial memory, was lower in the APPsw/WT group than in the WT/WT and APPsw/HSP70 cohorts (Fig. 2C). Again, there was no significant difference in these indices between WT/HSP70 and WT/WT mice (Fig. 2B,C). These results confirm the notion that overexpression of HSP70 ameliorates the spatial memory deficits of transgenic mice expressing APPsw.
Effect of overexpression of HSP70 on Aβ accumulation and neuronal and synaptic loss in transgenic mice expressing APPsw
The amounts of Aβ40 and Aβ42 in soluble and insoluble brain fractions were compared between APPsw/HSP70 and APPsw/WT mice by sELISA. As shown in Figure 3A, the levels of Aβ40 and Aβ42 in both brain fractions were lower in the APPsw/HSP70 group than in the APPsw/WT mice.
Figure 3.
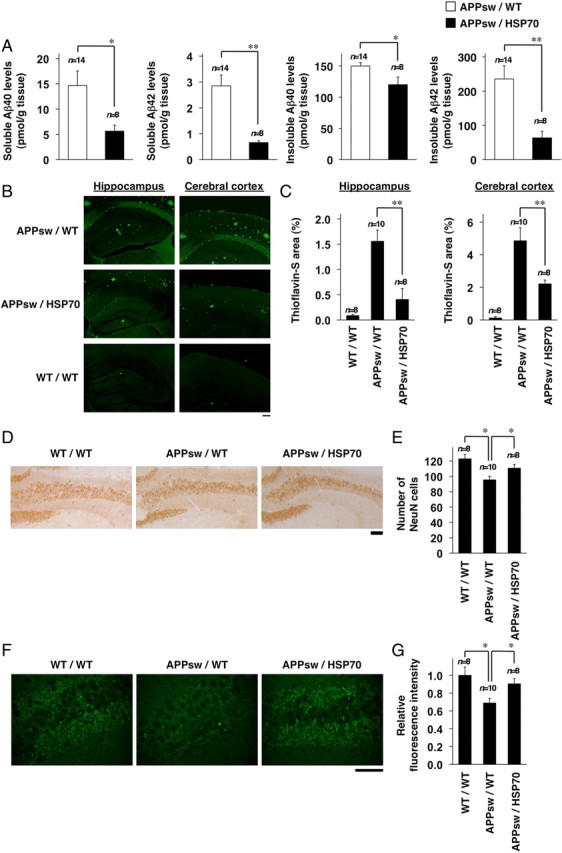
Effects of HSP70 overexpression on Aβ levels, Aβ plaque deposition, and neuronal and synaptic loss in the brain of transgenic mice expressing APPsw. A, Soluble and insoluble fractions were prepared from the brains of 12-month-old APPsw/WT (n = 14) and APPsw/HSP70 mice (n = 8). The amounts of Aβ40 and Aβ42 in each fraction were determined by sELISA as described in Materials and Methods. B, D, F, Brain sections were prepared from 18-month-old APPsw/WT (n = 10), APPsw/HSP70 (n = 8), and WT/WT mice (n = 8), and then subjected to thioflavin-S staining (B) and immunohistochemical analysis with an antibody to NeuN (D) or synaptophysin (F). Scale bars: B, 200 μm; D, 100 μm; F, 50 μm. C, E, G, Relative area stained with thioflavin-S (C), number of NeuN-positive cells in hippocampal CA3 region (E), and relative fluorescence intensity (synaptophysin) in hippocampal CA3 region (G) (3 sections per brain) were determined. Values are given as mean ± SEM. **p < 0.01; *p < 0.05.
We then examined the effect of overexpression of HSP70 on plaque deposition and neurotoxicity in APP23 mice. Because plaque deposition and neurotoxicity (neuronal and synaptic loss) were not observed in APP23 mice at the age of 12 months (data not shown), we used mice at the age of 18 months. At first, we compared the level of Aβ plaque deposition in the brain between APPsw/HSP70 mice and APPsw/WT mice by thioflavin-S staining. As shown in Figure 3, B and C, the level of Aβ plaque deposition in both the hippocampus and the cerebral cortex was much lower in the APPsw/HSP70 mice than in the APPsw/WT animals. Very little Aβ plaque deposition was observed in WT/WT mice (Fig. 3B,C).
We then compared the number of neurons in the hippocampal CA3 region of APPsw/HSP70 and APPsw/WT mice by NeuN staining. As shown in Figure 3, D and E, the number of NeuN-positive cells (neurons) was significantly higher in the WT/WT and APPsw/HSP70 brain sections than in the APPsw/WT tissue, suggesting that Aβ-induced neuronal loss was ameliorated by overexpression of HSP70. We also estimated the number of synapses based on synaptophysin staining. The level of synaptophysin staining was higher in sections from both WT/WT and APPsw/HSP70 mice than from APPsw/WT mice (Fig. 3F,G), indicating that overexpression of HSP70 suppresses Aβ-induced synaptic loss. Together, the results in Figure 3 suggest that overexpression of HSP70 decreases the level of Aβ and Aβ plaque deposition in the brain and protects it against Aβ-induced neurotoxicity.
Effect of overexpression of HSP70 on the production and oligomerization of Aβ
To understand the molecular mechanism governing the HSP70-mediated decrease in levels of Aβ and Aβ plaque deposition, we examined the effect of overexpression of HSP70 on the production of Aβ. In general, production of Aβ is regulated by either modification of APP or modulation of secretase activity. We first examined the effect of overexpression of HSP70 on the maturation of APP, an essential step in the production of Aβ. The mature (N- and O-glycosylated) and immature (N-glycosylated alone) forms of APP (mAPP and imAPP, respectively) can be separated by SDS-PAGE on the basis of molecular weight (Tomita et al., 1998a). As shown in Figure 4A, mAPP and imAPP were detected in transgenic mice expressing APPsw, and the total amount of APP and the ratio of mAPP and imAPP were indistinguishable between APPsw/HSP70 mice and APPsw/WT animals. We also found that overexpression of HSP70 did not affect the level of PS1 (Fig. 4A).
Figure 4.

Effects of HSP70 overexpression on the production of Aβ. A–C, Whole-cell extracts (A, B) and membrane fractions (C) were prepared from the brains of 12-month-old WT/WT, WT/HSP70, APPsw/WT, and APPsw/HSP70 mice. Whole-cell extracts were subjected to immunoblotting with an antibody to APP (A, B), HSP70 (A), PS1 (A), or actin (A). Membrane fractions were subjected to a β- or γ-secretase-mediated peptide cleavage assay as described in Materials and Methods [APPsw/WT (n = 10) and APPsw/HSP70 mice (n = 8)]. Values are given as mean ± SEM. n.s., Not significant (C). D, E, Primary neurons prepared from APPsw/WT mice (n = 3) and APPsw/HSP70 mice (n = 3) were incubated for 7 d. Whole-cell extracts were prepared and subjected to immunoblotting with an antibody to APP, HSP70, or actin (D). After incubation for 48 h, the amounts of Aβ40 and Aβ42 in the conditioned medium were determined by sELISA. Values are given as mean ± SD. n.s., Not significant (E).
Next, we tested the notion that overexpression of HSP70 affects production of Aβ through modulation of secretase activity by comparing the amount of CTFs, the secreted forms of APP that are generated by α or β-secretase (CTFα or CTFβ, respectively, known as an indirect index of the secretase activity), between APPsw/HSP70 and APPsw/WT mice. Under our experimental conditions, we could not detect the band of CTFγ. As shown in Figure 4B, CTFα and CTFβ were detected in transgenic mice expressing APPsw, and the amounts of CTFα and CTFβ were indistinguishable between the APPsw/HSP70 and APPsw/WT mice. We then directly measured β- and γ-secretase activity, using the APP-derived fluorescent substrate of each secretase (Hoshino et al., 2009). As shown in Figure 4C, the activity was indistinguishable between the APPsw/HSP70 and APPsw/WT groups. These results suggest that overexpression of HSP70 does not affect the production of Aβ.
To test this idea in vitro, we prepared primary cultures of neurons from APPsw/HSP70 and APPsw/WT mice and compared the level and modification of APP in the cells, as well as the level of Aβ40 and Aβ42 in the culture medium. We also confirmed the overexpression of HSP70 in primary neurons prepared from APPsw/HSP70 mice (Fig. 4D). As shown in Figure 4, D and E, no significant differences were observed in the total amount of APP, the ratio of mAPP and imAPP, and the levels of Aβ40 and Aβ42 in the primary neuronal cultures between the APPsw/HSP70 and APPsw/WT groups, supporting the idea that overexpression of HSP70 does not affect the production of Aβ.
We then compared the level of Aβ oligomer in the brain of APPsw/HSP70 and APPsw/WT mice by immunoblotting analysis. As shown in Figure 5, A and B, not only the level of Aβ monomer but also that of Aβ oligomers (dimer, trimer, and hexamer) was lower in the APPsw/HSP70 animals. The decrease in the level of Aβ oligomer in the brain of APPsw/HSP70 was confirmed by ELISA assay for Aβ oligomer (Fig. 5C) and dot-blotting assay with antibody that specifically recognizes oligomer form of Aβ (A11) (Fig. 5D,E). The specificity of this antibody (A11) to Aβ oligomer was suggested by the observation that this antibody did not give positive signals against samples from wild-type mice (without expression of APPsw) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.

Effects of HSP70 overexpression on the levels of Aβ oligomers in the brains of transgenic mice expressing APPsw. A–E, Whole-cell extracts (A, B) or soluble fractions (C–E) were prepared from the brains of 12-month-old APPsw/WT and APPsw/HSP70 mice, and then subjected to immunoblotting with an antibody to Aβ (6E10) (A), ELISA for Aβ oligomer [APPsw/WT (n = 5) and APPsw/HSP70 mice (n = 5)] (C), or dot-blotting assay with an antibody against Aβ oligomer (A11) (D). The band intensity of APP and each form of Aβ was determined, corrected with that of actin, and expressed relative to the control [APPsw/WT (n = 4) and APPsw/HSP70 mice (n = 4)] (B). The dot intensity was determined and expressed relative to the control (APP in APPsw/WT mice) [APPsw/WT (n = 5) and APPsw/HSP70 mice (n = 5)] (E). Values are given as mean ± SEM (B, C, E). **p < 0.01; *p < 0.05. n.s., Not significant.
We then examined the colocalization of Hsp70 with APP, Aβ, Aβ oligomer, and Aβ plaque. Intracellular colocalization of HSP70 with APP and Aβ was observed (supplemental Fig. 3A,C, available at www.jneurosci.org as supplemental material). However, extracellular colocalization of HSP70 with Aβ plaque was not observed so clearly (supplemental Fig. 3B,D, available at www.jneurosci.org as supplemental material). The intracellular colocalization of HSP70 with Aβ oligomer was also observed in both hippocampus and cerebral cortex (supplemental Fig. 3E,F, available at www.jneurosci.org as supplemental material).
Effect of overexpression of HSP70 on the expression of genes involving Aβ clearance
As described in Introduction, degradation by enzymes and phagocytosis by microglia and astrocytes are involved in the clearance of Aβ (Miners et al., 2008). We therefore next compared the expression of genes involved in this process. Real-time RT-PCR analysis of brain samples revealed that the mRNA expression of ide, but not that of neprilysin or ece-2, was higher in the APPsw/HSP70 mice than in the APPsw/WT animals (Fig. 6A). The upregulation of mRNA expression of ide by overexpression of HSP70 was also observed in wild-type mice (without expression of APPsw) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We also examined the effect of overexpression of HSP70 on the mRNA expression of ide in primary cultures of neurons, astrocytes, and microglia. As shown in Figure 6B, the mRNA expression of ide was higher in primary astrocyte and microglial cultures prepared from APPsw/HSP70 mice than in those from APPsw/WT mice. In contrast, no significant difference was seen in the neuronal cultures (Fig. 6B). These results suggest that the upregulation of ide expression is involved in the decrease in the level of Aβ observed in the brain of APPsw/HSP70 mice.
Figure 6.

Effects of HSP70 overexpression on the expression of Aβ-degrading enzymes. A, B, Total RNA was extracted from the brains of 12-month-old APPsw/WT (n = 10) and APPsw/HSP70 mice (n = 8) (A) or from primary cultures of neurons, astrocytes and microglia prepared from WT/WT (n = 3) and WT/HSP70 mice (n = 3) (B). RNA samples were subjected to real-time RT-PCR using a specific primer for each gene. Values were normalized to gapdh gene expression and expressed relative to the control sample. Values are given as mean ± SEM (A) or SD (B). **p < 0.01; *p < 0.05; n.s., not significant.
It has been suggested that inflammation stimulates the progression of the pathogenesis of AD (Wyss-Coray, 2006). However, inflammation also activates the phagocytotic activity of microglia and astrocytes, resulting in stimulation of Aβ clearance (Wyss-Coray et al., 2001). Thus, the relationship between inflammation and progression of AD is complicated, with some proinflammatory cytokines (such as TNF-α) being suggested to promote the progression of AD, whereas others (such as IL-1β and IL-6) have a negative effect in mouse models of the disease (Tesseur et al., 2006; He et al., 2007; Hoshino et al., 2007b; Shaftel et al., 2007). TGF-β1, a key cytokine regulating the response of the brain to injury and inflammation, has also been suggested to suppress the progression of AD. Therefore, we then examined the expression of cytokines that have been suggested to affect the phagocytosis of Aβ (IL-1β, IL-6, TNF-α, and TGF-β1) (Wyss-Coray et al., 2001; Tesseur et al., 2006; He et al., 2007; Hoshino et al., 2007b; Shaftel et al., 2007). As shown in Figure 7A, the mRNA expression of tgf-β1 in the brain was higher in the APPsw/HSP70 mice than in APPsw/WT animals. However, no differences were observed in the mRNA expression of the other cytokines (Fig. 7A). Similar results were obtained at the protein level, as judged by ELISA (Fig. 7B). The increased mRNA expression of tgf-β1 and protein level of TGF-β1 by overexpression of HSP70 was also observed in wild-type mice (without expression of APPsw) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Figure 7.

Effects of HSP70 overexpression on cytokine expression. A, B, Total RNA (A) or whole-cell extracts (B) were prepared from the brains of 12-month-old APPsw/WT (n = 10) and APPsw/HSP70 mice (n = 8). C, Total RNA was also extracted from primary culture of neurons, astrocytes, and microglia prepared from WT/WT (n = 3) and WT/HSP70 mice (n = 3). RNA samples were subjected to real-time RT-PCR as described in the legend of Figure 6 (A, C). The amounts of cytokine in the whole-cell extracts were determined by ELISA (B). Values are given as mean ± SEM (A, B) or SD (C). *p < 0.05. n.s., Not significant.
We also examined the effect of overexpression of HSP70 on the mRNA expression of tgf-β1 in primary cultures of neurons, astrocytes, and microglia. As shown in Figure 7C, tgf-β1 mRNA expression was higher in the neuron and astrocyte cultures prepared from APPsw/HSP70 mice than in those from APPsw/WT animals. However, no such difference was observed in the microglial cultures (Fig. 7C). Together, these findings suggest that the higher expression of tgf-β1 is responsible for the lower level of Aβ seen in the brains of APPsw/HSP70 mice, compared with their APPsw/WT counterparts.
Finally, we examined the effect of overexpression of HSP70 on microglial activation by immunohistochemical analysis with an antibody against Iba1 (a marker for activated microglia). As shown in supplemental Figure 5 (available at www.jneurosci.org as supplemental material), the level of Iba1 expression was higher in APPsw/HSP70 mice than APPsw/WT, suggesting that expression of HSP70 activates microglia in APP23 mice.
Discussion
HSPs have attracted considerable attention as AD drug targets because of their unique properties as molecular chaperones (i.e., their ability to unfold and refold abnormal proteins). Other activities of HSPs in vitro, such as cytoprotection against Aβ neurotoxicity, suppression of inflammation, and stimulation of Aβ phagocytosis, have increased this attention. Furthermore, the ameliorative effect of HSPs has also been suggested in the case of other neurodegenerative conditions, such as polyglutamine diseases and Parkinson's disease (Adachi et al., 2003; Katsuno et al., 2005; Lo Bianco et al., 2008). Despite this, no in vivo evidence exists to support the idea that HSPs are protective in vertebrate AD models. Therefore, in this study, we tested this idea using transgenic mouse strains expressing APPsw and HSP70.
Because AD results in cognitive impairment, it is important to find endogenous factors that affect not only the pathological but also the functional (cognitive) phenotypes in the animal models. This notion is supported by previous reports that some endogenous factors ameliorate cognitive dysfunction in AD model mice without affecting the pathological phenotypes (such as Aβ plaque deposition) (Roberson et al., 2007; Kanninen et al., 2009). In the present study, we found that APPsw/HSP70 mice display a higher level of cognitive function (spatial learning and memory) than APPsw/WT mice, suggesting that overexpression of HSP70 ameliorates the deficits in spatial learning and memory caused by expression of APPsw. Furthermore, we demonstrated that no significant differences in spatial learning and memory exist between WT/WT and WT/HSP70 mice, suggesting that overexpression of HSP70 does not affect the cognitive ability of wild-type mice.
We also found that there are lower levels of Aβ in both the soluble and insoluble fractions of APPsw/HSP70 brains than in those prepared from APPsw/WT mice, suggesting that overexpression of HSP70 suppresses production of Aβ. However, no such differences were observed in the levels of CTFα and CTFβ, indicators of proteolysis by α- or β-secretase, respectively. Furthermore, the activities of β- and γ-secretase in the brain were also indistinguishable between the two strains of mice. These in vivo results suggest that overexpression of HSP70 does not affect the production of Aβ. In fact, we showed that the level of Aβ in the conditioned medium was similar between HSP70-overexpressing neurons and control neurons. Inflammatory factors, such as PGE2 (prostaglandin E2) and proinflammatory cytokines, are known to enhance the production of Aβ (Wyss-Coray, 2006; Hoshino et al., 2007b, 2009), and the antiinflammatory activity of HSP70 has recently been reported (Chan et al., 2004; Sun et al., 2005; Tang et al., 2007). However, since expression of proinflammatory cytokines was not affected by overexpression of HSP70 either in vivo or in vitro, the antiinflammatory activity of HSP70 does not appear to contribute to the ameliorative effect of HSP70 in AD model mice.
As described in Introduction, Aβ is cleared from the brain through enzyme-mediated degradation, phagocytosis by microglia and astrocytes, and transport into the blood and lymph nodes (Miners et al., 2008; Zlokovic, 2008; Rodríguez et al., 2009). The inability of HSP70 to affect the production of Aβ suggests that the clearance of Aβ is enhanced in transgenic mice expressing HSP70. We found that the expression of IDE, an Aβ-degrading enzyme, is enhanced by overexpression of HSP70 both in vitro and in vivo. We also demonstrated that the expression of TGF-β1 is increased by overexpression of HSP70. Given that it has been reported that TGF-β1 stimulates Aβ clearance through activation of phagocytic microglia (Wyss-Coray et al., 2001) and we here suggested that overexpression of HSP70 activates microglia, we consider that the lower level of Aβ observed in the brains of transgenic mice expressing HSP70 is attributable to the stimulation of Aβ clearance through upregulation of expression of IDE and TGF-β1. In terms of the mechanism underpinning the upregulation of TGF-β1 expression because of overexpression of HSP70, we consider a possibility that extracellular HSP70 is responsible, as it has previously been reported that extracellular HSP70 stimulates the expression of TGF-β1 (Kimura et al., 1998).
However, there is no direct evidence to show that lower levels of Aβ in mice with overexpression of HSP70 is mediated by Aβ-degrading enzyme and TGF-β1, and thus another mechanism may be involved in the phenomenon. For example, Hsp70 might increase the degradation of Aβ by stimulating autophagy or Hsp70 might directly modulate the activity of microglia to degrade Aβ.
The current findings show that the level of Aβ plaque deposition in the brain is reduced in APPsw/HSP70 mice compared with APPsw/WT animals. We also found that the level of Aβ oligomers is lower in the APPsw/HSP70 mouse brain. One explanation of these phenotypes is the lower level of monomeric Aβ in the APPsw/HSP70 mice than in the APPsw/WT animals. However, we believe that the antiaggregation activity of HSP70 for Aβ that was suggested by previous in vitro studies (Muchowski and Wacker, 2005; Evans et al., 2006; Kumar et al., 2007; Yoshiike et al., 2008) is involved in these phenotypes. We also found that APPsw/HSP70 mice have less neuronal and synaptic loss than their APPsw/WT counterparts. Again, the phenotype can be explained by the lower level of Aβ. However, we consider that the cytoprotective activity of HSP70 against Aβ-induced neuronal apoptosis that was suggested by previous in vitro studies (Magrané et al., 2004) is involved in the phenotype.
As outlined in Introduction, the beneficial effects of HSP70 in animal models of various diseases suggest the potential therapeutic benefit of HSP70 inducers for these conditions (Tanaka et al., 2007, 2010; Asano et al., 2009; Suemasu et al., 2009; Matsuda et al., 2010; Yamashita et al., 2010), a possibility that can now be expanded to include AD based on the results of the present study. A number of recent studies have revealed new molecules that induce HSPs (Kieran et al., 2004; Westerheide et al., 2004; Yan et al., 2004). However, the development of new candidate drugs requires them to pass through the clinical trials process, with the prospect of encountering side effects. In light of this, we have focused our attention on geranylgeranylacetone (GGA). GGA, a leading antiulcer drug on the Japanese market, has been reported to be a nontoxic HSP inducer (Hirakawa et al., 1996). GGA has also been shown to upregulate the expression of HSP70 in various tissues, including the stomach, intestine, liver, heart, eye, ear, skin, lung, and brain (Hirakawa et al., 1996; Ooie et al., 2001; Katsuno et al., 2005; Asano et al., 2009). We and another group have previously reported that GGA suppresses not only gastric lesions but also lesions of the small intestine and inflammatory bowel disease-related colitis (Ohkawara et al., 2005; Asano et al., 2009; Suemasu et al., 2009). Furthermore, we have demonstrated that GGA ameliorates the phenotype in an animal model of spinal and bulbar muscular atrophy (a polyglutamine disease) by suppressing the aggregation of pathogenic proteins (Katsuno et al., 2005). Compared with new molecules that induce HSPs, GGA has an advantage, given that its safety has already been clinically demonstrated. We therefore consider that animal and clinical studies should be performed to prove the effectiveness of GGA for preventing and treating AD.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Health, Labour, and Welfare of Japan, as well as the Japan Science and Technology Agency, Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Drs. M. Staufenbiel (Novartis Institutes for BioMedical Research, Basel, Switzerland), and C. E. Angelidis and G. N. Pagoulatos (University of Ioannina, Ioannina, Greece) for providing transgenic mice.
References
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, Tanaka K, Yamakawa N, Adachi H, Sobue G, Goto H, Takeuchi K, Mizushima T. HSP70 confers protection against indomethacin-induced lesions of the small intestine. J Pharmacol Exp Ther. 2009;330:458–467. doi: 10.1124/jpet.109.152181. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chan JY, Ou CC, Wang LL, Chan SH. Heat shock protein 70 confers cardiovascular protection during endotoxemia via inhibition of nuclear factor-kappaB activation and inducible nitric oxide synthase expression in the rostral ventrolateral medulla. Circulation. 2004;110:3560–3566. doi: 10.1161/01.CIR.0000143082.63063.33. [DOI] [PubMed] [Google Scholar]
- Evans CG, Wisén S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J Biol Chem. 2006;281:33182–33191. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- Farmery MR, Tjernberg LO, Pursglove SE, Bergman A, Winblad B, Näslund J. Partial purification and characterization of gamma-secretase from post-mortem human brain. J Biol Chem. 2003;278:24277–24284. doi: 10.1074/jbc.M211992200. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology. 1996;111:345–357. doi: 10.1053/gast.1996.v111.pm8690199. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Tsutsumi S, Tomisato W, Hwang HJ, Tsuchiya T, Mizushima T. Prostaglandin E2 protects gastric mucosal cells from apoptosis via EP2 and EP4 receptor activation. J Biol Chem. 2003;278:12752–12758. doi: 10.1074/jbc.M212097200. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Araki W, Suzuki K, Suzuki T, Mizushima T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem J. 2007a;402:581–589. doi: 10.1042/BJ20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Homan T, Tanaka K, Sugimoto Y, Araki W, Narita M, Narumiya S, Suzuki T, Mizushima T. Involvement of prostaglandin E2 in production of amyloid-beta peptides both in vitro and in vivo. J Biol Chem. 2007b;282:32676–32688. doi: 10.1074/jbc.M703087200. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Namba T, Takehara M, Nakaya T, Sugimoto Y, Araki W, Narumiya S, Suzuki T, Mizushima T. Prostaglandin E2 stimulates the production of amyloid-beta peptides through internalization of the EP4 receptor. J Biol Chem. 2009;284:18493–18502. doi: 10.1074/jbc.M109.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, Staufenbiel M, Noda Y, Yamaguchi H, Nabeshima T, Saido TC, Iwata N. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K, Saido TC. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J Neurosci. 2004;24:991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, Ylä-Herttuala S, Tanila H, Levonen AL, Koistinaho M, Koistinaho J. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Yamada K, Sakai T, Mishima K, Nishimura H, Matsumoto Y, Singh M, Yoshikai Y. The regulatory role of heat shock protein 70-reactive CD4+ T cells during rat listeriosis. Int Immunol. 1998;10:117–130. doi: 10.1093/intimm/10.2.117. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Noda Y, Matsushita N, Nishii K, Sawada H, Nagatsu T, Nakahara D, Fukabori R, Yasoshima Y, Yamamoto T, Miura M, Kano M, Mamiya T, Miyamoto Y, Nabeshima T. Modest neuropsychological deficits caused by reduced noradrenaline metabolism in mice heterozygous for a mutated tyrosine hydroxylase gene. J Neurosci. 2000;20:2418–2426. doi: 10.1523/JNEUROSCI.20-06-02418.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Shorter J, Régulier E, Lashuel H, Iwatsubo T, Lindquist S, Aebischer P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest. 2008;118:3087–3097. doi: 10.1172/JCI35781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed β-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, Hoshino T, Yamashita Y, Tanaka K, Maji D, Sato K, Adachi H, Sobue G, Ihn H, Funasaka Y, Mizushima T. Prevention of UVB radiation-induced epidermal damage by expression of heat shock protein 70. J Biol Chem. 2010;285:5848–5858. doi: 10.1074/jbc.M109.063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima S, Tsutsumi S, Ushijima H, Takeda M, Fukuda I, Yokomizo K, Suzuki K, Sano K, Nakanishi T, Tomisato W, Tsuchiya T, Mizushima T. Induction of claudin-4 by nonsteroidal anti-inflammatory drugs and its contribution to their chemopreventive effect. Cancer Res. 2005;65:1868–1876. doi: 10.1158/0008-5472.CAN-04-2770. [DOI] [PubMed] [Google Scholar]
- Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI, Santoro MG. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol. 1998;16:833–838. doi: 10.1038/nbt0998-833. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Ohkawara T, Nishihira J, Takeda H, Miyashita K, Kato K, Kato M, Sugiyama T, Asaka M. Geranylgeranylacetone protects mice from dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 2005;40:1049–1057. doi: 10.1080/00365520510023161. [DOI] [PubMed] [Google Scholar]
- Ooie T, Takahashi N, Saikawa T, Nawata T, Arikawa M, Yamanaka K, Hara M, Shimada T, Sakata T. Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation. 2001;104:1837–1843. doi: 10.1161/hc3901.095771. [DOI] [PubMed] [Google Scholar]
- Perez N, Sugar J, Charya S, Johnson G, Merril C, Bierer L, Perl D, Haroutunian V, Wallace W. Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer's disease. Brain Res Mol Brain Res. 1991;11:249–254. doi: 10.1016/0169-328x(91)90033-t. [DOI] [PubMed] [Google Scholar]
- Plumier JC, Ross BM, Currie RW, Angelidis CE, Kazlaris H, Kollias G, Pagoulatos GN. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumier JC, Krueger AM, Currie RW, Kontoyiannis D, Kollias G, Pagoulatos GN. Transgenic mice expressing the human inducible Hsp70 have hippocampal neurons resistant to ischemic injury. Cell Stress Chaperones. 1997;2:162–167. doi: 10.1379/1466-1268(1997)002<0162:tmethi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 2009;16:378–385. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- Saito Y, Sano Y, Vassar R, Gandy S, Nakaya T, Yamamoto T, Suzuki T. X11 proteins regulate the translocation of amyloid beta-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by beta-site-cleaving enzyme in brain. J Biol Chem. 2008;283:35763–35771. doi: 10.1074/jbc.M801353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemasu S, Tanaka K, Namba T, Ishihara T, Katsu T, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai A, Mizushima T. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J Biol Chem. 2009;284:19705–19715. doi: 10.1074/jbc.M109.006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Chen D, Du B, Pan J. Heat shock response inhibits NF-kappaB activation and cytokine production in murine Kupffer cells. J Surg Res. 2005;129:114–121. doi: 10.1016/j.jss.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Namba T, Arai Y, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai A, Mizushima T. Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J Biol Chem. 2007;282:23240–23252. doi: 10.1074/jbc.M704081200. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ishihara T, Azuma A, Kudoh S, Ebina M, Nukiwa T, Sugiyama Y, Tasaka Y, Namba T, Ishihara T, Sato K, Mizushima Y, Mizushima T. Therapeutic effect of lecithinized superoxide dismutase on bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;298:L348–L360. doi: 10.1152/ajplung.00289.2009. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Xiao W, Wang H, Calderwood SK, Xiao X. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol. 2007;179:1236–1244. doi: 10.4049/jimmunol.179.2.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Kirino Y, Suzuki T. Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem. 1998a;273:6277–6284. doi: 10.1074/jbc.273.11.6277. [DOI] [PubMed] [Google Scholar]
- Tomita S, Kirino Y, Suzuki T. A basic amino acid in the cytoplasmic domain of Alzheimer's beta-amyloid precursor protein (APP) is essential for cleavage of APP at the alpha-site. J Biol Chem. 1998b;273:19304–19310. doi: 10.1074/jbc.273.30.19304. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Hoshino T, Matsuda M, Kobayashi C, Tominaga A, Nakamura Y, Nakashima K, Yokomizo K, Ikeda T, Mineda K, Maji D, Niwano Y, Mizushima T. HSP70 inducers from Chinese herbs and their effect on melanin production. Exp Dermatol. 2010;19:e340–e342. doi: 10.1111/j.1600-0625.2009.01061.x. [DOI] [PubMed] [Google Scholar]
- Yan D, Saito K, Ohmi Y, Fujie N, Ohtsuka K. Paeoniflorin, a novel heat shock protein-inducing compound. Cell Stress Chaperones. 2004;9:378–389. doi: 10.1379/CSC-51R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiike Y, Minai R, Matsuo Y, Chen YR, Kimura T, Takashima A. Amyloid oligomer conformation in a group of natively folded proteins. PLoS One. 2008;3:e3235. doi: 10.1371/journal.pone.0003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]