

Abstract
Dopamine is one of the major neuromodulators in the CNS, which is involved in learning and memory processes. A nonlinear, inverted U-shaped dose–response curve of its effects on cognition has been observed in animal studies. The basis for this nonlinear effect might be a similar effect of dopamine on neuroplasticity. Whereas it has been shown that dopamine affects paired associative stimulation (PAS)-induced plasticity, which might reflect learning-related processes to a larger degree than other noninvasive plasticity induction protocols in the human motor cortex in principle, its dose-dependency has not been explored previously. We studied the effect of different dosages of the dopamine precursor l-DOPA on motor cortex plasticity induced by facilitatory and inhibitory PAS of the motor cortex in 12 healthy humans. They received 25, 100, or 200 mg of l-DOPA or placebo medication combined with either excitability-enhancing or -diminishing PAS. Cortical excitability level was monitored before and for up to 2 d after plasticity induction by assessment of transcranial magnetic stimulation-induced motor-evoked potentials. Low-dose l-DOPA abolished the aftereffects of PAS and medium-dose l-DOPA prolonged facilitatory plasticity. High-dose l-DOPA reversed the excitability enhancement accomplished by facilitatory PAS to diminution. Thus, the results show a clear nonlinear effect of l-DOPA dosage on associative plasticity, different from that on nonfocal plasticity. This might help to explain dopaminergic effect on cognition and could be relevant for understanding the pathophysiology and treatment of neuropsychiatric diseases accompanied by alterations of the dopaminergic system.
Introduction
Dopamine influences neuroplasticity heterogeneously, depending on neuronal activity, dosage, and subreceptor specificity (Seamans and Yang, 2004). In neuropsychiatric diseases such as Parkinson's disease, Lewy body dementia, and schizophrenia, dopamine is associated with cognitive impairment (Molloy et al., 2005; Iversen and Iversen, 2007; Poewe et al., 2010). In the cognitive domain, dopamine improves learning and memory formation (Knecht et al., 2004; Shohamy et al., 2005; Molina-Luna et al., 2009). The likely neurophysiological basis of these effects is its impact on neuroplasticity, namely long term-potentiation (LTP) and -depression (LTD), as shown by animal experimentation (Seamans and Yang, 2004; Calabresi et al., 2007; Kung et al., 2007) and in humans (Kuo et al., 2008; Lang et al., 2008a). Spike-timing-dependent plasticity is a candidate mechanism for memory formation (Hebb, 1949; Letzkus et al., 2007). Similar plasticity can be induced in humans by paired associative stimulation (PAS) (Stefan et al., 2000), where combined somatosensory stimulation of a peripheral nerve and the motor cortex via transcranial magnetic stimulation (TMS) is performed. Synchronous activation of motor cortical neurons by somatosensory afferents and motor cortex TMS enhance cortical excitability, whereas asynchronous stimulation diminishes it. Because plasticity induced by PAS is thought to be largely restricted to the somatosensory–motor cortex synaptic connections activated by both the peripheral nerve and motor cortex TMS, it is termed associative, and suggested to induce a focal and synapse-specific kind of plasticity (Stefan et al., 2000, Weise et al., 2006). Excitability alterations induced by PAS can last for approximately an hour and are NMDA receptor- and calcium-dependent (Stefan et al., 2002; Wolters et al., 2003). In contrast, transcranial direct current stimulation (tDCS) induces a relatively nonfocal kind of neuroplasticity, which is not restricted to specific synaptic connections, but is thought to affect the majority of neurons under the stimulation electrodes (Purpura and McMurtry, 1965; Nitsche et al., 2007).
The dopamine precursor l-DOPA, at 100 mg oral dose, enhances PAS-induced and suppresses tDCS-induced facilitatory plasticity in humans, revealing a focusing effect of the drug on plasticity (Kuo et al., 2008). This relates well with its positive effect on learning and memory formation (Knecht et al., 2004; Floel et al., 2005a; Shohamy et al., 2005). However, l-DOPA did not improve performance in all studies testing its effects on cognition (Gotham et al., 1988; Shohamy et al., 2006; Ghilardi et al., 2007). This could be due to a nonlinear dosage-dependent effect of l-DOPA on cognition and neuroplasticity. Animal studies have revealed an inverted U-shaped dose–response curve of dopamine on cognition (Williams and Goldman-Rakic, 1995; Granon et al., 2000; Seamans and Yang, 2004). Interestingly, PAS-induced plasticity is absent in Parkinson′s disease patients off medication, but restituted by dopaminergic agents (Ueki et al., 2006). However, only dosage dependency of dopamine on tDCS-induced plasticity (Monte-Silva et al., 2010), but not associative plasticity, has been explored so far.
In this study, we aimed to explore a nonlinear effect of l-DOPA on associative plasticity in humans, using the human motor cortex as a model system.
Materials and Methods
Subjects.
Twelve healthy human volunteers (six females; age, 29.67 ± 8.04 years) participated in the study. All of them were right-handed according to the Edinburgh handedness inventory (Oldfield, 1971). The eligible age of subjects to participate in the study was between 18 and 65 years. None of the subjects had any metallic implant in the body or a history of neurological/psychiatric or chronic or acute medical disease. They took no acute/chronic medication during or up to 2 weeks before participating in the study. Pregnancy was ruled out in female subjects. All participants signed an informed consent form before participating in the experiment. The experiment conforms to the guidelines stated in the Declaration of Helsinki and was approved by the local Ethics Committee.
Monitoring motor cortex excitability.
Motor cortex excitability was monitored by the peak-to-peak amplitudes of motor-evoked potentials (MEP) generated by transcranial magnetic stimulation. Single magnetic pulses were delivered from a Magstim 200 stimulator (Magstim) at a frequency of 0.25 Hz. A figure-of-eight coil (diameter of one winding 70 mm; peak magnetic field 2.2T) was held tangentially on the scalp at an angle of 45° to the midsagittal plane with the handle pointing laterally and posteriorly to deliver the pulses. This coil position induces a posteroanterior directed current flow in the brain. The exact point for TMS was the motor cortex representation of the right abductor digiti minimi (ADM) muscle. This motor hot spot, as determined by TMS, was defined as the point where a magnetic stimulus of constant, slightly suprathreshold intensity consistently elicited MEPs of the highest amplitude. Surface electromyography (EMG) electrodes (Ag–AgCl) were placed over the right ADM in a belly–tendon montage for recording the MEPs. The signals from the EMG electrodes were amplified (gain, 1000), bandpass filtered (2 Hz–2 KHz), digitized at a frequency of 5 KHz, and stored in a laboratory computer for later offline analysis by Signal software and CED 1401 hardware (Cambridge Electronic Design). The intensity of the magnetic stimulus required to elicit ∼1 mV MEP amplitudes (SI1mV) was determined. At this intensity, 25–30 MEPs were recorded before and at several time points after the intervention. The change in the mean MEP amplitude over time was considered as measure of cortical excitability alterations caused by the intervention. Table 1 shows the mean values of the basic neurophysiological parameters measured during the experiment.
Table 1.
Comparison of baseline TMS parameters for different l-DOPA dosage conditions
| Parameter | Drug dosage |
|||
|---|---|---|---|---|
| Placebo | 25 mg l-DOPA | 100 mg l-DOPA | 200 mg l-DOPA | |
| Baseline S1mV (% MSO) | 47.64 ± 6.8 | 47.5 ± 6.8 | 47.68 ± 6.4 | 47.27 ± 6.8 |
| S1mV after drug (% MSO) | 47.82 ± 7.0 | 48.27 ± 7.3 | 48.64 ± 6.8 | 48.09 ± 6.7 |
| Mean baseline 1 (mV) | 1.14 ± 0.09 | 1.14 ± 0.11 | 1.04 ± 0.08 | 1.09 ± 0.08 |
| Mean baseline 2 (mV) | 1.08 ± 0.17 | 1.01 ± 0.35 | 1.01 ± 0.19 | 1.02 ± 0.22 |
| Mean baseline 3 (mV) | 1.09 ± 0.10 | 1.14 ± 0.14 | 1.10 ± 0.11 | 1.15 ± 0.13 |
Shown are mean ± SD values of the baseline TMS parameters for the different l-dopa dosage conditions. There was no significant difference between the parameters across the different conditions (Student's t test, paired, two-tailed, p ≤ 0.05). MSO, Maximum stimulator output.
Pharmacological intervention.
The subjects received a low (25 mg), medium (100 mg), or high (200 mg) dose of l-DOPA in combination with the dopamine decarboxylase inhibitor benserazide (one-fourth the dose of l-DOPA) or a placebo medication at each experimental session. One hour before the intake of this medication, the subjects received a 20 mg oral tablet of domperidone to counteract the systemic side effects of l-DOPA. For the high-dose sessions, the subjects were asked to take 20 mg of domperidone orally three times daily for 2 d before the experiment. The rationale for using the above-mentioned dosages of l-DOPA is that these had prominent nonlinear effects on another plasticity induction protocol in a recently published study (Monte-Silva et al., 2010).
PAS.
For PAS, an electrical pulse was delivered to the ulnar nerve at the wrist, followed by a magnetic pulse to the motor hot spot of the ADM. The intensity of the electrical pulse was three times the sensory perceptual threshold, delivered from a Digitimer D185 multipulse stimulator (Digitimer). The magnetic pulse had an intensity that resulted in SI1mV. Both stimuli were separated by an interval of either 10 or 25 ms, with the peripheral nerve pulse always followed by the TMS stimulus. These paired pulses were administered 90 times at a frequency of 0.05 Hz for 30 min over the motor hot spot of the ADM. Here, the interstimulus interval (ISI) determines the direction of plasticity that is induced. When the ISI is 10 ms (PAS-10), excitability diminution occurs, whereas an ISI of 25 ms (PAS-25) induces excitability enhancement (Stefan et al., 2000, 2002; Wolters et al., 2003). The reason for these different effects is that in PAS-10, the somatosensory stimulus reaches the primary motor cortex some milliseconds before the TMS stimulus (asynchronous stimulation), whereas in case of PAS-25, both stimuli reach the motor cortex simultaneously and such synchronous activation results in facilitation at the synapse.
Course of the experiment.
The study design was single-blinded, complete crossover, and placebo-controlled. Between the experimental sessions (eight sessions per subject), an interval of at least 1 week was essential to avoid interference effects.
The participants received domperidone or equivalent placebo medication before the start of the neurophysiological part of experiments, as outlined above. They were seated comfortably on a reclining chair with head and arm rests, and asked to relax completely. EMG electrodes were placed at the right ADM, and the motor cortex hotspot was determined. Both the position of the EMG electrodes and the motor hotspot were marked with a permanent skin marker to ensure their constant positioning throughout the experimental session. SI1mV was determined and at least 25 MEPs were recorded as baseline 1 at this stimulus intensity. Immediately after the baseline measurement, the participants received low/medium/high dose l-DOPA or placebo medication. The combination of drug dose and PAS was given in a randomized order for all the subjects. Baseline 2 (25 MEPs) was obtained after 1 h, because at this time l-DOPA has reached its maximal plasma concentration (Crevoisier et al., 1987) and has prominent effects on brain function (Floel et al., 2005a; Kuo et al., 2008), to reveal an influence of the medication on cortical excitability. In case of any drug-induced MEP amplitude changes, another set of MEPs was recorded at the adjusted SI1mV (baseline 3). Subsequently, either PAS-10 or PAS-25 was administered as described above. Following PAS, 25–30 MEPs were recorded at 0, 5, 10, 15, 20, 25, 30, 60, 90, and 120 min for all sessions. Since the aftereffects of PAS have not been reported to last for >90 min, we recorded MEPs in the placebo condition only until 120 min after cessation of stimulation. Further after-measurements were conducted the evening of the same day, next morning, next afternoon, next evening, and on the morning of the third day for all sessions except the placebo sessions (Fig. 1).
Figure 1.

Course of the experiment. MEPs elicited from single-pulse TMS over the motor hot spot were recorded at 1 mV intensity before drug intake [baseline 1 (BL1)]. One hour after drug intake, baseline 2 (BL2) was recorded to look for an effect of the drug on cortical excitability. In case of any individual MEP alterations from baseline 1, baseline 3 (BL3) was recorded by adjusting the stimulator output to obtain a mean of 1 mV. Then PAS-10 or PAS-25 was administered and was immediately followed by MEP after-measurements that lasted 120 min. For all sessions except the placebo medication sessions, after-measurements were performed until the morning of the third day following the stimulation.
Data analysis and statistics.
Individual mean MEP amplitudes for each subject for baselines 1, 2, and 3 and each time point following intervention were calculated. The postintervention MEP amplitudes were normalized to the mean baseline 3. In most of the individual measurements, l-DOPA did not alter baseline MEP amplitudes. In these cases, there was no need to change the SI1mV and baseline 3 was identical to baseline 2. The normalized MEP amplitudes from all subjects were pooled together session-wise by calculating the grand average across subjects for each condition and time point.
A repeated-measures ANOVA was performed for the normalized data. MEP amplitude served as dependent variable. We included only the data until 120 min after PAS in the analysis, which were obtained from all sessions. PAS (PAS-10/PAS-25) and drug (low dose/medium dose/high dose/placebo) served as within-subjects factors. Mauchly's sphericity test was performed and Greenhouse–Geisser correction was applied when necessary. If the ANOVA yielded significant results, we performed post hoc comparisons using Student's t test (paired, two-tailed, p < 0.05, not adjusted for multiple comparisons). Here we compared (1) the mean MEP amplitudes at all time points after PAS versus baseline 3 and (2) the means at a specific time point for the various drug conditions against the placebo medication condition. Baseline MEP amplitudes of all drug/PAS combinations were compared by Student′s t tests to exclude a priori differences between conditions, and baseline 1 and 2 MEP amplitudes to test for any influence of the drug alone on cortical excitability.
Results
All except one male subject tolerated the experimental procedures well. This subject developed nausea and vomiting ∼90 min after the intake of 200 mg of l-DOPA. We had to exclude the data of this participant from the analysis because of artifacts of MEP measures caused by insufficient relaxation. Mean absolute baseline MEP amplitudes and percentage of maximal stimulator output to achieve baseline amplitudes of ∼1 mV did not differ significantly between sessions (Student's t test, paired, two-tailed, p > 0.05). Baseline MEP amplitudes were not affected by any of the drug dosages significantly (Student's paired t test, two-tailed, p > 0.05). There was no significant difference of baseline 3 between the different sessions (Student's paired t test, two-tailed, p > 0.05). Also, baseline 1 and baseline 3 did not differ significantly (Student's paired t test, two-tailed, p > 0.05).
The repeated-measures ANOVA resulted in significant main effects of drug dosage (F(3,30) = 2.990; p = 0.047) and PAS (F(1,10) = 11.261; p = 0.007). There was no significant effect of time though (F(10,100) = 0.948; p = 0.426). Two-way interactions of drug dosage × PAS (F(3,30) = 12.182; p < 0.001) and PAS × time (F(10,100) = 2.761; p = 0.005) were significant. There was no significant interaction between drug dosage and time (F(30,300) = 1.176; p = 0.247). The three-way interaction of drug dosage × PAS × time was significant (F(30,300) = 2.245; p < 0.001).
Dose-dependent effect of l-DOPA on PAS-induced neuroplasticity
In the placebo medication condition, PAS-10 diminished excitability and PAS-25 enhanced it until at least 30 min following the stimulation. Under low dose (25 mg) l-DOPA, the excitability diminution induced by PAS-10 as well as the excitability enhancement induced by PAS-25 (Fig. 2A) were diminished; that is, following both PAS-10 and PAS-25, there was no significant difference in the MEP amplitudes compared with baseline values. Furthermore, MEP amplitudes in the low-dose condition differed significantly from those in the placebo condition at the initial time points after PAS-25 and at later time points after PAS-10. Following medium-dose (100 mg) l-DOPA, the excitability changes induced by both PAS-10 and PAS-25 were preserved (Fig. 2B). We observed that the excitability enhancement caused by PAS-25 was no longer significant between 60 and 120 min after PAS, but then recovered later and remained significantly enhanced until the afternoon of the next day after PAS, and thus for ∼24 h. More prominently enhanced and prolonged facilitation with 100 mg of l-DOPA was observed in a previous study by Kuo et al. (2008). The MEP amplitudes at identical time points did not differ significantly between the medium dose and placebo conditions. After intake of high-dose (200 mg) l-DOPA, the excitability diminution induced by PAS-10 lasted longer compared with that under placebo medication, whereas the facilitatory aftereffects of PAS-25 were converted into inhibition (Fig. 2C). Such inhibition was significant compared with the baseline until 20 min following the stimulation. Differences between the MEP amplitudes in the high-dose and placebo conditions were significant only for PAS-25.
Figure 2.
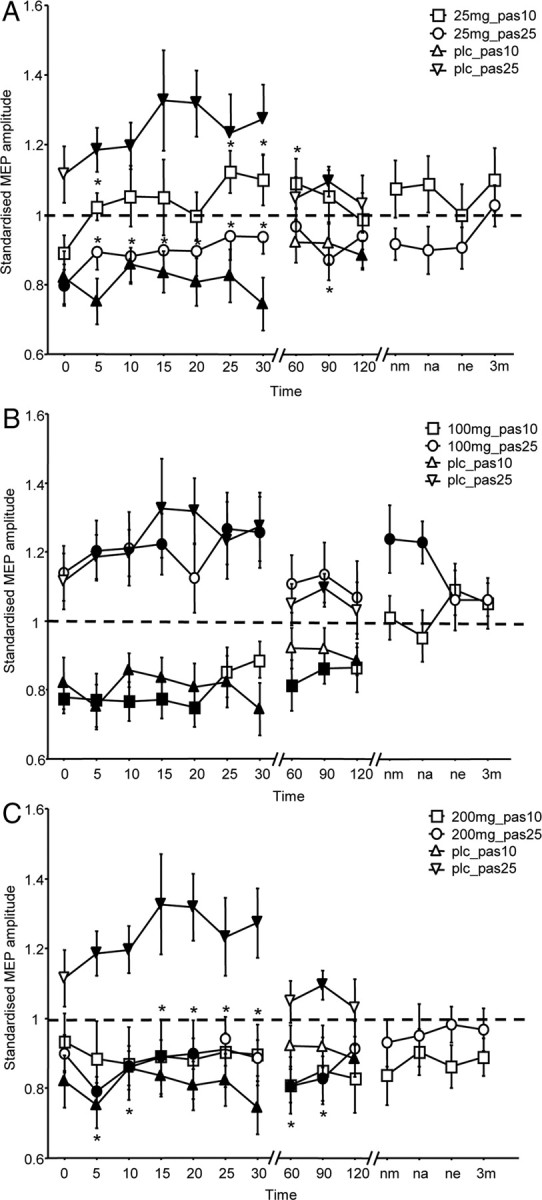
Dose-dependent effect of l-DOPA on PAS-induced neuroplasticity. The x-axis displays the time points (in minutes) of after-measurements during the experiment. MEP amplitudes standardized to the corresponding baseline values (mean ± SEM) are plotted on the y-axis. The graphs show that under placebo (plc) medication, facilitatory PAS-25 induces an excitability enhancement lasting for at least 30 min, whereas PAS-10 diminishes excitability for a similar duration following stimulation. A, Effect of 25 mg (low dose) of l-DOPA on the PAS-induced aftereffects. Low-dose l-DOPA reduces or abolishes the aftereffects of both PAS-25 and PAS-10. B, Medium-dose (100 mg) l-DOPA prolongs facilitatory PAS-induced plasticity, but does not alter the excitability-reducing aftereffects of PAS-10. C, High-dose (200 mg) l-DOPA reverses the facilitation induced by PAS-25 to inhibition while trendwise prolonging the inhibitory effect of PAS-10. Filled symbols indicate statistically significant deviation of the post-PAS values compared with the baseline. Asterisks indicate significantly different values in the l-DOPA condition compared with the placebo medication conditions at the same time points after the respective PAS protocols (Student's t test, paired, two-tailed, p ≤ 0.05). nm, Next morning; na, next afternoon; ne, next evening; 3m, morning of the third day.
Discussion
The results of this study reveal a nonlinear effect of l-DOPA dosage on associative plasticity in the human motor cortex. Low-dose l-DOPA reduces or abolishes the aftereffects of facilitatory and inhibitory PAS, medium dosage prolongs PAS-25-generated facilitation and preserves inhibition resulting from PAS-10, and high-dose l-DOPA reverses PAS-25-induced facilitation into inhibition, and trendwise prolongs the inhibition induced by PAS-10 (Fig. 3).
Figure 3.

Dose-dependent effect of l-DOPA on cortical excitability of the human motor cortex until 30 min following PAS. Shown is the change of the standardized mean MEP amplitudes pooled for time points until 30 min following PAS. PAS-25 and PAS-10 show maximum excitability enhancement and diminution, respectively, following medium-dose (100 mg) l-DOPA. Thus, optimal levels of PAS-induced aftereffects are observed with 100 mg of l-DOPA. LD, Low dose; MD, medium dose; HD, high dose.
Proposed mechanisms of action
For the medium-dose l-DOPA, the results are in principal accordance with those of a former study regarding the prolongation of the facilitatory aftereffects of PAS. Minor differences in the magnitude of the effects are probably caused by the different groups of participants. The prolongation of facilitatory plasticity by l-DOPA might be primarily caused by enhanced D1 receptor activation, because D2-like receptor block does not abolish this kind of plasticity (Nitsche et al., 2009). Specifically the NMDA receptor-enhancing function of moderate D1 activation (Seamans and Yang, 2004) is a likely candidate mechanism, since PAS is known to induce NMDA receptor-dependent plasticity (Stefan et al., 2002). In accordance with both D1- and D2-like activation (Monte-Silva et al., 2009; Nitsche et al., 2009), inhibitory plasticity was not enhanced or diminished by medium l-DOPA dosage. This does not, however, mean that dopamine does not affect inhibitory associative plasticity per se, as it was abolished by D2 receptor block (Nitsche et al., 2009). It is likely that a balanced activation of both receptors is needed for this kind of plasticity.
The plasticity-diminishing or -abolishing effects of low-dose l-DOPA is in accordance with a similar effect of low-dose ropinirole, a D2/D3 dopaminergic agonist, on PAS-induced facilitatory plasticity (Monte-Silva et al., 2009). The proposed mechanism of action is a preferential activation of presynaptic autoreceptors by low-dose dopaminergic activation, which reduces dopamine release (Yamada and Furukawa, 1980; Schmitz et al., 2003). This might also explain the plasticity-abolishing effect of low-dose l-DOPA on inhibitory plasticity. However, this kind of plasticity was not affected by low-dose ropinirole. The reason for this might be that ropinirole as a dopaminergic agonist also activates postsynaptic D2 receptors independent from dopamine excretion. This minor activation of postsynaptic D2 receptors might have been sufficient to preserve inhibitory plasticity in cases of low-dose ropinirole application.
For high-dose l-DOPA, the conversion of the facilitatory aftereffects of PAS-25 into inhibition is different than that of high-dose ropinirole, which abolished this kind of plasticity at high dosages. Thus, the D1 receptor might have contributed to this effect. Because a high grade of D1 receptor activation, in contrast to low or medium D1 activation, inhibits NMDA receptors (Seamans and Yang, 2004), this inhibition might reduce NMDA receptor activation to a level inducing LTD-like plasticity. The reason for this is that a low enhancement of intracellular calcium concentration induces LTD, whereas a larger enhancement generates LTP (Lisman, 2001), and NMDA receptor activity controls the amount of calcium influx. Alternatively, NMDA receptors are not inhibited by the level of dopaminergic activation accomplished by 200 mg of l-DOPA, but a major enhancement of NMDA receptor activity will result in an intracellular calcium concentration sufficiently large enough to activate hyperpolarizing potassium channels (Misonou et al., 2004), which will convert facilitatory plasticity into inhibition. For the only trendwise effect of high-dose l-DOPA medication on PAS-10-generated inhibition, this might hint for a larger range of dopaminergic activation compatible with inhibitory plasticity.
The results of the present study not only show some similarities with, but also differences from, a recently conducted study, where the effects of identical l-DOPA dosages on motor cortex plasticity induced by tDCS were explored (Monte-Silva et al., 2010) (Fig. 4). tDCS induces plasticity by a tonic modulation of resting membrane potentials (Nitsche and Paulus, 2000, 2001; Nitsche et al., 2003a, 2008). The aftereffects of tDCS, like those of PAS, depend on NMDA receptor and calcium channel activity (Nitsche et al., 2003b, 2004). However, contrary to PAS, which is thought to induce plasticity at somatosensory–motor cortical synapses activated by both peripheral nerve and transcranial magnetic stimulation of the motor cortex, plasticity induction by tDCS is thought not to be restricted to specific synaptic subgroups because of the completely different stimulation protocol: in tDCS, the relatively large stimulation electrodes, which deliver a continuous current flow for some minutes, are thought to affect the majority of neurons beneath the electrodes (Nitsche et al., 2007, Purpura and McMurtry, 1965) and thus the effects are much less restricted or focal. This difference in focality of the plasticity induction might account for some of the different results obtained by dopaminergic medication on plasticity induced by PAS and tDCS. For low dosages of l-DOPA, plasticity induced by tDCS was also prevented. Since the induction of aftereffects of tDCS, similar to those accomplished by PAS, need dopaminergic activity (Nitsche et al., 2006), this result is compatible with a presynaptic effect. However, for medium-dose l-DOPA, tDCS-induced facilitatory aftereffects were converted into inhibition, thus mimicking the high-dose l-DOPA effects on PAS-25-induced plasticity. This pattern of results can be explained by a larger calcium increase induced by the less selective and tonic stimulation induced by tDCS, compared with the more specific and phasic PAS procedure. Thus, facilitatory tDCS, in concert with medium-dose l-DOPA, may have enhanced intracellular calcium sufficiently to activate hyperpolarizing potassium channels. With high-dose l-DOPA, facilitatory as well as inhibitory plasticity were abolished, which could be explained by an NMDA receptor-inactivating effect of large D1 activation. Figure 4 summarizes the dose-dependent effect of l-DOPA on both focal PAS-induced and nonfocal tDCS-generated plasticity and hence illustrates the focusing effect of the drug on plasticity.
Figure 4.

Summary of the effect of different dosages of l-DOPA coupled with tDCS/PAS on motor cortex excitability. Shown are the effects of low-dose (LD; 25 mg), medium-dose (MD; 100 mg), and high-dose (HD; 200 mg) l-DOPA on motor cortex excitability level. Nonfocal stimulation by tDCS abolishes stimulation aftereffects at low and high doses but preserves inhibitory plasticity induced by cathodal tDCS and reverses the facilitatory aftereffects of anodal tDCS to inhibition at medium dosage (Monte-Silva et al., 2010). Focal, associative, and synapse-specific stimulation by PAS, when coupled with low-dose l-DOPA, abolishes aftereffects of both facilitatory and inhibitory PAS. Medium-dose l-DOPA preserves/prolongs the aftereffects of PAS, whereas high-dose l-DOPA preserves only inhibitory aftereffects of PAS-10 and reverses facilitatory aftereffects of PAS-25 to inhibition. White arrows indicate excitability-diminishing stimulation protocols (cathodal tDCS or PAS-10); black arrows indicate excitability-enhancing stimulation protocols (anodal tDCS or PAS-25).
It should be stressed that this mechanism of action of dopamine on PAS- and tDCS-induced plasticity is hypothetical at present, and should be addressed more directly in future studies. Apart from the proposed local mechanisms, dopamine might also affect motor cortex plasticity by an indirect effect on remote regions. Here, the well known impact of dopamine on striatal activity and excitability (Shen et al., 2008) might be an attractive candidate, which should be explored in future studies.
A complex picture of dopaminergic effects on plasticity emerges. The results of the present study and other studies suggest that the effect of l-DOPA on plasticity depends on its dosage, subreceptor specificity, and type of plasticity. Interestingly, a focusing effect of l-DOPA on facilitatory plasticity, as revealed by decreased nonfocal tDCS-induced facilitation and preserved focal PAS-induced facilitation, seems to be restricted to medium enhancement of dopaminergic activation.
General remarks
In the present study, we explored the impact of l-DOPA on focal associative plasticity as induced by PAS. Previous studies showed an improvement of cognitive performance in humans after administration of <100 mg (medium dose) of l-DOPA (Knecht et al., 2004; Floel et al., 2005a). However, some studies revealed heterogeneous effects of the drug on cognition (Gotham et al., 1988; Kulisevsky, 2000; Cools et al., 2001). It can be speculated that the nonuniformity of the effects of l-DOPA on cognition is partly explained by its nonlinear dose-dependent effects on plasticity. This might be relevant because dopamine levels are altered in many neuropsychiatric conditions where cognitive performance is impaired (Floel et al., 2005b; Liepert, 2008). Alterations of plasticity in these patients might correlate with impaired cognition and restoring plasticity might help them to regain cognitive performance. Although there is no evidence so far that PAS directly influences neurons in subcortical regions like the striatum, an indirect influence via the thalamo-cortico-basal ganglia circuits cannot be ruled out. This is potentially relevant because dopamine-dependent synaptic plasticity is well known to occur in this region (Surmeier et al., 2009). Specifically, it has been shown that the intrinsic excitability of striatal neurons depends on dopaminergic balance (Shen et al., 2008). Thus, the dopaminergic impact on plasticity could have been influenced by the drug's impact on striatal excitability. This possibility should be explored to a larger degree in future studies. Some limitations of the present study should be mentioned. Since plasma levels of dopamine were not obtained, we could not control for interindividual variability caused by differences in the bioavailability of the drug. However, we studied a fairly homogenous group of subjects. Blinding might have been somewhat compromised by different durations of the after-measures, but with the multitude of sessions (eight per subject) and blinded PAS protocols, an expectancy effect should not have been relevant. We only studied young healthy subjects, and it is important to be aware that the results might not directly translate to elderly subjects or patient populations. Further studies are needed to explore the effects in these subject groups. Moreover, apart from other confounding factors like age (Floel et al., 2008), tobacco smoking (Lang et al., 2008b), and genetics (Cheeran et al., 2008), altered dopamine levels in patients should be borne in mind when brain stimulation protocols are applied for diagnostic or therapeutic purposes.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant NI 683/6–1.
References
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Cheeran B, Talelli P, Mori F, Koch G, Suppa A, Edwards M, Houlden H, Bhatia K, Greenwood R, Rothwell JC. A common polymorphism in the brain-derived neurotrophic factor gene (BDNF) modulates human cortical plasticity and the response to rTMS. J Physiol. 2008;586:5717–5725. doi: 10.1113/jphysiol.2008.159905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, Barker RA, Sahakian BJ, Robbins TW. Enhanced or impaired cognitive function in Parkinson's disease as a function of dopaminergic medication and task demands. Cereb Cortex. 2001;11:1136–1143. doi: 10.1093/cercor/11.12.1136. [DOI] [PubMed] [Google Scholar]
- Crevoisier C, Hoevels B, Zürcher G, Da Prada M. Bioavailability of l-dopa after Madopar HBS administration in healthy volunteers. Eur Neurol. 1987;27(Suppl 1):36–46. doi: 10.1159/000116173. [DOI] [PubMed] [Google Scholar]
- Floel A, Breitenstein C, Hummel F, Celnik P, Gingert C, Sawaki L, Knecht S, Cohen LG. Dopaminergic influences on formation of a motor memory. Ann Neurol. 2005a;58:121–130. doi: 10.1002/ana.20536. [DOI] [PubMed] [Google Scholar]
- Floel A, Hummel F, Breitenstein C, Knecht S, Cohen LG. Dopaminergic effects on encoding of a motor memory in chronic stroke. Neurology. 2005b;65:472–474. doi: 10.1212/01.wnl.0000172340.56307.5e. [DOI] [PubMed] [Google Scholar]
- Floel A, Vomhof P, Lorenzen A, Roesser N, Breitenstein C, Knecht S. Levodopa improves skilled hand functions in the elderly. Eur J Neurosci. 2008;27:1301–1307. doi: 10.1111/j.1460-9568.2008.06079.x. [DOI] [PubMed] [Google Scholar]
- Ghilardi MF, Feigin AS, Battaglia F, Silvestri G, Mattis P, Eidelberg D, Di Rocco A. L-Dopa infusion does not improve explicit sequence learning in Parkinson's disease. Parkinsonism Relat Disord. 2007;13:146–151. doi: 10.1016/j.parkreldis.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Gotham AM, Brown RG, Marsden CD. ‘Frontal’ cognitive function in patients with Parkinson's disease ‘on’ and ‘off’ levodopa. Brain. 1988;111:299–321. doi: 10.1093/brain/111.2.299. [DOI] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci. 2000;20:1208–1215. doi: 10.1523/JNEUROSCI.20-03-01208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO. The organization of behavior. New York: Wiley; 1949. [Google Scholar]
- Iversen SD, Iversen LL. Dopamine: 50 years in perspective. Trends Neurosci. 2007;30:188–193. doi: 10.1016/j.tins.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Knecht S, Breitenstein C, Bushuven S, Wailke S, Kamping S, Flöel A, Zwitserlood P, Ringelstein EB. Levodopa: faster and better word learning in normal humans. Ann Neurol. 2004;56:20–26. doi: 10.1002/ana.20125. [DOI] [PubMed] [Google Scholar]
- Kulisevsky J. Role of dopamine in learning and memory: implications for the treatment of cognitive dysfunction in patients with Parkinson's disease. Drugs Aging. 2000;16:365–379. doi: 10.2165/00002512-200016050-00006. [DOI] [PubMed] [Google Scholar]
- Kung VW, Hassam R, Morton AJ, Jones S. Dopamine-dependent long term potentiation in the dorsal striatum is reduced in the R6/2 mouse model of Huntington's disease. Neuroscience. 2007;146:1571–1580. doi: 10.1016/j.neuroscience.2007.03.036. [DOI] [PubMed] [Google Scholar]
- Kuo MF, Paulus W, Nitsche MA. Boosting focally-induced brain plasticity by dopamine. Cereb Cortex. 2008;18:648–651. doi: 10.1093/cercor/bhm098. [DOI] [PubMed] [Google Scholar]
- Lang N, Speck S, Harms J, Rothkegel H, Paulus W, Sommer M. Dopaminergic potentiation of rTMS-induced motor cortex inhibition. Biol Psychiatry. 2008a;63:231–233. doi: 10.1016/j.biopsych.2007.04.033. [DOI] [PubMed] [Google Scholar]
- Lang N, Hasan A, Sueske E, Paulus W, Nitsche MA. Cortical hypoexcitability in chronic smokers? A transcranial magnetic stimulation study. Neuropsychopharmacology. 2008b;33:2517–2523. doi: 10.1038/sj.npp.1301645. [DOI] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Does spike timing-dependent synaptic plasticity underlie memory formation? Clin Exp Pharmacol Physiol. 2007;34:1070–1076. doi: 10.1111/j.1440-1681.2007.04724.x. [DOI] [PubMed] [Google Scholar]
- Liepert J. Pharmacotherapy in restorative neurology. Curr Opin Neurol. 2008;21:639–643. doi: 10.1097/WCO.0b013e32831897a3. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol. 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Molina-Luna K, Pekanovic A, Röhrich S, Hertler B, Schubring-Giese M, Rioult-Pedotti MS, Luft AR. Dopamine in motor cortex is necessary for skill learning and synaptic plasticity. PLoS One. 2009;4:e7082. doi: 10.1371/journal.pone.0007082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy S, McKeith IG, O'Brien JT, Burn DJ. The role of levodopa in the management of dementia with Lewy bodies. J Neurol Neurosurg Psychiatry. 2005;76:1200–1203. doi: 10.1136/jnnp.2004.052332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte-Silva K, Kuo MF, Thirugnanasambandam N, Liebetanz D, Paulus W, Nitsche MA. Dose-dependent inverted U-shaped effect of dopamine (D2-like) receptor activation on focal and nonfocal plasticity in humans. J Neurosci. 2009;29:6124–6131. doi: 10.1523/JNEUROSCI.0728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte-Silva K, Liebetanz D, Grundey J, Paulus W, Nitsche MA. Dosage-dependent non-linear effect of l-DOPA on human motor cortex plasticity. J Physiol. 2010;588:3415–3424. doi: 10.1113/jphysiol.2010.190181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol. 2000;527:633–639. doi: 10.1111/j.1469-7793.2000.t01-1-00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W. Sustained excitability elevations induced by transcranial DC motor cortex stimulation in humans. Neurology. 2001;57:1899–1901. doi: 10.1212/wnl.57.10.1899. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Nitsche MS, Klein CC, Tergau F, Rothwell JC, Paulus W. Level of action of cathodal DC polarisation induced inhibition of the human motor cortex. Clin Neurophysiol. 2003a;114:600–604. doi: 10.1016/s1388-2457(02)00412-1. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Fricke K, Henschke U, Schlitterlau A, Liebetanz D, Lang N, Henning S, Tergau F, Paulus W. Pharmacological modulation of cortical excitability shifts induced by transcranial direct current stimulation in humans. J Physiol. 2003b;553:293–301. doi: 10.1113/jphysiol.2003.049916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W. Consolidation of human motor cortical neuroplasticity by d-cycloserine. Neuropsychopharmacology. 2004;29:1573–1578. doi: 10.1038/sj.npp.1300517. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Lampe C, Antal A, Liebetanz D, Lang N, Tergau F, Paulus W. Dopaminergic modulation of long-lasting direct current induced cortical excitability changes in the human motor cortex. Eur J Neurosci. 2006;23:1651–1657. doi: 10.1111/j.1460-9568.2006.04676.x. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Doemkes S, Karaköse T, Antal A, Liebetanz D, Lang N, Tergau F, Paulus W. Shaping the effects of transcranial direct current stimulation of the human motor cortex. J Neurophysiol. 2007;97:3109–3117. doi: 10.1152/jn.01312.2006. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Cohen LG, Wassermann EM, Priori A, Lang N, Antal A, Paulus W, Hummel F, Boggio PS, Fregni F, Pascual-Leone A. Transcranial direct current stimulation: state of the art 2008. Brain Stimul. 2008;1:206–223. doi: 10.1016/j.brs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Kuo MF, Grosch J, Bergner C, Monte-Silva K, Paulus W. D1-receptor impact on neuroplasticity in humans. J Neurosci. 2009;29:2648–2653. doi: 10.1523/JNEUROSCI.5366-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield RC. The assessment and analysis of handedness: The Edinburgh Inventory. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson's disease: an old drug still going strong. Clin Interv Aging. 2010;5:229–238. doi: 10.2147/cia.s6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura DP, McMurtry JG. Intracellular activities and evoked potential changes during polarization of motor cortex. J Neurophysiol. 1965;28:166–185. doi: 10.1152/jn.1965.28.1.166. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Benoit-Marand M, Gonon F, Sulzer D. Presynaptic regulation of dopaminergic neurotransmission. J Neurochem. 2003;87:273–289. doi: 10.1046/j.1471-4159.2003.02050.x. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Grossman S, Sage J, Gluck MA. The role of dopamine in cognitive sequence learning: evidence from Parkinson's disease. Behav Brain Res. 2005;156:191–199. doi: 10.1016/j.bbr.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Geghman KD, Sage J, Gluck MA. L-dopa impairs learning, but spares generalization, in Parkinson's disease. Neuropsychologia. 2006;44:774–784. doi: 10.1016/j.neuropsychologia.2005.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123:572–584. doi: 10.1093/brain/123.3.572. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol. 2002;543:699–708. doi: 10.1113/jphysiol.2002.023317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol. 2009;19:621–628. doi: 10.1016/j.conb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y, Mima T, Kotb MA, Sawada H, Saiki H, Ikeda A, Begum T, Reza F, Nagamine T, Fukuyama H. Altered plasticity of the human motor cortex in Parkinson's disease. Ann Neurol. 2006;59:60–71. doi: 10.1002/ana.20692. [DOI] [PubMed] [Google Scholar]
- Weise D, Schramm A, Stefan K, Wolters A, Reiners K, Naumann M, Classen J. The two sides of associative plasticity in writer's cramp. Brain. 2006;129:2709–2721. doi: 10.1093/brain/awl221. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG, Benecke R, Classen J. A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol. 2003;89:2339–2345. doi: 10.1152/jn.00900.2002. [DOI] [PubMed] [Google Scholar]
- Yamada K, Furukawa T. Direct evidence for involvement of dopaminergic inhibition and cholinergic activation in yawning. Psychopharmacology (Berl) 1980;67:39–43. doi: 10.1007/BF00427593. [DOI] [PubMed] [Google Scholar]