

Abstract
Death receptor (DR) signaling has a major impact on the outcome of numerous neurological diseases, including ischemic stroke. DRs mediate not only cell death signals, but also proinflammatory responses and cell proliferation. Identification of regulatory proteins that control the switch between apoptotic and alternative DR signaling opens new therapeutic opportunities. Fas apoptotic inhibitory molecule 2 (Faim2) is an evolutionary conserved, neuron-specific inhibitor of Fas/CD95-mediated apoptosis. To investigate its role during development and in disease models, we generated Faim2-deficient mice. The ubiquitous null mutation displayed a viable and fertile phenotype without overt deficiencies. However, lack of Faim2 caused an increase in susceptibility to combined oxygen–glucose deprivation in primary neurons in vitro as well as in caspase-associated cell death, stroke volume, and neurological impairment after cerebral ischemia in vivo. These processes were rescued by lentiviral Faim2 gene transfer. In summary, we provide evidence that Faim2 is a novel neuroprotective molecule in the context of cerebral ischemia.
Introduction
The influence of death receptors (DRs) on the fate of neuronal networks is complex, depending not only on cell type and disease pathology, but also on the stage of a disease (Reich et al., 2008). Fas/CD95, the best-studied DR in the CNS (Choi and Benveniste, 2004), gives an example of how divergent the DR-mediated biological effects on neurons are. In addition to its eponymous apoptotic signaling, an increasing number of nonapoptotic functions emerge (Lambert et al., 2003; Wajant et al., 2003; Peter et al., 2005; Reich et al., 2008). Recent evidence that Fas signaling positively influences processes such as malignant brain tumor progression (Kleber et al., 2008) as well as induction of neurogenesis (Ceccatelli et al., 2004; Ricci-Vitiani et al., 2004) and neuritogenesis (Desbarats et al., 2003; Zuliani et al., 2006; Ruan et al., 2008) indicates its neuroplastic potential. Therefore, controlled modulation of Fas signaling seems a promising therapeutic strategy for many neurological diseases.
Several proteins with Fas-proximal apoptotic inhibitory properties exist (Reich et al., 2008), among them Fas apoptotic inhibitory molecule 2 (Faim2), an evolutionarily strongly conserved, predominantly neuronally expressed 35.1 kDa membrane protein. Its mechanism of cell death inhibition is a direct interaction with Fas/CD95 upstream of Fas-associated death domain-containing protein (FADD) (Somia et al., 1999), since Faim2 coimmunoprecipitates with Fas and not with FADD. Furthermore, Faim2 does not downregulate Fas or FADD expression, and it also does not interfere with binding of Fas agonists. Studies in rat revealed that postsynaptic membranes and dendrites are the subcellular predilection sites of the Faim2 homolog neuronal membrane protein 35 (NMP35) (Schweitzer et al., 1998). This is in line with the observation that postnatal upregulation of Faim2 coincides with terminal differentiation and synapse formation in the brain (Schweitzer et al., 2002). Faim2 expression is mediated by the phosphatidylinositol 3-kinase (PI 3-kinase)–Akt/protein kinase B (PKB) pathway (Beier et al., 2005)—a cascade with proven Fas-antagonistic capacity (Häusler et al., 1998) and association to protective effects in different neurological disease models (Noshita et al., 2001; Wagey et al., 2001; Henshall et al., 2002). The constitutive localization in lipid microdomains (Fernández et al., 2007) serves its function as an upstream modulator of receptor-mediated signaling. By now the action of Faim2 could only be linked to Fas signaling and not to other potential candidates like TNF-R. Sequence analysis suggests that Faim2 topology consists of seven conserved transmembrane domains and variable cytoplasmic N termini (Reimers et al., 2006). This makes Faim2 a member of the evolving ubiquitous eukaryotic gene family termed LFG (“Lifeguard”) (Hu et al., 2009). From an evolutionary point of view, the LFG gene family represents the expansion of one anti-apoptotic “founder” protein with general function and distribution to proteins with specialized expression profiles and subcellular localizations paralleling the increasingly complex apoptotic control. Although the neuronal Fas/CD95-antagonizing antiapoptotic function of Faim2 has been demonstrated in vitro (Beier et al., 2005; Fernández et al., 2007), the biological paradox of why differentiated cells simultaneously express a DR and its inhibitor is not solved.
To analyze the biological function of Faim2, a null mutant (Faim2−/−) was generated. Because embryonic lethality could not be excluded a priori, a Cre/loxP-system was used. Challenging the null mutant and a lentiviral Faim2-expressing system under ischemic–hypoxic conditions tested the neuroprotective potential of Faim2 in vitro and in vivo.
Materials and Methods
Faim2−/− mouse line
Faim2 gene targeting construct.
All necessary genomic elements were identified in clones of the 129/ola-cosmid library RZPD No. 121 (German Resource Center for Genome Research, Berlin, Germany) cloned via PCR into the targeting vector pCom-True. This ampicillin resistance-based vector contains 5′ and 3′ multiple cloning sites (MCS 1 and 2), one central loxP site flanked MCS 3, and one neomycin resistance gene (NeoR) flanked by frt sites. A 1488 bp genomic region containing Faim2 target exons was cloned as AscI/MfeI PCR-fragment (5′-ACAGGAGGGAAGGGACATTTG-3′ and 5′-TACC AGCTCTCTCCATCCCA-3′) into MCS 3, the flanking 5′ 531 bp arm of homology as NdeI/SacII PCR-fragment (5′-GAGGGAAAGATTAGCCGGAC-3′ and 5′-GTTCCCATCTC TCTCCCTGG-3′) into MSC 2 and the flanking 3′ 4514 bp arm of homology as XhoI/ClaI PCR-fragment (5′-AGAGCTGGTACCTTGACCCC-3′ and 5′-CTCGAAGCTACCCTATTG CG-3′) into MCS 1. All genomic elements of the targeting construct were verified by sequencing. Before electroporation, the targeting construct was linearized with NdeI.
Mouse genetics.
Genetic mouse experiments were performed in compliance with animal policies of the Max Planck Institute of Experimental Medicine. RI mouse embryonic stem (ES) cells were propagated on mitomycin-treated primary mouse embryonic fibroblasts. Ten million ES cells were electroporated (240 V, 500 μF; Gene pulser; Bio-Rad) with 50 μg of the linearized targeting vector. Transfected ES cells were cultured on gelatinized dishes (Falcon) in the presence of 103 U/ml leukemia inhibitory factor (Invitrogen). After 24 h, we started selection with 300 μg/ml G418 (Invitrogen). Screening and identification of homologously recombined ES cell clones were performed by seminested PCR using the forward primer P3′ (5′-GAAGAGCTC TAACCGGGCA-3′) and P3″ (5′-CGCCATGCTCATCTCTGA-3′) and the reverse primer P4 (5′-TCGCCTTCTTGACGAGTTCT-3′), which amplify a 746 bp fragment. Microinjection of positively screened ES cell clones was performed by standard procedures. Before crossing to C57BL/6J females to test for germline transmission, the chimerical animal of choice was reevaluated for homologous recombination by Southern blotting (HindIII digest, 5′ external 506 bp hybridization probe) and PCR analysis (primers 3′, 3″, and 4) from tail DNA. To generate the 5′ external Southern probe, a 514 bp genomic fragment was amplified via PCR (5′-CAAGGACCAAGCTTTGCTGT-3′ and 5′-GAGTGCC AGGCGAGTTTAAG-3′), subcloned into pcDNA3.1(+) (Invitrogen), sequenced, and double digested with HindIII and XbaI. Radioactive labeling was achieved by using 32P-α-dCTP according to the manufacturer's instructions (Quick Prime Kits, GE Healthcare). Ten micrograms of total HindIII-digested DNA per lane was size-separated on an agarose gel and transferred to a nylon membrane (Hybond-N+, GE Healthcare). Heterozygous offspring was crossed to a transgenic EIIa cre mouse line. Thus, the consecutive generation displayed genotypes that were heterozygous for permanently disrupted a3-mutant Faim2 alleles. The genotyping PCR from tail DNA to identify a 202 bp a3-specific fragment used the primers P5 (5′-TGAAAACC ACACTGCTCGAA-3′) and P6 (5′-GGGGTCAAGGTACCAGCTCT-3′), and the seminested PCR to identify a 300 bp a1-specific fragment used the forward primer P1 (5′-GAGCCTCTACCC ACCTACCC-3′) and the reverse primers P2′ (5′-CTCCCAGGGACTCATTTGAA-3′) and P2″ (5′-CTGGGTGAGACCTCAGAAGC-3′). The specificity of the a3 amplicon was verified by sequencing. By interbreeding heterozygous offspring, homozygous a3 mutant genotypes as well as a1 wild-type littermates were created.
Faim2−/− analysis.
To provide evidence that disrupted a3 genotypes are Faim2 null mutants, reverse transcriptase (RT)-PCR, Northern blot, and Western blot analyses were performed on samples prepared from total brain homogenates of homozygous a3 and a1 genotypes (Fig. 1B–F). All sample preparations followed standard procedures.
Figure 1.
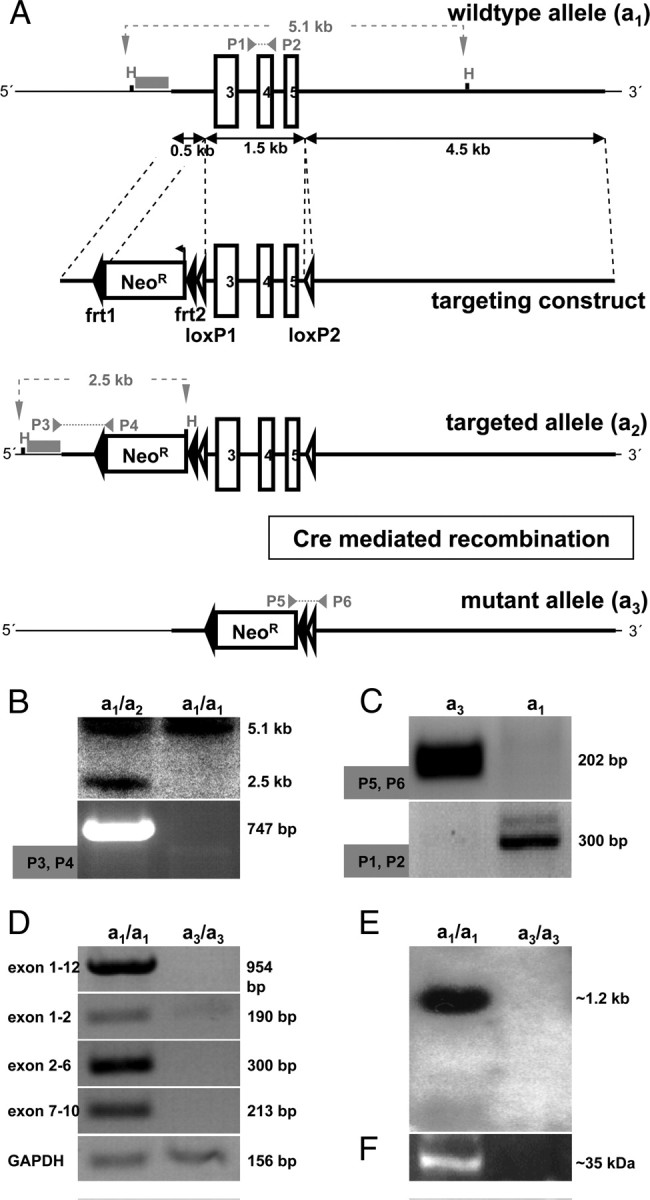
Generation of a Faim2−/− mouse line by targeted gene disruption via Cre-mediated recombination in vivo. A, Targeting strategy. First, Faim2 wild-type allele a1 displaying a 9 kb region of the minus strand of chromosome 15 containing the region of interest with exons 3–5 (white boxes), two HindIII restriction sites (H), an external 5′ 0.5 kb Southern hybridization probe (gray bar) with a predicted 5.1 kb a1-specific fragment (gray dashed line), and the annealing loci of PCR primers 1 and 2 (gray arrows) with a predicted a1-specific 300 bp amplicon (gray dashed line). Second, Targeting construct displaying a 5′ 0.5 kb arm of homology, an antisense-directed and directly repeated frt site (black arrowheads)-flanked neomycin-resistance gene (Neo box) including an exogenous HindIII restriction site (H), a 1.5 kb genomic region containing exons 3–5 flanked by directly repeated loxP sites (white arrowheads), and a 3′ 4.5 kb arm of homology. Third, Targeted Faim2 allele a2 after homologous recombination displaying the 5′ Southern probe with its predicted a2-specific 2.5 kb fragment (gray dashed line) and the annealing loci of PCR primers 3 and 4 (gray arrows), which amplify an a2-specific 746 bp product (gray dotted line). Fourth, Disrupted Faim2 allele a3 after Cre-mediated recombination in vivo displaying the remaining frt-flanked Neo-cassette and a single loxP site as well as the annealing loci of PCR primers 5 and 6 with their predicted a3-specific 202 bp amplicon (gray dotted line). B, Southern blot (top) (HindIII restriction enzyme digest, 5′ external probe) and PCR (bottom) (primers 3 and 4) on tail DNA verifying homologous recombination on the a2 allele. C, PCR [primers 1 and 2 (bottom) and 5 and 6 (top)] on tail DNA identifying a3-mutant and a1-wild-type allele, respectively. D, RT-PCR on brain homogenates verifying the absence of Faim2-specific transcripts in a3-null mutants; amplicons span from exon 1 to 12, 1 to 2, 2 to 6, and 7 to 10; GAPDH was used as loading control. E, F, Northern blot (E) and Western blot (F) prepared from brain homogenates documenting the absence of Faim2-specific mRNA as well as of the full-length protein in a homozygous a3-disrupted Faim2 genotype compared with homozygous a1-wild-type mice and thereby verifying an in vivo Faim2 null mutant.
Northern blot and RT-PCR analysis.
RNA was isolated by using RNeasy Mini kit (Qiagen). For cDNA synthesis, RNA was digested with RQ1 RNase Free DNase, protected against RNases by adding RNase Inhibitor Rnasin and reverse transcribed by M-MLV reverse transcriptase (all: Promega). The following primers were used to amplify various regions of the Faim2 cDNA: exons 1–12 (954 bp, complete cDNA): 5′-ATGACCCAGGGAAAGCTCTCTG-3′ and 5′-TCATTCCCGGTTGGTGCC-3′; exons 1–2 (190 bp): 5′-GGGAAAGCTCTCTGTGGCTA-3′ and 5′-CCAGCTTGGATGGAGTGG-3′; exons 2–6 (300 bp): 5′-GTGCCACTCCATCCAAGC-3′ and 5′-ACAGCAAGCCA GAGTCA GGT3′; exons 7–10 (231 bp): 5′-TCCCTTGGACCTGATTCTG-3′ and 5′-CGAGGAGGAGT CCACTGAAG-3. The primers for the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) loading control (156 bp) were as follows: 5′-TGGCAAAGTGGAGATTGTTGCC-3′ and 5′-AAGATG GTGATGGGCTTCCCG-3′. For Northern blot analysis, the amplicons of exons 1–2 (190 bp) and of exons 7–10 (231 bp) were used as templates for the 5′ and 3′ hybridization (not shown) probes, respectively. Radioactive labeling and further analysis followed standard procedures.
Western blot analysis.
Murine brain tissue was homogenized in 10 mm Tris, pH 7.4, 2% SDS, 150 mm NaCl, and 5 mm EDTA, including protease and kinase inhibitor Complete (Roche), by using a standard protocol (6500 rpm, 3 × 30 s) of Precellys 24 bead grinder homogenizer with ceramic grinding beads (Bertin Technologies). Protein lysates (60 μg of total protein per lane) were subjected to 12% SDS-PAGE and transferred to nitrocellulose membranes (Whatman). After 1 h of blocking at room temperature in TBS (5% milk powder), blots were incubated with a polyclonal rabbit antiserum raised against an N-terminal 15 aa epitope of murine Faim2 (APPSYEEATSGEGLK, amino acids 35–49) (Eurogentec; 1:500) at 4°C overnight and detected with HRP-coupled secondary antibodies.
Faim2 lentivirus
Constructs.
Full-length murine Faim2 cDNA (Faim2full length) [clone: MGC:40667 (IMAGE:5400222), clone sequence: BC032278.1, vector: pCMV-SPORT6, RZPD German Resource Center for Genome Research] and a truncated version encoding the first 104 aa (Faim2truncated) were subcloned into third-generation lentiviral vector (pRRLSIN.cPPT.PGK/GFP.WPRE, Tronolab) excluding the green fluorescent protein (GFP) gene cassette and introducing an N-terminal FLAG tag [common forward: 5′-GCGGATCCATGGATTACAAGGATGACGACGATAAG ACCCAGGGAAA GCTCTCTGTGG-3′, reverse (Faim2full length): 5′-CCGCTCGAGTCATTCCCGGTTGGTGC CAA-3′, reverse (Faim2truncated): 5′-CCGCTCGAGTCACTTTCTGATGA AGAGCC-3′]. The GFP-expressing virus served as control. The correct nature of all cloned sequences was confirmed by automated sequencing (Metabion).
Lentivirus generation and transgene expression.
Third-generation lentiviral particles were generated as described previously (Dull et al., 1998). After purification of the modified pRRLSIN.cPPT.PGK.WPRE vectors and cotransfection with packaging vectors (Invitrogen) into 293FT cells (Invitrogen) for 48 h, the supernatant was collected, concentrated by PEG-it Virus Precipitation Solution (System Biosciences), and resuspended in X-Vivo Medium (Bio Whittaker). The measurement of transgene expression in infected human embryonic kidney (HEK) 293 cells were performed by woodchuck hepatitis virus posttranscriptional regulatory element (WPRE)-based real-time quantitative reverse transcriptase-PCR using SYBR Green detection as described previously (Lizée et al., 2003). The relative copy numbers of lentivirus expressing full-length Faim2 protein (LV-Faim2full length), amino acids 1–105 truncated Faim2 protein (LV-Faim2truncated), and green fluorescent protein (LV-GFP) were 697, 1208, and 1973 WPRE molecules per 10,000 actin molecules/μl, respectively. Virus were used equimolarly in all applications, stored at −80°C, and kept on ice during stereotactic procedures.
Oxygen–glucose deprivation of primary mouse cortical neuronal cells
Primary mouse cortical neuronal (MCN) cells were derived from E16 embryos of Faim2+/+ and Faim2−/− mice and cultured for 10 d in vitro (DIV). Oxygen–glucose deprivation (OGD) was performed for 2.5 h with 24 h reoxygenation time as described previously (Harms et al., 2000). The release of lactate dehydrogenase (LDH) was determined as a marker for cell membrane disintegrity in the supernatant medium. Propidium iodide (PI) was added to naive cultures and pictures were taken as described recently (Harms et al., 2007). Cell counts of at least 2000 neurons per condition were performed in at least triplicate experiments. For Faim2 gene transfer, neurons were infected on day 3 DIV with two different amounts of lentiviral particles according to mRNA levels of WPRE (number of copies per 10,000 actin copies/μl): ∼700 (low) and ∼2100 (high). Efficiency of infection as measured by GFP fluorescence was >90% for all lentiviruses (data not shown).
Stereotaxic injections
Stereotaxic injections were performed as described recently (Krenz et al., 2009). Briefly, 3 weeks before middle cerebral artery occlusion (MCAo), mice were anesthetized with ketamine/xylazine solution (100/5 mg/kg BW), placed in flat skull position in a stereotactic frame (World Precision Instruments), and injected with 1 μl of equimolar (∼700 copies per 10,000 actin copies/μl) lentivirus into the ipsilateral striatum at the following two anterior, lateral, and ventral coordinates (in mm relative to bregma): 0.2, 2.0, 3.75 and 0.2, 2.0, 2.75. The injections were done along one needle tract using a Nanoliter 2000 microinjector (Word Precision Instruments) at a rate of 250 nl/min. Efficiency of infection was monitored histologically by anti-GFP and anti-Faim2 immunostaining on three animals per virus.
Cerebral ischemia
Model and measurement of physiological parameters.
Mice were housed and handled according to institutional and national guidelines. Male Faim2−/− and littermate Faim2+/+ mice were anesthetized with 1.0 volume percent isoflurane in 69% N2O and 30% O2 and subjected to filamentous MCAo for 30 min followed by reperfusion (Katchanov et al., 2001). Regional cerebral blood flow (CBF) was measured using laser Doppler flowmetry (Perimed) and fell to <20% during ischemia and returned to ∼80–100% within 5 min after reperfusion in all groups (p < 0.05). Core temperature was maintained at 36.5 + 0.5°C. In some animals, the left femoral artery was cannulated. Mean arterial blood pressure was recorded and arterial blood samples were analyzed for blood gases as described previously (Endres et al., 2003).
Lesion determination.
Brains were cut into 20 μm cryostat sections. After standard hematoxylin staining, direct cerebral lesion volumes were determined via computer-assisted infarct volumetry (Endres et al., 2003).
Faim2 and Fas/CD95 mRNA expression in the time course following MCAo.
Total RNA was isolated out of infarcted (ipsilateral), noninfarcted (contralateral), and sham-operated hemispheres of five animals per time point (0, 3, 18, and 48 h) after 30 min of MCAo, respectively, and reverse transcribed (2.5 μg of total RNA) with 250 units of M-MLV reverse transcriptase as described above. Two microliters of a 1:5 diluted cDNA sample was amplified by real-time PCR by using SYBR Green (Thermo Scientific, Epsom) and PCR primers for Faim2 (5′-AGAAGACATCATGACCCAGGG-3′ and 5′-CTTTCTGGTCATCC CAGCTG-3′), Fas/CD95 (5′-CTGCGATGAAGAGCATGGTTT-3′ and 5′-CCATAGGCGA TTTCTGGGA C-3′), and murine glyceraldehyde 3-phosphate dehydrogenase (5′-TGGCAAA GTGGAGATTGTTGCC-3′ and 5′-AAGATGGTGATGGGCTTCCCG-3′) as reference gene. Reactions were performed in a Mx3000P sequence detection system (Stratagene). Quantitative real-time PCR analysis was performed by using the 2−ΔΔCt method.
Caspase-3 and caspase-8 activity ex vivo after MCAo.
Faim2+/+ and Faim2−/− mice were subjected to 30 min of MCAo followed by reperfusion. At 20 h, animals were perfused with ice-cold physiological saline in deep anesthesia. Coronal brain slices of 2 mm thickness were obtained using a brain matrix. Ipsilateral and contralateral hemispheres were separated and incubated with ice-cold lysis buffer (100 μl per mg tissue) and homogenized briefly using an Ultraturax for 3 s. Tissue debris was spin down at maximum speed for 30 min at 4° after 20 min lysis on ice. Caspase activities were determined according to the manufacturer's instructions (ApoAlert Caspase Assay Kits, Clontech Laboratories) with the corresponding inhibitor to prove specificity.
Histochemistry and immunohistochemistry.
After reperfusion, nucleoside analog 5-bromo-2-deoxyuridine (BrdU) was administered to the mice twice daily by intraperitoneal injections at a dose of 50 μg/g of body weight at a concentration of 10 mg/ml. Animals were transcardially perfused with physiological saline followed by 4% paraformaldehyde. Brains were cut in the coronal plane in 40 μm thick sections. Sections were stained using free-floating immunohistochemistry as described in detail previously (Kronenberg et al., 2003). Some brains were embedded in paraffin, sectioned (4 μm), and stained according to standard procedures for activated caspase-8, activated caspase-3, Gallyas, NeuN, GFAP, Mac-3, Iba1, APP, GFP, and Faim2.
Primary antibodies were applied in the following concentrations: anti-BrdU (rat, 1:500, Harlan Seralab), anti-NeuN (mouse, 1:100, Millipore Bioscience Research Reagents), anti-Caspase 3 (rabbit, 1:200, Abcam), anti-Caspase 8 (rat, 1:200, ALEXIS), anti-GFAP (rabbit, 1:300, DakoCytomation), anti-APP (mouse, 1:2000, Millipore Bioscience Research Reagents), anti-GFP (rabbit, 1:500, Santa Cruz Biotechnology), anti-Faim2 (rabbit, 1:1000, Eurogentec), and anti-Iba1 (rabbit, 1:300, Wako). Immunohistochemistry followed the peroxidase method with biotinylated secondary antibodies (all: 1:500, Jackson ImmunoResearch Laboratories), ABC Elite reagent (Vector Laboratories), and diaminobenzidine (Sigma) as chromogen. For immunofluorescence, FITC-, RhodX-, or Cy5-conjugated secondary antibodies were all used at a concentration of 1:250. TUNEL staining was performed with the In Situ Cell Death Detection Kit (Roche Applied Science) according to the manufacturer's protocol.
Quantification and imaging.
The number of BrdU-positive cells per volume was assessed using StereoInvestigator (MicroBrightfield). In defined reference sections (i.e., interaural +4.9, +4.1, and +3.3 mm), the ischemic lesion and corresponding area in the contralateral hemisphere was delineated at ×100 magnification, and cells were counted at ×200 magnification. Phenotypic analyses of NeuN/TUNEL- and NeuN/caspase-positive cells were performed using a spectral confocal microscope (TCS SP2, Leica).
Simple neuroassessment of asymmetric impairment.
Simple neuroassessment of asymmetric impairment (SNAP) was performed according to Shelton et al. (2008) before and 2 h after MCAo. The following eight categories were scored (0–5): interactions, cage grasp, visual placing, pacing or circling, gait or posture, head tilt, visual field, and baton.
Results
Generation of Faim2−/− mice
Exons 3–5 (amino acids 72–146) of the Faim2 gene locus were chosen as the target region of the mutant strategy, because they cluster conveniently and code for the first 1.5 predicted transmembrane domains (Fig. 1A). The vector pConKO-True was used to produce a targeting allele, in which the region of interest remained sensitive to Cre recombinase-mediated deletion. Genetic targeting was performed by the standard blastocyst injection method. ES cells were identified via seminested PCR, the genotype of the founder mice by PCR and Southern blotting (Fig. 1B). Successful germline transmission was followed by crossing heterozygous offspring to the transgenic deleter EIIa cre mouse line, which is known to result in ubiquitous deletion of the loxP-flanked gene sequences and efficient and stable germline transmission of the mutation to subsequent generation (Lakso et al., 1996). After reaching homozygosity, the analyses confirmed the complete inactivation of Faim2 on DNA, RNA, cDNA, and protein levels, respectively (Fig. 1C–F). When interbreeding, litter size was regular and Mendelian genetic distribution showed equal numbers. In comparison to wild-type and heterozygous littermates, null mutants developed normally without overt phenotype, especially without neurological signs or symptoms, with regular weight gain and normal life spans. Furthermore, behavioral test batteries (open field, accelerod, tight rope test) as well as macroscopical and histological (NeuN, GFAP, Gallyas, Mac-3, APP) analyses did not reveal significant differences (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Increased susceptibility of Faim2−/− primary MCN to OGD rescued by lentiviral Faim2 gene transfer
To evaluate the influence of Faim2 on neuronal survival under ischemic conditions, MCN (DIV 10) were subjected to 2.5 h of OGD with 24 h reoxygenation time. Cell injury was assessed by LDH release assay and neuronal viability by PI exclusion counterstaining (Fig. 2A). Ischemic tolerance of Faim2−/− cortical neurons was significantly lower than under the wild-type condition. As proof of principle, Faim2−/− (Fig. 2B) and wild-type (Fig. 2C,D) MCN were infected with different amounts (low and high, see Materials and Methods) of murine Faim2-expressing lentivirus under the control of the phosphoglycerate kinase (PGK) promoter prior (DIV 3) to OGD. This resulted in a dose-dependent decrease in LDH release, so that the Faim2−/− OGD phenotype was similar to the wild-type level. Although not as effective, infection with truncated Faim2 lentivirus, which expresses the first 104 N-terminal exclusively cytosolic amino acids of Faim2, also yielded a dose-dependent reduction of OGD-induced cell death. In the wild-type condition, infection with high amounts of either Faim2 lentivirus led to significant reduction in polynucleated PI-positive nuclei in MCN after OGD (Fig. 2C,D). The reduction in LDH release yielded comparable results [units/ml of medium, mean ± SEM, LV-GFP: 206.9 ± 6.646 (n = 8), LV-Faim2full length: 121.5 ± 7.833 (n = 8), LV-Faim2truncated: 152.3 ± 7.278, one-way ANOVA with Tukey's multiple-comparison test, LV-GFP vs LV-Faim2full length and Faim2truncated p < 0.001].
Figure 2.
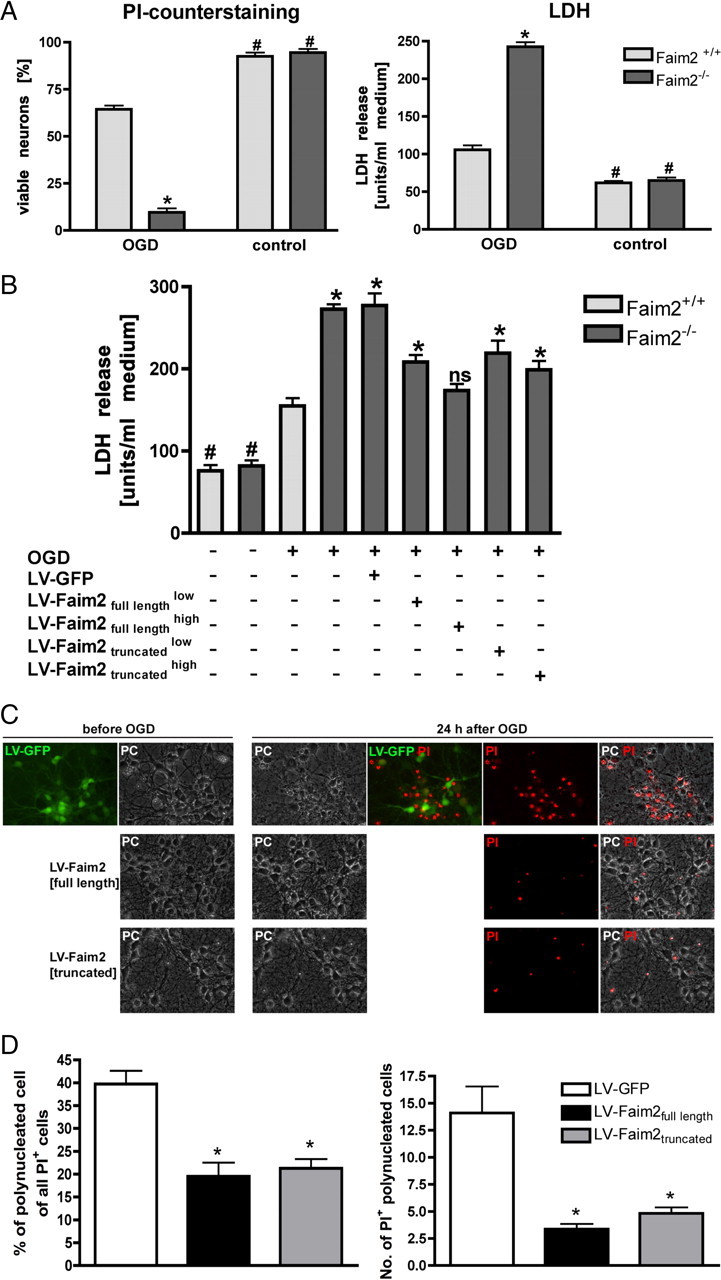
OGD and lentiviral Faim2 gene transfer of primary MCN derived from Faim2−/− and littermate Faim2+/+ mice. A, Viable neurons are shown as percentage of all cells (PI-negative cells + PI-positive cells except old dead cells with no visible cell membrane). PI was applied directly before pictures were taken in phase contrast and fluorescence. Approximately 500 neurons were analyzed per condition from at least 8 microscopic fields and repeated in triplicate independent experiments for quantification. Two-way ANOVA followed by Tukey post hoc analysis (Prism Software, SigmaStat): genotype, F(1,28) p < 0.001; side, p < 0.001; and genotype × OGD interaction, p < 0.001. *p < 0.001 versus OGD Faim2+/+. #p < 0.001 versus corresponding OGD in Faim2−/− and Faim2+/+ neuronal cultures. LDH was determined 24 h after OGD. Data are shown as mean ± SEM pooled from 4 independent experiments. Two-way ANOVA followed by Tukey post hoc analysis: genotype, F(1,47) p < 0.001; side, p < 0.001; and genotype × OGD interaction, p < 0.001. *p < 0.001 versus OGD Faim2+/+. #p < 0.001 versus corresponding OGD in Faim2−/−and Faim2+/+ neuronal cultures. B, Neurons were transduced with lentiviral particles on day 3 DIV with a low or high titer according to mRNA levels of WPRE as described. LDH was taken 24 h after 2.5 h OGD. Data are shown as mean ± SEM; n = 6–8 analyzed in triplicate experiments. One-way ANOVA followed by Tukey post hoc test (Prism Software, SigmaStat). *p < 0.05 versus OGD Faim2+/+. n.s., Not significant versus OGD Faim2+/+. #p < 0.01 versus corresponding OGD in Faim2−/− and Faim2+/+ neuronal cultures. C, D, Wild-type neurons were transduced with lentiviral particles as described using the high-titer preparations in the case of LV-Faim2full length and LV-Faim2truncated. Pictures in fluorescence and phase-contrast modes were taken before and after OGD using a Merzheuser stage and an inverted Olympus IX51 microscope with Cell M-based automated picture acquisition with defined stage positions. PI staining was followed by picture acquisition of the exact positions taken before OGD. Absolute number and percentage of PI-stained polynucleated cells were quantified. Data are shown as mean ± SEM. In each condition, 11 randomly selected visual fields were analyzed. One-way ANOVA followed by Tukey post hoc test (Prism Software, SigmaStat) was used. *p < 0.001 versus LV-GFP.
Faim2 and Fas/CD95 mRNA expression in the time course following transient focal ischemia
Quantitative Faim2 and Fas/CD95 mRNA (Fig. 3) real-time PCR expression analyses of ischemic brain tissue at 3, 18, and 48 h of reperfusion after transient focal ischemia revealed differential inverse regulation: whereas Fas/CD95 displayed ∼50% upregulation at 3 and 18 h after MCAo, Faim2 was significantly downregulated only at 18 h. At 48 h of reperfusion, the expression of both molecules reached baseline again.
Figure 3.

Time course of Faim2 and Fas/CD95 mRNA expression after focal brain ischemia. RNA was extracted at 0, 3, 18, and 48 h after 30 min MCAo out of the core region of infarct. Five animals per time point were analyzed with 1–3 repeated measurements. Data are represented as means ± SEM. One-way ANOVA (Prism Software, SigmaStat). *p < 0.001 versus control.
Increased infarct volume, neuronal cell death, and neurological defects in Faim2−/− mice after transient focal brain ischemia and its attenuation by lentiviral Faim2 gene transfer
To test whether lack of Faim2 influences disease pathology where Fas-dependent cell death plays a role, a model of mild transient brain ischemia was applied. After 30 min of MCAo followed by 72 h of reperfusion, we found significantly enlarged lesion volumes (mm3) in Faim2−/− mice compared with littermate Faim2+/+ controls [mean ± SEM, Faim2+/+: 20.81 ± 2.970 (n = 8), Faim2−/−: 42.21 ± 3.990 (n = 11), unpaired t test: p = 0.0009] (Fig. 4A). The difference in stroke volume was accompanied by significant increase of total numbers of TUNEL-positive cells as well as their density within the ipsilateral hemisphere and the ischemic lesion [TUNEL-positive cells/hemisphere (mean ± SEM), Faim2+/+: 10,030 ± 815.1 (n = 7), Faim2−/−: 17,550 ± 890.3 (n = 11), unpaired t test: p < 0.0001; TUNEL-positive cells/mm3 infarct × 104 (mean ± SEM), Faim2+/+: 0.3364 ± 0.02872 (n = 7), Faim2−/−: 0.4252 ± 0.01290 (n = 11), unpaired t test: p = 0.0057]. In both groups, a considerable amount of dying cells double stained for TUNEL-positive nuclei and the neuronal marker NeuN (Fig. 4B). Striatal injection of lentiviral Faim2 3 weeks before MCAo showed stable neuronal expression in most parts of the anatomic core region of the infarct (caudate–putamen) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This resulted in significant reduction of lesion volume in both genotypes after MCAo (Fig. 4C). Virus-mediated overexpression of Faim2 also reduced neurological deficits in Faim2−/− mice as measured by SNAP 2 h after MCAo (Fig. 4C), demonstrating the clinical significance of Faim2 neuroprotection.
Figure 4.
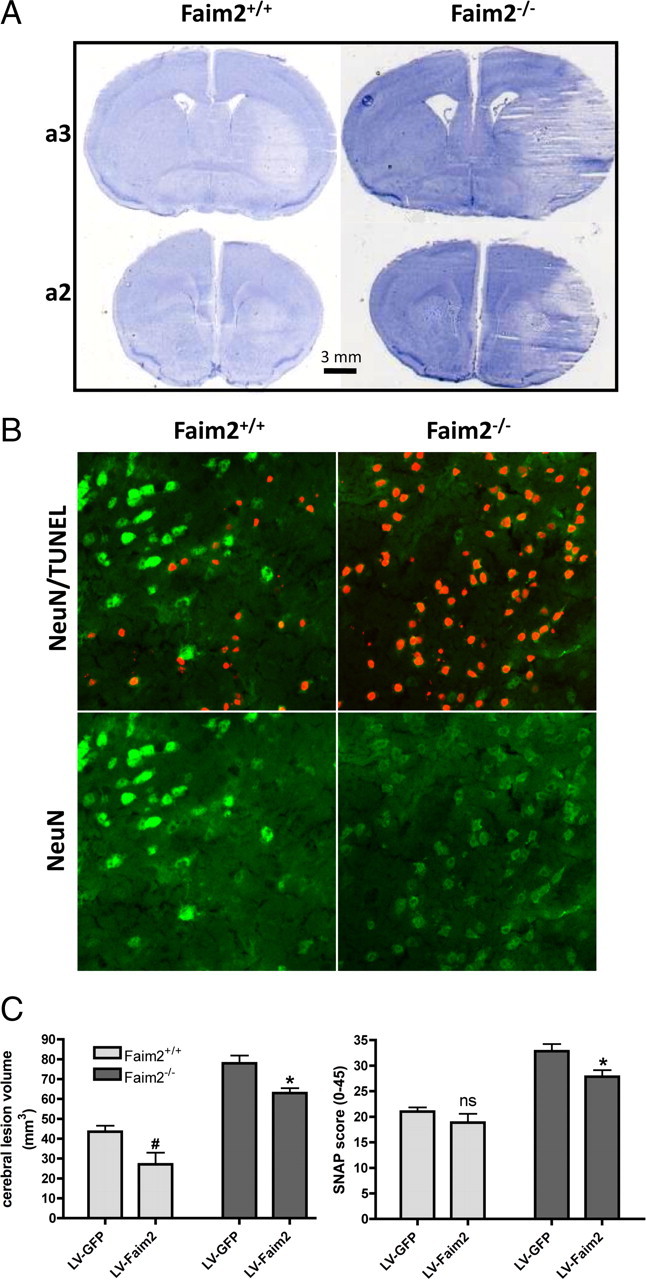
Effects of transient brain ischemia in Faim2−/− and littermate Faim2+/+ mice. Analyses were performed after 30 min of MCAo and 72 h of reperfusion. n = 8–11 male mice per group, age-matched. Data are expressed as mean ± SEM. Unpaired t test (Prism Software, SigmaStat). A, Examples of proximate (a2, a3) hematoxylin-stained 20 μm coronal brain sections taken from anterior–posterior serial coronal cryostat sections (20 μm). B, Examples of NeuN and NeuN/TUNEL-immunofluorescence staining in infarcted area. C, Stereotaxic injections of LV-Faim2 into the striatum 3 weeks before MCAo reduced lesion size and clinical deficits. SNAP score was evaluated before and 2 h after MCAo. Two-way ANOVA followed by unpaired t test (Prism Software, SigmaStat) was used. Data are shown as mean ± SEM. N = 5–8 animals per group. Lesion volume, Genotype p < 0.0001, virus treatment p = 0.0013, genotype × virus treatment interaction p = 0.8686. #p = 0.0218 versus Faim2+/+ littermates with lentiviral GFP infection, *p = 0.0126 versus Faim2−/− mice with lentiviral GFP infection. SNAP, Genotype p < 0.0001, virus treatment p = 0.0135, genotype × virus treatment interaction p = 0.2955. n.s. (p = 0.54) versus Faim2+/+ littermates with lentiviral GFP infection, *p = 0.0258 versus Faim2−/− mice with lentiviral GFP infection.
We measured physiological parameters in randomly selected animals during MCAo (Table 1). No differences in mean arterial blood pressure and arterial blood gas analysis (pH, partial pressure of carbon dioxide [PaCO2] and oxygen [PaO2]) were found in either genotype. Therefore, significant genotype-dependent influence of physiologic parameters on stroke outcome was unlikely.
Table 1.
Physiological parameters of Faim2−/− and Faim2+/+ mice before (basal) and during MCAo
| Parameter | Faim2+/+ | Faim2−/− |
|---|---|---|
| MABP basal (mmHg) | 90.8 ± 1.9 | 93.4 ± 4 (n.s.) |
| MABP MCAo (mmHg) | 95.3 ± 3.7 | 92.1 ± 4.3 (n.s.) |
| pH MCAo | 7.3 ± 0.01 | 7.31 ± 0.02 (n.s.) |
| PaCO2 MCAo (mmHg) | 41.4 ± 2.9 | 41.7 ± 2.2 (n.s.) |
| PaO2 MCAo (mmHg) | 97.9 ± 7.4 | 94.4 ± 5 (n.s.) |
MABP, Mean arterial blood pressure; PaCO2: arterial carbon dioxide partial pressure; PaO2, arterial oxygen partial pressure. No statistical differences were found between groups (n = four mice per group) (unpaired t test, Prism Software, GraphPad).
Together, loss of Faim2 resulted in enhanced vulnerability to cerebral ischemia in vivo with increased neuronal cell death, ischemic lesion volume, and neurological impairment. Preceding stereotaxic lentiviral Faim2 gene transfer attenuated lesion volume independently from genotype, whereas it significantly improved functional outcome only in the mutant.
Sustained and increased caspase-8 and caspase-3 activity in Faim−/− mice after transient focal brain ischemia
Fas-mediated apoptosis essentially depends on caspase activation. Whereas caspase-8 activation is an early initiating event, caspase-3 activation represents the end of the signal amplification and the beginning of the irreversible execution phase. To relate caspase activity to neuronal ischemic cell death, immunofluorescence microscopy and ex vivo caspase activity assays were performed at different reperfusion time points after 30 min of MCAo. At 20 h of reperfusion (Fig. 5A) a significant difference in ex vivo caspase-8 and caspase-3 activity in favor of the mutant was detectable. At 16 h of reperfusion morphological evidence for the presence of activated caspase-8 and caspase-3 in NeuN-positive cells was present in both genotypes (Fig. 5B,C). Whereas signal intensities were already fading in core regions of the infarcts (striatum), penumbra regions (cortex) displayed ongoing activities. Although genotype-dependent quantitative difference of double-stained cells could not be measured, qualitative difference with more irregular-shaped, pyknotic caspase-positive, and NeuN-positive cell bodies was apparent in the mutant.
Figure 5.
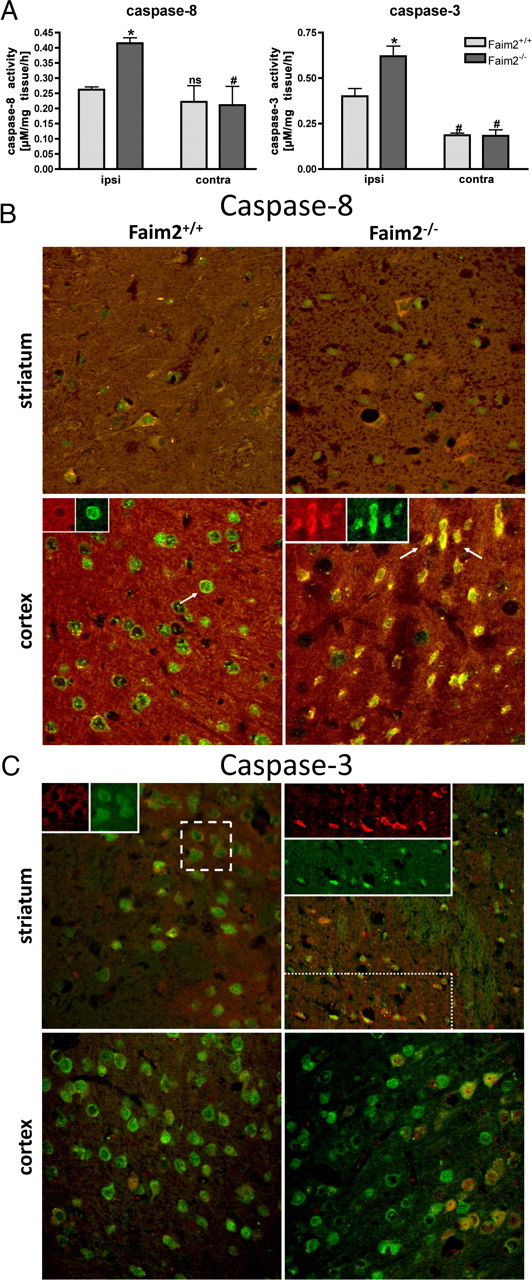
Caspase-8 and caspase-3 activity in Faim2−/− and littermate Faim2+/+ mice after focal brain ischemia. A, Caspase activities ex vivo in Faim2+/+ and Faim2−/− mice 20 h after 30 min MCAO. Data are shown as means ± SEM pooled from n = 3 animals per condition. Caspase-8, Two-way ANOVA followed by Tukey post hoc analysis (Prism Software, SigmaStat): genotype, F(1,11) p = 0.024; side, p < 0.001; genotype × side interaction, p = 0.021 (caspase-8). *p = 0.004 versus ipsilateral Faim2+/+. #p = 0.01 versus corresponding ipsilateral in Faim2−/− and Faim2+/+ mice. Caspase-3, Two-way ANOVA followed by Tukey post hoc analysis (Prism Software, SigmaStat): genotype, F(1,11) p = 0.129; side, F(1,11) p = 0.020; genotype × side interaction, p = 0.087 (caspase-3). *p = 0.033 versus ipsilateral Faim2+/+. #p = 0.009 versus ipsilateral Faim2−/−. n.s., Not significant versus ipsilateral Faim2+/+. B, Confocal immunofluorescence 16 h after 30 min MCAo. Costaining of NeuN (green) and cleaved caspase-8/subunit 18 (red) (picture size: 200 × 200 μm). C, Confocal immunofluorescence 16 h after 30 min MCAo. Costaining of NeuN (green) and cleaved caspase-3 (red) (picture size: 275 × 275 μm).
Discussion
The presented data give the first in vivo evidence that extracellular receptor-mediated cell death signaling during and after the acute phase of transient focal brain ischemia is significantly influenced by expression of Faim2, an intrinsic membrane protein that has a specificity for differentiated neurons and is dispensable for neuronal development. Given the frequency of cerebrovascular events in humans and the strong evolutionary conservation of Faim2, the therapeutic potential of this novel endogenous neuroprotectant is intriguing.
In mice, the model of transient MCAo reliably reproduces delayed neuronal cell death within the striatum (Katchanov et al., 2001), mimicking embolic focal brain ischemia in humans. Depending on the duration of MCAo and the extent of reperfusion, neurons in the central core of infarction die by energy failure, whereas the fate of the hypoperfused tissue around the ischemic core is additionally influenced by other deleterious factors produced by neighboring cells, such as glutamate excitotoxicity, reactive oxygen species, inflammatory cytokines, adhesion molecules, metalloproteinases, and endothelins (Ferrer and Planas, 2003). Although the nature of ischemic neuronal death remains complex (Fujikawa, 2000), it is commonly accepted that necrosis prevails in the infarct core and apoptosis in the penumbra (Charriaut-Marlangue et al., 1996). Therefore, application of mild and transient MCAo (30 min) is suitable to examine both processes, early core injury and delayed penumbral pathophysiology.
The relevance of Fas/CD95 apoptotic signaling for the outcome of ischemic stroke was shown in several mouse models: direct signal disruption, either genetically (Martin-Villalba et al., 1999; Rosenbaum et al., 2000; Graham et al., 2004) or via antibody-mediated neutralization of FasL (Martin-Villalba et al., 2001), resulted in significant reduction of stroke-related damage in models of cerebral ischemia-hypoxia. The current results under Faim2-deficient conditions display the other side of the coin: genetic disinhibition of Fas/CD95 signaling increases cell death, lesion volume, and clinical defects after transient focal brain ischemia. The reversibility by lentiviral gene transfer proved Faim2 specificity of these effects. In vitro with close to 90% efficacy of infection Faim2 gene transfer restored physiological levels of cell death in mutant mice. Under the wild-type condition, an additional neuroprotective effect was observed. In vivo, only the main part of the ischemic core—the caudatoputamen, but not cortical areas—was infected, resulting in partial rescue of both mutant and wild-type Faim2 mice. Together, these results support a strong and specific apoptotic inhibitory effect of Faim2 in hypoxia–ischemia with therapeutic potential.
As demonstrated by detection of neuronal TUNEL-positive nuclei as well as of neuronal activated caspase-3, not only the spatial expansion, but also the density of ischemic cell death is significantly increased in Faim2 mutant mice. This supports the idea of a quantitative and qualitative shift in the transition between dying, endangered, and rescued neuronal cells in disfavor of penumbral tissue and in favor of ischemic cell death due to the lack of Faim2.
The ex vivo measurements of caspase-3 and caspase-8 activity 20 h after MCAo suggests a similar difference: at this time of reperfusion after focal brain ischemia, the known activity peak of the early initiator caspase-8 is exceeded, whereas the one of the late effector caspase-3 impends (Velier et al., 1999). Accordingly, under the wild-type condition, an increase in caspase-3 activity was registered, whereas caspase-8 displayed baseline activity. Lack of Faim2 not only led to much stronger caspase-3 activation, but also to sustained caspase-8 activation, again arguing for an increased recruitment of penumbral into cell death prone tissue. Furthermore, confocal imaging at an earlier time point of reperfusion suggested genotype-dependent qualitative difference in caspase-8 activity on cellular level.
Although not explored systematically, the lentiviral in vitro experiments identified the 104 aa N-terminal cytoplasmic tail of Faim2 as the functional domain-containing part, since it displayed neuroprotective function in a dose-dependent manner nearly as effectively as the full-length protein. This is in line with the detailed sequence and phylogenetic analysis of the LFG protein family, which comprises apoptosis-modulating proteins defined by a seven transmembrane scaffold and conserved sequence motifs within its cytoplasmic segments (Hu et al., 2009). Whether these cytoplasmic regions represent some kind of targeting or signal is not known.
With high neuron-specific rates at basal level and during the acute phase (3 h), the expression profile of Faim2 fulfills criteria of a brain protectant. On the other hand, downregulation of Faim2 in the postacute phase (18 h) could enable induction of neuroregeneration, for example, by allowing for alternative Fas/CD95 signaling. This interpretation is supported by the postischemic expression profile of Fas: upregulation during initiation of cell death (3 h) and persistent expression at times when regenerative processes commence (18 h). Furthermore, Faim2-inducing PI 3-kinase–Akt/PKB signaling cascade (Beier et al., 2005) can be activated by a number of growth factors and neurotrophins, among them insulin-like growth factor (IGF-1), nerve growth factor (NGF), and erythropoietin (Sirén et al., 2001; Ruscher et al., 2002; Zheng et al., 2002; Kilic et al., 2005). All of them potentially mediated neuroprotection after ischemic insults.
Whether Faim2 regulates the switch from apoptotic to regenerative Fas/CD95 signaling in neurons prone to ischemic cell death remains speculative and has not been the focus of this study, but it fulfills a number of essential preconditions: (1) nearly exclusive expression by the cellular target (differentiated neurons), (2) strategic subcellular location to modulate receptor-mediated signals (membranous lipid rafts), (3) strong Fas/CD95 antiapoptotic properties in in vitro and in vivo ischemic–hypoxic cell death paradigms, (4) high constitutive expression level (neuroprotection in the acute phase of damage), and (5) downregulation of expression in the postacute phase of damage after transient ischemia (possibly initiation of Fas/CD95-induced neuroregeneration). The fact that Faim2 represents an endogenous neuroprotectant possibly inducible by known drugs, such as erythropoietin, might open a new therapeutic window in the treatment of ischemic stroke.
In the end, it has to be emphasized that the eponymous interaction between Faim2 and Fas does not rule out the possibility that Faim2 acts at least partially via yet unidentified modulation of other extracellular receptor-mediated signaling pathways. Although evidence of direct Faim2–Fas/CD95 interaction in vitro and of influence on apoptotic Fas/CD95 downstream signaling molecules by Faim2 in vitro and in vivo exists (Somia et al., 1999; Beier et al., 2005; Fernández et al., 2007) (Fig. 5), to date there is no proof for a direct association of these two molecules in cerebral ischemia in vivo. This issue needs to be addressed in the future to completely understand the neuroprotective potential of Faim2 in the context of neurological diseases.
Footnotes
The work done in Göttingen and Aachen was partially funded by the German Research Council (DFG) through the DFG-Research Center for Molecular Physiology of the Brain and the RWTH Aachen University, Faculty of Medicine (AG START, 690945/145/09). The work done in Berlin was funded by BMBF (Center for Stroke Research), DFG (SFB-TR), Volkswagen Foundation (Lichtenberg Program to M.E.), and European Union (ARISE and EUStroke Consortia). We thank Ulrike Bode (Department of Neurogenetics, Max Planck Institute for Experimental Medicine, Göttingen, Germany) for technical support on procedures of mouse genetics.
References
- Beier CP, Wischhusen J, Gleichmann M, Gerhardt E, Pekanovic A, Krueger A, Taylor V, Suter U, Krammer PH, Endres M, Weller M, Schulz JB. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci. 2005;25:6765–6774. doi: 10.1523/JNEUROSCI.1700-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccatelli S, Tamm C, Sleeper E, Orrenius S. Neural stem cells and cell death. Toxicol Lett. 2004;149:59–66. doi: 10.1016/j.toxlet.2003.12.060. [DOI] [PubMed] [Google Scholar]
- Charriaut-Marlangue C, Margaill I, Represa A, Popovici T, Plotkine M, Ben-Ari Y. Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J Cereb Blood Flow Metab. 1996;16:186–194. doi: 10.1097/00004647-199603000-00002. [DOI] [PubMed] [Google Scholar]
- Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev. 2004;44:65–81. doi: 10.1016/j.brainresrev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Desbarats J, Birge RB, Mimouni-Rongy M, Weinstein DE, Palerme JS, Newell MK. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol. 2003;5:118–125. doi: 10.1038/ncb916. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schröck H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U. Mechanisms of stroke protection by physical activity. Ann Neurol. 2003;54:582–590. doi: 10.1002/ana.10722. [DOI] [PubMed] [Google Scholar]
- Fernández M, Segura MF, Solé C, Colino A, Comella JX, Ceña V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem. 2007;103:190–203. doi: 10.1111/j.1471-4159.2007.04767.x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Confusion between neuronal apoptosis and activation of programmed cell death mechanisms in acute necrotic insults. Trends Neurosci. 2000;23:410–411. doi: 10.1016/s0166-2236(00)01601-5. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O'Riordan DP, Northington FJ. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Freyer D, Weih M, Dirnagl U, Weber JR, Hörtnagl H. Melatonin is protective in necrotic but not in caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures. FASEB J. 2000;14:1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hübner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hörtnagl H. Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci. 2007;27:4562–4571. doi: 10.1523/JNEUROSCI.5110-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusler P, Papoff G, Eramo A, Reif K, Cantrell DA, Ruberti G. Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur J Immunol. 1998;28:57–69. doi: 10.1002/(SICI)1521-4141(199801)28:01<57::AID-IMMU57>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, Simon RP. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22:8458–8465. doi: 10.1523/JNEUROSCI.22-19-08458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Smith TF, Goldberger G. LFG: a candidate apoptosis regulatory gene family. Apoptosis. 2009;14:1255–1265. doi: 10.1007/s10495-009-0402-2. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, Thiemann M, Mueller W, Sykora J, Kuhn A, Schreglmann N, Letellier E, Zuliani C, Klussmann S, Teodorczyk M, Gröne HJ, Ganten TM, Sültmann H, Tüttenberg J, von Deimling A, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 2008;13:235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Krenz A, Falkenburger BH, Gerhardt E, Drinkut A, Schulz JB. Aggregate formation and toxicity by wild-type and R621C synphilin-1 in the nigrostriatal system of mice using adenoviral vectors. J Neurochem. 2009;108:139–146. doi: 10.1111/j.1471-4159.2008.05755.x. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G. Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol. 2003;467:455–463. doi: 10.1002/cne.10945. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C, Landau AM, Desbarats J. Fas-beyond death: a regenerative role for Fas in the nervous system. Apoptosis. 2003;8:551–562. doi: 10.1023/A:1026113222478. [DOI] [PubMed] [Google Scholar]
- Lizée G, Aerts JL, Gonzales MI, Chinnasamy N, Morgan RA, Topalian SL. Real-time quantitative reverse transcriptase-polymerase chain reaction as a method for determining lentiviral vector titers and measuring transgene expression. Hum Gene Ther. 2003;14:497–507. doi: 10.1089/104303403764539387. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin KM. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Noshita N, Lewén A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Peter ME, Legembre P, Barnhart BC. Does CD95 have tumor promoting activities? Biochim Biophys Acta. 2005;1755:25–36. doi: 10.1016/j.bbcan.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Reich A, Spering C, Schulz JB. Death receptor Fas (CD95) signaling in the central nervous system: tuning neuroplasticity? Trends Neurosci. 2008;31:478–486. doi: 10.1016/j.tins.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Reimers K, Choi CY, Mau-Thek E, Vogt PM. Sequence analysis shows that Lifeguard belongs to a new evolutionarily conserved cytoprotective family. Int J Mol Med. 2006;18:729–734. [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pedini F, Mollinari C, Condorelli G, Bonci D, Bez A, Colombo A, Parati E, Peschle C, De Maria R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J Exp Med. 2004;200:1257–1266. doi: 10.1084/jem.20040921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Gupta G, D'Amore J, Singh M, Weidenheim K, Zhang H, Kessler JA. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J Neurosci Res. 2000;61:686–692. doi: 10.1002/1097-4547(20000915)61:6<686::AID-JNR12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Ruan W, Lee CT, Desbarats J. A novel juxtamembrane domain in tumor necrosis factor receptor superfamily molecules activates Rac1 and controls neurite growth. Mol Biol Cell. 2008;19:3192–3202. doi: 10.1091/mbc.E08-02-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci. 2002;22:10291–10301. doi: 10.1523/JNEUROSCI.22-23-10291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer B, Taylor V, Welcher AA, McClelland M, Suter U. Neural membrane protein 35 (NMP35): a novel member of a gene family which is highly expressed in the adult nervous system. Mol Cell Neurosci. 1998;11:260–273. doi: 10.1006/mcne.1998.0697. [DOI] [PubMed] [Google Scholar]
- Schweitzer B, Suter U, Taylor V. Neural membrane protein 35/Lifeguard is localized at postsynaptic sites and in dendrites. Brain Res Mol Brain Res. 2002;107:47–56. doi: 10.1016/s0169-328x(02)00445-x. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Pettigrew DB, Hermann AD, Zhou W, Sullivan PM, Crutcher KA, Strauss KI. A simple, efficient tool for assessment of mice after unilateral cortex injury. J Neurosci Methods. 2008;168:431–442. doi: 10.1016/j.jneumeth.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirén AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–4049. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somia NV, Schmitt MJ, Vetter DE, Van Antwerp D, Heinemann SF, Verma IM. LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci U S A. 1999;96:12667–12672. doi: 10.1073/pnas.96.22.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ. Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagey R, Lurot S, Perrelet D, Pelech SL, Sagot Y, Krieger C. Phosphatidylinositol 3-kinase activity in murine motoneuron disease: the progressive motor neuropathy mouse. Neuroscience. 2001;103:257–266. doi: 10.1016/s0306-4522(00)00551-0. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14:53–66. doi: 10.1016/s1359-6101(02)00072-2. [DOI] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Quirion R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J Neurochem. 2002;80:1049–1061. doi: 10.1046/j.0022-3042.2002.00783.x. [DOI] [PubMed] [Google Scholar]
- Zuliani C, Kleber S, Klussmann S, Wenger T, Kenzelmann M, Schreglmann N, Martinez A, del Rio JA, Soriano E, Vodrazka P, Kuner R, Groene HJ, Herr I, Krammer PH, Martin-Villalba A. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell Death Differ. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720. [DOI] [PubMed] [Google Scholar]