

Abstract
We developed a new protocol that induces long-term adaptation of horizontal optokinetic response (HOKR) eye movement by hours of spaced training and examined the role of protein synthesis in the cerebellar cortex in the formation of memory of adaptation. Mice were trained to view 800 cycles of screen oscillation either by 1 h of massed training or by 2.5 h to 8 d of training with 0.5 h to 1 d space intervals. The HOKR gains increased similarly by 20–30% at the end of training; however, the gains increased by 1 h of massed training recovered within 24 h, whereas the gains increased by spaced training were sustained over 24 h. Bilateral floccular lidocaine microinfusions immediately after the end of training recovered the gains increased by 1 h of massed training but did not affect the gains increased by 4 h of spaced training, suggesting that the memory trace of adaptation was transferred from the flocculus to the vestibular nuclei within 4 h of spaced training. Blockade of floccular protein synthesis, examined by bilateral floccular microinfusions of anisomycin or actinomycin D 1–4 h before the training, impaired the gains increased by 4 h of spaced training but did not affect the gains increased by 1 h of massed training. These findings suggest that the transfer of the memory trace of adaptation occurs within 4 h of spaced training, and proteins synthesized in the flocculus during training period may play an important role in memory transfer.
Introduction
The memory acquired through spaced learning, i.e., repetitions of learning with appropriate intervals, remains longer than the memory acquired through massed learning. The spacing effect, originally referred by Ebbinghaus (1885), is widely recognized in both the nondeclarative and declarative memory throughout the animal kingdom (Squire and Kandel, 2000). The spacing effect is considered to be closely related to memory consolidation; however, the physiological studies of the underlying neural mechanisms are few, particularly on the mammalian brain. Here, using the spacing effect, we developed a new eye movement training paradigm for mice that induces a long-term adaptation in the horizontal optokinetic response (HOKR) eye movement within 4 h of training, and we examined the role of protein synthesis in the cerebellar cortex in the formation of motor memory.
The HOKR is a compensatory eye movement to the horizontal motion of the visual field, and its gain is quantified by comparing the evoked eye movement and external surrounding motion. The neural circuitry of the HOKR is composed of the accessory optic tract, cerebellar flocculus, vestibular nuclei, and extraocular muscular motor nuclei. Despite its simple neural circuitry, the HOKR is under learning control (Ito, 1984). Hours of massed training with a sustained exposure to a sufficient amount of retinal slip, i.e., image motion on the retina, adaptively induce an increase in HOKR gains, which recovers within 24 h, in rabbits (Collewijn and Grootendorst, 1979; Nagao, 1983) and mice (Katoh et al., 1998, 2000; Shutoh et al., 2002, 2003, 2006). Results of lesion (Nagao, 1983; Katoh et al., 1998), unit recording (Nagao, 1988), and pharmacological and gene knock-out (Katoh et al., 2000; Shutoh et al., 2002, 2003, 2006) experiments consistently suggest that the flocculus and long-term depression (LTD) of parallel fiber-Purkinje cell synapses (Ito et al., 1982; Ito, 1989, 2001) play a key role in the HOKR adaptation induced by hours of massed training. Moreover, our recent study on mice suggested that a long-term HOKR gain increase, which remains for >24 h, is induced when 1 h of massed training is repeated for several days and, concomitantly, the memory trace of adaptation is transferred from the flocculus to the vestibular nuclei (Shutoh et al., 2006). Here, to further reveal the neural mechanisms underlying the transfer of memory trace of adaptation, we first examined the duration of memory of HOKR adaptation induced by training at various spacing intervals and developed a new protocol that induces adaptation of long-term memory by only 4 h of spaced training in mice. We then confirmed that the transfer of memory trace of adaptation occurs within 4 h of spaced training by pharmacological shutdown experiments using lidocaine. Finally, we examined the role of protein synthesis in the transfer of memory trace of adaptation using protein synthesis inhibitors (anisomycin or actinomycin D). The results suggest that the protein synthesis localized in the cerebellar cortex during the training period may play an important role in the transfer of memory trace that underlies the memory consolidation.
Materials and Methods
Eye movement measurements.
The experimental protocols followed the principles of laboratory animal care (National Institutes of Health publication no. 86-23, revised in 1996) and were approved by the Research Ethics Section of RIKEN. C57BL/6J male mice (12–16 weeks old) obtained from Clea Japan were used in all experiments. All efforts were made to minimize the number of mice used and their suffering throughout the course of experiments. Under isoflurane (Escain, Mylan Japan) anesthesia and aseptic conditions, a platform for head fixation was made on the cranial bone of a mouse using synthetic resin (Superbond C&B, Sun Medical) and one 15 mm stainless bolt. After recovery from surgery, the mouse was mounted on the table surrounded by a checked-pattern cylindrical (diameter, 60 cm) screen (check size, 4°) with its head fixed and its body loosely restrained in a plastic tube at the center of a cylindrical screen. Eye movements were recorded using an infrared TV camera (Nagao, 1990; Katoh et al., 1998). The frontal view of the right eye was monitored using a CCD TV camera (SSC-M350; SONY), which was fixed above the mouse, through a cold mirror. The right eye was illuminated by an infrared (wavelength, 920 nm) LED and displayed on a 13-inch LCD monitor (magnification, ×55). The area of the pupil was determined from the difference in brightness between the pupil and the iris. The real-time position of the eye was measured by calculating the central position of the pupil using an IMAQ (version 6, LabVIEW, National Instruments)-equipped system (Sakatani and Isa, 2004) and stored in a personal computer. Pupil size was kept constant by exposing the mouse to white noise sounds. The mean effective diameter of the mouse eyeball was estimated to be 2.3 mm. The spatial resolution of the TV camera system was 0.25°. The HOKR was examined by 50 cycles of sinusoidal screen oscillation by 15° (peak-to-peak) at 0.22 Hz (maximum screen velocity, 10.4°/s) on the horizontal plane in the light. Over 10 cycles of the evoked eye movements, free from blinks and saccades, were selected for averaging. No correction methods were used to delete quick eye movements. Mean amplitudes and phases were calculated on the averaged eye position traces by a modified Fourier analysis (Jastreboff, 1979). HOKR gain was defined as the ratio of the peak-to-peak amplitude of eye movements to that of screen oscillation.
HOKR training protocols.
The mice were trained to view 800 cycles of 0.22 Hz, 15° screen oscillation using five different training protocols (Fig. 1). In the massed training (M), mice received 800 cycles of 1 h continuous screen oscillation. In the spaced training (S0.5 h), mice received 200 cycles (15 min) of continuous screen oscillation at 0.5 h intervals four times. In S1 h training, mice received 200 cycles (15 min) of continuous screen oscillation at 1 h intervals four times. In S1d-1 training, the mice received 200 cycles (15 min) of continuous screen oscillation at 1 d intervals four times. In S1d-2 training, the mice received 100 cycles (7.5 min) of continuous screen oscillation at 1 d intervals eight times. Note that it took 1 h for M, 2.5 h for S0.5 h and 4 h for S1 h to complete training. Throughout these five sets of training protocols, the mice were kept in the dark in their home cages except for the sessions of training until they finished training. The HOKR gains were measured on the eye position traces of the initial and final 50 cycles of screen oscillation during each training session and by 50 cycles of screen oscillation from 24 h to 14 d after the end of S1d-1 training. These mice were reared in their home cages under normal light condition (12 h light/dark) after the end of training.
Figure 1.

Massed and spaced training protocols for HOKR adaptation. Mice received 800 cycles of 0.22 Hz, 15° screen oscillation by massed (M) training or spaced training at intervals of 0.5 h (S0.5 h), 1 h (S1 h), and 24 h (S1d-1 and S1d-2). Total time of 2.5 h, 4 h, 4 d, and 8 d for S0.5 h, S1 h, S1d-1, and S1d-2, respectively, is required to complete each training protocol. Except for sessions of training, the mice were kept in the dark.
Lesion experiments.
Under isoflurane anesthesia, tiny holes (diameter, 0.5 mm) were made on the bilateral temporal bones over the paraflocculus. Bilateral flocculi were lesioned by injecting high-frequency electrical currents (500 kHz, 3–5 mA) for 20 s through a small probe (RFG-4A, Radionics) positioned within the flocculus through the holes made on the temporal bones in six mice. Three days after lesioning, these mice were trained using the spaced training at 1 h intervals (S1 h). One week later, four of them were again trained using the spaced training at 1 d intervals (S1d-1). At the end of experiments, under deep general anesthesia induced by intraperitoneal administration of 60 mg/body weight sodium pentobarbital (Nembutal, Dainippon-Sumitomo Pharma), all the mice were perfused with PBS and 4% paraformaldehyde. The extents of lesions were examined histologically on Nissl-stained, 20-μm-thick coronal sections.
Pharmacological experiments.
The effects of flocculus shutdown were compared in 17 mice. They received massed training (M, n = 11) or spaced training at 1 h intervals (S1 h, n = 6). At the end of the training, 0.4 μl of 2.5% lidocaine chloride (Sigma-Aldrich) dissolved in standard Ringer's solution was infused into the bilateral flocculi (0.2 μl for each side) using two 1 μl microsyringes (80135, Hamilton) mounted on standard micromanipulators. Infusion was finished within 10 min. The HOKR was measured 0.5, 1, and 1.5 h after infusions. Except for the session for the training and measurements of HOKR, the mice were kept in the dark in their home cages. Infusions of lidocaine were also examined in six additional mice that received S0.5 h training.
The role of protein synthesis in HOKR adaptation was examined by infusion of 0.8 μl (0.4 μl for each side) of anisomycin (125 μg/μl, Sigma-Aldrich) dissolved in Ringer's solution at pH 7 by referring to Nader et al. (2000) on the study of rat amygdala. The drugs were infused into the bilateral flocculi similarly as lidocaine 4 h before the initiation of massed training (M, n = 6) or 1 h before the initiation of spaced training at 1 h intervals (S1 h, n = 6). As a control, the same amount of Ringer's solution was infused into the bilateral flocculi before the same training sessions (M, n = 5; S1 h, n = 5). In 6 mice, HOKR gains were measured 6 h and 24 h after infusions of anisomycin without any training. We also infused 0.8 μl (0.4 μl for each side) of actinomycin D (6.25 μg/μl, Sigma-Aldrich) dissolved in Ringer's solution into the bilateral flocculi (n = 6) 1 h before the initiation of S1 h training by referring to Lin et al. (2003) on the study of rat amygdala.
After the end of experiments, 1% fluorescein isothiocyanate hydrochloride (FITC, Research Organics) dissolved in dimethyl sulfoxide (Merck Japan) was infused into the bilateral flocculi similarly as infusions of lidocaine, anisomycin, or actinomycin D to estimate the extent of drug diffusion. The mice were perfused under pentobarbital anesthesia (60 mg /kg body weight, Nembutal, Dainippon Sumitomo Pharma). Drug diffusions were examined histologically on coronal sections under a fluorescence microscope (BX50, Olympus).
Histological inspections of the flocculus after anisomycin infusions were carried out in seven mice. For four mice, 0.4 μl of anisomycin and the same amount of Ringer's solution was infused into the right and left flocculi, respectively. The other three mice were used for noninfusion control. They were kept in the dark without any training and then perfused under the pentobarbital anesthesia 24 h after infusions. The total number of Purkinje cells and the general cortical structure were compared on Nissl-stained coronal sections (thickness, 12 μm) for the anisomycin-infused, Ringer's solution-infused, and noninfused flocculi. The significance of the data obtained in the whole series of experiments was evaluated using Statview (version 5, SAS Institute).
Results
Spacing effect in HOKR adaptation
To examine the spacing effect in HOKR adaptation, we trained C57BL/6J mice to view 800 cycles of 0.22 Hz, 15° screen oscillation in the head-fixed position using five different training protocols (Fig. 1). One group of mice received 800 cycles of massed training in 1 h (M, n = 11), whereas the three spaced training groups received 4 × 200 cycles of training at 0.5 h (S0.5 h, n = 5), 1 h (S1 h, n = 9), and 1 d (S1d-1, n = 7) intervals, and the remaining group received 100 cycles of training at 1 d intervals for 8 d (S1d-2, n = 5). These mice were kept in the dark during intervals and thus they received no additional visual inputs. We first measured the HOKR gain and then trained the mice to induce adaptation in gain. The HOKR gains (mean ± SE) at the start of training were similar among the five groups (from 0.28 ± 0.01 to 0.32 ± 0.02, p > 0.25, one-factorial ANOVA) (Table 1). The HOKR gain increase (%) is normalized to the mean gain at the start of training. Figure 2 shows a comparison of the gain increase among five training protocols. The gain increase fluctuated among training sessions, and a tendency of small gain increase was often observed in the spacing intervals (Fig. 2B,C). At the end of five training sessions, the HOKR gains increased similarly by 22.1–31.2%. The retention of memory of adaptation was examined 24 h after the end of the training sessions. The HOKR gain increased by 1 h of massed training (Fig. 2A,F) decreased by one-half (p < 0.02, paired t test), whereas the gain increased by all four spaced training protocols (S0.5 h–S1d-2), remained unchanged for 24 h (Fig. 2B–F, p > 0.5–0.9, paired t test). Thus, the spacing effect is clearly observed in the retention of HOKR adaptation.
Table 1.
Mean HOKR gain (SE) to 0.22 Hz, 15° (peak-to-peak) screen oscillation at the start of 5 training sessions
| Training | M (n = 11) | S0.5 h (n = 5) | S1 h (n = 9) | S1d-1 (n = 7) | S1d-2 (n = 5) |
|---|---|---|---|---|---|
| Gain (SE) | 0.30 (0.01) | 0.28 (0.01) | 0.28 (0.02) | 0.32 (0.02) | 0.32 (0.02) |
The gains at the start of training were similar among the 5 groups (p > 0.25, one-factorial ANOVA).
Figure 2.

HOKR adaptation induced by massed and spaced training protocols. Mice were trained using 800 cycles of 0.22 Hz, 15° screen oscillation using five different protocols (Fig. 1). A, Eight hundred cycles of massed training (M, n = 11). B, Spaced training with 4 × 200 cycles at 0.5 h intervals (S0.5 h, n = 5). C, Spaced training with 4 × 200 cycles at 1 h intervals (S1 h, n = 9). D, Spaced training with 4 × 200 cycles at 1 d intervals (S1d-1, n = 7). E, Spaced training with 8 × 100 cycles at 1 d intervals (S1d-2, n = 5). F, Comparison of gain increase immediately after (open columns) and 24 h after (filled columns) the end of training among the five protocols. The HOKR gain increase (%) is normalized to the mean gain at the start of training. Error bars indicate SE; *p < 0.05; ns, p > 0.05 (paired t test).
We examined the recovery of HOKR adaptation for the five mice that received S1d-1 training every 3 d up to 14 d after the end of training. These mice were reared under normal light condition (12 h, light/dark) without any other optokinetic training after the end of S1d-1 training, except for the brief time for the measurements of HOKR. The gain increase remained for 10 d after the end of S1d-1 training (16.2–24.1%, p < 0.05, Dunnett's test; compared with the gains before the training), and a tendency of small gain increase (10.6 ± 3.3%, p > 0.05) was observed 14 d after the end of S1d-1 training, which was similar to our previous study using 5 d of 600 cycles of 0.17 Hz-15° screen oscillation at 1 d intervals (Shutoh et al., 2006). The HOKR phases were not affected by any of the five training protocols, and are not discussed in this article.
Effects of floccular lesions on spaced training-induced HOKR adaptation
We previously reported that the HOKR adaptation induced by 1 h (600 cycles) of massed training with 0.17 Hz, 15° screen oscillation is abolished by bilateral cerebellar flocculus lesions in mice (Katoh et al., 1998; Shutoh et al., 2006). To confirm these findings in the HOKR adaptation induced by the spaced training, we carried out lesion experiments. We made bilateral floccular lesions in six mice by the electric coagulation method 3 d before the training. In these mice, >75% of the paraflocculus and flocculus region was destroyed bilaterally, whereas the vestibular nerve and nuclei remained intact (Fig. 3A). We used data obtained with normal mice shown in Figure 2, C and D, as the control as in our previous studies (Katoh et al., 1998; Shutoh et al., 2006). The HOKR gain at 0.22 Hz-15° screen oscillation after floccular lesions (0.24 ± 0.01, n = 10) was small (p < 0.01, Student's t test) compared with the control group (0.30 ± 0.01, n = 16). We trained the mice using 4 × 200 cycles of 0.22 Hz, 15° screen oscillation at 1 h (S1 h) or 1 d (S1d-1) intervals. The HOKR gain decreased slightly after S1 h (−7.1 ± 5.0% n = 6) (Fig. 3B) or S1d-1 (−8.5 ± 4.5% n = 4) (Fig. 3C) training. These gain changes were both statistically significant compared with the data for the control group shown in Figure 1, C and D (p < 0.01, Student's t test). Thus, the flocculus is necessary for the HOKR adaptation induced by spaced training through S1 h and S1d-1.
Figure 3.

Effects of bilateral floccular lesions on HOKR adaptation induced by spaced training. A, Photograph of coronal section of right paraflocculus (PFL) and flocculus (FL) after lesioning. Scale bar, 500 μm. B, HOKR gain increase after 4 × 200 cycles of screen oscillation at 1 h intervals (S1 h) for lesioned mice (filled column, n = 6). Control data (open column, n = 9) are those shown in Figure 2C. C, Similar to B but after 4 × 200 cycles of screen oscillation at 1 d intervals (S1d, n = 4). Control data (n = 7) are those shown in Figure 2D. **p < 0.01 (Student's t test). Error bars indicate SE.
Effects of reversible flocculus shutdown on HOKR adaptation after training
On the basis of findings of our previous study (Shutoh et al., 2006) in which the effects of pharmacological floccular shutdown on HOKR adaptation were examined, we suggested that the memory trace of HOKR adaptation induced by 1 h (600 cycles) of 0.17 Hz, 5° screen oscillation training is located in the flocculus and that the memory trace is transferred to the vestibular nuclei after repetition of 1 h of training for 3 d. In the present study, we found that the duration of memory for adaptation induced by S0.5 h and S1 h spaced training protocols is longer than that of 1 h of M training. It took 2.5 and 4 h for S0.5 h and S1 h training protocols to be completed, respectively (Fig. 1). Thus, it is possible that the memory trace of adaptation is transferred from the flocculus to the vestibular nuclei within 2.5 to 4 h during these spaced training protocols. Then, we examined the location of the memory trace of adaptation induced by the spaced training (S1 h) by bilateral pharmacological floccular shutdown immediately after the end of training sessions.
We compared the effects of lidocaine infusions on the HOKR adaptation between M and S1 h training protocols. The mean HOKR gains before training were similar between M (0.30 ± 0.01, n = 11) and S1 h (0.29 ± 0.02, n = 6; p > 0.2, Student's t test) training sessions, and the gain increase induced by these training protocols were also similar (23.7 ± 1.9% for M vs 27.9 ± 5.7% for S1 h, p > 0.3, Student's t test). We infused 0.4 μl (0.2 μl for each side) of 2.5% lidocaine into the bilateral flocculi immediately after the end of training. The infused lidocaine covered the entire flocculus 1 h after infusion, and this was confirmed by 0.4 μl (0.2 μl for each side) infusions of FITC carried out similarly as lidocaine infusions (Fig. 4A). We did not observe any nystagmus-like or slow-drift eye movements when we examined the HOKR 30 min after the end of infusions. The mean number of eye position traces free from saccades in 50 cycles of screen oscillation did not differ before and after infusions (20 ± 1 vs 22 ± 1, n = 23 infusions; p > 0.1, paired t test). The gain increase induced by M training decreased 0.5 and 1 h after infusions (p < 0.05, Dunnett's test) (Fig. 4B,D,G), and recovered to the level of end of training 1.5 h after infusions (p > 0.05). We consider that the recovery at 1.5 h may be due to the decline of anesthetic action of lidocaine. By contrast, the gain increase induced by S1 h training was not affected 0.5–1.5 h after infusions (p > 0.05) (Fig. 4C,E,H). These findings indicate that the memory trace of adaptation induced by M training was maintained within the flocculus at the end of training, but that induced by S1 h training was no longer located there at the end of training. We also examined the effects of flocculus shutdown on the HOKR adaptation induced by S0.5 h training and obtained similar results as those for S1 h training (Fig. 4F,I). Thus, the transfer of memory trace of adaptation occurs rather fast, within 2.5–4 h of training, when space intervals are inserted.
Figure 4.
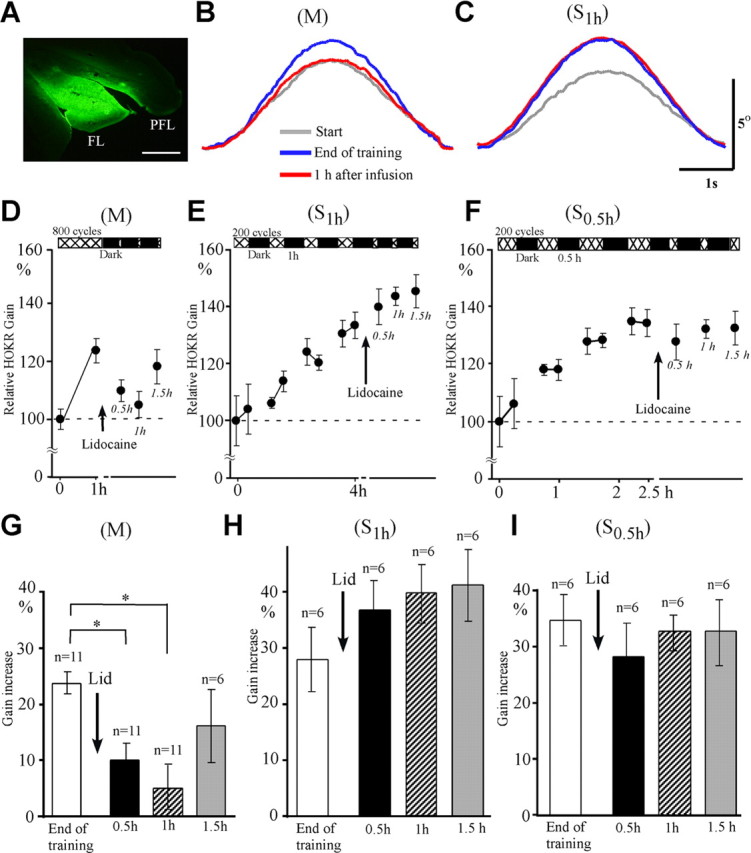
Effects of reversible shutdown of bilateral flocculi on adaptation induced by 800 cycles of massed training (M) and spaced training using 4 × 200 cycles at 1 h intervals (S1 h). A, Extension of lidocaine diffusion estimated by FITC infusion. FL, Flocculus. PFL, paraflocculus. Scale bar, 500 μm. B, Example of eye position traces before training (gray curve), at the end of training (blue curve), and 1 h after infusion of lidocaine (red curve) during M training. Averaged eye position traces from 16 to 25 cycles obtained from the same mouse are shown. C, Similar to B but for those during S1 h training. Averaged eye position traces from 16 to 27 cycles obtained from the same mouse are shown. Scale bars, 1 s and 5°. D–F, Time course of HOKR adaptation and effects of floccular lidocaine infusions for M (D), S1 h (E), and S0.5 h (F) training. G, Comparison of gain increase at the end of M training, 0.5 h, 1 h, and 1.5 h after lidocaine (Lid) infusions (n = 6–11). H, I, Similar to G but for those of S1 h training (H, n = 6) and S0.5 h training (I, n = 6). *p < 0.05 (Dunnett's test). Error bars indicate SE.
Effects of floccular protein synthesis blockade on HOKR adaptation
We then examined pharmacologically whether the de novo floccular protein synthesis plays a role in the HOKR adaptation induced by M and S1 h training protocols. We infused 0.8 μl (0.4 μl for each side) of anisomycin (125 μg/μl) or control Ringer's solution into the bilateral flocculi by referring to the experiments on rat fear conditioning (Nader et al., 2000; Debiec et al., 2002). We measured the nonadapted HOKR gains at 1, 4, 6, and 24 h after bilateral floccular anisomycin infusions. No differences were observed on HOKR gains for 24 h after infusions, compared with those before infusions (p > 0.05, Dunnett's test) (Table 2). The total number of floccular Purkinje cells, which were counted histologically 24 h after infusions, was around 700, and no differences were observed among anisomycin-infused, Ringer's solution-infused, or noninfused flocculi (p > 0.05, Tukey–Kramer test) (Table 3). Furthermore, the general cortical structures were not altered by infusions of anisomycin except for the track of the infusion needle tip (Fig. 5A). The diffusion of drugs was examined using 0.4 μl of FITC infusions similarly as in Figure 4A. The infused FITC diffused within the flocculus and did not reach the brainstem region, both 1 and 5 h after infusions (data not shown in figures).
Table 2.
Mean HOKR gain (SE) to 0.22 Hz, 15° (peak-to-peak) screen oscillation before and 1–24 h after floccular infusions of anisomycin
| Before infusions (n = 18) | 1 h (n = 6) | 4 h (n = 6) | 6 h (n = 6) | 24 h (n = 6) | |
|---|---|---|---|---|---|
| Gain (SE) | 0.28 (0.01) | 0.25 (0.01) | 0.28 (0.03) | 0.30 (0.01) | 0.30 (0.02) |
The gains after infusions were similar to those before infusions (p > 0.05, Dunnett's test).
Table 3.
Mean (SE) number of floccular Purkinje cells 24 h after floccular drug infusions and noninfused control
| Anisomycin (n = 4) | Ringer's solution (n = 4) | Control (n = 3) | |
|---|---|---|---|
| Number of Purkinje cells (SE) | 678 (22) | 696 (22) | 730 (47) |
The numbers of floccular Purkinje cells were counted in the anisomycin-infused right side and Ringer's solution-infused left side on 25–30 Nissl-stained coronal serial sections (thickness, 12 μm) in four mice, and those for noninfused control were counted similarly from three naive mice.
Figure 5.
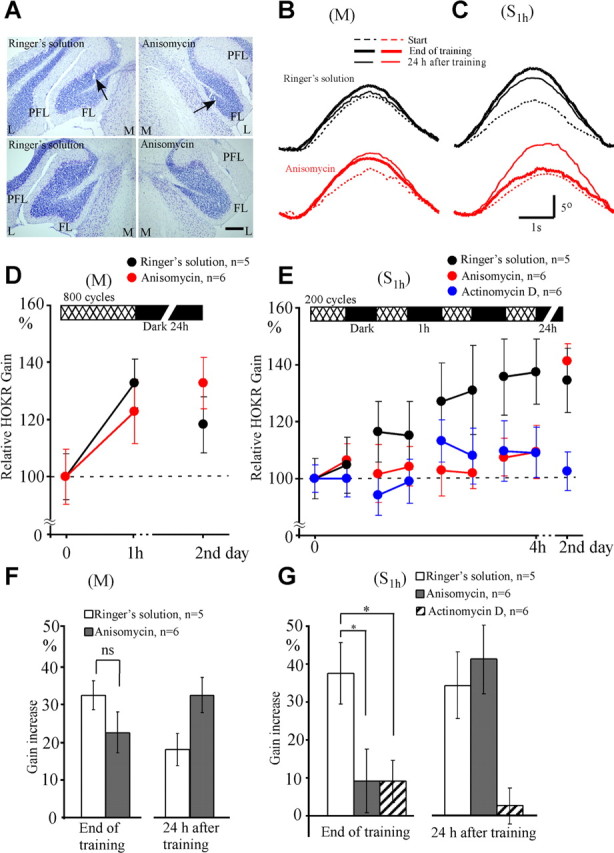
Effects of protein synthesis inhibition on adaptation induced by massed (M) and spaced (S1 h) training. Anisomycin or control Ringer's solution was infused 4 h before the initiation of M training and 1 h before the initiation of S1 h training. A, Photographs of Nissl-stained flocculus. The left two photographs show examples 24 h after infusions of Ringer's solution, and the right two photographs show examples 24 h after anisomycin infusions. Arrows show positions of the tip of infusion needle. The distance between the upper and lower photographs is 80 μm. Note that tissue damage by infusion was minimal, and the general cortical structure was not affected by infusions. FL, Flocculus; L, lateral; M, medial; PFL, paraflocculus. Scale bar, 200 μm. B, Averaged eye position traces obtained from the same mouse in M training. Black dotted, thick, and thin curves show eye position traces at the start, end, and after 24 h of training, respectively, after infusion of Ringer's solution. Red curves similarly show eye position traces after anisomycin infusion. C, Similar to B but for the data obtained during S1 h training. Eye position traces for 10–21 cycles were averaged in B and C. Scale bars, 1 s and 5°. D, E, Time course of HOKR adaptation and its retention after infusions of Ringer's solution (black circles), anisomycin (red circles), and actinomycin D (blue circles). F, Comparison of gain increase at the end and 24 h after the end of M training. Unfilled and filled columns indicate data for Ringer's solution (n = 5) and anisomycin (n = 6) infusions, respectively. G, Similar to F, but for the gain increase after S1 h training. Data obtained after actinomycin D infusions (n = 6) were also shown by striped columns. *p < 0.05; ns, p > 0.05 (Dunnett's test or Student's t test). Error bars indicate SE.
M training started 4 h after the infusions of drugs, S1 h training started 1 h after the infusions, and both finished 5 h after the infusions. The HOKR gains at the start of training were similar between Ringer's solution (0.26 ± 0.02, n = 12) and anisomycin (0.27 ± 0.02, n = 10) infusions. The gain increase induced by M training was similar between Ringer's solution (32.5 ± 3.7%, n = 5) and anisomycin (22.5 ± 5.4%, n = 6; p > 0.1, Student's t test) infusions (Fig. 5B,D,F) and was within the range of gain increase shown in Figure 2. The increased gain declined 24 h after the end of training in Ringer's solution-infused mice (18.0 ± 4.3% vs 32.5 ± 3.7%, n = 5, p < 0.05, paired t test) but did not decline in anisomycin-infused mice (22.5 ± 5.4% vs 32.5 ± 4.6%, n = 6, p > 0.05) (Fig. 5F). Thus, anisomycin did not impair the adaptation induced by massed training but affected its retention.
In contrast, the gain increase induced by S1 h training in anisomycin-infused mice (9.2 ± 8.4%, n = 6) was significantly smaller than that in Ringer's solution-infused mice (37.5 ± 8.0%, n = 5; p < 0.05, Dunnett's test) (Fig. 5C,E,G). However, the HOKR gain increase measured 24 h after the end of S1 h training was similar between anisomycin-infused (41.2 ± 9.1%, n = 6) and Ringer's solution-infused (34.4 ± 8.7%, n = 5, p > 0.05, Dunnett's test) (Fig. 5C,E) mice. Thus, anisomycin infusions inhibited the adaptation induced by S1 h training, but very little affected the adaptation induced by M training. The inhibitory effect of anisomycin on the adaptation induced by S1 h recovered 24 h after training.
We also infused 0.8 μl (0.4 μl for each side) of actinomycin D (6.25 μg/μl) into the bilateral flocculi 1 h before S1 h training in 6 mice (Fig. 5E,G). The HOKR gains were not different before (0.27 ± 0.01) and after (0.26 ± 0.01) the infusions. The gain increase at the end of S1 h training in actinomycin D-infused mice (9.1 ± 5.6%, n = 6; p < 0.05, Dunnett's test) was also small compared with that in Ringer's solution-infused mice (34.4 ± 8.7%, n = 5). The depression of gain increase continued 24 h after the end of training (2.7 ± 4.7%, n = 6; p < 0.05), unlike anisomycin infusions. Thus, the actinomycin D infusions impaired the HOKR adaptation induced by S1 h training, without any recovery 24 h after training.
Discussion
We compared the duration of memory of HOKR adaptation in five groups of mice that were trained by 800 cycles of screen oscillation with or without intervals (Fig. 1). After the end of training sessions, the HOKR gains increased similarly in all the groups, and the spacing effect was demonstrated in term of the retention of adapted gains after 24 h (Fig. 2). By using the spacing effect, we developed a protocol that produces a long-term memory in HOKR adaptation by 4 h of spaced training. We confirmed that the cerebellar flocculus is necessary for the adaptation induced by 4 h of spaced training by lesion experiments (Fig. 3) and that the memory trace of the adaptation is transferred from the flocculus to vestibular nuclei within 2.5 to 4 h of spaced training by pharmacological experiments (Fig. 4). We revealed that the pharmacological blockade of floccular protein synthesis did not affect the adaptation induced by 1 h of massed training, but it impaired the adaptation induced by 4 h of spaced training (Fig. 5), suggesting that the de novo floccular protein synthesis may be necessary for the transfer of the memory trace that underlies the consolidation of motor memory. The retention of memory induced by 1 h of massed training was affected by blockade of floccular protein synthesis, which is not discussed further in this article.
Neural mechanisms for memory trace transfer
The training history-dependent transfer of memory trace of adaptation has been demonstrated by pharmacological shutdown experiments in the HOKR in mice (Shutoh et al., 2006) and horizontal vestibulo-ocular reflex (HVOR) in cats (Kassardjian et al., 2005) and monkeys (Anzai et al., 2010). One may speculate that flocculus shutdown would increase the spontaneous discharges of flocculus target vestibular nuclear neurons, alter the general performance of the HOKR/HVOR neural circuitry, and consequently may affect HOKR/HVOR gains. However, we consider that such a possibility is unlikely, because effects of floccular lidocaine infusions were specific to the gains changed by adaptation (Nagao and Kitazawa, 2003; Shutoh et al., 2006; Anzai et al., 2010), and floccular CNQX infusions, which would only mildly affect the spontaneous discharges of Purkinje cells, induced similar effects on the adaptation as those of lidocaine (Kassardjian et al., 2005). These studies consistently suggest that the memory trace of adaptation induced by 1–2 h of training is located in the flocculus and that memory trace is transferred to the vestibular nuclei after days of training. However, the results of the present study suggest that the memory transfer could occur within 2.5 to 4 h, when appropriate intervals are inserted to training.
Two possible mechanisms are listed for the transfer of the memory trace of adaptation. One is that the memory of adaptation may be initially formed in the floccular Purkinje cell synapses, which may secondarily induce plastic changes in the vestibular nuclei. Alternatively, the memory of adaptation may be formed independently in parallel within the flocculus and vestibular nuclei with different time courses. Results of the present study (Fig. 5) and those of the following four studies consistently suggest that the metabolic or electrical activity of Purkinje cells strongly influences the induction of plasticity of the vestibular or cerebellar nuclei that underlies memory transfer. Floccular infusions of NG-monomethyl-l-arginine, which blocks LTD/long-term potentiation (LTP) of parallel fiber-Purkinje cell (pf-PC) synapses, depress both the acquisition and transfer of the memory of HOKR adaptation in mice (Shutoh et al., 2006). In mice genetically devoid of the substrate for the cyclic GMP-dependent protein kinase, which is uniquely concentrated in Purkinje cells, the acquisition of memory of HOKR adaptation is normal, but the memory transfer is specifically attenuated (Endo et al., 2009). In mice in which feedforward inhibitory inputs to Purkinje cells are removed genetically, the transfer of memory trace of HVOR adaptation is specifically impaired (Wulff et al., 2009). In rabbit eyeblink conditioning, the consolidation of conditioned responses is inhibited by temporal inactivation of the cerebellar cortex by muscimol infusion immediately following each training session, whereas the inactivation of cerebellar nuclei after training induced no inhibitory effect (Attwell et al., 2002; Cooke et al., 2004; Kellett et al., 2010).
Pharmacological actions of anisomycin and actinomycin D
In the present study, the mice received massed training (M) 4–5 h after floccular infusions of anisomycin and spaced training (S1 h) 1–5 h after infusions of anisomycin or actinomycin D. Because inhibition of protein synthesis was demonstrated to reach maximum 1–5 h after infusions and then gradually decline (Rosenblum et al., 1993), the mice were trained at the time when the protein synthesis inhibition was maximum in both training protocols. It is also suggested that anisomycin may induce other depressant effects, e.g., facilitation of apoptosis, on neurons (Rudy et al., 2006) (but see Canal et al., 2007). We, however, consider that the nonspecific depressant effects of anisomycin were unlikely to explain the results of the present study for several reasons. First, anisomycin infused before the training did not alter the nonadapted HOKR gains (Table 2), floccular structure (Fig. 5), or number of floccular Purkinje cells (Table 3) for 24 h after infusions. Second, inhibitory effects of anisomycin were observed specifically in the adaptation induced by S1 h and not observed in the adaptation induced by M (Fig. 5), although both training protocols were carried out at similar period of time after infusions. Third, it is reported that both anisomycin and actinomycin D induce no appreciable changes in the general excitability of Purkinje cells, i.e., membrane resistance, resting membrane potentials, parallel fiber-induced EPSPs, or climbing fiber-induced Ca2+ spikes (Karachot et al., 2000, 2001).Together, we consider that a contribution of nonspecific depressant effects might be small in the inhibitory actions of anisomycin and actinomycin D on the adaptation induced by S1 h. Thus, we suggest that the floccular protein synthesis inhibition caused by local drug application led to the reduced adaptation by S1 h. Results of in vitro experiments suggest that the protein synthesis plays a role in LTD of pf-PC synapses, but whether it is related to induction (Karachot et al., 2001) or consolidation (Linden, 1996) is unclear.
A slight difference was recognized in the effects on adaptation between anisomycin, a translation inhibitor, and actinomycin D, a transcription inhibitor. The depressive actions of anisomycin on memory transfer recovered 24 h after the training, while those of actinomycin D remained for 24 h (Fig. 5). One possibility for the different actions of these two drugs is the long-lasting effects of actinomycin D, which not only inhibits transcription but also binds to DNA. Another possibility is the necessity of transcription-dependent protein synthesis for memory transfer. A specific inhibition by transcription inhibitors (actinomycin D and α-amanitin) was reported in Aplysia long-term behavioral sensitization (Castellucci et al., 1986; Sweatt and Kandel, 1989). The initial trigger of the gene cascade that induces long-term changes might not be activated in the presence of actinomycin D.
Role of protein synthesis in memory transfer
A number of pharmacological studies have suggested that protein synthesis is important in memory retention, but not necessary for memory acquisition (e.g., Squire, 1987; Squire and Kandel, 2000; Costa-Mattioli et al., 2009). Protein synthesis is necessary specifically for the long-term facilitation of the sensory-motor synapses and behavioral sensitization of gill-withdrawal reflex in Aplysia (Castellucci et al., 1986; Montarolo et al., 1986; Schacher et al., 1988). In the fear conditioning of rats, anisomycin infusion in amygdala did not affect the acquisition of conditioned response but impaired its reconsolidation when anisomycin was infused 24 h after the conditioning (Nader et al., 2000; Debiec et al., 2002). Similar results were obtained in the inhibitory avoidance task of rats after hippocampal infusion of the amanitin (Katche et al., 2010). In rabbit eyeblink conditioning, infusions of anisomycin into the cerebellar interpositus nuclei depress both the acquisition and retention of the conditioned response (Bracha et al., 1998; Gomi et al., 1999). The role of protein synthesis has been suggested in the memory acquisition induced by the spaced training in the conditioning of the proboscis extension reflex of honey bees (Menzel et al., 2001) and in the contextual learning of fear conditioning in rats (Scharf et al., 2002).
The results of present study suggest that protein synthesis in the cerebellar cortex during the training period may play an important role for the induction of plasticity in the vestibular or cerebellar nuclei. What links the protein synthesized in the cerebellar cortex with the plasticity in the vestibular or cerebellar nuclei (Fig. 6)? One possibility is that proteins synthesized in Purkinje cells during training may be transported to their axon terminals and act postsynaptically on vestibular nuclear neurons. The destruction of the inferior olive induces depressive effects on the synaptic transmission in vestibular nuclei, which is assumed to be mediated through axonal transport (Ito et al., 1979; Karachot et al., 1987). Another possibility is that protein synthesis in the cerebellar cortex may be related to the plasticity of electrical properties of cerebellar Purkinje and other neurons. Protein synthesis is suggested to be involved in the late phase of LTD/LTP (Frey et al., 1988; Huang et al., 1996; Linden, 1996). Altered excitability of cerebellar cortical neurons caused by the absence of the late LTD/LTP may induce some depressant effects on the vestibular nuclear neurons that are closely connected with the cerebellar cortex. The other possibility is that the protein synthesis may be related to restructuring of cerebellar cortical neural circuitry. A decrease in the number of pf-PC synapses has been suggested in the rabbit cerebellar hemisphere after acquisition of eyeblink conditioning (Connor et al., 2009) and in the mouse flocculus after long-term HOKR adaptation (Nakadate et al., 2004). The reduced pf-PC synapses may decrease the tonic inhibitory drive of Purkinje cells on vestibular or cerebellar nuclear neurons. It has been suggested that the intrinsic excitability (Nelson et al., 2003) or responsiveness to mossy fiber inputs (Pugh and Raman, 2006; McElvain et al., 2010) of nuclear neurons is regulated by the inhibitory actions of Purkinje cells.
Figure 6.

Neural circuitry for mouse HOKR and memory trace of adaptation induced by massed and spaced training. The optokinetic signals that drive the HOKR are mediated to the vestibular nuclei (VN) directly through mossy fiber (mf) collateral inputs from the accessory optic tract (AOT) or indirectly through the axons of the flocculus (FL) Purkinje cells (PC). The retinal slip signals, which are necessary for the induction of HOKR adaptation, are mediated to the flocculus Purkinje cells through climbing fiber inputs (cf). Our study reveals that the memory induced by massed training is maintained in FL, whereas that induced by spaced training is maintained, most presumably, in VN. GC, Granule cells; IO, inferior olive; OMN, oculomotor neurons; pf, parallel fibers.
Footnotes
This study was supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (No. 22300112), the research funds of RIKEN, the Naito Foundation, and Japan Foundation for Aging and Health. We thank Dr. Masao Ito (RIKEN Brain Science Institute, Wako, Saitama, Japan) for critical reading of our manuscript. We also thank Drs. Fumihiro Shutoh (Laboratory of Neuroendocrinology, Graduate School of Comprehensive Human Science, University of Tsukuba, Tsukuba, Ibaraki, Japan) and Tadashi Yamazaki (RIKEN Brain Science Institute-TOYOTA Collaboration Center, Wako, Saitama, Japan) for helpful suggestions.
References
- Anzai M, Kitazawa H, Nagao S. Effects of reversible pharmacological shutdown of cerebellar flocculus on the memory of long-term horizontal vestibulo-ocular reflex adaptation in monkeys. Neurosci Res. 2010;68:191–198. doi: 10.1016/j.neures.2010.07.2038. [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Cooke SF, Yeo CH. Cerebellar function in consolidation of motor memory. Neuron. 2002;34:1011–1020. doi: 10.1016/s0896-6273(02)00719-5. [DOI] [PubMed] [Google Scholar]
- Bracha V, Irwin KB, Webster ML, Wunderlich DA, Stachowiak MK, Bloedel JR. Microinjections of anisomycin into the intermediate cerebellum during learning affect the acquisition of classically conditioned responses in the rabbit. Brain Res. 1998;788:169–178. doi: 10.1016/s0006-8993(97)01535-7. [DOI] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intraamygdala injections of a protein synthesis inhibitor. Proc Natl Acad Sci U S A. 2007;104:12500–12505. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci VF, Frost WN, Goelet P, Montarolo PG, Schacher S, Morgan JA, Blumenfeld H, Kandel ER. Cell and molecular analysis of long-term sensitization in Aplysia. J Physiol (Paris) 1986;81:349–357. [PubMed] [Google Scholar]
- Collewijn H, Grootendorst AF. Adaptation of optokinetic and vestibulo-ocular reflexes to modified visual input in the rabbit. Prog Brain Res. 1979;50:771–781. doi: 10.1016/S0079-6123(08)60874-2. [DOI] [PubMed] [Google Scholar]
- Connor S, Bloomfield J, LeBoutillier JC, Thompson RF, Petit TL, Weeks AC. Eyeblink conditioning leads to fewer synapses in the rabbit cerebellar cortex. Behav Neurosci. 2009;123:856–862. doi: 10.1037/a0016370. [DOI] [PubMed] [Google Scholar]
- Cooke SF, Attwell PJ, Yeo CH. Temporal properties of cerebellar-dependent memory consolidation. J Neurosci. 2004;24:2934–2941. doi: 10.1523/JNEUROSCI.5505-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus HE. Über das Gedächtnis. In: Ruger HA, Bussenius C, translators. Reprinted as Memory: a contribution to experimental psychology. New York: Teachers College-Columbia UP; 1885. 1913. [Google Scholar]
- Endo S, Shutoh F, Dinh TL, Okamoto T, Ikeda T, Suzuki M, Kawahara S, Yanagihara D, Sato Y, Yamada K, Sakamoto T, Kirino Y, Hartell NA, Yamaguchi K, Itohara S, Nairn AC, Greengard P, Nagao S, Ito M. Dual involvement of G-substrate in motor learning revealed by gene deletion. Proc Natl Acad Sci U S A. 2009;106:3525–3530. doi: 10.1073/pnas.0813341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Gomi H, Sun W, Finch CE, Itohara S, Yoshimi K, Thompson RF. Learning induces a CDC2-related protein kinase, KKIAMRE. J Neurosci. 1999;19:9530–9537. doi: 10.1523/JNEUROSCI.19-21-09530.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- Ito M. The cerebellum and neural control. New York: Raven; 1984. [Google Scholar]
- Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Ito M, Nisimaru N, Shibuki K. Destruction of inferior olive induces rapid depression in synaptic action of cerebellar Purkinje cells. Nature. 1979;277:568–569. doi: 10.1038/277568a0. [DOI] [PubMed] [Google Scholar]
- Ito M, Sakurai M, Tongroach P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J Physiol. 1982;324:113–134. doi: 10.1113/jphysiol.1982.sp014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastreboff PW. Evaluation and statistical judgment of neural responses to sinusoidal stimulation in cases with superimposed drift and noise. Biol Cybern. 1979;33:113–120. doi: 10.1007/BF00355259. [DOI] [PubMed] [Google Scholar]
- Karachot L, Ito M, Kanai Y. Long-term effects of 3-acetylpyridine-induced destruction of cerebellar climbing fibers on Purkinje cell inhibition of vestibulospinal tract cells of the rat. Exp Brain Res. 1987;66:229–246. doi: 10.1007/BF00243301. [DOI] [PubMed] [Google Scholar]
- Karachot L, Shirai Y, Vigot R, Yamamori T, Ito M. Rapidly turned over protein maintains metabotropic synaptic transmission in Purkinje cells. Neuroreport. 2000;11:2903–2906. doi: 10.1097/00001756-200009110-00015. [DOI] [PubMed] [Google Scholar]
- Karachot L, Shirai Y, Vigot R, Yamamori T, Ito M. Induction of long-term depression in cerebellar Purkinje cells requires a rapidly turned over protein. J Neurophysiol. 2001;86:280–289. doi: 10.1152/jn.2001.86.1.280. [DOI] [PubMed] [Google Scholar]
- Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM. The site of a motor memory shifts with consolidation. J Neurosci. 2005;25:7979–7985. doi: 10.1523/JNEUROSCI.2215-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katche C, Bekinschtein P, Slipczuk L, Goldin A, Izquierdo IA, Cammarota M, Medina JH. Delayed wave of c-Fos expression in the dorsal hippocampus involved specifically in persistence of long-term memory storage. Proc Natl Acad Sci U S A. 2010;107:349–354. doi: 10.1073/pnas.0912931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh A, Kitazawa H, Itohara S, Nagao S. Dynamic characteristics and adaptability of mouse vestibulo-ocular and optokinetic response eye movements and the role of flocculo-olivary system revealed by chemical lesions. Proc Natl Acad Sci U S A. 1998;95:7705–7710. doi: 10.1073/pnas.95.13.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh A, Kitazawa H, Itohara S, Nagao S. Inhibition of nitric oxide synthesis and gene knockout of neuronal nitric oxide synthase impaired adaptation of mouse optokinetic response eye movements. Learn Mem. 2000;7:220–226. doi: 10.1101/lm.7.4.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellett DO, Fukunaga I, Chen-Kubota E, Dean P, Yeo CH. Memory consolidation in the cerebellar cortex. PLoS One. 2010;5:e11737. doi: 10.1371/journal.pone.0011737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu HY, Gean PW. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci. 2003;23:8310–8317. doi: 10.1523/JNEUROSCI.23-23-08310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ. A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron. 1996;17:483–490. doi: 10.1016/s0896-6273(00)80180-4. [DOI] [PubMed] [Google Scholar]
- McElvain LE, Bagnall MW, Sakatos A, du Lac S. Bidirectional plasticity gated by hyperpolarization controls the gain of postsynaptic firing responses at central vestibular nerve synapses. Neuron. 2010;68:763–775. doi: 10.1016/j.neuron.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel R, Manz G, Menzel R, Greggers U. Massed and spaced learning in honeybees: the role of CS, US, the intertrial interval and the test interval. Learn Mem. 2001;8:198–208. doi: 10.1101/lm.40001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nagao S. Effects of vestibulocerebellar lesions upon dynamic characteristics and adaptation of vestibulo-ocular and optokinetic responses in pigmented rabbits. Exp Brain Res. 1983;53:36–46. doi: 10.1007/BF00239396. [DOI] [PubMed] [Google Scholar]
- Nagao S. Behavior of floccular Purkinje cells correlated with adaptation of horizontal optokinetic eye movement response in pigmented rabbits. Exp Brain Res. 1988;73:489–497. doi: 10.1007/BF00406606. [DOI] [PubMed] [Google Scholar]
- Nagao S. A non-invasive method for real-time eye position recording with an infrared TV-camera. Neurosci Res. 1990;8:210–213. doi: 10.1016/0168-0102(90)90022-7. [DOI] [PubMed] [Google Scholar]
- Nagao S, Kitazawa H. Effects of reversible flocculus shutdown of the monkey flocculus on the retention of adaptation of the horizontal vestibulo-ocular reflex. Neuroscience. 2003;118:563–570. doi: 10.1016/s0306-4522(02)00991-0. [DOI] [PubMed] [Google Scholar]
- Nakadate K, Shutoh F, Nagao S, Shigemoto R. Reduction of synapse density after long-term adaptation of horizontal optokinetic response in the mouse flocculus. Neurosci Res. 2004;50(Suppl 1):P2–P092. [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron. 2006;51:113–123. doi: 10.1016/j.neuron.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Meiri N, Dudai Y. Taste memory: the role of protein synthesis in gustatory cortex. Behav Neural Biol. 1993;59:49–56. doi: 10.1016/0163-1047(93)91145-d. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learn Mem. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Sakatani T, Isa T. PC-based high-speed video-oculography for measuring rapid eye movements in mice. Neurosci Res. 2004;49:123–131. doi: 10.1016/j.neures.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Schacher S, Castellucci VF, Kandel ER. cAMP evokes long-term facilitation in Aplysia sensory neurons that requires new protein synthesis. Science. 1988;240:1667–1669. doi: 10.1126/science.2454509. [DOI] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T. Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol. 2002;87:2770–2777. doi: 10.1152/jn.2002.87.6.2770. [DOI] [PubMed] [Google Scholar]
- Shutoh F, Katoh A, Kitazawa H, Aiba A, Itohara S, Nagao S. Loss of adaptability of horizontal optokinetic response eye movements in mGluR1 knockout mice. Neurosci Res. 2002;42:141–145. doi: 10.1016/s0168-0102(01)00308-x. [DOI] [PubMed] [Google Scholar]
- Shutoh F, Katoh A, Ohki M, Itohara S, Tonegawa S, Nagao S. Role of protein kinase C family in the cerebellum-dependent adaptive learning of horizontal optokinetic response eye movements in mice. Eur J Neurosci. 2003;18:134–142. doi: 10.1046/j.1460-9568.2003.02717.x. [DOI] [PubMed] [Google Scholar]
- Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S. Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience. 2006;139:767–777. doi: 10.1016/j.neuroscience.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and brain. Oxford: Oxford UP; 1987. [Google Scholar]
- Squire LR, Kandel ER. Memory: From mind to molecules. New York: Holt; 2000. [Google Scholar]
- Sweatt JD, Kandel ER. Persistent and transcriptionally-dependent increase in protein phosphorylation in long-term facilitation of Aplysia sensory neurons. Nature. 1989;339:51–54. doi: 10.1038/339051a0. [DOI] [PubMed] [Google Scholar]
- Wulff P, Schonewille M, Renzi M, Viltono L, Sassoè-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI. Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci. 2009;12:1042–1049. doi: 10.1038/nn.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]