

Abstract
The release of hormones and neurotransmitters, mediated by regulated exocytosis, can be modified by regulation of the fusion pore. The fusion pore is considered stable and narrow initially, eventually leading to the complete merger of the vesicle and the plasma membranes. By using the high-resolution patch-clamp capacitance technique, we studied single vesicles and asked whether the Sec1/Munc18 proteins, interacting with the membrane fusion-mediating SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins, affect fusion pore properties. Munc18-1 mutants were transfected into lactotrophs to affect the interaction of Munc18-1 with syntaxin1 (Synt1) (R39C), Rab3A (E466K), and Mints (P242S). Compared with wild-type, Munc18-1 E466K increased the frequency of the fusion event. The latter two mutants increased the fusion pore dwell-time. All the mutants stabilized narrow fusion pores and increased the amplitude of fusion events, likely via preferential fusion of larger vesicles, since overexpression of Munc18-1 R39C did not affect the average size of vesicles, as determined by stimulated emission depletion (STED) microscopy. Single-molecule atomic force microscopy experiments revealed that wild-type Munc18-1, but not Munc18-1 R39C, abrogates the interaction between synaptobrevin2 (Syb2) and Synt1 binary trans-complexes. However, neither form of Munc18-1 affected the interaction of Syb2 with the preformed binary cis-Synt1A-SNAP25B complexes. This indicates that Munc18-1 performs a proofing function by inhibiting tethering of Syb2-containing vesicles solely to Synt1 at the plasmalemma and favoring vesicular tethering to the preformed binary cis-complex of Synt1A-SNAP25B. The association of Munc18-1 with the ternary SNARE complex leads to tuning of fusion pores via multiple and converging mechanisms involving Munc18-1 interactions with Synt1A, Rab3A, and Mints.
Introduction
Regulated exocytosis is a spatiotemporally tightly controlled process ending with the fusion of the vesicle and the plasma membranes. This leads to the formation of the fusion pore, through which vesicle content is released (Burgoyne and Morgan, 2003). Fusion pore diameter is considered stable and narrow initially (Spruce et al., 1990), and subsequently increases upon stimulation (Vardjan et al., 2007). After formation, the fusion pore either fully expands (full fusion exocytosis) or reversibly closes (transient exocytosis) (Heuser and Reese, 1973). In the latter case, the release of vesicle cargo depends on the fusion pore dwell-time and diameter (Vardjan et al., 2007). However, the molecular events mediating transitions between stages of exocytosis are unclear.
Vesicle fusion with the plasma membrane is likely mediated by the formation of the soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) complex, consisting of vesicle synaptobrevin2 (Syb2), plasma membrane syntaxin1 (Synt1), and synaptosome-associated protein 25 (SNAP25) (Jahn et al., 2003). It was suggested that SNARE complexes are briefly associated with full fusion exocytosis (An et al., 2010). How are the SNARE complexes affecting the dynamics and diameter of fusion pores? What are the interactions between single SNARE molecules during these events? Studying the role of other SNARE-interacting proteins, such as Sec1/Munc18 (Brenner, 1974; Novick and Schekman, 1979) in the fusion pore dynamics, and how they affect single SNARE molecule interactions, could help answer these questions.
Munc18-1 can interact with SNAREs (Zilly et al., 2006; Toonen and Verhage, 2007; Südhof and Rothman, 2009) by: (1) binding to “closed” or “ajar” Synt1A, or (2) clasping the four-helix bundle of the assembled ternary SNARE complex to form a “hypercomplex.” Munc18-1-null mutant mice inhibited neurotransmission (Verhage et al., 2000) and exocytosis in chromaffin cells (Voets et al., 2001). In Drosophila neurons (Wu et al., 1998), expression of Munc18-1 orthologs inhibited exocytosis; in bovine chromaffin and PC12 cells, this effect was absent (Graham et al., 1997). These contrasting results may be due to Munc18-1 affecting different stages of exocytosis or due to interactions of Munc18-1 with other proteins, such as Rab3A (Graham et al., 2008) and Mints (Okamoto and Südhof, 1997), which are upstream regulators of SNARE-dependent membrane fusion. Amperometric studies in the presence of different Munc18-1 mutants that exhibit impairments in binding to individual binding proteins (Fisher et al., 2001; Ciufo et al., 2005; Barclay, 2008; Graham et al., 2008) indicate that Munc18-1 may regulate the fusion pore dynamics. In this study, a force spectroscopy approach (Liu et al., 2006) has been used to investigate single-molecule interactions between Munc18-1 and SNARE proteins (SNAREs). The results show that Munc18-1 can prevent the interactions between Synt1A-Syb2 while favoring the formation of the Synt1A-SNAP25B-Syb2 SNARE ternary complex. Stimulated emission depletion (STED) microscopy (Hell and Wichmann, 1994) was also used to determine the size of secretory vesicles. We correlate these results with cell-attached high-resolution membrane capacitance (Cm) measurements (Neher and Marty, 1982), where we directly monitored Munc18-1 mutant effects on exocytotic events.
Materials and Methods
Cell cultures and transfection.
Lactotroph cell cultures were prepared from adult male Wistar rats as described previously (Ben-Tabou et al., 1994; Jorgačevski et al., 2008). Cells were placed on poly-l-lysine-coated coverslips and maintained in high-glucose DMEM (Invitrogen) supplemented with 10% newborn calf serum and 2 mm l-glutamine in an atmosphere of humidified air (95%) and CO2 (5%).
The experimental animals were cared for in accordance with the International Guiding Principles for Biomedical Research Involving Animals, developed by the Council for International Organizations of Medical Sciences, and the Directive on Conditions for Issue of License for Animal Experiments for Scientific Research Purposes (Official Gazette of the Republic of Slovenia 40/85 and 22/87). The procedures using animals were approved by the Veterinary Administration of the Republic of Slovenia (approval no. 3440-29/2006).
One day after isolation, lactotrophs were cotransfected by plasmids (1 μg per coverslip) encoding Munc18-1 (WT) or a Munc18-1 mutant (R39C, P242S, and E466K) (Fisher et al., 2001; Ciufo et al., 2005) and with the plasmid (1 μg per coverslip) encoding enhanced green fluorescence protein (EGFP). Cells were transfected by lipofection (Invitrogen). All experiments were performed within 4 d of isolation.
Single-molecule studies: atomic force microscopy.
Generation of plasmids encoding recombinant proteins used in this study has been described previously (Liu et al., 2006). Briefly, the cytosolic tails of rat Syb2 (aa 1–94) or rat Synt1 (aa 1–266) were tagged with 6 histidines (H6) at their C termini (Syb2-H6 and Synt1A-H6); the full-length rat SNAP25B or rat Munc18-1 were tagged with H6 at their N termini (H6-SNAP25B and H6-Munc18-1). These plasmids were generously provided by Dr. Edwin R. Chapman, University of Wisconsin, Madison, WI. Similarly, we constructed a plasmid encoding the N terminus H6-tagged truncated form of rat Synt1 (H6-Synt1A178-266) containing the SNARE domain (aa 178–266), but lacking the N-terminal part of the molecule. Plasmids encoding the full-length rat Munc18-1 and its mutated form (R39C) were tagged with glutathione S-transferase (GST) on their N termini (Fisher et al., 2001), and were provided by Dr. Jonathan Pevsner, Johns Hopkins University, Baltimore, MD. All plasmids were used in recombinant protein production. Proteins containing the H6 tag were purified using Ni-NTA agarose beads (Qiagen); GST-tagged proteins were purified using glutathione columns (GE Healthcare Life Sciences). Purified proteins were quantified using the Bradford reagent (Pierce Biotechnology) and bovine serum albumin as a standard (Liu et al., 2006). Expression of the recombinant proteins was confirmed by Western blotting as we described previously (Ciufo et al., 2005; Montana et al., 2009). Immunoreactivity was detected using enhanced chemiluminescence (GE Healthcare Life Sciences), with all proteins showing single immunoreactive bands with appropriate molecular weights. We find no evidence that the R39C mutation leads to a loss of stability or misfolding (Fisher et al., 2001; Ciufo et al., 2005).
Nickel films ∼150 nm in thickness were deposited on atomic force microscope (AFM) triangular silicon nitride cantilevers (320 μm long; Digital Instruments) with integral tips or glass coverslips (Fisher Scientific) using a thermal evaporator (Quorum Technologies Emitech; model E6500). After nickel oxidation in air for ∼24 h, the tips were functionalized with solutions of Synt1A-H6 (0.1 g/L), H6-Synt1A178–266 (0.22 g/L), or H6-SNAP25B (0.1 g/L) recombinant proteins for 3 h at room temperature. In some experiments, after rinsing, tips coated with Synt1A-H6 were subsequently co-functionalized with H6-SNAP25B for 30 min. Nickel-coated glass coverslips were functionalized with Syb2-H6 (0.17 g/L) or H6-Munc18-1 (0.1 g/L) by applying a solution containing proteins for 1 h at room temperature. Following functionalization, coverslips and tips were rinsed three times with internal solution containing the following (in mm): 140 potassium gluconate, 10 NaCl, and 10 HEPES, pH 7.35, and then used in experiments within a few hours. Before experiments, the glass coverslips were mounted on metal disc AFM sample holders.
Our directional approach for protein deposition allows attached proteins to freely mechanically interact. The limitation of the method is that only protein interactions whose strength is less than the strength of binding between Ni2+ and H6 can be investigated. This was the case for all interactions reported in the present study (see Fig. 2), since the mean value of the single molecule unbinding force between H6 and Ni2+ is 525 ± 41 pN at ∼ 20 nN/s loading rate, as we reported previously (Montana et al., 2008). To accommodate for variations in the success of the protein deposition on the tips and coverslips, we performed matching controls with all of the treatments to allow for day-to-day comparison of the data.
Figure 2.

The force values for dissociation of proteins as a function of the force loading rate. The mean rupture force increases exponentially with the increase in the loading rate. The solid lines represent fits according to the equation Y = a + b × ln(X), where Y is the rupture force (in pN), and X is the force loading rate (pN/s); a and b are fitting parameters. Extrapolating the force loading rate to zero force enables us to estimate spontaneous dissociation lifetimes (Table 1; t0). Points represent mean ± SEM. Interacting proteins are listed according to their location (tip/coverslip, solution). A, Syt1A/Munc18-1 (47–67 events; r = 0.95). B, Synt1A/Syb2 (45–65 events; r = 0.98). C, Synt1A/Syb2, Munc18-1 R39C (51–59 events; r = 0.97). D, Synt1A+SNAP25B/Syb2 (49–63 events; r = 1.00). E, Synt1A+SNAP25B/Syb2, Munc18-1 (44–61 events; r = 0.99). F, Synt1A+SNAP25B/Syb2, Munc18-1 R39C (45–66 events; r = 0.99). r is the fitting correlation coefficient.
We used an AFM (LFM-3) equipped with an E scanner (Digital Instruments) in force spectroscopy mode. All experiments were performed at room temperature (20–24°C) in a fluid cell filled with internal solution. In some experiments, an internal solution contained either GST–Munc18-1 (0.15 g/L) or GST–Munc18-1(R39C) (0.9 g/L). Spring constants, ranging from 10 to 20 mN/m, were determined for each cantilever, using a previously described method (Hutter and Bechhoefer, 1993), and used for calculation of force. The bending of the cantilever was taken into account in the calculation of the extension (Harris et al., 2000). The piezoelectric tube extension, including nonlinearities, was calibrated using an interferometer for all force-loading rates used (Chen and Mohideen, 2001). All extension and force measurements are expressed as mean ± SEM. All distributions of the extension and force displayed only a single (unitary) peak/mode, since our method was optimized for the measurements of single intermolecular bonds (Liu et al., 2006). As we previously reported (Montana et al., 2009), force distributions containing peaks that were quantized in integer multiples of the unitary peak (for details, see Florin et al., 1994) could be observed using a thicker layer of nickel, resulting in a corrugated surface of this metal, for deposition of proteins, but such tips and coverslips were not used in the present study.
The contact force between functionalized tips and coverslips was between 1 and 4 nN; the contact time varied between 50 and 200 ms. Because of the forces applied to the proteins on contact, there was an exponential enhancement in the rate of assembly of the protein–protein complex by a factor of 1010–1035 (Bell, 1978; Evans, 2001; Hummer and Szabo, 2001; Liu et al., 2006). We report on the probability and the extension for dismantling of the protein–protein interactions using tips and coverslips that were pulled away at a single retraction velocity of 1.5 μm/s (corresponding to ∼20 nN/s force loading rate). To report on stability of the protein–protein interactions, spontaneous dissociation times were estimated from the dismantling/rupture force measurements obtained using various force loading rates, ranging from 538 pN/s to ∼68 nN/s.
Immunocytochemistry, confocal microscopy, and image analysis.
Lactotrophs were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Nonspecific immunoreactive sites were blocked with 3% bovine serum albumin and 10% goat serum. We used the following antibodies: rabbit (1:20, Millipore Bioscience Research Reagents) and guinea pig (1:1000, Dr A. F. Parlow, National Hormone and Peptide Program, Torrance, CA) anti-prolactin (Prl) polyclonal antibodies, rabbit anti-Munc18-1 polyclonal antibodies (1:200, Synaptic Systems), and mouse anti-Synt1 monoclonal antibodies (1:100, Synaptic Systems) in combination with the appropriate secondary antibodies (1:600, Invitrogen), conjugated to fluorescent dyes Alexa Fluor 546 (red) and 488 (green). For double staining, the two primary antibodies were added sequentially. Before confocal microscopy, cells were treated using the Slow Fade Gold Antifade kit (Invitrogen).
The fluorescent images were acquired with an inverted confocal microscope (LSM 510; Zeiss) with an oil-immersion plan apochromatic objective [63×, 1.4 numerical aperture (NA)] using 488 nm Ar-ion and 543 nm He-Ne laser excitation. The fluorescence was collected through the bandpass emission filter (505–530 nm) and through the long-pass emission filter (cutoff below 560 nm).
The colocalization of Munc18-1, prolactin, and Synt1 was analyzed by the program CellAn, written for MATLAB (MathWorks) software (Kreft et al., 2004), that counts red, green, and colocalized pixels. The colocalization was calculated for all pixels above the threshold, which was set at 25% of maximum green and red fluorescence intensity, and was expressed as the percentage of colocalized red pixels.
To evaluate the average rate of transfection, we transfected lactotrophs with the plasmid encoding WT Munc18-1 as described above. Two days after the transfection (85% of electrophysiological recordings were made 2 d after the isolations or later), transfected and nontransfected cells were fixed and subsequently labeled with anti-Munc18-1 antibody in combination with Alexa Fluor 488 secondary antibody. Fluorescence of labeled-transfected and nontransfected cells was then visualized using identical image acquisition settings. For quantification of fluorescence, the stained cell area was defined and the fluorescence intensity per area determined using LSM confocal software. The level of immunostaining with Munc18-1 antibody was determined as the average fluorescence intensity per stained area rather than total fluorescence, to avoid errors arising from variations in size of the selected regions of interest.
Electrophysiology.
Capacitance recordings were performed at room temperature on cells cotransfected with EGFP and the mutant of choice. Cells were bathed in external solution containing the following (in mm): 130 NaCl, 5 KCl, 8 CaCl2, 1 MgCl2, 10 d-glucose, 10 HEPES, pH 7.2/NaOH. Cells expressing EGFP were identified using an excitation wavelength of 470 nm provided by a monochromator (Polychrome IV, Till Photonics). Emission light was collected through the 515–565 nm filter (Zeiss) and captured by a Till IMAGO CCD camera connected to a 12-bit frame grabber (Till Photonics).
Capacitance in attached cells was measured as described previously (Vardjan et al., 2007; Jorgačevski et al., 2008) with a dual-phase lock-in patch-clamp amplifier (1591 Hz, 111 mV rms; SWAM IIC, Celica). We used thick-walled glass pipettes, which were fire polished and heavily coated with Sylgard 184. When filled with and immersed in the external solution, their resistance was 2–4 MΩ. The pipette potential was held at 0 mV. The criteria for the correct phase settings were as described previously (Neher and Marty, 1982; Zorec et al., 1991).
Transient fusion events in the imaginary part of the admittance signal (Im) that exhibited projections to the real part of the admittance signal (Re) were used to calculate fusion pore conductance (Gp = (Re2 + Im2)/Re) (Lollike and Lindau, 1999) and vesicle capacitance (Cv = [(Re2 + Im2)/Im]/ω, where ω denotes angular frequency). Gp was used to calculate fusion pore diameter: Gp = (πr2)/(ρλ), where r denotes fusion pore radius, ρ is the estimated resistivity of the saline (100 Ω cm), and λ is the estimated length of a junction channel (15 nm) (Spruce et al., 1990). The vesicle diameter was calculated by assuming spherical geometry of vesicles and by using specific membrane capacitance (Cm) of 8 fF/μm2.
We considered a fusion event to be transient if an on-step in Cv was followed by an off-step within 15 s and the off-step amplitude did not differ from the preceding on-step amplitude by >15%.
STED microscopy.
STED microscopy was performed with a custom-built setup. The laser beams for excitation (570 ± 2 nm) and STED laser beam (720 ± 10 nm) were all provided by the same laser source (Wildanger et al., 2008). The pulse energies of the excitation and STED pulses at the sample were 1 pJ and 1.5 nJ, respectively. The objective was a 1.4 NA, 100× lens (PL APO 100×/1.40–0.7 OIL, Leica Microsystems). STED-imaging was obtained by piezo-scanning the sample at a pixel dwell time of 1 ms with a pixel size of 20 × 20 nm.
Fluorescence labeling was performed as described in Immunocytochemistry, confocal microscopy, and image analysis (Materials and Methods). We used mouse anti-prolactin polyclonal antibodies (1:600, Thermo Fisher Scientific) in combination with the red-emitting dye Atto 590 (1:100, Enzo Biochem), for which emission was detected at 600–640 nm.
Data analysis.
The analysis of force spectroscopy data was performed using Multimode SPM software (Digital Instruments). We used Matlab (MathWorks) to analyze the electrophysiological data. The analysis of STED images was performed using NIH ImageJ (http://rsbweb.nih.gov/ij/index.html; Open Source). Figures were assembled for display using Adobe Photoshop. Values are presented as mean ± SEM of average values in respective recordings. Normality of the data distribution was tested using the Shapiro–Wilk test. The Mann–Whitney U test was used to compare electrophysiological data. Student's t test was used to compare data obtained with STED microscopy. One-way ANOVA followed by Fisher's least significant difference test was used for comparisons of extension data obtained using force spectroscopy.
Results
Single-molecule mechanical interactions between the SNARE partners and Munc18-1
We assessed the mechanical interactions between Munc18-1 and the SNAREs using single-molecule force spectroscopy (Table 1; Fig. 1). The recombinant proteins had H6 tags attached on either their C termini (cytosolic domains of Syb2 and Synt1A) or their N termini (full length of SNAP25B and Munc18-1, and truncated Synt1A178–266). These proteins were directionally attached to the nickel-coated AFM cantilevers and coverslips through histidine residues–Ni2+ coordinate bonds (Fig. 1Ai) to assure appropriate orientation of proteins (Liu et al., 2006, 2008; Montana et al., 2008, 2009). We investigated the probability and extension of the interactions between proteins using a single, relatively fast force loading rate (∼ 20 nN/s). To investigate stability of the interactions, reported as spontaneous dissociation lifetimes, we measured interaction forces at various force loading rates (∼0.5–68 nN/s). First, we investigated interactions between Munc18-1 and one of the SNAREs. When the proteins that can interact are brought into contact, they can form an intermolecular bond, the presence of which can be observed as the proteins are pulled away from each other using the force–distance (F-D) plot (Liu et al., 2006, 2008, 2009; Montana et al., 2008, 2009). Figure 1Aii displays the retraction part of a typical F-D plot recorded for individual protein pairs of Munc18-1 and Synt1A. The initial downward deflection in the trace (asterisk, Fig. 1Aii) represents a nonspecific interaction (Liu et al., 2006). The second downward deflection in the F-D plot (arrow) indicates a specific protein–protein interaction. We recorded such interactions in ∼32% of F-D plots when probing Munc18-1 deposited on the coverslip with a Synt1A-coated tip; this probability is similar to that previously observed for various pairs of SNAREs (Liu et al., 2006, 2008, 2009; Montana et al., 2009). Extensions for the Synt1A-Munc18-1 pair were 16.4 ± 1.1 nm, exceeding the length of Synt1A SNARE domain (aa 180–262; 83 aa × 0.15 nm = 12.45 nm, where 0.15 nm is the axial distance between two amino acid residues in the α helix) (Stryer, 1995) combined with the uncoiled sequence from the SNARE domain to the anchor of the cytosolic Synt1A C terminus to the coverslip (aa 263–266; 4 aa × 0.25 nm = 1 nm, where the axial distance between two amino acid residues in the uncoiled peptide is 0.2–0.3 nm) (Stryer, 1995). As previously indicated, we observed only single mode/peak of extension and force (see below) measurements. The measured extension is inconsistent with solely observing a Munc18-1 interaction with the very N-terminal part of Synt1A, which would be much larger. Rather, Synt1A-Munc18-1 extension was significantly shorter than that measured for the Synt1A-Syb2, but longer than that of ternary Synt1A+SNAP25B-Syb2 complex interactions (Table 1). Thus, it most likely reports on stretching the entire SNARE domain and a limited portion of Synt1A N-terminally from it, perhaps a part of a linker region to the Habc domain, and/or interacting domains of Munc18-1. Forces necessary to rupture the intermolecular interactions between Synt1A and Munc18-1 were 246 ± 11 pN at ∼ 20 nN/s force loading rate, consistent with previous measurements obtained for pairs of SNAREs involving Synt1A (Liu et al., 2006, 2008, 2009; Montana et al., 2009). However, this pulling rate, optimized to detect differences between the probability and extension of various interacting exocytotic proteins (Table 1), cannot offer meaningful comparison of rupture forces (one-way ANOVA, F(5,366) = 0.8). Consequently, we performed force loading rate experiments to investigate the stability of the interactions between Munc18-1 and the SNAREs (Fig. 2), as described previously (Liu et al., 2006). As the force loading rate is increased, the mean rupture force to take apart interacting proteins displays an exponential relationship. Spontaneous dissociation lifetime of the interactions can be calculated by extrapolating the mean rupture force to zero loading rate; interaction between Synt1A and Munc18-1 had a spontaneous dissociation lifetime of 880 ms (Table 1). Together, the combined data do not support the existence of Munc18-1 bound to the very N-terminal portion of Synt1A via a low-affinity site alone, but rather show evidence only for Munc18-1 strongly interacting with four coiled coils of Synt1A. To assess the specificity of Synt1A-Munc18-1 interactions, we also probed Munc18-1 with truncated Synt1A178–266, which lacks the Munc18-1 Habc binding sites. We did not detect protein–protein interactions. Similarly, Munc18-1 did not interact with Syb2 or SNAP25B (Table 1).
Table 1.
The probability, extension, and spontaneous dissociation lifetimes of interactions between various SNARE proteins and Munc18-1
| Tip | Coverslip | Solution | Tested | Positive | Positive (%) | Extension (nm) | t0 (s) |
|---|---|---|---|---|---|---|---|
| Synt1A-H6 | H6-Munc18-1 | 210 | 67 | 32 | 16.4 ± 1.1 | 0.88; A | |
| H6-Synt1A178–266 | H6-Munc18-1 | 190 | 9 | 5 | |||
| H6-SNAP25B | H6-Munc18-1 | 185 | 6 | 3 | |||
| Syb2-H6 | H6-Munc18-1 | 190 | 8 | 4 | |||
| Synt1A-H6 + H6-SNAP25B | H6-Munc18-1 | 205 | 8 | 4 | |||
| Synt1A-H6 | Syb2-H6 | 200 | 65 | 33 | 22.9 ± 0.9* | 0.17; B | |
| Synt1A-H6 | Syb2-H6 | GST-Munc18-1 | 175 | 6 | 3 | ||
| Synt1A-H6 | Syb2-H6 | GST-Munc18-1(R39C) | 175 | 59 | 34 | 22.7 ± 0.9* | 0.18; C |
| Synt1A-H6 + H6-SNAP25B | Syb2-H6 | 180 | 63 | 35 | 12.3 ± 0.7† | 2.00; D | |
| Synt1A-H6 + H6-SNAP25B | Syb2-H6 | GST-Munc18-1 | 170 | 57 | 34 | 12.5 ± 1.0† | 2.09; E |
| Synt1A-H6 + H6-SNAP25B | Syb2-H6 | GST-Munc18-1(R39C) | 180 | 61 | 34 | 12.6 ± 0.8† | 2.04; F |
The probability (percentage) and extension (nanometer) values were obtained at force loading rate of ∼20 nN/s; extension values are reported only for interactions that display significant incidence (>5%). Significance was established using one-way ANOVA followed by Fisher's least significant difference test: significantly larger (*) or smaller (†) than other groups, p < 0.01. Spontaneous dissociation lifetimes [t0 (s)] were estimated from force loading experiments; letters indicate matching graphs in Fig. 2 containing force measurements at various force loading rates which were used to obtain estimates.
Figure 1.

Munc18-1 reduces the probability of interactions between Synt1A (green) and Syb2 (red) in the absence of SNAP25B (blue). Ai, The experimental approach. Recombinant full-length Munc18-1 (H6-Munc18-1; orange) is attached to the nickel-coated coverslip surface through H6 at its N terminus; the recombinant cytosolic tail Synt1A-H6 (green) is similarly attached by means of a C terminus histidine tag to the nickel-coated cantilever tip. These two proteins are brought into contact (approach; arrow pointing down) and then taken apart (retract, arrow pointing up). Aii, The retraction part of a typical force–distance (F-D) plot using a Synt1A-H6 functionalized tip and a H6-Munc18-1 functionalized coverslip. Asterisk indicates the segment of the plot while the coverslip and cantilever are still in contact; the arrow indicates the rupture of the protein–protein intermolecular bond. These two marks are omitted from the remaining F-D plots for clarity. Bi, Synt1A-H6 functionalized tips were used to probe Syb2–H6 functionalized coverslips (double arrow). Bii, The retraction part of a typical F-D plot showing interactions between Synt1A and Syb2. These interactions are absent when Munc18-1 is present in the recording solution (Biii; boxed area), but not in the presence of mutated Munc18-1 R39C protein (Biv). Bv, Synt1A-H6+H6-SNAP25B co-functionalized tips were used to probe Syb2–H6 functionalized coverslips (double arrow). Bvi, The retraction part of a typical F-D plot showing interactions between the proteins of the ternary SNARE complex. Bvii, Bviii, These interactions were unaffected by the presence of Munc18-1 (Bvii) or its R39C mutated form (Bviii). Drawings are not to scale. Position of Munc18-1 is arbitrary, as well as is the display of Synt1A in “ajar” form.
Next, we studied the probability of Munc18-1 interactions with binary SNARE protein pairs. In the first experiments, the AFM cantilevers were co-functionalized with Synt1A-H6 and H6-SNAP25B to form a binary SNARE cis-complex, yielding 1:1 stoichiometry, that readily interacted with Syb2 to form the ternary Synt1A+SNAP25B-Syb2 complex (Liu et al., 2006; Montana et al., 2009) (Table 1). The binary Synt1A+SNAP25B cis-complex was probed using nickel-coated coverslips functionalized with H6-Munc18-1 (Table 1). Synt1A, containing the entire cytosolic domain that alone interacted with Munc18-1, did not show such interactions when precomplexed with SNAP25B, indicating that the binary cis-complex does not interact with Munc18-1. Furthermore, our data do not support the existence of a possible intermediate complex in which Munc18-1 is bound to the weak interactive site at the extreme N terminus of open Synt1A 1:1 complexed with SNAP25B.
The second set of experiments assessed interactions between binary SNARE trans-complexes as previously observed (Liu et al., 2006). Here, we probed Syb2-H6 functionalized coverslips with Synt1A-H6 functionalized tips (Fig. 1Bi,ii). Individual Syb2-Synt1A trans-complexes were formed at contact, and then dismantled as the tip and coverslips were pulled apart. The probability of Syb2-Synt1A interactions, that occur via their SNARE domains, was ∼33% with measured extension of 22.9 ± 0.9 nm and estimated spontaneous dissociation time of 170 ms (Table 1), consistent with previous reports (Liu et al., 2006, 2008, 2009; Montana et al., 2009). Synt1-Syb2 interactions were abrogated in the presence of Munc18-1, but not in the presence of Munc18-1 R39C mutant (exhibiting reduced Synt1A binding activity; Fisher et al., 2001) (Table 1; Fig. 1Biii,iv). In the presence of R39C mutant, the probability, extension, and spontaneous dissociation time of Synt1A-Syb2 interactions were similar to those measured in control Synt1A-Syb2 interactions in the absence of any other protein (Table 1; Fig. 1Bii). These data indicate that binding of Munc18-1 to Synt1A prevents formation of the binary Syb2-Synt1A trans-complex.
To study the effect that Munc18-1 may exert on the ternary SNARE complex, we used the AFM cantilevers co-functionalized with the binary cis-complexes containing Synt1A-H6+H6-SNAP25B; the nickel-coated coverslips were functionalized with Syb2-H6 (Fig. 1Bv). At the contact site with the coverslip, a binary Synt1A-SNAP25B cis-complex at the tip binds Syb2 on the coverslip to form a ternary Syb2-Synt1A-SNAP25B core SNARE complex. Retracting the coverslip dissociated this complex. We measured the probability for this type of single intermolecular interaction in 35% of attempts (Table 1; Fig. 1Bv,vi), with measured extension of 12.3 ± 0.7 nm and estimated spontaneous dissociation time of 2 s (Table 1), consistent with previous reports (Liu et al., 2006, 2009; Montana et al., 2009); extension was shorter than that of both Synt1A-Syb2 and Synt1A-Munc18-1 interactions (Table 1). When Munc18-1 was present in solution at the contact site with the coverslip, a binary Synt1A-SNAP25B cis-complex at the tip binds Syb2 on the coverslip and Munc18-1 in solution to form a hypercomplex. The presence of Munc18-1 or its R39C mutant did not affect the probability, extension, and spontaneous dissociation time of interactions within the ternary SNARE complex (Table 1; Fig. 1Bvii,viii). Thus, once SNAP25B binds Synt1A, Munc18-1 does not prevent Synt1A interaction with Syb2. Furthermore, the unchanged extension measurements and spontaneous dissociation time for the disassembly of the ternary complex indicate either that Munc18-1 is not bound to the ternary complex or that Munc18-1 clasping of this complex does not make a major contribution to the docking otherwise accomplished by the ternary complex alone. Furthermore, we have observed only a single mode of force and (short) extension measurements, which suggests that Munc18-1 could not associate with the assembled ternary SNARE complex solely via N terminus of the open and complexed Synt1A.
Capacitance steps in lactotrophs transfected with Munc18-1 mutants
Having determined mechanical interactions between Munc18-1 and SNAREs that likely play a role in vesicular predocking, we studied the postdocking role of Munc18-1, more specifically, its effect in vesicle fusion using live cells, rat lactotrophs, a suitable cell model to study unitary exocytic events (Vardjan et al., 2007; Jorgačevski et al., 2008). We expressed the Munc18-1 mutants and studied the fusion pore properties, using overexpression of WT protein as a control. In addition to the Munc18-1 R39C mutant, we also tested the mutant Munc18-1 E466K, which exhibits an increased direct binding of Munc18-1 to Rab3A (Graham et al., 2008), and the mutant P242S with an apparent impairment in Mint binding (Ciufo et al., 2005).
Immunocytochemical labeling by Prl antibodies in 3 different cell preparations showed fluorescent signal in 89 ± 4% of cells (n = 16 microscope fields), indicating the purity of our preparation. To verify that lactotrophs endogenously express Munc18-1 and Synt1, we performed double immunostaining with antibodies against Munc18-1, Synt1, and Prl (see Materials and Methods; Fig. 3A,B), which confirmed that Munc18-1, Synt1, and Prl are endogenously expressed in lactotrophs, as previously shown (Jacobsson and Meister, 1996).
Figure 3.

Discrete irreversible and reversible capacitance steps in lactotrophs transfected with Munc18-1. A, The colocalization of Synt1 (green) and Prl (red) (i), and Munc18-1 (green) and Prl (red) (ii). The colocalization of red and green channels is represented in yellow (overlay). B, The average colocalization of Munc18-1 with Synt1 was 90 ± 2% (with respect to Synt1). The colocalization of Prl with Synt1 and Munc18-1 was 46 ± 3% and 53 ± 11%, respectively (both with respect to Prl). Numbers above the error bars indicate the number of fluorescent cells analyzed. The colocalization between Prl-Synt1 and Prl-Munc18-1 was not significantly different. C, Confocal fluorescent micrographs showing the immunochemical localization of anti-Munc18-1 antibody in control cells (i) and in cells overexpressing WT Munc18-1 (ii). Note the increased fluorescence intensity of transfected lactotroph compared with the wild-type cell. Di, A DIC image of the cell. Successfully transfected lactotrophs were identified by cotransfecting respective Munc18-1 mutants with EGFP as shown in Dii, where the same cell is viewed in epifluorescence. Diii, The same cell as in Di and Dii in the cell-attached configuration. E, Representative irreversible downward and upward steps (arrowheads) in the Im trace, likely reflecting irreversible events of endocytosis and exocytosis, respectively. F, Representative reversible steps, observed in the Im trace, were used to calculate vesicle capacitance (Cv) and, if exhibiting a measurable projection to the Re trace (reversible steps on the right side of the panel), fusion pore conductance (Gp). Arrows in Gp traces indicate immeasurable projections. Asterisks denote calibration pulses. Scale bars, 10 μm.
The efficiency of transfection was tested by transfecting lactotrophs with the plasmid encoding Munc18-1 WT and immunostaining these cells with anti-Munc18-1 antibodies. The average fluorescence of randomly chosen immunostained transfected lactotrophs (Fig. 3Cii) was significantly higher than that recorded in nontransfected cells (Fig. 3Ci; 8.5 ± 1.0 a.u./μm2 vs 6.1 ± 0.2 a.u./μm2; n = 10 cells in each group; p < 0.05). Thus, the expression level of Munc18-1 can be increased in single cells by transfection.
We then monitored changes in Cm in cells expressing Munc18-1 mutants by cotransfecting the plasmid encoding the green fluorescent protein, EGFP (Fig. 3D; Graham et al., 2004). Twenty percent of coverslips from each cell preparation were controls (cells transfected with EGFP and Munc18-1 WT). We observed irreversible steps in Cm (Fig. 3E), most likely because of full fusion/fission events, and reversible steps in Cm, likely representing transient exocytotic fusion events (Fig. 3F) (Neher and Marty, 1982; Alvarez de Toledo et al., 1993). Discrete steps in Cm were observed in 46 of 165 cells (29%), as reported previously (Vardjan et al., 2007; Jorgačevski et al., 2008).
In some of the transient fusion events recorded, in the imaginary part of the admittance signal (Im, reflecting changes in Cm), a cross talk to the real part of the admittance signal (Re) was detected (Fig. 3F, right). In these events, the cross talk was used to calculate vesicle capacitance (Cv) and fusion pore conductance (Gp) (Fig. 3F; see Materials and Methods). For transient fusion events devoid of cross talk, Cv equals the change in Im; Gp is too large to be determined with this experimental setup (Debus and Lindau, 2000). The limit of our experimental setup was ∼500 pS, depending on the noise in the recording (Jorgačevski et al., 2010).
Amplitudes of the observed upward (n = 1577) and downward (n = 1537) steps in Cv from controls and treated cells were similar (p = 0.68; Fig. 4A). Most of these events (n = 1470) consisted of transient fusion events, where upward steps in Cv are followed by a downward step of similar amplitude within a few seconds (Fig. 4B). The high correlation between the amplitudes of upward and downward discrete steps in Cv (r = 0.97) and the slope near 1 (0.95 ± 0.01; n = 1470) support the view that reversible events represent transient fusion pore openings (Alvarez de Toledo et al., 1993). Most transient fusion events in individual patches appeared in bursts, and the ensuing events had similar amplitudes (Fig. 4C, inset), likely due to repetitive interactions of the same vesicle with the plasma membrane (Jorgačevski et al., 2010). The calculated vesicle diameters (see Materials and Methods) were in the range 100–500 nm (Fig. 4A), with an average of 233 ± 2 nm (n = 1577), as reported for Prl-containing vesicles (Angleson et al., 1999; Huerta-Ocampo et al., 2005).
Figure 4.
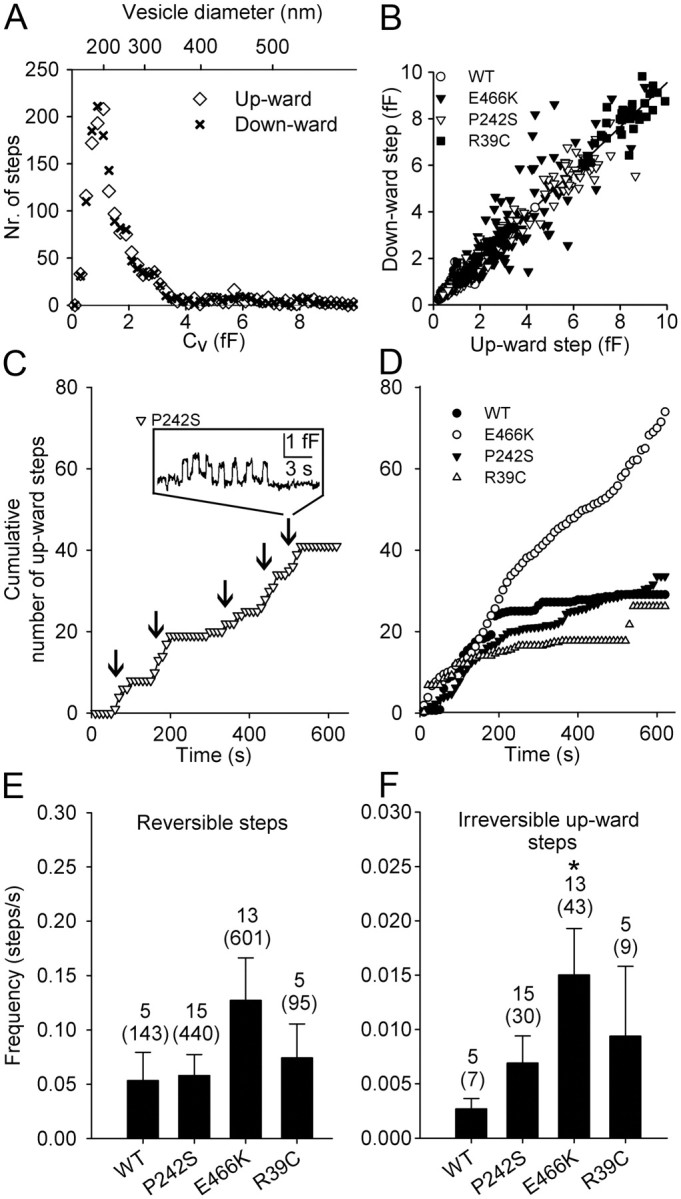
The amplitude and the frequency of appearance of irreversible and reversible fusion events. A, The frequency distribution of all amplitudes of Cv upward (open symbols; n = 1577) and downward (closed symbols; n = 1537) steps (left) was similar (p = 0.68). B, The amplitudes of Cv upward and ensuing downward steps within 15 s were best fitted with a linear regression line: y = (0.95 ± 0.01) × x + (0.06 ± 0.01) (r = 0.97; n = 1470), indicating the upward step denotes fusion pore opening and the downward steps denotes subsequent closure of the fusion pore. C, The representative cumulative time course of fusion events in a cell transfected by Munc18-1 P242S, analyzed in 10 s epochs. The inset shows the reversible steps in membrane capacitance that usually occurred in bursts. Five were present in this recording (arrows). D, The average cumulative number of fusion events in cells cotransfected with EGFP and Munc18-1 WT and in cells cotransfected with EGFP and a mutant of Munc18-1 (E466K, P242S and R39C). E, F, The average frequency of reversible (E) and irreversible (F) fusion events. The reversible fusion events occurred approximately tenfold more frequently than the irreversible fusion events (note the one order of difference in the scales of the graphs). Expression of E466K increased the frequency of irreversible fusion events sevenfold compared with Munc18-1 WT (p = 0.02). Cells transfected with P242S or R39C exhibited similar frequencies for both types of fusion events as cells transfected with Munc18-1 WT. Numbers above the error bars indicate the number of analyzed recordings (samples in statistical analysis) and fusion events (in parentheses).
Munc18-1 E466K increases the frequency of fusion events
Previous amperometric studies suggested that the Munc18-1E466K mutant increases the frequency of individual release events (Ciufo et al., 2005). Therefore, we studied whether the expression of Munc18-1 mutants in nonstimulated lactotrophs affects the frequency of discrete changes in Cm. In the presence of the E466K mutant, the average number of fusion events increased versus controls (Fig. 4D). In contrast, the cumulative time course of increase in the number of fusion events following expression of P242S and R39C remained similar to that of controls (Fig. 4D). The expression of E466K mutant significantly increased the frequency of irreversible fusion events (p = 0.02). The expression of P242S and R39C elicited no changes, compared with controls (Fig. 4E,F). Transient reversible fusion pore events were approximately tenfold more frequent than the irreversible fusion events (Fig. 4E,F, compare scales), as reported for nontransfected lactotrophs (Jorgačevski et al., 2008).
Munc18-1 E466K and P242S increase fusion pore dwell-time
The kinetics of vesicle cargo release in transient exocytosis can be regulated by the fusion pore dwell-time—the effective duration of the connection (i.e., fusion pore) between the vesicle lumen and the cell exterior. Therefore, we analyzed the time between the upward and the subsequent downward Cv steps of transient events, corresponding to the fusion pore dwell-time, which was unaffected by the expression of R39C compared with the overexpression of the WT protein (Fig. 5A,B). In contrast, transfection with P242S and E466K mutants increased the average fusion pore dwell-time ∼5-fold, respectively (Fig. 5A,B), which was independent of transient fusion event amplitude (data not shown).
Figure 5.
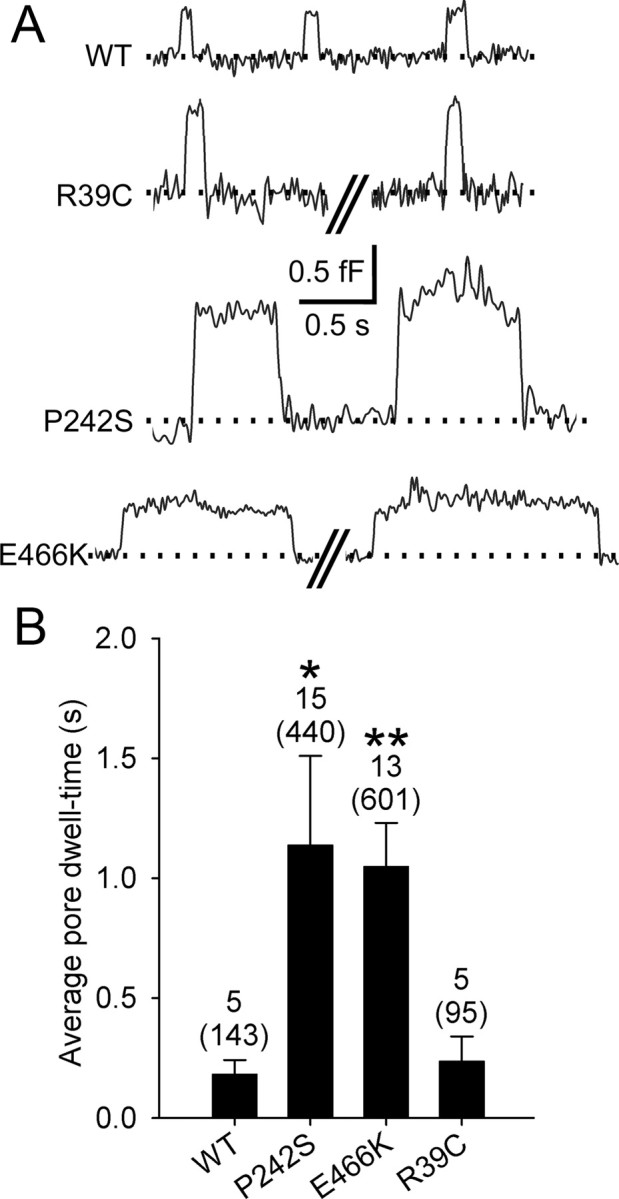
The effect of expression of Munc18-1 mutants on the fusion pore dwell-time. A, Representative reversible transient fusion events, observed in the Im trace. B, The average fusion pore dwell-time increased significantly in cells transfected with Munc18-1 P242S and E466K, compared with controls (p = 0.04 and 0.005, respectively). Cells transfected with Munc18-1 R39C exhibited no differences in the average pore dwell-time, compared with cells transfected with Munc18-1 WT. Numbers above the error bars indicate the number of analyzed patches (samples in statistical analysis) and reversible steps (in parentheses).
Munc18-1 mutants stabilize narrow fusion pores
Vesicle cargo discharge in transient exocytosis can be regulated not only by the fusion pore dwell-time but also by the geometry (effective diameter) of the fusion pore, which has been shown to have initial conductance similar to ion channels (Lollike et al., 1995). Most fusion pores then expand, although some remain narrow (Fig. 3F) (Vardjan et al., 2007). Transient fusion events with a narrow fusion pore can be determined by measuring the cross talk between the Im and Re (Lollike et al., 1995).
Analyses showed that ∼50% of the transient events in controls exhibit measurable fusion pores. With Munc18-1 P242S and E466K mutants, the percentage of narrow fusion pores increased to 60% and 67%, respectively. In cells transfected with the Munc18-1 R39C, nearly all transient fusion events (99%) exhibited narrow fusion pores. The average Cv in individual patches of cells transfected with E466K and P242S mutants approximately doubled compared with controls; the Cv in cells transfected with R39C increased fourfold (Fig. 6A). This may mean that larger vesicles become more competent to enter the fusion process. However, we have not detected any correlation between the frequency of occurrence of transient fusion events and their Cv (Fig. 6B).
Figure 6.
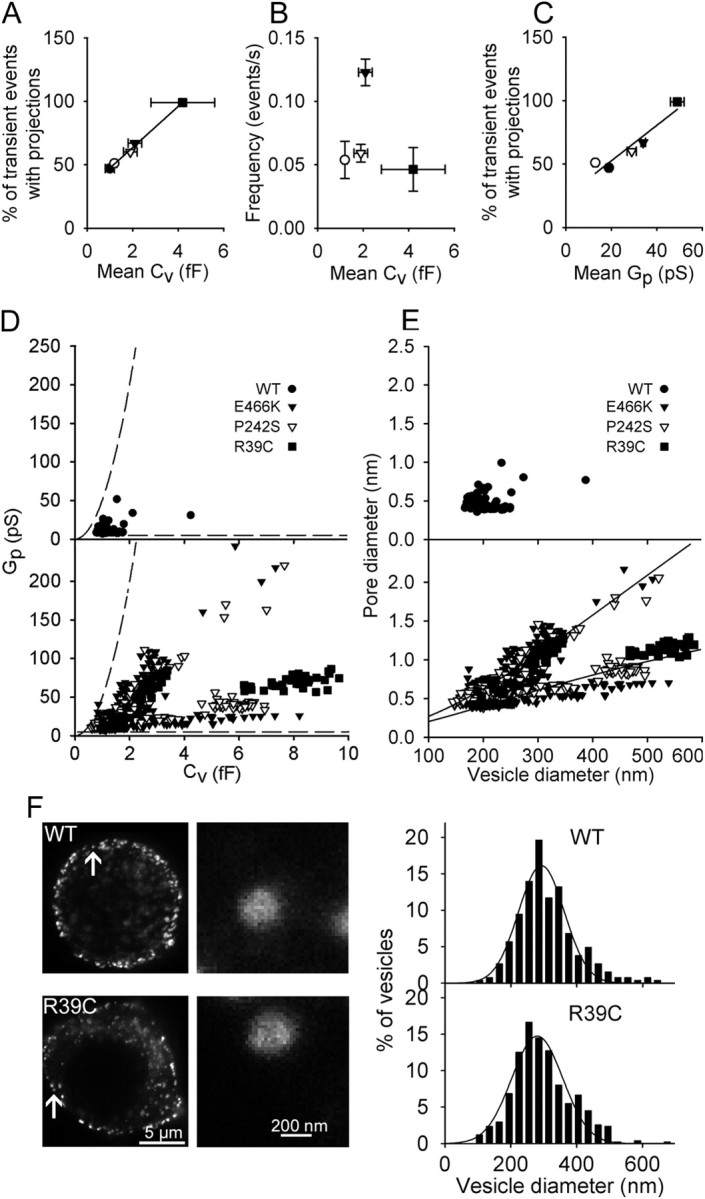
Munc18-1 mutants stabilize the fusion pores at narrower diameters. A, The percentage of transient events exhibiting projections to the Re (%) was higher in experiments where the mean Cv was larger. The regression line is of the form: (%) = (16 ± 1%/fF) × Cv + (31 ± 1%). B, The average frequency of transient events was uncorrelated with the mean Cv. Correlation coefficient was 0.24, not significantly different from zero. C, The percentage of transient events exhibiting projections to the Re (%) was correlated with the mean Gp. The line represents a linear fit: (%) = (1.4 ± 0.3%/pS) × Gp + (24 ± 9%). D, Scatter plot of Gp versus Cv of cells transfected with Munc18-1 WT (top) and with Munc18-1 mutants, where data can be divided into two distinct subgroups (bottom). Dashed lines indicate the limits of detection of signals related to the Gp calculation (Debus and Lindau, 2000). E, Data from D were recalculated into fusion pore and vesicle diameters (see Materials and Methods). Experimental points of cells transfected with Munc18-1 mutants (bottom) were divided into two subgroups by a linear regression line, fitted to all data. The lower and the upper subgroups were best fitted with the curves d(pore in nm) = 3.2 × exp{−846/[(d(vesicle)/2) + 205]} and d(pore in nm) = 13.9 × exp{−1458/[(d(vesicle)/2) + 270]}, respectively. F, STED images of prolactin-labeled vesicles in lactotrophs transfected with Munc18-1 WT and Munc18-1 R39C and corresponding distributions of vesicle diameters. Arrows in STED images mark enlarged vesicles on the right panels. Distributions of vesicle diameters were fitted with Gaussian functions: f = (16 ± 1) × exp(−0.5 × ((d(vesicle) − (292 ± 5 nm))/(70 ± 5))∧2) (Munc18-1 WT; n = 5 cells, 265 vesicles) and f =(15 ± 1) × exp(−0.5 × ((d(vesicle) − (280 ± 5 nm))/(78 ± 5))∧2) (Munc18-1 R39C; n = 7 cells, 384 vesicles).
To assess the diameters of narrow fusion pores and the possible effect of Munc18-1 mutants on them, we analyzed the Gp (from 6.7 to 245.6 pS, corresponding to fusion pore diameters of 0.36–2.16 nm). The average Gp was higher in cells expressing Munc18-1 E466K, P242S, and R39C mutants, compared with controls. However, the average Gp was also correlated with the incidence of measured narrow fusion pores (the percentage of transient fusion events exhibiting a cross talk between the Im and Re) (Fig. 6C). While these data indicate that in the presence of Munc18-1 mutants the occurrence of observing a narrow fusion pore is augmented, one cannot rule out the possibility that the higher incidence of observed events with cross talk may be influenced by the signal-to-noise properties of the recording system, which determines the range of measurable Gp and depends on Cv (Debus and Lindau, 2000). To further analyze the apparent increase in the average Gp in cells transfected with Munc18-1 (Fig. 6C), we plotted the ranges at which our setup faithfully discriminates Gp as a function of Cv (dashed lines, Fig. 6D), together with measured Gp versus Cv for all fusion events with measurable Gp. Figure 6D shows that the resolution of our setup meets the discrimination properties for detecting Gp as a function of recorded Cv. The top graph of Figure 6D shows the relationship between Gp and Cv in controls and the bottom one the relationship between Gp and Cv in cells transfected with Munc18-1 mutants, where the experimental points were clearly divided into two subgroups (Fig. 6D, bottom graph), which were not observed in controls (Fig. 6D, top), consistent with a previous report (Jorgačevski et al., 2010). The lower subgroup of data (Fig. 6D, bottom) shows that Munc18-1 mutants induce a reduction in Gp relative to the Cv, which can be taken to reflect that vesicles may exhibit a relatively low fusion pore conductance. Both patterns remained after recalculation of Gp into fusion pore diameters and Cv into vesicle diameters (Fig. 6E) (see Materials and Methods). Experimental points of subgroups in Munc18-1 mutants were fitted with curves of the form d = Δ0 × exp{−κ/[(d(vesicle)/2) + B]} (Jorgačevski et al., 2010), where Δ0 (the maximal fusion pore diameter at equilibrium), and κ and B (constants) were determined by the least-squares method, whereas d represents the fusion pore diameter at equilibrium (Jorgačevski et al., 2010). The data points for the two apparent subgroups were divided by linear regression, fitted to all experimental points. The lower subgroup yielded the maximum equilibrium diameter of 3.2 nm, much smaller than the maximum equilibrium diameter of the upper subgroup, which was 13.9 nm (Fig. 6E, bottom), comparable to the nontransfected cells (Fig. 6E) (Jorgačevski et al., 2010). In the same patch (n = 4), we observed transient fusion pore events belonging to the two subgroups; therefore, it is likely that the expression of Munc-18-1 mutants stimulates the transitions between the discrete narrow fusion pore states observed.
To study whether the increased amplitude of Cv in transfected cells with the Munc18-1 mutant R39C affects the vesicle size distribution, we labeled lactotrophs with Prl antibodies and used STED microscopy to determine the diameters of vesicles (for details, see Materials and Methods). The distributions of vesicle diameters of vesicles in cells transfected with Munc18-1 WT and R39C were practically identical (Fig. 6F; p > 0.05), indicating that the transfection with Munc18-1 WT and R39C did not affect the average vesicle size. The average diameter of Prl-containing vesicles that we measured is, by taking into account the size of primary and secondary antibodies (Osborne, 1998), in agreement with previous reports (Huerta-Ocampo et al., 2005). However, the percentage of vesicles that were larger than 400 nm (including the size of antibodies) was higher in cells transfected with Munc18-1 R39C compared with the cells transfected with Munc18-1 WT (22% vs 16%). It is possible that overexpression of Munc18-1 R39C led to the enhanced compound vesicular fusion (Vardjan et al., 2009) before exocytosis, which would explain the increased amplitude of Cv in cells transfected with this mutant.
Discussion
We propose that the SM protein Munc18-1 tunes the interactions between SNAREs by proofing vesicular tethering/docking to occur via the ternary SNARE complex, and by permitting the transition of vesicle stages between states with a narrow fusion pore to those with wider pores leading to full fusion (Fig. 7).
Figure 7.

A model describing interactions between Munc18-1 and SNARE proteins together with the role of Munc18-1 in regulating the fusion pore. A, Munc18-1 binds to Synt1A and reduces the probability (crossed arrow) of Syb2–Synt1A trans interaction (i). When SNAP25B is bound to Synt1A at the plasma membrane to form the cis-complex (ii), Syb2 can interact with this complex to form the ternary SNARE complex (ii, arrow; iii) or hypercomplex in the presence of Munc18-1 (ii, dotted arrow; iii). Lighter appearance of Munc18-1 (in iii) indicates its possible presence or absence. In the hypercomplex Munc18-1 could display its postdocking functions (iv). B, The vesicle stages from docking/priming, hemifusion state, leading to fusion pore formation and full fusion, with the action of respective Munc18-1 mutants.
It was previously proposed that Munc18-1 bound to Synt1 might be susceptible to the action of Rab/Rab effectors or other associated proteins, leading to a conformational change that allows Munc18-1 and Synt1A to interact with SNAP25B and Syb2, and initiation of ternary SNARE complex formation; once the ternary SNARE complex is formed, Munc18-1 detaches from it (Misura et al., 2000; Zilly et al., 2006). Recent alternative models propose (non)productive parallel pathways, the presence of Munc18-1 in the hypercomplex, and the involvement of the Ca2+-sensor protein synaptotagmin 1 in vesicular docking (Zilly et al., 2006; Toonen and Verhage, 2007; Verhage and Sorensen, 2008; de Wit et al., 2009; Südhof and Rothman, 2009). Based on our results, we propose some refinements of the preexisting models.
The results suggest that within the cell there could be two modes of vesicle–plasma membrane interactions depending on the presence of cytosolic Munc18-1 (Fig. 7A). If trans Synt1A-Syb2 interactions are possible in vivo, they could be tuned by Munc18-1 interacting with Synt1A and preventing binding of vesicular Syb2 (Fig. 7Ai). The presence of Munc18-1 would favor the formation of Synt1A-SNAP25B cis-complex at the plasma membrane and interactions of vesicular Syb2 with it (Fig. 7Aiii). We suggest that Synt1A-SNAP25B cis-complex would be formed subsequent to, but also in parallel with, Synt1A-Munc18-1 interaction. If this is a subsequent event, then the arrival of SNAP25B to the Synt1A-Munc18-1 cis-complex could displace Munc18-1, thus diminishing its ability to uncouple Synt1A-Syb2. Alternatively, the formation of dead-end Synt1A-Munc18-1 complex would promote the formation of 1:1 Synt1A-SNAP25B cis-complexes. Subsequent engagement of Syb2 with the Synt1A+SNAP25B cis-complex in the presence or absence of Munc18-1 (Fig. 7Aii–iv) would allow more stable tethering of vesicles than if tethering was based on Synt1A-Syb2 interaction alone (Liu et al., 2006). In electrophysiology experiments, both such events followed by vesicular fusions might be detectable when Munc18-1 mutants are expressed. Munc18-1 in the cellular milieu can interact with several proteins, so that, for example, the R39C mutant would compete with Munc18-1 WT for binding sites, perhaps even to Synt1A, to which it shows much reduced binding (Fisher et al., 2001). This would (in)directly lead to reduced availability of Munc18-1 WT to interact with Synt1A, and consequently allow both vesicular tethering/fusion modes to occur, one based on Syb2-Synt1A alone and the other on Syb2-Synt1A+SNAP25B interactions. Perhaps one display of such a dichotomy of tethering/fusion events is evident in our finding of two distinct fusion pore states in Figure 6D,E, where mutant proteins were expressed in lactrotrophs with native Munc18-1.
Given that all tested Munc18-1 mutants (E466K, P242S, and R39C) resulted in two distinct fusion pore states, one can conclude that the Munc18-1, the Synt1-interacting molecule, is involved in determining the fusion pore properties. Synt1 has been implicated to represent the lining of the fusion pore (Han et al., 2004); however, direct observation of whether the fusion pore lining part of Synt1 affects the fusion pore states (Fig. 6D,E) remains to be studied. Although Munc18-1 binding occurs at the proximal part of Synt (Habc domain and a part of the H3 domain), some of its binding/dissociation energy may transfer via the α coil of the SNARE domain to affect the distal part of the Synt molecule, which may contribute to the pore lining. Such tuning of the fusion pore characteristics would be absent in the presence of R39C because of its much reduced Synt1-binding capabilities. Alternatively, fusion pore modulation could be due to additional Munc18-1 interactions with proteins within, or as an accessory to, the ternary SNARE complex.
Munc18-1 E466K increases the frequency of exocytotic fusion pore events (Graham et al., 2008), and we expressed this and other mutants in lactotrophs, which endogenously express Munc18-1 (Fig. 3A) (Jacobsson and Meister, 1996). Step-like changes in Cm (Fig. 3E,F) recorded in these cells (Fig. 3C,D) were taken to mirror unitary fusion/fission events of secretory vesicles (Fig. 4A,B) (Neher and Marty, 1982).
Overexpression of Munc18-1 E466K increased the frequency of full fusion exocytotic events (Fig. 4D,F), in agreement with amperometric studies (Ciufo et al., 2005). The E466K mutation increases direct binding of Rab3A to Munc18-1, but does not affect binding to other known interacting proteins (Graham et al., 2008). Based on these findings, and because Rab3A stimulates vesicle docking in the presence of Munc18-1 (van Weering et al., 2007), we can attribute the increase in the frequency of full fusion exocytotic events in cells transfected with E466K to enhanced vesicle docking (Fig. 7B).
Not all effects of E466K mutant can be attributed to changes in the docking step, because E466K was seen to increase the average pore dwell-time (Fig. 5). This effect could be mediated through Synt1, because it was shown that functional effects of E466K require mode 1 Munc18-1-Synt1 interaction (Graham et al., 2008). If such a scenario occurs, then serial interactions between Munc18-1, Synt1, and Rab3A are required, because these three proteins were not found in the same complex (Graham et al., 2008). Alternatively, binding of the three proteins in the same complex may have been missed because of the poor stability of the complex.
One of the least explored Munc18-1 binding interactions is that with Mint1 and Mint2, although it is known that Mints bind Munc18-1, regardless of whether Munc18-1 is in complex with Synt1 (Okamoto and Südhof, 1997). We observed that in cells transfected with Munc18-1 P242S, the average fusion pore dwell-time increased as with Munc18-1 E466K mutant (Fig. 5), consistent with the amperometric study (Ciufo et al., 2005). The expression of P242S did not affect the frequency of exocytotic events (Fig. 3C–F). Therefore, we suggest that these effects may be mediated through the N terminus of Synt1 in mode 2 interaction, when the SNARE complex is already fully assembled (Dulubova et al., 2007) (see also Fig. 7).
Transfection of distinct Munc18-1 mutants shared a common effect of enhancing the fusion of larger vesicles. Fusion of larger vesicles could be the consequence of enhanced compound exocytosis (Vardjan et al., 2009). However, lactotrophs contain at least two populations of large dense-core vesicles (Huerta-Ocampo et al., 2005), and the expression of Munc18-1 mutants may lead to preferential fusion of larger vesicles. Results obtained with STED microscopy show that in cells transfected with Munc18-1 WT and R39C (the mutant in which we observed fusion events with the largest Cv), the distribution of vesicle diameters was unchanged (Fig. 6F). This result supports the view that Munc18-1 mutants lead to preferential fusion of larger vesicles. In addition, several studies suggest an important role of Munc18-1 in vesicle docking (Voets et al., 2001; van Weering et al., 2007), a step that could lead to the discrimination of differently sized vesicles. Compound exocytosis would likely be accompanied by an increase in the frequency of exocytotic events (Vardjan et al., 2009), which was observed only in one of the tested mutants (Munc18-1 E466K; Fig. 5).
Previously we observed that in nontransfected lactotrophs, the narrow fusion pore diameter depends on the vesicle size; larger vesicles exhibit relatively wider pores (Jorgačevski et al., 2010). In contrast with this monotonous relationship between vesicle size and fusion pore diameter, we now show that the relationship between vesicle size and narrow fusion pore diameter in lactotrophs transfected with Munc18-1 mutant shows two subpopulations (Fig. 6E). The upper subpopulation has a relationship similar to that of the nontransfected lactotrophs (Jorgačevski et al., 2010). We assume that fusion events in this subpopulation had molecular machinery similar to that of the nontransfected cells. On the other hand, in the lower subpopulation, the fusion events had considerably narrower fusion pore diameters at the given vesicle size. In this case, molecular machinery likely differed from the one in nontransfected cells, including the Munc 18-1 mutants. Since Munc18-1 E466K, P242S, and R39C mutants result in altered binding affinity to different proteins, but all converging in their interaction to regulating the SNAREs, it is not unexpected that all of them alter similarly the relationship between the vesicle diameter and the narrow fusion pore diameter. It is proposed that the reduction in steepness of the relationship between vesicle and the narrow fusion pore diameters (Fig. 7B) could be the consequence of binding or changed dynamics of cytosolic Munc18-1 (mutants) in tuning the binding of Syb2 with the binary 1:1 Synt1A-SNAP25B cis-complex (Fig. 7A).
This work suggests two new considerations on the mechanism of exocytosis. First, single-molecule interaction studies show directly that Munc18-1 significantly affects the interactions between SNARE partners. Second, by studying the fusion pore properties directly by electrophysiological cell-attached Cm measurements in the presence of Munc18-1 mutants, we were able to correlate single-molecule interactions with changes observed in the fusion pore frequency, conductance, and kinetics.
Footnotes
This work was supported by Grants P3 0310 0381, J3-3632-1683-10, and CipKeBip (to R.Z.) from the Ministry of Higher Education, Sciences and Technology of the Republic of Slovenia, and by a grant from the National Science Foundation (CBET 0943343 to V.P.). We thank Vedrana Montana and Randy F. Stout for production of proteins used for AFM studies.
The authors declare no conflicts of interest.
References
- Alvarez de Toledo G, Fernández-Chacón R, Fernández JM. Release of secretory products during transient vesicle fusion. Nature. 1993;363:554–558. doi: 10.1038/363554a0. [DOI] [PubMed] [Google Scholar]
- An SJ, Grabner CP, Zenisek D. Real-time visualization of complexin during single exocytic events. Nat Neurosci. 2010;13:577–583. doi: 10.1038/nn.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angleson JK, Cochilla AJ, Kilic G, Nussinovitch I, Betz WJ. Regulation of dense core release from neuroendocrine cells revealed by imaging single exocytic events. Nat Neurosci. 1999;2:440–446. doi: 10.1038/8107. [DOI] [PubMed] [Google Scholar]
- Barclay JW. Munc-18-1 regulates the initial release rate of exocytosis. Biophys J. 2008;94:1084–1093. doi: 10.1529/biophysj.107.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- Ben-Tabou S, Keller E, Nussinovitch I. Mechanosensitivity of voltage-gated calcium currents in rat anterior pituitary cells. J Physiol. 1994;476:29–39. [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Secretory granule exocytosis. Physiol Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- Chen F, Mohideen U. Fiber optic interferometry for precision measurements of the voltage and frequency dependence of the displacement of piezoelectric tubes. Rev Sci Instrum. 2001;72:3100–3102. [Google Scholar]
- Ciufo LF, Barclay JW, Burgoyne RD, Morgan A. Munc18-1 regulates early and late stages of exocytosis via syntaxin-independent protein interactions. Mol Biol Cell. 2005;16:470–482. doi: 10.1091/mbc.E04-08-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debus K, Lindau M. Resolution of patch capacitance recordings and of fusion pore conductances in small vesicles. Biophys J. 2000;78:2983–2997. doi: 10.1016/S0006-3495(00)76837-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, Rizo J. Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci U S A. 2007;104:2697–2702. doi: 10.1073/pnas.0611318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E. Probing the relation between force–lifetime–and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- Fisher RJ, Pevsner J, Burgoyne RD. Control of fusion pore dynamics during exocytosis by Munc18. Science. 2001;291:875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]
- Florin EL, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- Graham ME, Sudlow AW, Burgoyne RD. Evidence against an acute inhibitory role of nSec-1 (munc-18) in late steps of regulated exocytosis in chromaffin and PC12 cells. J Neurochem. 1997;69:2369–2377. doi: 10.1046/j.1471-4159.1997.69062369.x. [DOI] [PubMed] [Google Scholar]
- Graham ME, Barclay JW, Burgoyne RD. Syntaxin/Munc18 interactions in the late events during vesicle fusion and release in exocytosis. J Biol Chem. 2004;279:32751–32760. doi: 10.1074/jbc.M400827200. [DOI] [PubMed] [Google Scholar]
- Graham ME, Handley MT, Barclay JW, Ciufo LF, Barrow SL, Morgan A, Burgoyne RD. A gain-of-function mutant of Munc18-1 stimulates secretory granule recruitment and exocytosis and reveals a direct interaction of Munc18-1 with Rab3. Biochem J. 2008;409:407–416. doi: 10.1042/BJ20071094. [DOI] [PubMed] [Google Scholar]
- Han X, Wang CT, Bai J, Chapman ER, Jackson MB. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 2004;304:289–292. doi: 10.1126/science.1095801. [DOI] [PubMed] [Google Scholar]
- Harris BW, Chen F, Mohideen U. Precision measurement of the Casimir force using gold surfaces. Phys Rev A. 2000;62 052109. [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Heuser J, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Ocampo I, Christian HC, Thompson NM, El-Kasti MM, Wells T. The Intermediate lactotroph: a morphologically distinct, ghrelin-responsive pituitary cell in the dwarf (dw/dw) rat. Endocrinology. 2005;146:5012–5023. doi: 10.1210/en.2005-0335. [DOI] [PubMed] [Google Scholar]
- Hummer G, Szabo A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci U S A. 2001;98:3658–3661. doi: 10.1073/pnas.071034098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter JL, Bechhoefer J. Calibration of atomic-force microscope tips. Rev Sci Instrum. 1993;64:1868–1873. [Google Scholar]
- Jacobsson G, Meister B. Molecular components of the exocytotic machinery in the rat pituitary gland. Endocrinology. 1996;137:5344–5356. doi: 10.1210/endo.137.12.8940356. [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Südhof TC. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- Jorgačevski J, Stenovec M, Kreft M, Bajić A, Rituper B, Vardjan N, Stojilkovic S, Zorec R. Hypotonicity and peptide discharge from a single vesicle. Am J Physiol Cell Physiol. 2008;295:C624–C631. doi: 10.1152/ajpcell.00303.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgačevski J, Fosnaric M, Vardjan N, Stenovec M, Potokar M, Kreft M, Kralj-Iglic V, Iglic A, Zorec R. Fusion pore stability of peptidergic vesicles. Mol Membr Biol. 2010;27:65–80. doi: 10.3109/09687681003597104. [DOI] [PubMed] [Google Scholar]
- Kreft M, Milisav I, Potokar M, Zorec R. Automated high through-put colocalization analysis of multichannel confocal images. Comput Methods Programs Biomed. 2004;74:63–67. doi: 10.1016/S0169-2607(03)00071-3. [DOI] [PubMed] [Google Scholar]
- Liu W, Montana V, Bai J, Chapman ER, Mohideen U, Parpura V. Single molecule mechanical probing of the SNARE protein interactions. Biophys J. 2006;91:744–758. doi: 10.1529/biophysj.105.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Montana V, Parpura V, Mohideen U. Comparative energy measurements in single molecule interactions. Biophys J. 2008;95:419–425. doi: 10.1529/biophysj.107.127886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Montana V, Parpura V, Mohideen U. Single molecule measurements of interaction free energies between the proteins within binary and ternary SNARE complexes. J Nanoneurosci. 2009;1:120–129. doi: 10.1166/jns.2009.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lollike K, Lindau M. Membrane capacitance techniques to monitor granule exocytosis in neutrophils. J Immunol Methods. 1999;232:111–120. doi: 10.1016/s0022-1759(99)00169-6. [DOI] [PubMed] [Google Scholar]
- Lollike K, Borregaard N, Lindau M. The exocytotic fusion pore of small granules has a conductance similar to an ion channel. J Cell Biol. 1995;129:99–104. doi: 10.1083/jcb.129.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- Montana V, Liu W, Mohideen U, Parpura V. Single molecule probing of exocytotic protein interactions using force spectroscopy. Croat Chem Acta. 2008;81:31. [PMC free article] [PubMed] [Google Scholar]
- Montana V, Liu W, Mohideen U, Parpura V. Single molecule measurements of mechanical interactions within ternary SNARE complexes and dynamics of their disassembly: SNAP25 vs. SNAP23. J Physiol. 2009;587:1943–1960. doi: 10.1113/jphysiol.2009.168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Marty A. Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci U S A. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Schekman R. Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1979;76:1858–1862. doi: 10.1073/pnas.76.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Südhof TC. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J Biol Chem. 1997;272:31459–31464. doi: 10.1074/jbc.272.50.31459. [DOI] [PubMed] [Google Scholar]
- Osborne M. Immunofluorescence microscopy of cultured cells. In: Celis JE, editor. Cell biology: a laboratory handbook. San Diego: Academic; 1998. pp. 462–468. [Google Scholar]
- Spruce AE, Breckenridge LJ, Lee AK, Almers W. Properties of the fusion pore that forms during exocytosis of a mast cell secretory vesicle. Neuron. 1990;4:643–654. doi: 10.1016/0896-6273(90)90192-i. [DOI] [PubMed] [Google Scholar]
- Stryer L. Biochemistry. New York: WH Freeman; 1995. [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. 2007;30:564–572. doi: 10.1016/j.tins.2007.08.008. [DOI] [PubMed] [Google Scholar]
- van Weering JR, Toonen RF, Verhage M. The role of Rab3a in secretory vesicle docking requires association/dissociation of guanidine phosphates and Munc18-1. PLoS One. 2007;2:e616. doi: 10.1371/journal.pone.0000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardjan N, Stenovec M, Jorgacevski J, Kreft M, Zorec R. Subnanometer fusion pores in spontaneous exocytosis of peptidergic vesicles. J Neurosci. 2007;27:4737–4746. doi: 10.1523/JNEUROSCI.0351-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardjan N, Jorgačevski J, Stenovec M, Kreft M, Zorec R. Compound exocytosis in pituitary cells. Ann N Y Acad Sci. 2009;1152:63–75. doi: 10.1111/j.1749-6632.2008.04008.x. [DOI] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Wildanger D, Rittweger E, Kastrup L, Hell SW. STED microscopy with a supercontinuum laser source. Opt Express. 2008;16:9614–9621. doi: 10.1364/oe.16.009614. [DOI] [PubMed] [Google Scholar]
- Wu M, Littleton JT, Bhat MA, Prokop A, Bellen HJ. ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. EMBO J. 1998;17:127–139. doi: 10.1093/emboj/17.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly FE, Sørensen JB, Jahn R, Lang T. Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol. 2006;4:e330. doi: 10.1371/journal.pbio.0040330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorec R, Sikdar SK, Mason WT. Increased cytosolic calcium stimulates exocytosis in bovine lactotrophs. Direct evidence from changes in membrane capacitance. J Gen Physiol. 1991;97:473–497. doi: 10.1085/jgp.97.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]