

Abstract
Unraveling the mechanisms by which the molecular manipulation of genes of interest enhances cognitive function is important to establish genetic therapies for cognitive disorders. Although CREB is thought to positively regulate formation of long-term memory (LTM), gain-of-function effects of CREB remain poorly understood, especially at the behavioral level. To address this, we generated four lines of transgenic mice expressing dominant active CREB mutants (CREB-Y134F or CREB-DIEDML) in the forebrain that exhibited moderate upregulation of CREB activity. These transgenic lines improved not only LTM but also long-lasting long-term potentiation in the CA1 area in the hippocampus. However, we also observed enhanced short-term memory (STM) in contextual fear-conditioning and social recognition tasks. Enhanced LTM and STM could be dissociated behaviorally in these four lines of transgenic mice, suggesting that the underlying mechanism for enhanced STM and LTM are distinct. LTM enhancement seems to be attributable to the improvement of memory consolidation by the upregulation of CREB transcriptional activity, whereas higher basal levels of BDNF, a CREB target gene, predicted enhanced shorter-term memory. The importance of BDNF in STM was verified by microinfusing BDNF or BDNF inhibitors into the hippocampus of wild-type or transgenic mice. Additionally, increasing BDNF further enhanced LTM in one of the lines of transgenic mice that displayed a normal BDNF level but enhanced LTM, suggesting that upregulation of BDNF and CREB activity cooperatively enhances LTM formation. Our findings suggest that CREB positively regulates memory consolidation and affects memory performance by regulating BDNF expression.
Introduction
Understanding the mechanisms that regulate cognitive function at the molecular level could enable the development of genetic therapies for cognitive deficits caused by neuronal disorders (Lee and Silva, 2009). Therefore, it is important to identify genes that modulate cognitive functions such as learning and memory. These candidate genes should display gain and loss of function with opposite effects (impairment or improvement) on cognitive function. Moreover, it is necessary to unravel the mechanisms by which the molecular manipulation of the gene of interest enhances cognitive function.
Previous studies have demonstrated that new gene expression is required for memory consolidation (Flexner et al., 1965; Davis and Squire, 1984; Abel et al., 1997; Silva et al., 1998; Martin et al., 2000; McGaugh, 2000). Indeed, pharmacological studies have shown that inhibition of mRNA or protein synthesis disrupted the formation of long-term memory (LTM) without affecting short-term memory (STM) (Abel et al., 1997; Silva et al., 1998; Kida et al., 2002; Suzuki et al., 2004; Duvarci et al., 2008). These studies have shown that LTM, but not STM, depends on new gene expression.
Activity-dependent gene expression plays a central role in memory consolidation and long-term potentiation (LTP) (Nguyen et al., 1994; Silva et al., 1998; Martin et al., 2000). Increases in the intracellular concentration of Ca2+ and cAMP activate the serine threonine kinase CaMKIV and protein kinase A (PKA), respectively (Jensen et al., 1991; Matthews et al., 1994; Nakamura et al., 1995). These activated protein kinases phosphorylate the transcription factor cAMP responsive element binding protein (CREB) at serine 133 (S133) (Nakamura et al., 1995; Bito et al., 1996; Silva et al., 1998). Phosphorylated CREB at S133 interacts with the transcription coactivator CREB-binding protein (CBP), resulting in the activation of activity-dependent CRE-mediated transcription through the recruitment of CBP to the promoter (Chrivia et al., 1993; Bito et al., 1996; Silva et al., 1998). This interaction of CREB–CBP is an essential step of CREB-mediated transcription.
Importantly, previous genetic studies have demonstrated that loss of function of CREB disrupts memory consolidation and impairs certain forms of LTP (Bourtchuladze et al., 1994; Oike et al., 1999; Kida et al., 2002; Pittenger et al., 2002; Korzus et al., 2004). Therefore, it was proposed that CREB functions as a positive molecular switch for memory formation. However, little is known about the impacts of increased levels of CREB activity on memory performance. Indeed, recent studies using viruses to overexpress CREB suggested that the upregulation of CREB activity improves memory formation (Josselyn et al., 2001; Brightwell et al., 2007; Han et al., 2008; Zhou et al., 2009). In contrast, the memory performance of transgenic mice expressing a dominant active CREB mutant (VP16-CREB) in the forebrain displayed contradictory results (i.e., the impairment and weak improvement of memory performance) (Viosca et al., 2009a,b). In this study, we generated transgenic mice expressing dominant active CREB (CREB-Y134F or CREB-DIEDML) in the forebrain that exhibited moderate upregulation of CREB activity (1.5- to 2.5-fold) at the basal level and examined the effects of upregulating CREB activity after memory performance and LTP.
Materials and Methods
Mice.
Mice were housed in cages of five or six, maintained on a 12 h light/dark schedule, and allowed ad libitum access to food and water in their home cages. All experiments were conducted during the light phase of the cycle in an illuminated testing room, according to the Guide for the Care and Use of Laboratory Animals, Japan Neuroscience Society and Tokyo University of Agriculture. All experiments were conducted blind to the treatment condition of the mouse. Animal behavior was recorded using a video camera.
Plasmid constructions.
Full-length cDNA encoding rat CREB-Y134F was obtained by PCR using Rous sarcoma virus (RSV)-CREB (kindly provided by M. Montminy, Salk Institute, La Jolla, CA), as described previously (Kida et al., 2002). To generate CREB-Y134F cDNA, two separate fragments of CREB-Y134F cDNA (nucleotides 1–390 and 391–1030) were amplified by PCR using the following primers: 1/390 primers (sense, gggaagcttgccgccaccatgacgcgtagatctatgaccatggactctgga; antisense, gggggatcctctagaaaggatttcccttcgttt); 391/1030 primers (sense, gggaagctttctagaaggccttccttcaggaaaattttgaatga; antisense, gggggatccacgcgtgaattcttaatctgacttgtggcagtaaaggtctttaagtgcttttagctcc; the point mutations are underlined). To generate CREB-DIEDML cDNA, two separate fragments of the CREB-DIEDML cDNA (nucleotides 1–408 and 409–1030) were amplified by PCR using the following primers as previously described (Hosoda et al., 2009): 1/408 primers (sense, gggaagcttgccgccaccatgacgcgtagatctatgaccatggactctgga; antisense, gggggatcccttaagcatgtcttctatgtctgaaaggatttcccttcg); 409/1030 primers (sense, gggaagcttcttaagattttgaatgacttatct; antisense, gggggatccacgcgtgaattcttaatctgacttgtggcagtaaaggtctttaagtgcttttagctcc; the DIEDML sequences are underlined). The resulting PCR fragments were subcloned into the HindIII–BamHI sites of pBluescript II (SK−) (Stratagene), generating pBS-CREB-Y134F and pBS-CREB-DIEDML. Furthermore, these fragments were subcloned into the BglII–EcoRI sites of the pEYFP-C1 vector (Clontech), generating pEYFP-CREB-Y134F and pEYFP-CREB-DIEDML. To generate plasmids expressing GAL4-CREB-Y134F or -DIEDML (pM-CREB-Y134F or CREB-DIEDML), fragments of the CREB-Y134F and -DIEDML cDNA were amplified by PCR using the primers (sense, gggggattcgaattcgtatgaccatggactctgga; antisense, gggaagcttttaatctgacttgtggca). These fragments were subcloned into the HindIII–BamHI sites of the pM vector (Clontech). pM-CREB-wild type (WT), pM-CREB-S133A, pEYFP-CREB-WT, and pEYFP-CREB-S133A were generated as described above.
The KIX domain of CBP and one copy of the nuclear localization signal (nls) was amplified by PCR using RSV-human CBP (kindly provided by R. H. Goodman, Oregon Health and Science University, Portland, OR) as a template and the primers (sense, gggaagctttccggaggcggcggaattcaaaacacaatt; antisense, ggggaatccttagccgtcctccaccttgcgcttcttcttgggcccttgagaaacctgcat). To generate a plasmid expressing ECFP-KIXnls (pECFP-KIXnls), the resulting PCR fragment was subcloned into the BspEI and BamHI sites of pECFP-C1 (Clontech).
To generate the plasmids expressing EYFP-CREB-WT, -S133A, -Y134F, or -DIEDML and ECFP-KIXnls (pCKIXnls-YCREB-WT, -S133A, -Y134F, or -DIEDML), the MluI fragment containing the CMV promoter and the coding region of EYFP-CREB-WT, -S133A, -Y134F, or -DIEDML was subcloned into the MluI site of pECFP-KIXnls. These expression plasmids (pCKIXnls-YCREB-WT or -mutants) facilitate the expression of ECFP-KIXnls and EYFP-CREB-WT or -mutants from a single vector.
To generate the constitutively active CaMKIV (1–317 nucleotide) expression plasmid (pCMV-acCaMKIV), cDNA encoding acCaMKIV (kindly provided by Dr. Anthony R. Means, Duke University, Durham, NC) was amplified by PCR using the primers (sense, gggggtaccgccgccaccatgacgcgtatgctcaaagtcacggtg; antisense, gggctcgagacgcgtttagagcttcttttgagcggt). The resulting PCR fragment encoding CaMKIV was subcloned into the KpnI-XhoI sites of pc-DNA3 (Invitrogen).
The GAL4-dependent luciferase reporter plasmid (pGAL4RE-Luc) was generated as described previously (Iwamoto et al. 2005). The β-galactosidase expression plasmid (pCMV-β-gal) and PKA catalytic subunit expression plasmid (pCMV-PKA) were purchased from Clontech or Stratagene, respectively.
Generation of transgenic mice.
MluI fragments encoding CREB mutants from pBS-CREB-Y134F and CREB-DIEDML were ligated with an MluI fragment from pMM-403-CaMKIV (Fukushima et al., 2008), generating pMM-403-CREB-Y134F or -DIEDML, respectively. pMM-403-CREB-Y134F and -DIEDML were digested with SfiI, and transgenic mice were generated by injecting the purified insert into the pronuclei of C57BL/6N zygotes. Transgenic founders were crossed with C57BL/6N mice (Charles River Japan). Genotyping was performed as described previously (Kida et al., 2002; Fukushima et al., 2008; Hasegawa et al., 2009).
Biochemical experiments.
Northern blotting using a 32P-labeled SV40 poly(A) probe and in situ hybridization using a DIG-labeled SV40 poly(A) probe were performed as described previously (Kida et al., 2002; Uchida et al., 2006). Hippocampal extracts were prepared as described previously (Kida et al., 2002). The expression of the brain-derived neurotrophic factor (BDNF) protein was assessed using an ELISA-Emax Immunoassay System kit as indicated in the manufacturer's protocol (Promega). The BDNF expression levels in CREB mutant mice were expressed as a ratio of WT mice. Western blotting, using a rabbit polyclonal anti-CREB antibody (1:1000; Cell Signaling Technology) and a mouse monoclonal anti-α-tubulin antibody (1:1000; Santa Cruz Biotechnology), was performed as described previously (Hosoda et al., 2004). Equal protein loading was confirmed by reprobing with the anti-α-tubulin antibody. For immunoprecipitation, nuclear extracts from the hippocampus were prepared using a protocol described by Lubin et al. (2005). Nuclear proteins were precleared with protein-G Sepharose beads (50% slurry; GE Healthcare) at 4°C for 30 min. The beads were collected by centrifugation, and the precleared extracts were incubated with 3 μl of anti-CBP antibody (sc-369; Santa Cruz Biotechnology) at 4°C for 3 h. The precleared extracts were incubated with protein-G Sepharose beads at 4°C for 1 h. The beads were washed with TNE buffer (10 mm Tris-HCl, pH 7.5, 0.25% NP-40, 150 mm NaCl, and 1 mm EDTA) and collected by centrifugation. The beads were resuspended in an SDS-PAGE loading buffer.
Behavioral experiments.
Before the commencement of behavioral experiments, the mice were individually handled for 2 min each day for 1 week. All experiments were performed blind to the group of subjects. Individual mice were used for all experiments. We tested at each time point using different groups of animals.
Contextual fear conditioning.
Mice were trained and tested in conditioning chambers (17.5 × 17.5 × 15 cm) fitted with a stainless steel grid floor through which footshocks could be delivered as described previously (Suzuki et al., 2004; Fukushima et al., 2008). Training consisted of placing the mice in the chamber and delivering an electrical footshock [2 s duration; 0.2 mA (see Fig. 5C), 0.4 mA (see Figs. 1G, 3C,D, 4G, 8A,C)] 148 s later. Mice were returned to the home cage 30 s after the final footshock. Memory was assessed at 5 or 30 min or at 2 or 24 h later by calculating the percentage of time spent freezing during 5 min when re-placed in the training context. Freezing behavior (defined as the complete lack of movement, except for respiration) was measured automatically as described previously (Anagnostaras et al., 2000).
Figure 5.

Expression of CREB-DIEDML enhances STM and LTM. A, B, Social recognition test. A, Recognition index. *p < 0.05. B, Investigation time. At 5 min, 30 min, or 2 h after the first exposure, WT and DIEDML mice showed a significant reduction in investigation time (5 min, p < 0.05; 30 min, p < 0.05; 2 h, p < 0.05). In contrast, at 24 h after the first exposure, DIEDML but not WT mice showed a significant reduction in investigation time (WT, p > 0.05; α and β, p < 0.05). These observations indicated that DIEDML mice formed 5 min, 30 min, and 2 h STM and LTM, whereas WT mice formed only STMs but not LTM. *p < 0.05, compared with the first exposure. Data are shown for 5 min (WT, n = 32; α, n = 22; β, n = 17), 30 min (WT, n = 41; α, n = 16; β, n = 17), 2 h (WT, n = 27; α, n = 14; β, n = 20), and 24 h (WT, n = 22; α, n = 15; β, n = 9). C, Contextual fear-conditioning test. Data are shown for 5 min (WT, n = 24; α, n = 8; β, n = 20), 30 min (WT, n = 33; α, n = 19; β, n = 16), 2 h (WT, n = 22; α, n = 8; β, n = 13), and 24 h (WT, n = 70; α, n = 14; β, n = 24). Error bars represent SEM. *p < 0.05.
Figure 1.

Generation of CREB-Y134F expression mice. A, Activation of CREB-mediated transcription by the upstream signal transduction pathway in PC12 cells. B, CREB–CBP (KIX) complex formation in COS-1 cells in the acceptor bleaching test. We analyzed 34–54 cells in each group. ap < 0.05, compared with the other groups; bp < 0.05, compared with the other groups. C, Real-time imaging of CREB–CBP (KIX) complex formation in response to the activation of the PKA signaling pathway in PC12 cells. Emission ratios were measured before and after the application of FSK (0.1 or 1 μm). We analyzed 32–39 cells in each group. In the presence of 0.1 μm FSK, repeated ANOVA revealed significant effects of time, mutation (WT vs Y134F) and time versus mutation interaction (time, F(19,1254) = 29.050, p < 0.05; mutation, F(1,66) = 7.920, p < 0.05; interaction, F(19,1254) = 2.721, p < 0.05). In contrast, in the presence of 1 μm FSK, repeated ANOVA revealed a significant effect of time, but not effects of mutation and time vs mutation interaction (time, F(19,1406) = 52.206, p < 0.05; mutation, F(1,74) = 0.283, p > 0.05; interaction, F(19,1406) = 0.503, p > 0.05). D, Schematic representation of the CREB-Y134F transgenic construct. E, Northern blot analysis of the transgene in the hippocampus and cortex of WT, Y134F-A (A), and Y134F-C (C) mice. The SV40 poly(A) probe detected transgenic mRNA of 2.5 kb. F, In situ hybridization analyses of transgene mRNA expression in Y134F-A, Y134F-C, and WT mice. The SV40 poly(A) probe detected transgene mRNA in the forebrain of Y134F-A and Y134F-C mice. Scale bar, 500 μm. G, Immunohistochemical analyses of c-fos expression after fear conditioning. Scale bar, 100 μm. H, Quantification of the number of c-fos-positive cells after contextual fear conditioning in WT, Y134F-A, and Y134F-C mice. The conditioning group consisted of WT (n = 8), A (n = 8), and C (n = 8); the no-conditioning group consisted of WT (n = 8), A (n = 9), and C (n = 10). I, Quantification of the number of c-fos-positive cells at the basal condition. One-way ANOVA followed by the post hoc Newman–Keuls test showed significantly higher expression levels of c-fos in the CA1 (p < 0.05) but not the CA3 or dentate gyrus (DG) areas of the hippocampus in Y134F-A and -C mice compared with WT mice. Similarly, Y134F-C mice showed significantly higher expression levels of c-fos in the amygdala compared with WT mice (p < 0.05). Y134F-C mice displayed higher expression levels of c-fos in the hippocampal CA1 and amygdala compared with Y134F-A mice, although this difference was not significant. WT mice, n = 11; Y134F-A mice, n = 11; Y134F-C mice, n = 10. Error bars represent SEM. *p < 0.05. BLA, Basolateral amygdala; LA, lateral amygdala.
Figure 3.

Expression of CREB-Y134F enhances memory formation. A, B, Social recognition test. A, Recognition index. *p < 0.05. B, Investigation time. Thirty minutes or 2 h after the first exposure, WT and Y134F mice showed a significant reduction in investigation time (a paired t test, p < 0.05). In contrast, at 24 h after the first exposure, Y134F, but not WT, mice showed a significant reduction in investigation time (WT, p > 0.05; A and C, p < 0.05). *p < 0.05, compared with the first exposure. Data show 30 min STM (WT, n = 13; A, n = 14; C, n = 13), 2 h STM (WT, n = 25; A, n = 11; C, n = 11), and 24 h LTM (WT, n = 24; A, n = 8; C, n = 12). C, Contextual fear-conditioning test. Data show 30 min STM (WT, n = 22; A, n = 12; C, n = 15), 2 h STM (WT, n = 18; A, n = 11; C, n = 14), and 24 h LTM (WT, n = 44; A, n = 17; C, n = 20). *p < 0.05. D, Contextual discrimination test. Data show 1 d (WT-CXT-S, n = 10; WT-CXT-D, n = 11; Y134F-C-CXT-S, n = 13; Y134F-C-CXT-D, n = 12) or 1 month (WT-CXT-S, n = 18; WT-CXT-D, n = 10; Y134F-C-CXT-S, n = 17; Y134F-C-CXT-D, n = 12) totals. *p < 0.05. E–G, Morris water maze test. WT, n = 22; A, n = 12; C, n = 17. *p < 0.05, compared with the other three quadrants (a paired t test); #p < 0.05. H, Passive avoidance test. Data show 2 h (WT, n = 13; C, n = 13) and 24 h (WT, n = 14; C, n = 12) totals. *p < 0.05. Error bars represent SEM.
Figure 4.

Generation of CREB-DIEDML expression mice. A, Activation of CREB-mediated transcription in PC12 cells. Luc., luciferase. B, CREB–CBP (KIX) complex formation in COS-1 cells in the acceptor bleaching test. We analyzed 52–65 cells in each group. C, Schematic representation of the CREB-DIEDML transgenic construct. D, Northern blot analysis of the transgene in the hippocampus and cortex of WT mice, DIEDML-α (α), and DIEDML-β (β) mice. The SV40 poly(A) probe detected transgenic mRNA of 2.5 kb. E, In situ hybridization analyses of transgene mRNA expression in DIEDML-α, DIEDML-β, and WT mice. The SV40 poly(A) probe detected transgene mRNA in the forebrain of DIEDML-α and -β mice. Scale bar, 500 μm. F, Levels of total CREB in the hippocampus. Active CREB mice displayed higher levels of total CREB compared with WT mice. G, Immunohistochemical analyses of c-fos expression after fear conditioning. Scale bar, 100 μm. H, Quantification of the number of c-fos-positive cells after contextual fear conditioning in WT, DIEDML-α, and DIEDML-β mice. Data for the conditioning group (WT, n = 7; α, n = 9; β, n = 7) and no-conditioning group (WT, n = 6; α, n = 7; β, n = 6) are shown. I, Coimmunoprecipitation (I.P.) experiments were performed using an anti-CBP antibody, and Western blot analysis was performed using anti-CREB or anti-α tubulin antibodies. The input lanes represent 10% of the total cell lysates in the binding reaction. J, Quantification of CREB level (input). Both DIEDML transgenic lines displayed significant increases in the levels of CREB in the hippocampus compared with WT mice. K, Quantification of CREB levels (I.P.). DIEDML mice displayed significantly higher levels of CREB protein in the precipitates after incubation with the anti-CBP antibody compared with WT mice. WT, n = 7; α, n = 7; β, n = 7. Error bars represent SEM. *p < 0.05.
Figure 8.

Effects of the microinfusion of BDNF and K252a on STM and LTM in active CREB and WT mice. A, Effects of the microinfusion of BDNF into the dorsal hippocampus on 2 h short-term contextual fear memory in WT mice (VEH, n = 14; BDNF, n = 11). *p < 0.05, compared with the VEH group at test. B, D, G, J, M, P, S, V, Cannula tip placements from mice infused with each drug for A, C, E, H, K, N, Q, and T, respectively. Coronal sections are adapted from Paxinos and Franklin (1997) (1.94 mm posterior to bregma). C, Effects of the microinfusion of K252a into the dorsal hippocampus on 2 h short-term contextual fear memory in WT mice (VEH, n = 9; K252a, n = 8.). *p < 0.05, compared with the VEH group at test. E–G, Effects of the microinfusion of BDNF into the dorsal hippocampus on the 2 h short-term social recognition memory in WT mice (VEH, n = 8; BDNF, n = 8). E, Recognition index. *p < 0.05, compared with the VEH group. F, Investigation time. The VEH or BDNF group showed significant reductions in investigation time (p < 0.05), indicating that both groups formed 2 h STM. *p < 0.05, compared with the first exposure (left). H–J, Effects of the microinfusion of K252a into the dorsal hippocampus on the 2 h short-term social recognition memory in WT mice (VEH, n = 8; K252a, n = 8). H, Recognition index. *p < 0.05, compared with the VEH group. I, Investigation time. The VEH, but not K252a, group showed a significant reduction in investigation time (VEH, F(1,14) = 17.82, p < 0.05; K252a, F(1,14) = 0.727, p > 0.05), indicating that only the VEH group formed 2 h STM. *p < 0.05, compared with the first exposure. K–M, Effects of the microinfusion of a low dose of BDNF into the dorsal hippocampus on 30 min STM in WT and Y134F-C mice (WT-VEH, n = 8; WT-BDNF, n = 9; C-VEH, n = 9; C-BDNF, n = 8). K, Recognition index. *p < 0.05, compared with the other three groups. L, Investigation time. WT and Y134F-C mice treated with BDNF or VEH showed a significant reduction in investigation time (p < 0.05), indicated that all groups formed STM. *p < 0.05, compared with the first exposure. N–P, Effects of the microinfusion of low or high doses of K252a (3 or 9.35 ng per side) into the dorsal hippocampus on 2 h STM in WT and DIEDML-β mice (WT-VEH, n = 10; WT-low, n = 8; WT-high, n = 8; β-VEH, n = 8; β-low, n = 9; β-high, n = 10). N, Recognition index. #p < 0.05, compared with WT-VEH; *p < 0.05, compared with WT-low K252a. O, Investigation time. The WT-VEH, β-VEH, and β-low groups showed significant reductions in investigation time (p < 0.05), whereas the other groups did not (p > 0.05). These observations indicated that the WT-VEH, β-VEH, and β-low groups formed 2 h STM. *p < 0.05, compared with the first exposure. Q–S, Effects of the microinfusion of a low or high dose of anti-BDNF antibody (0.5 μg or 1 μg per side) into the dorsal hippocampus. A higher dose of anti-BDNF antibody is required to impair 2 h STM in DIEDML-β mice compared with WT mice (WT-VEH, n = 9; WT-low, n = 9; WT-high, n = 8; β-VEH, n = 11; β-low, n = 10; β-high, n = 10). Q, Recognition index. A significant interaction between genotype × drug treatment (low vs high dose of anti-BDNF antibody) was observed (F(1,33) = 8.594, p < 0.05). The β-VEH group exhibited enhanced 2 h STM compared with the WT-VEH group (p < 0.05). WT-low and -high groups displayed significantly worse recognition indices compared with the WT-VEH group (p < 0.05). The β-low group displayed a comparable recognition index (p > 0.05), but the β-high group displayed a significantly worse recognition index, compared with the WT-VEH group, respectively. #p < 0.05, compared with WT-VEH; *p < 0.05, compared with WT-low BDNF antibody. R, Investigation times. The WT-VEH, β-VEH, and β-low groups showed a significant reduction in investigation time (p < 0.05), whereas the other groups did not (p > 0.05). These observations indicated that the WT-VEH, β-VEH, and β-low groups formed 2 h STM. *p < 0.05, compared with the first exposure. T–V, Effects of the microinfusion of BDNF into the dorsal hippocampus on 48 h LTM in Y134F-A and WT mice (WT-VEH, n = 9; WT-BDNF, n = 10; A-VEH, n = 9; A-BDNF, n = 11). T, Recognition index. *p < 0.05, compared with the other three groups. U, Investigation time. The WT-BDNF and A-BDNF groups (p < 0.05), but not the WT-VEH and A-VEH groups (p > 0.05), showed significant reductions in investigation time, indicating that the WT-BDNF and A-BDNF groups formed LTM, whereas the other groups did not. *p < 0.05, compared with the first exposure. Error bars represent SEM.
For the contextual discrimination test, the mice were trained in the conditioning chamber described above. The mice assigned to the CXT-same (CXT-S) condition were tested in the same conditioning chamber as in training. The mice in the CXT-different (CXT-D) condition were tested in a different chamber (11 × 11 × 13 cm), which was made of smoked Plexiglas. For testing, the mice were assigned to a CXT-S or CXT-D condition and tested for 1 d (short delay) or 28 d (long delay) after fear conditioning. Mice were randomly assigned to the CXT or delay conditions. Memory was assessed as the percentage of time the mice spent freezing during 5 min.
Social recognition test.
The social recognition test was performed as described previously (Kogan et al., 2000; Fukushima et al., 2008). Adult mice were placed into individual plastic cages in an experimental room under dim light. The cages were identical to those in which mice were normally housed (plastic, 30 × 17× 12 cm). After a period of 60 min, a juvenile mouse was placed into a cage with a subject for a first-exposure trial lasting 45 s (see Fig. 8T) or 1 min (see Fig. 6A,C), 1.5 min (see Figs. 3A, 5A, 6E,G, 8E,K), or 3 min (see Fig. 8H,N,Q). The duration of social investigation behavior exhibited by the adult mouse was determined with a hand-held stopwatch. Social investigation was measured as described previously (Thor and Holloway, 1982). Memory was reassessed at 5 min, 30 min, 2 h, 24 h, or 48 h later by recording the length of investigation time exhibited by the subject to the same juvenile (second exposure). To evaluate the differences of ability to form social memory between the groups of mice, we calculated a recognition index, the ratio of the duration of the second and first investigation times. To examine effects of protein synthesis inhibition on 2 h STM (see Fig. 6E) and 24 h LTM (see Fig. 6G), mice received a systemic injection of anisomycin (ANI; 150 mg/kg, i.p.) immediately after the first exposure. ANI (Wako) was dissolved in saline (pH adjusted to 7.0–7.4 with NaOH).
Figure 6.
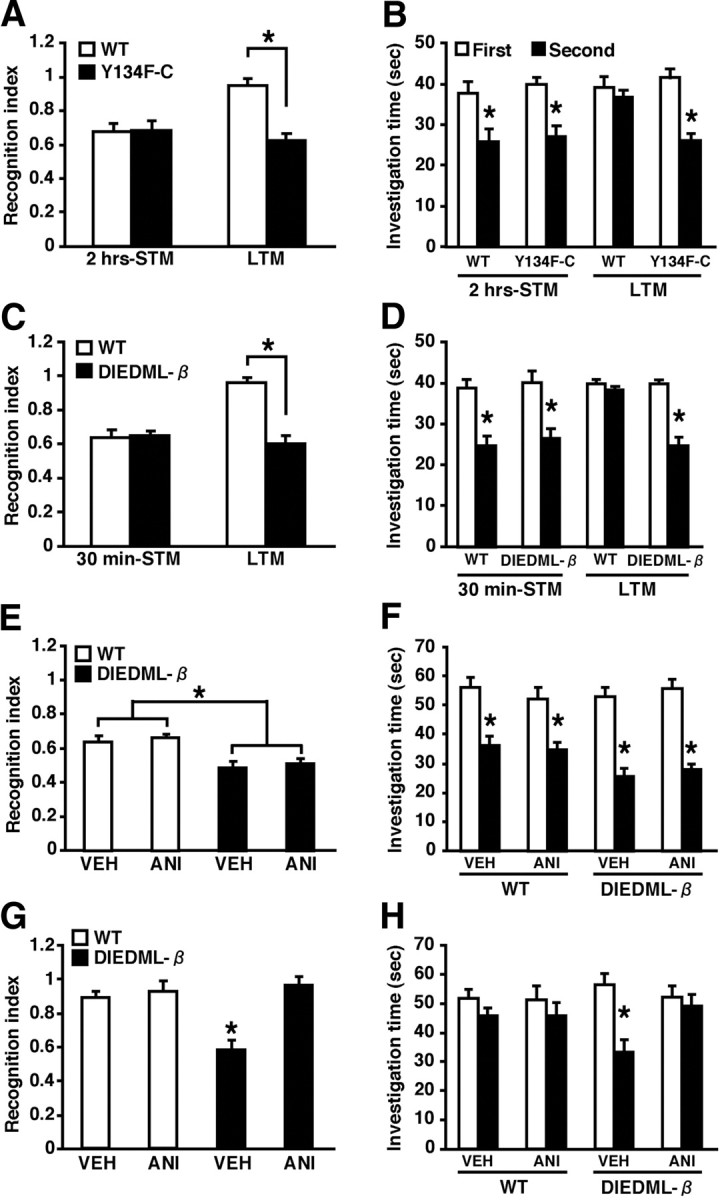
Dissociation of mechanisms for the enhancement of short-term and long-term social recognition memory in active CREB mice. A, B, Two-hour STM (WT, n = 8; C, n = 8) and 24 h LTM (WT, n = 8; C, n = 8) in Y134F-C and WT mice. C, D, Thirty-minute STM (WT, n = 8; β, n = 8) and 24 h LTM (WT, n = 8; β, n = 8) in DIEDML-β and WT mice. A, C, Recognition index. *p < 0.05. B, D, Investigation time. At 30 min (B) or 2 h (D) after the first exposure, WT and active CREB mutant mice showed a significant reduction in investigation time (p < 0.05). In contrast, 24 h after the first exposure (B, D), active CREB mutant but not WT mice showed a significant reduction in investigation time (WT, p > 0.05; C, p < 0.05; β, p < 0.05). These observations indicated that active CREB mice formed STM and LTM, whereas WT mice formed only STM but not LTM. *p < 0.05, compared with the first exposure. E, F, Effect of protein synthesis inhibition on 2 h STM in DIEDML-β and WT mice (WT-VEH, n = 8; WT-ANI, n = 9; β-VEH, n = 10; β-ANI, n = 9). E, Recognition index. *p < 0.05. F, Investigation time. Two hours after the first exposure, WT and DIEDML-β mice treated with ANI or VEH showed significant reductions in investigation time (p < 0.05), indicating that all groups formed STM. *p < 0.05, compared with the first exposure. G, H, Effect of protein synthesis inhibition on 24 h LTM in DIEDML-β and WT mice (WT-VEH, n = 11; WT-ANI, n = 10; β-VEH, n = 8; β-ANI, n = 8). G, Recognition index. *p < 0.05. H, Investigation time. Twenty-four hours after the first exposure, the β-VEH group showed a significant reduction in investigation time (p < 0.05), whereas the other groups of mice did not (p > 0.05), indicating that only the β-VEH group formed LTM. *p < 0.05, compared with the first exposure. Error bars represent SEM.
Morris water maze test.
The Morris water maze test was performed as described previously (Suzuki et al., 2004; Kim et al., 2011). Mice were trained with two trials per day for 10 d. The mice were tested at approximately the same time everyday. In the probe test at 24 h after 5 and 10 d of training, the platform was removed, and the mice were allowed to swim for 60 s. We measured the time the mice spent in each quadrant.
Passive avoidance test.
The passive avoidance test was performed as described previously (Fukushima et al., 2008; Zhang et al., 2011). The step-through passive avoidance apparatus (O'Hara & Co. Ltd.) consisted of a box with separate light and dark compartments. During the training sessions, each mouse was allowed to habituate to the light compartment for 30 s, and the guillotine door was raised to allow access to the dark compartment. As soon as the mice had entered the dark compartment, the guillotine door was closed. After 5 s, a footshock (0.2 mA) was delivered for a total period of 2 s. Memory was assessed at 2 or 24 h later as the crossover latency for the mice to enter into the dark compartment when re-placed in the light compartment, as in training. The test ended if the mice did not enter the shock chamber after 1200 s.
Surgery for drug microinfusion.
Surgeries were performed as described previously (Suzuki et al., 2008; Mamiya et al., 2009; Kim et al., 2011). Under Nembutal anesthesia and using standard stereotaxic procedures, stainless steel guide cannulae (22 gauge) were implanted into the dorsal hippocampus (−1.8 mm, ±1.8 mm, −1.9 mm). Mice were allowed to recover for at least 1 week after surgery. After this, they were handled for 1 week before the commencement of contextual fear conditioning or social recognition. BDNF (Promega), the BDNF inhibitor (K252a; Wako), and the anti-BDNF antibody (AB1513P; Millipore) were dissolved in vehicle solution [artificial CSF (ACSF)]. Mice were trained as described above and received intracranial injections of BDNF or K252a before or after the training as indicated. Infusions into each brain region (0.5 μl) were made at a rate of 0.25 μl/min. The injection cannula was left in place for 2 min after the infusion.
Immunohistochemistry.
Mice were contextual fear conditioned as described above. The no-conditioned group [conditioned stimulus (CS) only] received a training session in the absence of footshocks. The c-fos-positive cells were analyzed at 90 min after the training, as described previously (Zhang et al., 2011), using a polyclonal rabbit primary antibody for anti-c-fos (1:5000, Ab-5; Calbiochem). Quantification of c-fos-positive cells in sections (100 × 100 μm) of the amygdala (−1.22 and −1.34 mm relative to bregma) and dorsal hippocampus (−1.46 and −1.82 mm relative to bregma) were analyzed (Suzuki et al., 2008; Mamiya et al., 2009; Zhang et al., 2011). Data for the c-fos-positive cells in each group were expressed as a percentage of the averaged values in the no-conditioning WT control group (Kee et al., 2007).
Brain slice electrophysiology.
Brain slice electrophysiology was performed as described previously (Zhuo et al., 1999; Fukushima et al., 2008). Coronal brain slices (300 μm) containing the hippocampus from 6- to 8-week-old WT and Y134F mice were prepared using standard methods (Zhao et al., 2005). Coronal brain slices were transferred to a submerged recovery chamber with oxygenated (95% O2 and 5% CO2) ACSF containing (in mm) 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgSO4, 25 NaHCO3, 1 NaH2PO4, and 10 glucose at room temperature for at least 1 h. Whole-cell patch-clamp recordings were performed under visual guidance using an Olympus microscope fitted with infrared differential interference contrast optics (BX51WI; Olympus). EPSCs were recorded from CA1 pyramidal neurons with an Axon 200B amplifier (Molecular Devices), and stimulations were delivered by a bipolar tungsten stimulating electrode placed in the striatum radium. EPSCs were induced by repetitive stimulations at 0.02 Hz, and neurons were voltage clamped at −70 mV. The recording pipettes (3–5 MΩ) were filled with a solution containing (in mm) 145 K-gluconate, 5 NaCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Mg-ATP, and 0.1 Na3-GTP (adjusted to pH 7.2 with KOH). The induction protocol (referred to as the spike-timing protocol) for LTP involved three paired presynaptic stimuli that caused three EPSPs (10 ms ahead) with three postsynaptic action potentials (APs) at 30 Hz, paired 15 times every 5 s. The NMDA receptor-mediated component of EPSCs was pharmacologically isolated in ACSF containing CNQX (20 μm) and picrotoxin (100 μm). The patch electrodes for measuring NMDA receptor-mediated EPSCs contained (in mm) 102 CsMeSO3, 5 tetraethylammonium (TEA) chloride, 3.7 NaCl, 11 BAPTA, 0.2 EGTA, 20 HEPES, 2 MgATP, 0.3 NaGTP, and 5 QX-314 Cl (adjusted to pH 7.2 with CsOH). Neurons were voltage clamped at −30 mV, and NMDA receptor-mediated EPSCs were evoked at 0.05 Hz. Picrotoxin (100 μm) was always present to block GABAA receptor-mediated inhibitory currents. Access resistance was 15–30 MΩ and was monitored throughout the experiment. Data were discarded if access resistance changed >15% during an experiment.
For hippocampal field recording, male WT and CREB-Y134F-C mice at 8–10 weeks of age were anesthetized with inhaled halothane. Transverse slices of hippocampus (350 μm) were rapidly prepared and maintained in an interface chamber at 28°C, where they were subfused with ACSF consisting of (in mm) 124 NaCl, 4.4 KCl, 1 NaH2PO4, 1 MgSO4, 1.6 CaCl2, 25 NaHCO3, 10 glucose, and 25 sucrose, bubbled with 95% O2 and 5% CO2. The protocol of electrical stimulation and recordings has been described previously (Zhuo et al., 1999). Slices were kept in the recording chamber for at least 2 h before the experiments. A bipolar tungsten stimulating electrode was placed in the Schaffer collateral–commissural fibers, and extracellular field potentials were also recorded in the stratum radiatum using a glass microelectrode (3–12 MΩ, filled with ACSF). For LTP recording, stable field EPSPs (fEPSPs) were evoked by adjusting stimulation intensity so that a half-maximal fEPSP was elicited. Test responses were elicited at 0.02 Hz. LTP was induced using high-frequency stimulation (four 100 Hz, 1 s trains at 5 min intervals). Data were collected by Clampex 5.0 and analyzed using the pClamp 9.2 software. Data were normalized with respect to the mean values of the responses in the baseline period. For the statistical comparison of LTP in WT and CREB Y134F-C mice, data were calculated from 0 to 180 min after LTP induction.
Cell culture experiments.
COS-1 and PC12 cells were maintained and transiently transfected as described previously (Hosoda et al., 2004, 2009; Iwamoto et al., 2005). Luciferase and β-galactosidase activities were measured as described previously (Hosoda et al., 2004, 2009; Iwamoto et al., 2005). All reporter assays were performed in triplicate in three independent experiments. For the luciferase reporter assay, PC12 cells grown in 24-well plates were transiently transfected with pGAL4-Luc (100 ng) and pCMV-β gal (50 ng) with or without pM-CREB-WT, pM-CREB-Y134F, or pM-CREB-DIEDML (50 ng). pCMV-β-gal was cotransfected as a control for transfection efficiency. For the fluorescence resonance energy transfer (FRET)-based acceptor bleaching test, COS-1 cells grown in a 35 mm glass-bottom culture dish (MatTek) were transiently transfected with pCKIXnls-YCREB-WT, pCKIXnls-YCREB-S133A, or pCKIXnls-YCREB-Y134F (1000 ng) with or without pCMV-PKA or pCMV-acCaMKIV (100 ng). For the FRET-based real-time imaging, PC12 cells in a 35 mm glass-bottom culture dish were transiently transfected with pCKIXnls-YCREB-WT or pCKIXnls-YCREB-Y134F (1000 ng). The empty vector (pcDNA3) was used to standardize the total amount of transfected DNA. Solutions of forskolin (FSK) and ionomycin (Wako) were prepared in DMSO at 1 or 0.5 mm, respectively. Forty-eight hours after the transfection, imaging analyses were performed. Cells were imaged on an inverted microscope (IX-70; Olympus) with a cooled CCD camera and a w-view system (Hamamatsu Photonics) controlled by AUQACOSMOS software (Hamamatsu Photonics). We used a FRET filter set [excitation filter for cyan fluorescent protein (CFP), 440DF21; excitation filter for yellow fluorescent protein (YFP), 500AF25; emission filter for CFP, 480DF30; emission filter for YFP, 535DF25; dichroic mirror, 455DRLP] from Omega Optical for fluorescence imaging. For real-time imaging, the emission ratio (535 nm/480 nm) with excitation at 440 nm was measured. For the acceptor bleaching test, we used a 540AF30 filter (Omega Optical) for the photobleaching of YFP. Transfected cells were illuminated for 60 s without neutral density filters. FRET efficiency (E) is given by E = 1 − (Fda/Fd). Fd and Fda show the donor emission intensity before and after photobleaching, respectively. Each value represents the mean ± SD.
Data analysis.
Data were analyzed with ANOVA. One-way or two-way ANOVA followed by post hoc Newman–Keuls and post hoc Bonferroni's comparisons, respectively, were used to analyze the effects of groups, genotypes, times, treatments, and drugs. All values in the text and figure legends represent mean ± SEM. A paired t test was used to analyze differences of social investigation times within each group between first and second exposures in the social recognition test and to analyze differences of time spend in the target quadrant (TQ) compared with the other quadrants in Morris water maze test.
Results
Generation of CREB-Y134F expression mice
To generate transgenic mice expressing a dominant active CREB mutant, we used the CREB-Y134F mutant that displays a higher affinity with PKA compared with WT-CREB (Du et al., 2000). We first analyzed the function of CREB-Y134F in vitro using cultured cells. Two-way ANOVA followed by a post hoc Bonferroni's test revealed significant effects of mutation (WT-CREB and Y134F) and drug (0, 0.1, and 1 μm) (FSK: mutation, F(1,12) = 11.897, p < 0.05; drug, F(2,12) = 29.243, p < 0.05; ionomycin: mutation, F(1,12) = 29.844, p < 0.05; drug, F(2,12) = 22.41, p < 0.05). Consistent with a previous report, weak activation of PKA by the application of a low dose of FSK (0.1 μm) is sufficient to fully activate transcription by CREB-Y134F (Y134F vs WT, p < 0.05), whereas strong activation of PKA by the application of a high dose of FSK (1 μm) is required for WT-CREB (Y134F vs WT, p > 0.05) (Fig. 1A). CREB-Y134F also displayed significantly higher transcriptional activation compared with WT-CREB in response to activation of the Ca2+ signal transduction pathway by the application of ionomycin (Y134F vs WT, 0.1 μm, p < 0.05; 1 μm, p < 0.05) (Fig. 1A). We next compared the ability of Y134F and WT-CREB to form complexes with the transcriptional coactivator CBP using a FRET-based assay. We measured FRET efficiency using the acceptor bleaching approach in COS-1 cells transiently expressing YFP fused with CREB, CREB-S133A (a dominant-negative mutant of CREB), or CREB-Y134F (YFP-CREBs), and CFP fused with KIX, a domain of CBP that interacts with CREB (CFP-KIX). Two-way ANOVA followed by a post hoc Bonferroni's test revealed significant effects of mutation and enzyme expression (PKA or CaMKIV) (PKA: mutation, F(1,163) = 10.306, p < 0.05; enzyme, F(1,163) = 13.156, p < 0.05; CaMKIV: mutation, F(1,73) = 16.515, p < 0.05; enzyme, F(1,173) = 22.908, p < 0.05). CREB-Y134F formed more complexes with CBP compared with WT-CREB, with and without the overexpression of the catalytic subunit of PKA (p < 0.05) or a dominant active mutant of CaMKIV (acCaMKIV; p < 0.05), whereas CREB-S133A failed to form a complex with KIX (p > 0.05) (Fig. 1B). Furthermore, real-time image monitoring of the interaction between CREB and KIX in PC12 cells revealed that weak activation of PKA by the application of a low dose of FSK is sufficient to induce the formation of a complex between CREB-Y134F and KIX (Fig. 1C). These in vitro observations indicate that CREB-Y134F functions as an active CREB mutant that exhibits a higher sensitivity to the activation of upstream signal transduction compared with WT-CREB.
We next constructed a transgene that contained an αCaMKII promoter, a hybrid intron in the 5′ untranslated leader, the coding region of CREB-Y134F fused with the Flag tag sequence at the N terminus, and a polyadenylation signal (Fig. 1D). The αCaMKII promoter exhibits strong activity in regions of the forebrain including the hippocampus, cortex, and amygdala (Mayford et al., 1996; Kida et al., 2002; Fukushima et al., 2008; Hasegawa et al., 2009). In the present study, we generated two lines of CREB-Y134F transgenic mice and performed expression analyses. Northern blot analysis, using a specific probe for the transgene mRNA, showed that a transgene transcript was expressed in two lines and that transgenic line C (Y134F-C) mice display higher expression of transgene mRNA than line A (Y134F-A) mice (Fig. 1E). Consistent with this, in situ hybridization analyses revealed the expression of transgene mRNA in the CA1, CA3, and dentate gyrus of the hippocampal and cortical regions in both transgenic lines (Fig. 1F).
To examine the effects of CREB-Y134F expression in the forebrain, we analyzed the expression levels of the CREB target gene c-fos (Sheng et al., 1991) in the CA1 region of the hippocampus and amygdala after fear conditioning (Fig. 1G,H). We performed contextual fear conditioning, a form of associative memory between a context (CS) and an aversive stimulus [unconditioned stimulus (US)]. Transgenic mice and WT littermates were trained with a footshock (0.4 mA; CS–US) or no footshock (CS only), and the level of c-fos expression was assessed 90 min after conditioning using immunohistochemistry. Two-way ANOVA followed by a post hoc Bonferroni's test revealed significant effects of genotype [Y134F-A (A) or -C (C) vs WT] and conditioning (A: CA1, genotype, F(1,29) = 20.014, p < 0.05; conditioning, F(1,29) = 22.446, p < 0.05; amygdala, genotype, F(1,29) = 14.452, p < 0.05; conditioning, F(1,29) = 35.146, p < 0.05; C: CA1, genotype, F(1,30) = 36.42, p < 0.05; conditioning, F(1,30) = 12.834, p < 0.05; amygdala, genotype, F(1,30) = 28.494, p < 0.05; conditioning, F(1,30) = 16.199, p < 0.05). The expression of c-fos in the CA1 region and amygdala was significantly higher in conditioned WT and Y134F mice compared with unconditioned WT and Y134F mice (p < 0.05), suggesting that contextual fear learning was associated with increased levels of c-fos in the CA1 region of the hippocampus and in the amygdala. However, these results could not exclude the possibility that CREB-mediated expression is activated by a footshock but not in response to learning. More importantly, Y134F-A and -C mice displayed significantly higher c-fos expression levels in the CA1 region and amygdala with or without contextual fear conditioning in a dose-dependent manner of transgene expression compared with WT mice (p < 0.05). These results suggest that the expression of CREB-Y134F was reflected by an increased level of CREB-mediated gene expression.
Consistently, the basal levels of c-fos expression in the hippocampus and the amygdala (without any treatment) were significantly greater in Y134F-A and -C mice compared with WT littermates in a dose-dependent manner of transgene expression (Fig. 1I).
Basal synaptic transmission and LTP in the hippocampus of CREB-Y134F expression mice
It is predicted that the expression of CREB-Y134F leads to changes in synaptic plasticity. To study the properties of synaptic transmission, we performed whole-cell patch-clamp recordings in the CA1 region of hippocampal slices using Y134F-C mice that display higher transgene expression compared with Y134F-A transgenic mice. We examined the input–output relationships of AMPA receptor- and NMDA receptor-mediated EPSCs evoked by various stimulus intensities. To record the AMPA receptor-mediated EPSCs, we applied 50 μm AP-5 in the recording solution. The input–output relationships of the AMPA receptor-mediated EPSCs in Y134F-C mice was comparable to that of WT mice (Fig. 2A). Next, we examined the NMDA receptor-mediated EPSCs in the presence of 20 μm CNQX. There was no difference in the NMDA receptor-mediated EPSCs between WT and Y134F-C mice (Fig. 2B). Similarly, no differences were found in the current–voltage relationship of EPSCs mediated by either the AMPA or NMDA receptors between WT and Y134F-C mice [AMPA I–V relationship (Fig. 2C), NMDA I–V relationship (Fig. 2D)].
Figure 2.

Enhanced LTP in the hippocampal CA1 neurons of CREB-Y134F mice. A, Input–output relationship of the AMPA receptor-mediated EPSCs. B, Input–output relationship of the NMDA receptor-mediated EPSCs. C, Current–voltage relationship of EPSCs mediated by the AMPA receptor. D, Current–voltage relationship of EPSCs mediated by the NMDA receptor. E, Paired-pulse facilitation. F, Miniature EPSCs in the presence of 1 μm tetrodotoxin. G, H, LTP was induced by three presynaptic stimuli that caused three EPSPs (10 ms ahead) with three postsynaptic APs at 30 Hz, paired 15 times every 5 s (EPSP–AP protocol). The insets show averages of six EPSCs at baseline response (a) and 25–30 min after induction of LTP (b). Normalized EPSC amplitudes at 25–30 min after LTP induction (bar graph) are shown. G, Y134F-C. H, Y134F-A. I, L-LTP induced by high-frequency stimulation [4 × 100 Hz, 1 s trains at 5 min intervals] in hippocampal slices. Inset, representative fEPSP traces before and 180 min after stimulation. Normalized fEPSC amplitudes at 0–180 min after LTP induction (bar graph) are shown. WT, n = 5–8 (A–I); A, n = 7 (H); C, n = 5–8 (A–G, I). Error bars represent SEM. *p < 0.05.
Next, we examined paired-pulse facilitation, a simple form of synaptic plasticity; however, we observed no differences between WT and Y134F-C mice (Fig. 2E). We also measured miniature EPSCs in the presence of 1 μm tetrodotoxin. We found no significant difference in the frequency (0.4 ± 0.1 vs 0.4 ± 0.1 Hz; t test, p > 0.05), amplitude (8.8 ± 0.7 vs 8.8 ± 0.9 pA; t test, p > 0.05), rise time (10–90% of peak amplitude: 2.2 ± 0.1 vs 2.4 ± 0.2 ms; t test, p > 0.05), or decay time (90–37% of peak amplitude: 9.0 ± 0.4 vs 10.2 ± 0.4 ms; t test, p > 0.05) between WT and Y134F-C mice (Fig. 2F). These results suggest that basal synaptic transmission is not altered by the expression of CREB-Y134F.
We also examined whether the expression of CREB-Y134F affected synaptic potentiation. LTP was induced using three presynaptic stimuli that caused three EPSPs (10 ms ahead) with three postsynaptic APs at 30 Hz, paired 15 times every 5 s (the EPSP-AP protocol) (Zhao et al., 2005). LTP was induced within 12 min after establishing the whole-cell configuration to avoid the washout of intracellular contents that are critical for the establishment of synaptic plasticity (Tsvetkov et al., 2002; Zhao et al., 2005). LTP was induced using the EPSP-AP protocol resulting in a significant, long-lasting synaptic potentiation in slices from WT mice (169.6 ± 11.8% at 25–30 min after LTP induction; t test, p < 0.05 compared with the baseline responses before the induction). The same protocol also induced LTP in Y134F-C mice (240.5 ± 29.2% at 25–30 min after LTP induction; t test, p < 0.05 compared with the baseline responses) (Fig. 2G) and in Y134F-A mice that express the transgene at a lower level than the Y134F-C mice (170.7 ± 12.4% at 25–30 min after LTP induction; t test, p < 0.05 compared with the baseline responses) (Fig. 2H). The magnitude of synaptic potentiation in Y134F-C mice was significantly greater at 25–30 min after LTP induction than those of WT mice (WT, 170.7 ± 12.4% of baseline; Y134F-C, 240.5 ± 29.2%; one-way ANOVA, p < 0.05) (Fig. 2G), and there was no significant difference in the magnitude of synaptic potentiation between WT and Y134F-A mice (one-way ANOVA, p > 0.05) (Fig. 2H). These results indicate that the expression of CREB-Y134F led to significant enhancement of synaptic potentiation in CA1 hippocampal neurons in a dose-related manner of transgene expression.
To further investigate the contribution of CREB to late-phase LTP (L-LTP) in the hippocampus, we have also performed field recordings of L-LTP in the CA1 region of the hippocampus using interface slice recording. As reported previously (Zhuo et al., 1999), L-LTP was induced by applying four trains of tetanic stimulation (100 Hz for 1 s) delivered at a 5 min interval. Consistently, we found that L-LTP measured at 0–180 min after the induction was significantly enhanced in hippocampal slices in Y134F-C mice compared with those in WT littermates (WT, 179.8 ± 2.3% of baseline; Y134F-C, 279.3 ± 2.2%; one-way ANOVA, p < 0.05) (Fig. 2I). These results indicate that enhancement of CREB activity contributes to increases in hippocampal L-LTP.
Expression of CREB-Y134F enhances memory formation
To examine the effects of expressing CREB-Y134F in the forebrain on learning and memory, we performed aversive and nonaversive memory tasks using transgenic mice and WT littermates. In these memory tests, the mice were trained using weak protocols that would increase the likelihood of observing enhanced memory formation in Y134F mice (Fukushima et al., 2008).
We first performed the social recognition test to examine the ability to form nonaversive social memory using Y134F and WT male littermates (Fig. 3A,B). This task measures the difference in time taken to investigate a juvenile male mouse by comparing between the first and second exposures for 1.5 min. The second exposure was performed at 30 min, 2 h, or 24 h after the first exposure. We assessed the recognition index (the ratio of the second investigation time relative to the first investigation time). To clarify effects of genotype (WT and Y134F-A and -C), we performed one-way ANOVAs with genotype at each time point. We observed effects of genotype when memory was tested 2 and 24 h, but not 30 min, after training (2 h, F(2,44) = 4.398, p < 0.05; 24 h, F(2,41) = 5.737, p < 0.05; 30 min, F(2,35) = 0.105, p > 0.05). The post hoc Newman–Keuls test revealed that Y134F-A and -C mice showed significantly better recognition indices of 24 h LTM (p < 0.05) but showed comparable recognition indices for 30 min STM (p > 0.05), compared with WT mice. These observations indicated that Y134F mice displayed comparable 30 min STM but enhanced 24 h LTM compared with WT mice. Importantly, the Y134F-C mice display a better recognition index at 24 h compared with Y134F-A mice although this difference was not statistically significant, suggesting that Y134F mice displayed enhanced LTM in a dose-dependent manner of transgene expression.
More interestingly, Y134F-C mice (p < 0.05), but not Y134F-A mice (p > 0.05), exhibited a significantly better recognition index for 2 h STM compared with WT mice, indicating that Y134F-C but not Y134F-A mice displayed an enhanced 2 h STM. Importantly, the Y134F-A mice display a better and worse recognition index at 2 h compared wit WT or Y134F-C mice, respectively, although these differences were not statistically significant, indicating that Y134F-C mice are different from WT but Y134F-A mice are not. Thus, these observations indicate that the Y134F-A mice display enhanced LTM, whereas Y134F-C mice, with higher transgene expression than Y134F-A mice, display enhanced STM and LTM.
Consistently, comparisons of the investigation times confirmed these observations (Fig. 3B); Y134F mice formed 30 min and 2 h STM and LTM, whereas WT mice formed only STMs but not LTMs.
We next performed the contextual fear-conditioning task to examine the ability to form aversive memories (Fig. 3C). Mice were trained with a single footshock (0.4 mA), and behavioral freezing was assessed at 30 min, 2 h, or 24 h after training. Consistent with the results of the social recognition experiment, one-way ANOVAs with genotype revealed effects of genotype at 2 and 24 h, but not 30 min (2 h, F(2,40) = 5.027, p < 0.05; 24 h, F(2,86) = 4.898, p < 0.05; 30 min, F(2,46) = 0.566, p > 0.05). The post hoc Newman–Keuls test revealed that Y134F-A and -C mice displayed enhanced 24 h LTM (p < 0.05) and normal 30 min STM (p > 0.05) compared with WT mice. Moreover, Y134F-C mice showed enhanced 2 h STM compared with WT and Y134F-A mice (p < 0.05), whereas Y134F-A mice showed comparable 2 h STM compared with WT mice. These observations confirmed our results that Y134F-C mice exhibit enhanced 2 h STM and LTM, whereas Y134F-A mice exhibit only enhanced LTM. However, we could not exclude the possibility that Y134F-A mice also show enhanced STM in other experimental conditions such as stronger training protocols. Importantly, Y134F mice displayed normal pain sensitivity (data not shown).
To further examine whether CREB-Y134F mice form stronger memories than WT mice, we compared the ability in context fear discrimination between Y134F-C and WT mice (Fig. 3D). Mice were conditioned in the contextual chamber A (training context). Behavioral freezing 1 d or 1 month after conditioning was assessed in the training context or a novel environment (novel chamber). Three-way ANOVA revealed significant genotype (WT vs Y134F-C) × time point (1 d vs 1 month) interaction (F(1,95) = 4.408, p < 0.05) and genotype × context (training chamber vs novel environment) interaction (F(1,95) = 13.198, p < 0.05). To further clarify the time point × context interaction, we performed ANOVAs on WT mice and Y134F-C mice, respectively, and observed significant time point × context interactions in WT mice but not Y134-F mice (WT, F(1,45) = 4.947, p < 0.05; Y134F-C, F(1,50) = 0.477 p > 0.05). Consistent with previous findings (Frankland et al., 1998; Wiltgen and Silva, 2007; Winocur et al., 2007), a post hoc Bonferroni's test revealed that WT mice displayed an impaired contextual discrimination when the memory was remote but not recent; conditioned freezing levels were significantly greater in the training context than in the novel environment (p < 0.05) when tested 1 d after the training, whereas these were similar in both contexts when tested 1 month after training (p > 0.05). In contrast, Y134F-C mice maintained the contextual discrimination of the training context and a novel environment; their conditioned freezing levels were greater in the training context than in the novel environment at both time points (p < 0.05). Importantly, Y134F-C mice froze significantly more often in the training context but not in the novel environment compared with WT mice at both time points (p < 0.05), confirming the observations in Figure 3C that Y134F-C mice display enhanced contextual fear memories. Thus, these observations indicated that Y134F-C mice form more precise contextual memories compared with WT mice.
We also performed the Morris water maze task to assess the formation of spatial memory (Fig. 3E–G). In this test, animals learned to escape to a hidden platform in a swimming pool using spatial cues located around the pool. WT and Y134F-A and -C mice were trained with two trials per day for 10 d (at 1 min intervals). Two-way repeated-measure ANOVA with genotype (WT and Y134F-A and -C) and time (days 1–10) revealed significant effects of time and genotype (time, F(9,432) = 96.909, p < 0.05; genotype, F(2,432) = 5.232, p < 0.05). All groups of mice improved their performance as indicated by the decreasing escape latencies to find the platform over successive days. To further clarify effects of genotype, we performed two-way ANOVAs with genotype (WT vs Y134F-A or -C) and time. Significant effects of genotype and time × genotype interactions were observed between WT and Y134F-C mice, but not between WT and Y134F-A mice (genotype, WT vs Y134F-A, F(1,32) = 3.103, p > 0.05; WT vs Y134F-C, F(1,37) = 10.26, p < 0.05; genotype vs time, WT vs Y134F-A, F(1,288) = 1.793, p > 0.05; WT vs Y134F-C, F(1,333) = 1.966, p < 0.05). These results indicated that Y134F-C mice displayed significantly superior performance during training. To measure the formation of spatial memory, spatial memory was evaluated in probe tests at 6 and 11 d after training for 5 and 10 d, respectively. In the probe trials, mice were allowed to swim for 60 s, and the time spent in each quadrant of the pool was scored. In probe test 1, χ2 test revealed no significant differences in time spent in four quadrants in all groups (WT and Y134F-A and -C, p > 0.05), suggesting that all groups displayed no preference for the TQ. However, planned comparisons using a paired t test revealed that Y134-A and -C mice displayed significantly more time spent in the TQ compared with all of the other quadrants [TQ vs opposite (OP), adjacent right (AR), and adjacent left (AL); paired t test, p < 0.05] but WT mice did not. Together, these observations suggest that Y134F-A and -C mice formed spatial memory faster than WT mice. In probe test 2, χ2 test revealed significant differences in time spent in the four quadrants in Y134F-C mice but not WT and Y134F-A mice (WT, p > 0.05; Y134F-A, p > 0.05; Y134F-C, p < 0.05), suggesting that Y134F-C mice displayed a preference for the TQ but WT and Y134F-A mice did not. Additionally, planned comparisons using a paired t test revealed that all groups spent time significantly in the TQ compared with all of the other quadrants (TQ vs OP, AR or AL; paired t test, p < 0.05), suggesting that all groups formed spatial memory. Furthermore, Y134F-C mice showed significantly more time spent in the TQ compared with WT mice (one-way ANOVA, F(1,37) = 6.617, p < 0.05). Together, these observations indicated that Y134F-C mice displayed enhanced spatial memory. Thus, Y134F-A and -C mice revealed enhanced spatial memory formation in a dose-related manner of transgene expression compared with WT mice.
Furthermore, we also performed a passive avoidance task and observed enhanced 2 h STM and 24 h LTM in Y134F-C mice (Fig. 3H). There were no significant differences in crossover latency observed across the groups during training. In contrast, one-way ANOVA revealed significant effects of genotype (2 h, F(1,26) = 4.251; 24 h, F(1,27) = 5.678, p < 0.05). Consistently, Y134F-C mice exhibited significantly higher crossover latency at 2 and 24 h after the training compared with WT mice (p < 0.05).
Our observations in Figure 3 indicate that the expression of CREB-Y134F in the forebrain improves the formation of LTM. Interestingly, the results from Y134F-C mice suggest that the higher expression of CREB-Y134F enhances STM and LTM.
Expression of CREB-DIEDML enhances STM and LTM
The CREB-DIEDML mutant contains mutations of a 6 amino acid cluster that enables it to constitutively interact with CBP (Cardinaux et al., 2000). Our in vitro analyses using cultured cells showed that CREB-DIEDML induced a higher level of CREB-meditated transcription at the basal condition and formed more complexes with CBP compared with CREB-Y134F and WT-CREB (Fig. 4A,B). To further investigate the effects of upregulating CREB activity on STM and LTM, we generated transgenic mice expressing CREB-DIEDML in the forebrain under the control of the αCaMKII promoter (Fig. 4C). Two lines (α and β) of CREB-DIEDML mutant mice displayed comparable transgene expression levels in the forebrain (Fig. 4D,E). Importantly, the results of Western blotting using an anti-CREB antibody showed that both transgenic lines displayed higher total CREB levels compared with Y134F mice (Fig. 4F). To examine the effects of CREB-DIEDML expression at the molecular level, we analyzed the expression levels of c-fos after contextual fear conditioning using the same procedure as in Figure 1 (Fig. 4G,H). Two-way ANOVA identified significant effects of genotype (DIEDML-α or -β vs WT) and conditioning (α, CA1, genotype, F(1,25) = 31.568, p < 0.05; conditioning, F(1,25) = 11.505, p < 0.05; amygdala, genotype, F(1,25) = 16.236, p < 0.05; conditioning, F(1,25) = 10.235, p < 0.05; β, CA1, genotype, F(1,22) = 33.85; conditioning, F(1,22) = 14.054, p < 0.05; amygdala, genotype, F(1,22) = 42.95, p < 0.05; conditioning, F(1,22) = 15.476, p < 0.05). DIEDML-α and -β mice revealed significantly higher c-fos expression levels in the hippocampal CA1 and amygdala with or without contextual fear conditioning compared with WT mice (p < 0.05), suggesting that expression of CREB-DIEDML contributes to the upregulation of CREB activity in the forebrain. Importantly, coimmunoprecipitation of hippocampal nuclear extracts with an anti-CBP antibody showed that DIEDML mice formed more complexes between CREB and CBP in the hippocampus compared with WT mice (Fig. 4I–K). These observations suggested that the upregulation of CREB-mediated gene expression in DIEDML mice reflects the increased formation of CREB–CBP complexes after the expression of CREB-DIEDML.
We then performed behavioral experiments using these transgenic mice. In social recognition paradigms, we observed similar results for DIEDML mice with those of Y134F-C mice (Fig. 5A,B). We measured 5 min STM because our pilot experiment suggested that DIEDML mice displayed enhancement of 30 min STM. One-way ANOVAs with genotype revealed effects of genotype at 30 min, 2 h, and 24 h but not at 5 min (30 min, F(2,65) = 11.564, p < 0.05; 2 h, F(2,40) = 5.327, p < 0.05; 24 h, F(2,105) = 8.972, p < 0.05; 5 min, F(2,49) = 0.031, p > 0.05). DIEDML-α and -β mice showed enhanced 2 h STM and 24 h LTM (p < 0.05) and normal 5 min STM (p > 0.05) compared with WT mice. However, in contrast to the results for Y134F-C mice, DIEDML-α and -β mice also displayed enhanced 30 min STM compared with WT mice (p < 0.05).
Consistent with these observations, in contextual fear-conditioning paradigms (Fig. 5C), one-way ANOVAs with genotype revealed effects of genotype at 30 min, 2 h, and 24 h but not at 5 min (30 min, F(2,71) = 4.492, p < 0.05; 2 h, F(2,58) = 5.257, p < 0.05; 24 h, F(2,43) = 6.039, p < 0.05; 5 min, F(2,68) = 0.242, p > 0.05). DIEDML-α and -β mice revealed normal 5 min STM (p > 0.05) but revealed enhanced 30 min and 2 h STMs and 24 h LTM (p < 0.05). Importantly, DIEDML mice displayed normal pain sensitivity (data not shown). These results confirmed and extended our observations that the upregulation of CREB activity leads to the enhancement of STM and LTM, even 30 min STM.
Dissociation of the mechanisms for the enhancement of STM and LTM in active CREB mice
We observed the enhancement of STM and LTM in three lines of active CREB mutant mice (Y134F-C and DIEDML-α and -β). To determine whether the mechanisms for the enhancement of STM and LTM are dissociable, we performed social recognition paradigms using a weaker training protocol than shown in Figures 3 and 5 (Fig. 6A–D). Y134F-C, DIEDML-β, and WT male mice were exposed to juvenile male mice twice for 1 min, but not 1.5 min, for the short (2 h or 30 min, respectively) and long (24 h) interval. In contrast to the results shown in Figures 3 and 5, two-way ANOVA between the active CREB mutants and WT mice revealed significant time point (30 min or 2 h STM vs LTM) × genotype (Y134F-C or DIEDML-β vs WT) interactions (C, 2 h STM vs LTM, F(1,28) = 9.772, p < 0.05; β, 30 min STM vs LTM, F(1,28) = 14.475, p < 0.05). Y134F-C and DIEDML-β mice showed comparable recognition indices for 2 h and 30 min STM (p > 0.05), respectively, but showed significantly better recognition indices for 24 h LTM (p < 0.05) compared with WT mice. These results indicated that in these experimental conditions, Y134F-C and DIEDML-β mice displayed enhanced LTM but showed normal STM. These observations indicate that expression of these active CREB mutants enhanced LTM without affecting STM in weaker training conditions and that, interestingly, enhancement of STM by the upregulation of CREB activity depends on the training condition; transgenic mice failed to enhance STM under the weaker training condition.
To further compare mechanisms for the enhancement of STM and LTM, we examined effects of inhibition of protein synthesis on enhanced 2 h STM (Fig. 6E,F) and 24 h LTM (Fig. 6G,H) in DIEDML-β mice. WT and DIEDML-β male mice were exposed to juvenile male mice twice for 1.5 min in 2 or 24 h intervals and received a systemic injection of ANI (150 mg/kg) immediately after the first exposure. Two-way ANOVA revealed a significant effect of genotype but not drug treatment and genotype × drug interaction when 2 h STM was assessed (genotype, F(1,32) = 14.656, p < 0.05; drug, F(1,32) = 0.396, p > 0.05; interaction, F(1,32) = 0.00018, p > 0.05). DIEDML-β mice treated with and without ANI showed comparable recognition indices for 2 h STM (p > 0.05), but these groups of mice showed significantly better recognition indices (p < 0.05) compared with both groups of WT mice. These results indicated that protein synthesis inhibition did not affect the enhancement of STM in DIEDML-β mice, suggesting that this enhanced STM does not depend on new gene expression. In contrast to these results, two-way ANOVA revealed a significant genotype × drug treatment interaction when 24 h LTM was assessed (F(1,33) = 8.781, p < 0.05). Consistent with previous observations, DIEDML-β mice treated with vehicle showed a significantly better recognition index (p < 0.05) compared with the other groups. However, DIEDML-β mice treated with ANI showed a significantly worse (p < 0.05) or comparable (p > 0.05) recognition index compared with DIEDML-β mice treated with vehicle and both groups of WT mice, respectively. These results indicated that DIEDML-β mice treated with ANI failed to enhance LTM formation.
Together, these observations suggest the dissociation of mechanisms for enhancement of STM and LTM in active CREB mice.
Roles of BDNF in the enhancement of STM
CREB-mediated transcription is required for memory consolidation (Bourtchuladze et al., 1994; Oike et al., 1999; Kida et al., 2002; Pittenger et al., 2002; Korzus et al., 2004). Therefore, the upregulation of CREB activity is thought to improve LTM by enhancing memory consolidation. However, the mechanism of enhanced STM by increased CREB activity is unclear; abundant studies have shown that CREB is not required for STM. The BDNF gene has been identified as a CREB-target gene (Finkbeiner et al., 1997). Previous studies have demonstrated that infusion of BDNF or a BDNF inhibitor into the dorsal hippocampus enhanced or impaired, respectively, STM in a passive avoidance task (Alonso et al., 2002, 2005). These findings raise the possibility that BDNF is upregulated by the constitutive activation of CREB-mediated transcription at the basal condition in active CREB mutant mice, thereby leading to enhanced STM. To test this, we first measured the levels of BDNF in the hippocampus of active CREB mutant and WT mice at the basal condition (Fig. 7). Compared with WT mice, Y134F-C mice exhibited significantly higher levels of BDNF (p < 0.05), whereas Y134F-A mice exhibited normal BDNF levels (p > 0.05). More importantly, DIEDML-α and -β mice exhibited significantly higher expression levels of BDNF even compared with Y134F-C mice (p < 0.05). It is important to note that DIEDML-β mice displayed comparable expression levels of TrkB and p75 receptor mRNAs in the hippocampus compared with WT mice (data not shown).
Figure 7.

BDNF expression levels in the hippocampus of active CREB mutant mice. Y134F-C (n = 6), DIEDML-α (n = 8), and DIEDML-β (n = 8) mice showed a higher level of BDNF expression in the hippocampus compared with WT (n = 15) and Y134F-A (n = 6) mice. Error bars represent SEM. #p < 0.05, compared with WT; *p < 0.05.
Together with our previous behavioral findings, these observations suggest a positive correlation between hippocampal BDNF expression levels and the enhancement of STM; higher expression of BDNF leads to enhancement of shorter-term memory.
It is important to note that we observed a decrease in BDNF expression levels in the hippocampus of CREBαδ−/− mice (data not shown) that displayed a decrease in CREB activity and impaired 1 h STM (Bourtchuladze et al., 1994). This result supports our hypothesis that BDNF expression levels affect STM.
We examined the effects of the microinfusion of BDNF or a BDNF inhibitor (TrkB blocker; K252a) into the dorsal hippocampus on memory performance. Mice were trained with a single footshock (0.4 mA), and behavioral freezing at 2 h after training was assessed. In agreement with previous findings (Alonso et al., 2002, 2005), microinfusion of BDNF at 3 h before training or K252a immediately after training into the dorsal hippocampus enhanced or impaired, respectively, the 2 h short-term contextual fear memory (BDNF, F(1,23) = 10.334, p < 0.05; K252a, F(1,17) = 8.861, p < 0.05) (Fig. 8A–D). Furthermore, we extended these observations using social recognition paradigms (Fig. 8E–J). WT mice received the dorsal hippocampal microinfusion of BDNF or vehicle (VEH) at 3 h before the first exposure, or K252a or VEH immediately after the first exposure, and were exposed to a juvenile male mouse twice for 1.5 or 3 min, respectively. The BDNF- or K252a-treated group showed a significantly better or worse, respectively, recognition index compared with the VEH group (BDNF, F(1,14) = 4.976, p < 0.05; K252a, F(1,14) = 5.757, p < 0.05), indicating that microinfusion of BDNF or K252a enhanced or impaired 2 h STM, respectively.
To further investigate whether the upregulation of BDNF contributes to the enhanced STM in active CREB mice, we examined the effects of the microinfusion of BDNF or K252a on short-term social recognition memory in active CREB mice.
Y134F-C mice expressing high levels of BDNF displayed normal 30 min STM [but showed enhanced 2 h STM (Fig. 3A)]. In contrast, enhanced 30 min STM was observed in DIEDML mice (Fig. 5A) expressing higher levels of BDNF than Y134F-C mice (Fig. 7). Therefore, it is possible that the microinfusion of a low dose of BDNF into the hippocampus is sufficient to enhance 30 min STM in Y134F-C mice. To test this hypothesis, WT and Y134F-C male mice were microinfused with a low dose of BDNF (50 ng/side) into the dorsal hippocampus at 3 h before first exposure and exposed to juvenile male mice twice for 1.5 min in 30 min intervals (Fig. 8K–M). Two-way ANOVA revealed a significant genotype × drug interaction (F(1,30) = 5.737, p < 0.05). Y134F-C mice treated with BDNF displayed a significantly better recognition index compared with the other three groups (p < 0.05) that displayed comparable recognition indices (p > 0.05). These results indicated that the infusion of a low dose of BDNF enhanced 30 min STM in Y134F-C, but not WT mice, suggesting that the increased levels of BDNF observed in Y134F-C mice contribute to the enhancement of 30 min STM.
We compared the effects of K252a infusion into the dorsal hippocampus on 2 h STM between DIEDML-β and WT mice (Fig. 8N-P). In this experiment, mice were trained using a stronger protocol (3 min exposure) that was expected to increase the likelihood of observing the impaired STM after K252a infusion. Male mice were exposed to juvenile male mice twice for 3 min with a 2 h interval and received an infusion of K252a (3 or 9.35 ng per side) immediately after the first exposure. Two-way ANOVA comparing recognition indices revealed significant interactions between genotype × drug treatment (low dose vs high dose of K252a; F(1,33) = 4.719, p < 0.05). Consistent with the previous observations, we observed that DIEDML-β mice exhibited enhanced 2 h STM compared with WT mice when both groups were treated with vehicle (p < 0.05). WT mice treated with low and high doses of K252a displayed significantly worse recognition indices compared with WT mice treated with vehicle (p < 0.05). Importantly, DIEDML-β mice displayed a significantly better recognition index when treated with a low dose of K252a (p < 0.05), but a comparable recognition index was observed when they were given injections of a high dose of K252a, compared with WT mice treated with a low dose of K252a. These results indicated that a low dose of K252a disrupted 2 h STM in WT mice and that a higher dose of K252a is required to impair 2 h STM in DIEDML-β mice compared with WT mice. To also confirm effects of inhibition of BDNF, we examined the effects of the microinfusions of anti-BDNF antibody into the dorsal hippocampus on 2 h STM between DIEDML-β and WT mice and obtained similar results with those using K252a (Fig. 8Q–S).
Together, these findings indicated a regulatory role of BDNF in STM, suggesting a contribution of the increased BDNF levels on the enhanced STM of active CREB mice.
Contribution of upregulated BDNF on LTM in active CREB mice
We finally asked whether increased levels of BDNF contribute to the enhancement of LTM and STM in active CREB mice. To test this, we examined the effects of a BDNF infusion (250 ng per side) on LTM in Y134F-A mice that displayed enhanced LTM but normal STM (Fig. 8T–V). In this experiment, mice were trained using a weaker protocol (45 s exposure and 48 h LTM) than in the previous experiments, which was expected to enable us to observe the effects of the interaction between the increase in CREB activity and BDNF infusion. Male mice were infused with BDNF (250 ng per side) into the dorsal hippocampus at 3 h before the first exposure and exposed to juvenile male mice twice for 45 s with a 48 h interval. Two-way ANOVA comparing the recognition indices revealed a significant genotype × drug treatment interaction (F(1,35) = 4.308, p < 0.05). WT and Y134F-A mice treated with BDNF displayed significantly better recognition indices compared with WT or Y134F-A mice, respectively, treated with vehicle (p < 0.05). Importantly, Y134F-A mice treated with BDNF displayed a significantly better recognition index compared with WT mice treated with BDNF (p < 0.05). These results indicated that BDNF infusion not only led to enhanced LTM in WT mice but also led to a further enhancement of LTM in Y134F-A mice, suggesting that the increased levels of BDNF and the upregulation of CREB activity cooperatively enhanced LTM formation. However, we could not exclude the possibility that the Y134F-A transgenic line does have an upregulation of BDNF, at a low level that falls below the sensitivity of the assay performed, thereby leading to further enhancement of LTM by the infusion of additional BDNF.
Discussion
Previous studies have shown that the loss of CREB function by inhibiting CREB-mediated transcription impairs LTM without affecting STM, indicating that CREB-dependent gene expression is required for memory consolidation (Bourtchuladze et al., 1994; Oike et al., 1999; Kida et al., 2002; Pittenger et al., 2002; Korzus et al., 2004). Thus, previous studies have suggested that CREB is a positive regulator of memory consolidation. In this study, to further understand the effects of the gain of function of CREB on memory performance, we generated and analyzed four lines of transgenic mice expressing dominant active CREB mutants (CREB-Y134F or CREB-DIEDML) in the forebrain. Expectedly, Y134F-A transgenic mice with the lowest transgene expression level among all transgenic lines exhibited enhanced LTM but normal STM (Fig. 3A,C). These observations indicated that the upregulation of CREB-mediated transcription resulted in the enhanced formation of LTM without affecting STM. Together with previous findings, our observations strongly suggested that CREB positively regulates memory consolidation.
Interestingly, Y134F-C mice displayed enhanced 2 h STM but normal 30 min STM (Fig. 3A,C). Similarly, two lines of DIEDML mice displayed an enhancement of 30 min STM but displayed normal 5 min STM (Fig. 5A,C). These observations indicate that the upregulation of CREB activity led to the enhancement of STM without affecting memory encoding or learning. Importantly, we found a dissociation of the mechanisms enhancing STM and LTM in active CREB mice; weak training only leads to the enhancement of LTM without affecting STM (Fig. 6). From this observation, similarly with the case of Y134F-A mice, the enhancement of LTM observed in Y134F-C and DIEDML mice is thought to attribute to enhanced memory consolidation.
By what mechanism is STM enhanced in active CREB mice? As STM is thought to be independent of new gene expression, it is suggested that CREB plays a regulatory role in STM, but this enhanced STM is not mediated by the transcriptional activation of CREB target genes immediately after training. BDNF gene is a target of CREB (Finkbeiner et al., 1997). Previous studies have shown that hippocampal BDNF positively regulates STM and LTM (Alonso et al., 2002, 2005). Importantly, we found that active CREB mice displaying enhanced STM had increased levels of BDNF in the hippocampus; transgenic lines displaying higher expression levels of BDNF exhibit enhanced STM (Fig. 7). More importantly, we found the following. (1) Infusion of BDNF or a BDNF inhibitor (K252a) into the hippocampus of WT mice enhanced or impaired STM, respectively (Fig. 8A–J). (2) Infusion of a low dose of BDNF into the hippocampus was sufficient to enhance 30 min STM in Y134F-C mice that display a normal 30 min STM and increased BDNF levels but not in WT mice that display a normal 30 min STM and normal BDNF levels (Fig. 8K–M). (3) Infusion of a higher dose of the BDNF inhibitor into the hippocampus is required to impair STM in DIEDML mice compared with WT mice (Fig. 8N–S). These observations suggest that the increased levels of BDNF observed at the basal condition as a result of the upregulation of CREB activity contribute to the enhancement of STM in active CREB mice. From these findings, CREB is thought to play a regulatory role in STM through the regulation of BDNF expression. Interestingly, a previous genetic study indicated that mutant mice lacking α/δ CREB isoforms exhibited impaired 1 h STM but normal 30 min STM (Bourtchuladze et al., 1994). This deficit of STM in CREBαδ−/− mutant mice may contribute to the downregulation of BDNF as a result of the decreased CREB activity (data not shown).
What is the impact of the upregulation of BDNF on LTM? We found that a hippocampal infusion of BDNF into Y134F-A mice displaying enhanced LTM, but normal BDNF levels, further improved LTM (Fig. 8T–V). This observation suggested that the increased levels of BDNF contribute to the enhancement of not only STM but also LTM in active CREB mice. Supporting this hypothesis, Y134F-C mice upregulating BDNF exhibited a superior ability in contextual discrimination even 1 month after training (Fig. 3D). Thus, the upregulation of CREB activity and BDNF expression may synergistically improve LTM.
In summary, our findings suggest that CREB not only regulates memory consolidation through the activation of activity-dependent transcription but also indirectly affects the status of ability in memory performance through the regulation of BDNF expression; CREB positively regulates memory consolidation through the transcriptional activation of target genes after training, whereas the regulation of BDNF expression levels by CREB appears to determine the degree of ability in memory performance, including STM and LTM.
Consistent with our observations, previous studies examining the effects of upregulating CREB using viral expression vectors revealed that the expression of WT-CREB or CREB-Y134F in the hippocampus or amygdala led to the enhancement of spatial or fear memory, respectively (Josselyn et al., 2001; Han et al., 2008; Restivo et al., 2009). However, transgenic mice expressing VP16-CREB in the forebrain under the control of the tetracycline-dependent transcription activator revealed contradictory observations (Viosca et al., 2009a,b). As VP-16 is an usually potent transcription activation domain that is not contained in CREB (Sadowski et al., 1988), there is a possibility that the fusion transcription factor with VP16 strongly activates transcription by recruiting other transcription cofactors that would not normally be recruited by nonfusion transcription factors. In contrast, the dominant active CREB mutants used in this study are thought to activate transcription in a similar manner to WT-CREB and to display lower thresholds of transcription activation in response to the activation of the upstream signal transduction pathway. Indeed, CREB-Y134F and -DIEDML are thought to enhance CREB-mediated transcription without changing the transcription factors recruited into the transcription machinery; CREB-Y134F displays a lower threshold for interacting with CBP in response to the activation of the upstream signal transduction pathway, whereas CREB-DIEDML constitutively interacts with CBP (Cardinaux et al., 2000). Therefore, it is possible that VP16-CREB abnormally activates CREB-mediated transcription and indirectly affects transcription of non-CREB target genes by interacting with transcription cofactors that are required for the transcription regulation of these genes.
In agreement with previous observations (Barco et al., 2002), we demonstrated that the upregulation of CREB activity enhances LTP evoked by a spike-timing protocol and L-LTP in CA1 neurons without affecting basal synaptic transmission (Fig. 2). It is possible that the enhancement of LTM observed in Y134F-C mice does not simply reflect enhanced LTP in CA1 neurons and/or that further enhancement of LTP in VP16-CREB mice (Barco et al., 2002) compared with Y134F-C mice has not been observed because of the ceiling affects. Additional studies are required to compare the relationship of enhanced LTP and memory in these mutant mice. Conversely, previous studies have shown that the genetic inhibition of CREB function impairs L-LTP in CA1 neurons (Huang et al., 1994; Pittenger et al., 2002; Korzus et al., 2004). Thus, similar loss-of-function and gain-of-function effects for CREB activity were observed on the formation of CA1-LTP and memory. However, it is possible that enhanced synaptic plasticity in areas other than the hippocampus CA1, such as the amygdala (Wei et al., 2002) or the anterior cingulate cortex (Wu et al., 2008), also contributes to the enhanced memory formation observed in the active CREB mice. It is necessary to further examine the effect of CREB-Y134F or -DIEDML expression on the synaptic plasticity in other such areas. Interestingly, a previous study has shown that the upregulation of CREB activity by the viral expression of CREB-Y134F not only enhances LTP but also modulates basal synaptic transmission by increasing NMDA receptor-mediated synaptic transmission (Marie et al., 2005). Levels of CREB-Y134F expression in excess of those produced by the transgenic mice used in our present study may be required to modulate basal synaptic transmission. In addition, it is important to note that there are no reports concerning the modulation of basal synaptic transmission in the CA1 hippocampal region associated with the genetic inhibition of the CREB gene.
CaMKIV functions as an upstream kinase of CREB and CBP (Chrivia et al., 1993; Matthews et al., 1994; Bito et al., 1996; Sun et al., 1996 Deisseroth et al., 1998; Korzus et al., 2004). Consistent with this view, the genetic inhibition of CaMKIV, CREB, or CBP disrupts memory consolidation and LTP (Yin et al., 1994; Lu et al., 1999; Ho et al., 2000; Ribar et al., 2000; Kang et al., 2001; Kida et al., 2002; Pittenger et al., 2002; Wei et al., 2002; Korzus et al., 2004; Mouravlev et al., 2006). In our present and previous studies (Fukushima et al., 2008), we showed that the upregulation of CREB or CaMKIV activity leads to the improvement of memory consolidation and LTP. Moreover, DIEDML mice with an enhanced interaction between CREB and CBP exhibited enhanced LTM, suggesting that facilitating the interaction between CREB and CBP improves memory formation. Together, these findings strongly support the critical contribution of the CaMKIV–CREB–CBP signaling pathway to memory consolidation and LTP.
Footnotes
S.K. was supported by Grants-in-Aid for Scientific Research, High Technology Research, and Priority Areas–Molecular Brain Science (18022038 and 22022039) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Core Research for Evolutional Science and Technology, Japan; a Research Grant for Nervous and Mental Disorders from the Ministry of Health, Labor, and Welfare, Japan; The Sumitomo Foundation, Japan; and the Takeda Science Foundation, Japan. M.Z. was supported by grants from the EJLB Foundation–Canadian Institutes of Health Research (CIHR) Michael Smith Chair in Neurosciences and Mental Health, Canada Research Chair, and CIHR operating grants (CIHR66975).
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus. 2002;12:551–560. doi: 10.1002/hipo.10035. [DOI] [PubMed] [Google Scholar]
- Alonso M, Bekinschtein P, Cammarota M, Vianna MR, Izquierdo I, Medina JH. Endogenous BDNF is required for long-term memory formation in the rat parietal cortex. Learn Mem. 2005;12:504–510. doi: 10.1101/lm.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostaras SG, Josselyn SA, Frankland PW, Silva AJ. Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice. Learn Mem. 2000;7:58–72. doi: 10.1101/lm.7.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseeroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Brightwell JJ, Smith CA, Neve RL, Colombo PJ. Long-term memory for place learning is facilitated by expression of cAMP response element-binding protein in the dorsal hippocampus. Learn Mem. 2007;14:195–199. doi: 10.1101/lm.395407. [DOI] [PubMed] [Google Scholar]
- Cardinaux JR, Notis JC, Zhang Q, Vo N, Craig JC, Fass DM, Brenna RG, Goodman RH. Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol Cell Biol. 2000;20:1546–1552. doi: 10.1128/mcb.20.5.1546-1552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Du K, Asahara H, Jhala US, Wagner BL, Montminy M. Characterization of a CREB gain-of-function mutant with constitutive transcriptional activity in vivo. Mol Cell Biol. 2000;20:4320–4327. doi: 10.1128/mcb.20.12.4320-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. De novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learn Mem. 2008;15:747–755. doi: 10.1101/lm.1027208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Flexner LB, Flexner JB, Stellar E. Memory and cerebral protein synthesis in mice as affected by graded amounts of protein. Exp Neurol. 1965;13:264–272. doi: 10.1016/0014-4886(65)90114-7. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Cestari V, Filipkowski RK, McDonald RJ, Silva AJ. The dorsal hippocampus is essential for context discrimination but not for contextual conditioning. Behav Neurosci. 1998;112:863–874. doi: 10.1037//0735-7044.112.4.863. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Maeda R, Suzuki R, Suzuki A, Nomoto M, Toyoda H, Wu LJ, Xu H, Zhao MG, Ueda K, Kitamoto A, Mamiya N, Yoshida T, Homma S, Masushige S, Zhuo M, Kida S. Upregulation of CaMKIV improves memory formation and rescues memory loss with aging. J Neurosci. 2008;28:9910–9919. doi: 10.1523/JNEUROSCI.2625-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Yiu AP, Cole CJ, Hsiang HL, Neve RL, Josselyn SA. Increasing CREB in the auditory thalamus enhances memory and generalization of auditory conditioned fear. Learn Mem. 2008;15:443–453. doi: 10.1101/lm.993608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S, Furuichi T, Yoshida T, Endoh K, Kato K, Sado M, Maeda R, Kitamoto A, Miyao T, Suzuki R, Homma S, Masushige S, Kajii Y, Kida S. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol Brain. 2009;2:6. doi: 10.1186/1756-6606-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissaian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Hoitzman DM, Linden DJ, Zhou M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP responsive element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda H, Motohashi J, Kato H, Masushige S, Kida S. A BMAL1 mutant with arginine 91 substituted with alanine acts as a dominant negative inhibitor. Gene. 2004;338:235–241. doi: 10.1016/j.gene.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Hosoda H, Kato K, Asano H, Ito M, Kato H, Iwamoto T, Suzuki A, Masushige S, Kida S. CBP/p300 is a cell type-specific modulator of CLOCK/BMAL1-mediated transcription. Mol Brain. 2009;2:34. doi: 10.1186/1756-6606-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Mamiya N, Masushige S, Kida S. PLCgamma2 activates CREB-dependent transcription in PC12 cells through phosphorylation of CREB at serine 133. Cytotechnology. 2005;47:107–116. doi: 10.1007/s10616-005-3763-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KF, Ohmstede CA, Fisher RS, Sahyoun N. Nuclear and axonal localization of Ca2+/calmodulin-dependent protein kinase type Gr in rat cerebellar cortex. Proc Natl Acad Sci U S A. 1991;88:2850–2853. doi: 10.1073/pnas.88.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Sun L, Atkins CM, Soderling TR, Wilson MA, Tonegawa S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell. 2001;106:771–783. doi: 10.1016/s0092-8674(01)00497-4. [DOI] [PubMed] [Google Scholar]
- Kee N, Teixeira CM, Wang AH, Frankland PW. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci. 2007;10:355–362. doi: 10.1038/nn1847. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Kim R, Moki R, Kida S. Molecular mechanisms for the destabilization and restabilization of reactivated spatial memory in the Morris water maze. Mol Brain. 2011;4:9. doi: 10.1186/1756-6606-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Johnston LD, Sweatt JD, Anderson AE. Kainate mediates nuclear factor-kappa B activation in hippocampus via phosphatidylinositol-3 kinase and extracellular signal-regulated protein kinase. Neuroscience. 2005;133:969–981. doi: 10.1016/j.neuroscience.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–413. doi: 10.1523/JNEUROSCI.4639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Baranes D, Podsypanina K, Kandel ER. The 3-untranslated region of CaMKIIα is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci U S A. 1996;93:13250–13255. doi: 10.1073/pnas.93.23.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Mouravlev A, Dunning J, Young D, During MJ. Somatic gene transfer of cAMP response element-binding protein attenuates memory impairment in aging rats. Proc Natl Acad Sci U S A. 2006;103:4705–4710. doi: 10.1073/pnas.0506137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Okuno S, Sato F, Fujisawa H. An immunohistochemical study of C2+/calmodulin-dependent protein kinase IV in the rat central nervous system: light and electron microscopic observations. Neuroscience. 1995;68:181–194. doi: 10.1016/0306-4522(95)00092-w. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Elsevier Academic; 1997. [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Restivo L, Tafi E, Ammassari-Teule M, Marie H. Viral-mediated expression of a constitutively active form of CREB in hippocampal neurons increases memory. Hippocampus. 2009;19:228–234. doi: 10.1002/hipo.20527. [DOI] [PubMed] [Google Scholar]
- Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR. Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci. 2000;20:RC107. doi: 10.1523/JNEUROSCI.20-22-j0004.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Sun P, Lou L, Maurer RA. Regulation of activating transcription factor 1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinase type I, II, and IV. J Biol Chem. 1996;271:3066–3073. doi: 10.1074/jbc.271.6.3066. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Mukawa T, Tsukagoshi A, Frankland PW, Kida S. Activation of LVGCCs and CB1 receptors required for destabilization of reactivated contextual fear memories. Learn Mem. 2008;15:426–433. doi: 10.1101/lm.888808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thor DH, Holloway WR. Social memory of the male laboratory rat. J Comp Physiol Psychol. 1982;96:1000–1006. [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300. doi: 10.1016/s0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- Uchida S, Sakai S, Furuichi T, Hosoda H, Toyota K, Ishii T, Kitamoto A, Sekine M, Koike K, Masushige S, Murphy G, Silva AJ, Kida S. Tight regulation of transgene expression by tetracycline-dependent activator and repressor in brain. Genes Brain Behav. 2006;5:96–106. doi: 10.1111/j.1601-183X.2005.00139.x. [DOI] [PubMed] [Google Scholar]
- Viosca J, Lopez de Armentia M, Jancic D, Barco A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learn Mem. 2009a;16:193–197. doi: 10.1101/lm.1254209. [DOI] [PubMed] [Google Scholar]
- Viosca J, Malleret G, Bourtchouladze R, Benito E, Vronskava S, Kandel ER, Barco A. Chronic enhancement of CREB activity in the hippocampus interferes with the retrieval of spatial information. Learn Mem. 2009b;16:198–209. doi: 10.1101/lm.1220309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Qiu CH, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calucium-calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002;5:573–579. doi: 10.1038/nn0602-855. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Silva AJ. Memory for context becomes less specific with time. Learn Mem. 2007;14:313–317. doi: 10.1101/lm.430907. [DOI] [PubMed] [Google Scholar]
- Winocur G, Moscovitch M, Sekeres M. Memory consolidation or transformation: context manipulation and hippocampal representations of memory. Nat Neurosci. 2007;10:555–557. doi: 10.1038/nn1880. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Zhang XH, Fukushima H, Zhang F, Wang H, Toyoda H, Li BM, Kida S, Zhuo M. Genetic enhancement of trace fear memory and cingulate potentiation in mice overexpressing Ca2+/calmodulin-dependent protein kinase IV. Eur J Neurosci. 2008;8:1923–1932. doi: 10.1111/j.1460-9568.2008.06183.x. [DOI] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fukushima H, Kida S. Induction and requirement of gene expression in the anterior cingulate cortex and medial prefrontal cortex for the consolidation of inhibitory avoidance memory. Mol Brain. 2011;4:4. doi: 10.1186/1756-6606-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. A selective role of calcineurin alpha in synaptic depotentiation in hippocampus. Proc Natl Acad Sci U S A. 1999;96:4650–4655. doi: 10.1073/pnas.96.8.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]