

Abstract
Central sensitization in inflammatory pain conditions results in behavioral mechanical hypersensitivity. Specifically, C-fiber-driven spinal hyperexcitability enables A fibers to gain access to specific spinal circuitry, via heterosynaptic facilitatory mechanisms, to mediate mechanical hypersensitivity. However, the precise circuitry engaged is not known. Lamina I neurokinin 1 (NK1) receptor expressing (NK1R+) dorsal horn neurons, many of which are projection neurons, are required for the development of this hypersensitivity and are therefore likely to be a component of this circuitry. To investigate, whole-cell patch-clamp recordings were made from lamina I NK1R+ neurons in the spinal cord slice preparation with attached dorsal root, obtained from rats with or without complete Freund's adjuvant (CFA) hindpaw inflammation. EPSCs were recorded in response to electrical stimulation of the dorsal root. Control neurons predominantly received monosynaptic C-fiber input (69%) with a smaller proportion receiving monosynaptic Aδ-fiber input (28%). In contrast, CFA inflammation significantly increased the incidence (by twofold) and magnitude (by 75% in a subset) of monosynaptic Aδ-fiber but not monosynaptic C-fiber-evoked responses. Aβ-fiber input to lamina I NK1R+ neurons was minimal, polysynaptic in nature, and unaltered by CFA inflammation. Additional examination of control neurons revealed that a proportion received silent monosynaptic Aδ-fiber input, suggesting that these may provide the substrate for the novel Aδ inputs observed in CFA inflammation. This inflammation induced unmasking and strengthening of monosynaptic Aδ drive to lamina I NK1R+ neurons may contribute to the heterosynaptic facilitatory mechanisms underlying mechanical hyperalgesia in inflammatory pain.
Introduction
Chronic pain conditions are characterized by behavioral hypersensitivity, consisting of exaggerated pain responses to noxious stimulation (hyperalgesia) and pain responses to innocuous stimulation (allodynia). These chronic pain symptoms arise from a multitude of underlying molecular and cellular changes that lead to increased excitability of both peripheral and central sensory pathways and are termed peripheral and central sensitization, respectively (Scholz and Woolf, 2002). The prevailing view is that peripheral sensitization primarily underlies thermal hypersensitivity, whereas central sensitization predominantly mediates mechanical hypersensitivity (Lewin et al., 2004).
Since the discovery of central sensitization, it has been known that activity in C-fiber nociceptors is the necessary “trigger” to elicit this enhanced excitability within spinal sensory pathways by inducing activity-dependent changes in dorsal horn neurons (Woolf, 1983). As a result of this C-fiber-driven spinal hyperexcitability, it is believed that Aβ- and Aδ-fiber primary afferents are now able to gain access to specific spinal circuitry, via heterosynaptic facilitatory mechanisms, enabling these afferents to “mediate” the sensations of mechanical allodynia and mechanical hyperalgesia, respectively (Treede and Magerl, 2000; Latremoliere and Woolf, 2009). However, the precise spinal circuitry that is engaged by Aβ and Aδ afferents and that mediates the sensations of allodynia and hyperalgesia is not specifically known.
A key player in spinal cord chronic pain mechanisms is the lamina I neurokinin 1 (NK1) receptor-expressing (NK1R+) neuron, a significant proportion of which are projection neurons and target sites in the brainstem and thalamus (Marshall et al., 1996; Todd et al., 2000; Spike et al., 2003; Al-Khater et al., 2008). These neurons are driven by nociceptive primary afferent input (predominantly C-fiber monosynaptic) (Ruscheweyh et al., 2004; Torsney and MacDermott, 2006) and are at the origin of a spino-bulbo-spinal loop that controls spinal cord excitability (Suzuki et al., 2002; Khasabov et al., 2005; Rahman et al., 2008). Importantly, selective ablation of superficial NK1R+ neurons, with a substance P and saporin conjugate, diminishes central sensitization (Mantyh et al., 1997; Khasabov et al., 2002) and attenuates allodynia and hyperalgesia in a variety of chronic pain models (Mantyh et al., 1997; Nichols et al., 1999).
Interestingly, it has been shown that lamina I projection neurons, expressing the NK1R, exhibit homosynaptic potentiation of C-fiber monosynaptic inputs when these C-fiber inputs are repetitively electrically stimulated in vitro, at rates that mimic their spontaneous firing pattern in inflammatory pain conditions (Ikeda et al., 2006). In addition, if the disrupted spinal inhibition that occurs in chronic pain conditions (Zeilhofer and Zeilhofer, 2008; Basbaum et al., 2009) is mimicked pharmacologically in vitro, polysynaptic A-fiber input to lamina 1 NK1R+ neurons is facilitated (Torsney and MacDermott, 2006). Given the potential relevance of these homosynaptic and heterosynaptic facilitatory central spinal mechanisms to the manifestation of mechanical hypersensitivity in chronic pain conditions, it is essential to investigate their likely contribution in chronic pain models. To address this question, primary afferent synaptic input to lamina 1 NK1R+ neurons has therefore been characterized in an inflammatory pain model exhibiting robust mechanical hypersensitivity.
Materials and Methods
Inflammatory pain model.
Peripheral inflammation was induced by intraplantar injection (1 μl/g body weight) of complete Freund's adjuvant (CFA) (0.5 mg/ml saline) into the left hindpaw of Sprague Dawley rats [postnatal day 16 (P16) to P19, both sexes] under isoflurane anesthesia. The animals were allowed to recover and were kept for 2–6 d, during which timeframe there is clear evidence of behavioral mechanical hypersensitivity (see Results), before being used at approximately P21 for electrophysiology. Control rats were untreated. All experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act, 1986 and were approved by the University of Edinburgh Ethical Review Committee.
Sensory testing.
Hindpaw edema was assessed, in a subgroup, by measuring the thickness of the dorsoventral paw with calipers daily, 2–6 d after intraplantar CFA injection. Mechanical hypersensitivity was also determined using von Frey filaments (Stoelting) over the same time course. The minimum force required to elicit a flexion withdrawal reflex on four of eight von Frey applications to the plantar surface of the hindpaw was defined as the mechanical threshold.
Isolated dorsal root preparation.
CFA-treated or naive non-inflamed rats (P19–P24) were anesthetized with isoflurane and decapitated, and lumbar (L4/L5) region of the spinal cord with attached dorsal roots was removed. The spinal cords and roots were initially placed in ice-cold dissection solution, and the L4/L5 dorsal roots were isolated (left side only, CFA treated). Roots were incubated in oxygenated recovery solution at 36–37°C for 1 h before recording. Roots were transferred to a recording chamber and continuously perfused with oxygenated Krebs' solution at a flow rate of 1–2 ml/min, and recordings were made at room temperature. Krebs' solution, saturated 95% O2/5% CO2, had the following composition (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 25 glucose, 1 MgCl2, and 2 CaCl2, pH 7.4. Recovery was the same as Krebs' solution but with 1.5 mm CaCl2 plus 6 mm MgCl2. Dissection was the same as recovery but with 1 mm kynurenic acid.
Compound action potential recording.
Two suction electrodes were used: one for electrical stimulation of the root and the other for recording compound action potentials. Electrical stimulation (10×) was performed at 0.2 Hz for 0.1 ms using an ISO-Flex Stimulus Isolator (A.M.P.I. Intracel). The stimulation intensities used were 2.5, 5, 10, 15, 20, 25, 30–100 (in increments of 10), and 150–500 μA (in increments of 50) which covers the Aβ-, Aδ-, and C-fiber range (Nakatsuka et al., 2000). Data were recorded and acquired using a Cygnus ER-1 differential amplifier (Cygnus Technology) and pClamp 10 software (Molecular Devices). Data were filtered at 10 kHz and sampled at 50 kHz.
Three main components of the compound action potentials could be distinguished: fast (Aα/β), medium (Aδ), and slow (C) conducting components, each having a triphasic (positive–negative–positive) profile. Sometimes small intermediate components were present among these three groups, but these were not analyzed. The amplitude of each component was calculated by measuring the distance between the negative and positive peaks, and the conduction velocity was calculated based on the latency to the negative peak at 25, 100, and 500 μA for Aα/β, Aδ, and C components, respectively. The effect of CFA inflammation on the threshold stimulation intensity, conduction velocity and amplitude of the afferent components was determined using two-way ANOVA.
Spinal cord slice preparation.
Spinal cords (L4/L5) with attached dorsal roots from CFA-treated or naive non-inflamed rats (P19–P24) were obtained as detailed above. The spinal cords were initially placed in ice-cold dissection solution, embedded in an agarose block, and cut into transverse slices (350 μm) with attached dorsal roots (left dorsal roots only, CFA treated). Slices were incubated in oxygenated recovery solution at 36–37°C for 1 h and then incubated with 20–35 nm tetramethylrhodamine conjugated substance P (TMR-SP) for 20 min at room temperature, as described previously (Labrakakis and MacDermott, 2003; Torsney and MacDermott, 2006), and allowed to recover for 1 h before recording. Slices were transferred to an upright microscope equipped with fluorescence for identification of positive neurons and infrared-differential interference contrast (IR-DIC) for electrophysiological recordings. Slices were continuously perfused with oxygenated Krebs' solution at a flow rate of 1–2 ml/min. Recordings were made at room temperature.
Patch-clamp recording.
Whole-cell patch configuration (holding potential of −70 mV unless otherwise stated) was used to record EPSCs from “identified” neurons while the dorsal root was stimulated with a suction electrode, as for the compound action potential recordings. Intracellular solution had the following composition (in mm): 120 Cs-methylsulfonate, 10 Na-methylsulfonate, 10 EGTA, 1 CaCl2, 10 HEPES, 5 QX-314-Cl [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine chloride], and 2 Mg2+-ATP, pH adjusted to 7.2 with CsOH (osmolarity 290). In most cases, 1 μm Alexa Fluor 488 hydrazide was included in the recording pipette. Junction potential was corrected before recording. Data were recorded and acquired using an Axopatch 200B amplifier and pClamp 10 software (Molecular Devices). Data were filtered at 5 kHz and digitized at 10 kHz.
To assess primary afferent input, the dorsal root was stimulated (three times) at 25, 100, and 500 μA to activate Aβ-, Aδ-, and C-fiber inputs, respectively, at a low stimulation frequency of 0.05 Hz (duration, 0.1 ms) using an ISO-Flex Stimulus Isolator (A.M.P. I. Intracel) as described previously (Torsney and MacDermott, 2006). An 0.1 ms pulse width was chosen to replicate the electrical stimuli established previously to activate the different afferent fiber types in this age of juvenile rats (Nakatsuka et al., 2000) and also, importantly, to replicate the pulse width used by Ikeda et al. (2006) to facilitate comparison between it and the present study. However, the possibility of an underestimation of the C-fiber contribution cannot be excluded given that longer pulse widths have also been used to stimulate C-fiber inputs (Baba et al., 1999). Response stability was used to discriminate between EPSCs that were “evoked” and therefore timed locked with the stimulus and responses that were spontaneous and therefore unconnected with the stimulus. At this low frequency of stimulation, both primary afferent monosynaptic and polysynaptic responses could be seen as stable responses.
Stable, afferent input was then dissected into monosynaptic and polysynaptic components, in the manner of Nakatsuka et al. (2000) as described in detail previously (Torsney and MacDermott, 2006). The dorsal root was stimulated (20 times) at the following frequencies and intensities [25 μA, 20 Hz; 100 μA, 2 Hz (and 10 Hz, see below); 500 μA, 1 Hz] to assess the monosynaptic/polysynaptic nature of the Aβ-, Aδ-, and C-fiber inputs, respectively. These frequencies are chosen because action potential firing in C fibers begins to fail at stimulation frequencies above 1 Hz, in Aδ fibers above 2 Hz but in Aβ fibers do not fail even at 20 Hz. If higher stimulation frequencies than those stated were used, it would not be possible to distinguish a failure of primary afferent fibers to fire action potentials from that of a synaptic failure. For A-fiber-evoked EPSCs, an absence of synaptic failures and a stable latency (≤2 ms) was considered monosynaptic, whereas synaptic failures and/or latency variability (>2 ms) was considered polysynaptic. In all neurons, the dorsal root was stimulated at a frequency of 10 Hz in addition to 2 Hz at the 100 μA stimulation intensity as many Aδ fibers can still fire action potentials at this rate. Testing at this frequency allows more rigorous testing of the monosynaptic/polysynaptic nature of the response. Interestingly, in more than half of the neurons tested, dorsal root stimulation at 10 Hz did not discount responses identified as monosynaptic at the 2 Hz stimulation rate, suggesting that, as expected, many of the recorded Aδ-fiber inputs can repetitively fire action potentials at 10 Hz. For C-fiber EPSCs evoked at 1 Hz, a lack of synaptic failures was considered evidence of monosynaptic input, even if there was a prolongation of latency (Nakatsuka et al., 2000), whereas a response showing synaptic failures was considered polysynaptic. Although a 1 Hz stimulation rate is a less rigorous test of monosynaptic versus polysynaptic, it was used to classify C-fiber inputs because higher frequencies would lead to the misidentification of monosynaptic C-fiber inputs as polysynaptic.
Silent synapse identification procedure.
Monosynaptic Aδ-fiber inputs mediated by NMDA receptors alone were identified in neurons lacking conventional monosynaptic Aδ-fiber inputs as follows. These neurons exhibit polysynaptic activity when the dorsal root is stimulated at 100 μA, Aδ-fiber stimulation strength when held at −70 mV. To establish the presence of monosynaptic Aδ-fiber inputs mediated by NMDA receptors alone, 10 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) was therefore applied to block this polysynaptic activity (Torsney and MacDermott, 2006), and 10 μm bicuculline and 1 μm strychnine were applied to block inhibition. The membrane was then depolarized to +50 mV, and the presence of monosynaptic Aδ-fiber input mediated by NMDA receptors alone was evidenced by the consistent appearance of a slow outward current in response to dorsal root stimulation (0.05 Hz, three times) at 100 μA, Aδ-fiber (but not 25 μA, Aβ-fiber) stimulation strength. To be classified as a silent monosynaptic Aδ-fiber input, this outward current response at +50 mV had to be evident on averaging when evoked by dorsal root stimulation (20 times) at 2 Hz, be blocked by 30 μm APV, and no longer be evident after return to a holding current of −70 mV.
Patch-clamp analysis.
To quantify the degree of polysynaptic Aβ-fiber input, synaptic activity was analyzed by measuring the area under the curve or EPSC area from the stimulus artifact to the end of the trace (900 ms), with respect to a baseline period before the stimulus artifact, for each of the three EPSCs evoked by dorsal root stimulation at 25 μA, 0.05 Hz using pClamp 10 Software (Molecular Devices), and the values were then averaged. Neurons without evoked Aβ-fiber input will therefore have EPSC areas close to zero, either positive or negative in value depending on the level of spontaneous activity present before or after the stimulus artifact. Primary afferent monosynaptic EPSC amplitudes and latencies were also measured using pClamp 10 Software for each of the three EPSCs evoked by dorsal root stimulation (0.05 Hz) at a given stimulation intensity and averaged. Silent synapse peak amplitudes, latencies, and 10–90% rise time values were measured from averaged EPSCs recorded at +50 mV in response to dorsal root stimulation (0.05 Hz, three times) and evaluated with pClamp 10 Software. Estimated conduction velocities of primary afferent monosynaptic responses were calculated using the length of dorsal root stimulated and the latency of the monosynaptic response (root length/latency).
Materials.
TMR-SP was purchased from Anaspec. Alexa Fluor 488 was purchased from Invitrogen. Bicuculline, d-APV, and NBQX were purchased from Tocris Bioscience. Strychnine and complete Freund's adjuvant were obtained from Sigma-Aldrich.
Results
Hindpaw inflammation
Intraplantar injection of CFA into the hindpaw, at P18, resulted in edema, erythema, and mechanical hypersensitivity. Figure 1, A and B, demonstrates changes in paw thickness (a measure of edema) and mechanical sensitivity, respectively, in the days after CFA injection. Figure 1A shows that, 2–6 d after CFA injection, there was a significant increase in the diameter of the inflamed hindpaw. Similarly, Figure 1B shows that, 2–6 d after CFA injection, there was a significant reduction in the mechanical threshold for evoking the flexion withdrawal reflex in the CFA-injected hindpaw. Based on these data, isolated dorsal roots and spinal cord slices were prepared 2–6 d after CFA injection (at ∼P18) to investigate the mechanisms underlying inflammatory mechanical hypersensitivity.
Figure 1.

A, B, Time course of paw edema (A) and mechanical hypersensitivity (B) after intraplantar injection of CFA into the hindpaw at P18 (n = 10). Paw edema was quantified by measuring the dorsoventral paw thickness, and mechanical hypersensitivity was assessed by determining the mechanical threshold of the flexion withdrawal reflex with von Frey filaments. Averaged data are represented as mean ± SE. The legend in A also applies to B. Two-way repeated measures ANOVA followed by Bonferroni's post hoc tests were used to compare ipsilateral and contralateral measurements at all time points (***p < 0.001).
CFA inflammation does not alter primary afferent thresholds or conduction velocities
Primary afferents could be divided into three distinct groups, corresponding to Aα/β, Aδ, and C fibers, on the basis of threshold and conduction velocity of compound action potentials recorded extracellularly from the dorsal root (Fig. 2A). Intraplantar CFA injection, 2–6 d before isolation of the dorsal roots did not significantly alter the threshold stimulation intensity required to activate the different afferent components (Fig. 2B). Similarly, CFA inflammation did not significantly alter the conduction velocity of the afferent components (Fig. 2C) or the amplitude of any of the afferent components (Aβ control, 2.5 ± 0.3 μA; Aβ CFA, 3.5 ± 0.7 μA; Aδ control, 0.12 ± 0.02 μA; Aδ CFA, 0.15 ± 0.03 μA; C control, 0.21 ± 0.01 μA; C CFA, 0.22 ± 0.01 μA). Notably, these values are similar to comparable in vitro recordings (Labrakakis et al., 2003; Géranton et al., 2009). The lack of effect of peripheral CFA inflammation on primary afferent thresholds and conduction velocities has previously been established in adult rats (Baba et al., 1999; Nakatsuka et al., 1999). However, it was important to confirm this finding in juvenile rats (∼P21) before commencing patch-clamp recording of primary-afferent-evoked activity in spinal slices from both CFA-pretreated and naive juvenile rats. Based on these data, stimulation intensity values previously used to activate the different afferent components in naive (∼P21) rat spinal slices (Torsney and MacDermott, 2006) were similarly used in the present study and are detailed in Figure 2B.
Figure 2.

CFA inflammation does not alter primary afferent thresholds or conduction velocities. A, Representative compound action potentials recorded from an isolated dorsal root, from a naive rat, illustrating the fast (Aα/β), medium (Aδ), and slow (C) conducting components evoked by 25, 100, and 500 μA stimulation in the top, middle, and bottom panels, respectively (average of 10 traces shown). Arrows denote the negative component of each triphasic (positive–negative–positive) afferent profile. B, C, Effect of CFA inflammation on the threshold stimulation intensity (B) and conduction velocity (C) of the different afferent components. Averaged data are represented as mean ± SE. Dotted lines in B indicate the dorsal root stimulation intensities (Aβ, 25 μA; Aδ, 100 μA; C, 500 μA) used in the subsequent patch-clamp electrophysiology studies. Two-way ANOVA reveals no significant effect of CFA inflammation. n = 8 in both groups.
Primary afferent synaptic input to lamina I NK1R+ neurons
Whole-cell patch configuration was used to record EPSCs from lamina I NK1R+ neurons in spinal slices from control untreated and CFA-pretreated rats (holding potential, −70 mV; control neurons, n = 56; CFA neurons, n = 41). The dorsal root was stimulated over a range of intensities and frequencies to characterize the primary afferent input and its monosynaptic/polysynaptic nature. Figure 3A shows an example of a lamina I NK1R+ neuron, in a control spinal slice, identified after incubation with 35 nm TMR-SP. The recording pipette often contained 1 μm Alexa Fluor 488 hydrazide to confirm that the neuron labeled with TMR-SP was that recorded under IR-DIC (Fig. 3Aiii). Lamina I NK1R+ neurons could easily be distinguished because of the lack of NK1R+ neurons in lamina II (Bleazard et al., 1994; Brown et al., 1995). Laminar location of the recording pipette was always confirmed at the end of the experiment using low magnification (4×), allowing confirmation that recordings were dorsal (lamina I) to the relatively translucent lamina II.
Figure 3.

Primary afferent synaptic input to lamina I NK1R+ neurons. A, TMR-SP labeling of a control lamina I neuron. The neuron is visualized with TMR-SP fluorescence (i), IR-DIC (ii), and filled with Alexa Fluor 488 hydrazide (iii). Recording pipette can be seen in iii. Scale bar, 20 μm. B, Characterization of primary afferent synaptic input to a control lamina I NK1R+ neuron receiving monosynaptic C-fiber input. C and D show characterization of primary afferent synaptic input to CFA lamina I NK1R+ neurons receiving monosynaptic Aδ-fiber and monosynaptic Aδ-fiber and monosynaptic C-fiber input, respectively. In B–D, i shows examples of EPSCs evoked by stimulation (0.1 ms) using Aβ-fiber (25 μA), Aδ-fiber (100 μA), and C-fiber (500 μA) stimulation intensities at low frequency. Each trace comprises three averaged traces evoked at 0.05 Hz. ii shows examples of EPSCs evoked by higher-frequency stimulation (25 μA/20 Hz; 100 μA/2 Hz; 500 μA/1 Hz). Each trace comprises 20 superimposed traces.
In this preparation, TMR-SP labels a small proportion of lamina I neurons (on average, 5–10 neurons per slice), clearly less than the 45% of lamina I neurons reported to express the NK1 receptor (Todd et al., 1998). Given that many lamina I projection neurons (∼5% of lamina I neurons) that express the NK1 receptor do so at moderate to strong levels (Spike et al., 2003), we are more apt to be targeting this projection neuron population rather than the population of weakly NK1R-immunoreactive neurons (Cheunsuang and Morris, 2000) that are likely to be interneurons (Al Ghamdi et al., 2009). It has been established that CFA inflammation localized to the hindpaw does not alter the numbers of NK1R-expressing neurons in lamina I (Abbadie et al., 1997; Honor et al., 1999), and therefore TMR-SP is expected to label the same neuronal population in spinal slices from control untreated and CFA-pretreated rats. Comparable cell capacitance values were obtained for lamina I TMR-SP-labeled neurons in both groups (control, 30.56 ± 2.38; CFA, 28.89 ± 2.21; Mann–Whitney test, p = 0.8, not significantly different), which supports this assertion, as shown previously in similar studies (Rycroft et al., 2007; Vikman et al., 2008).
Our technique for pre-identifying NK1R+ neurons is expected to have little impact on the synaptic activity we are studying. TMR-SP is one of the least biologically active of fluorescence-conjugated substance P analogs and does not inhibit neuronal M-type K+ current at the nanomolar concentration used in our study, although it does elevate calcium in Chinese hamster ovary cells expressing NK1 receptor (Bennett and Simmons, 2001). Moreover, this TMR-SP labeling approach has been validated in a previous study of spinal NK1R+ neurons (Tong and MacDermott, 2006).
Dorsal root stimulation at different intensities and frequencies was used to establish the nature of the primary afferent input to lamina I NK1R+ neurons. The most commonly occurring types of afferent input are shown in Figure 3. Figure 3B shows an example of EPSCs recorded from a lamina I NK1R+ neuron, in a control spinal slice, that received monosynaptic C-fiber input. Low-frequency dorsal root stimulation (0.1 ms, 0.05 Hz, three times) at Aβ-fiber (25 μA) and Aδ-fiber (100 μA) intensities evoked small EPSCs, whereas stimulation at C-fiber intensity (500 μA) clearly evoked a response (Fig. 3Bi). High-frequency stimulation revealed that the small A-fiber-evoked response was polysynaptic because the response sometimes failed. The C-fiber-evoked response was shown to be monosynaptic in nature because high-frequency stimulation reliably evoked EPSCs (Fig. 3Bii).
Figure 3C shows an example of EPSCs recorded from a lamina I NK1R+ neuron, in a CFA spinal slice, that received monosynaptic Aδ-fiber input. Dorsal root stimulation at Aβ intensity only evoked small EPSCs, whereas stimulation at Aδ-fiber intensity evoked a clear response. Increasing the stimulation intensity to C-fiber strength did not recruit any additional components (Fig. 3Ci). The small Aβ response was shown to be polysynaptic because the response showed failures at high-frequency stimulation rates, whereas the Aδ-fiber response was reliably evoked after high-frequency (2 Hz) stimulation with little variability in latency (≤2 ms) (Fig. 3Cii). This monosynaptic response profile was also generated by stimulating the dorsal root at the higher frequency of 10 Hz (data not shown).
Figure 3D shows an example of EPSCs recorded from a lamina I NK1R+ neuron, in a CFA spinal slice, that received monosynaptic Aδ-fiber and monosynaptic C-fiber input. Dorsal root stimulation at Aβ-fiber intensity only evoked small EPSCs, whereas stimulation at Aδ-fiber intensity evoked a clear response, and increasing the stimulation intensity to C-fiber strength revealed an additional longer-latency component (Fig. 3Di). Again the small Aβ response was shown to be polysynaptic because the response showed failures at high-frequency stimulation rates, whereas the Aδ- and C-fiber responses were reliably evoked after high-frequency stimulation with the Aδ-fiber response showing little variability in latency (≤2 ms) (Fig. 3Dii), indicating that both the Aδ- and C-fiber inputs were monosynaptic.
CFA inflammation does not facilitate polysynaptic Aβ-fiber input or alter disinhibition revealed polysynaptic Aβ-fiber input to lamina I NKIR+ neurons
It has been demonstrated previously that pharmacologically mimicking the disrupted spinal inhibition that occurs in chronic pain conditions can facilitate novel and enhanced polysynaptic Aβ-fiber input to lamina I NK1R+ neurons (Torsney and MacDermott, 2006). Given that disrupted spinal inhibition is a feature of inflammatory pain conditions (Zeilhofer and Zeilhofer, 2008), it was determined whether the amount of polysynaptic Aβ-fiber input to lamina I NK1R+ neurons was increased by CFA inflammation. Lamina I NK1R+ neurons, in both control and inflamed spinal slices, received limited amounts of polysynaptic Aβ-fiber input as can be seen in Figure 3B–D. Figure 4A shows that CFA inflammation did not significantly alter the degree of polysynaptic Aβ-fiber input to these neurons, as determined by quantifying the area of Aβ-fiber-evoked EPSCs and comparing between control and CFA conditions. This suggests that the spinal disinhibition that occurs in CFA inflammation does not impinge on the low-threshold polysynaptic drive to these neurons.
Figure 4.

Polysynaptic Aβ-fiber input to lamina I NK1R+ neurons, under control and disinhibition conditions, is not altered by CFA inflammation. A, Scatter plot shows the effect of CFA inflammation on Aβ-fiber-evoked EPSC area. Three neurons in each group were not included for analysis because they received monosynaptic Aβ-fiber input. Mann–Whitney test reveals no significant effect of CFA inflammation (control, n = 53; CFA, n = 38). B displays data from a CFA neuron with polysynaptic Aβ-fiber input, which is facilitated during disinhibition. Bi, Examples of EPSCs evoked by Aβ stimulation intensity (0.1 ms, 25 μA) at low frequency in Krebs' solution. Three superimposed traces evoked at 0.05 Hz are shown. ii shows EPSCs evoked by the same stimulation protocol but in the presence of bicuculline (bic; 10 μm) and strychnine (strych; 300 nm). C, Scatter plot shows the effect of CFA inflammation on disinhibition induced fold changes in Aβ-fiber-evoked EPSC area. Mann–Whitney test reveals no significant effect of CFA inflammation (n = 11, each group).
To further characterize the integrity of the inhibitory control that regulates this low-threshold polysynaptic drive onto lamina I NK1R+ neurons, the consequence of disinhibition was assessed, in a subset of both control and CFA neurons and compared. Figure 4Bi shows the Aβ-fiber-evoked response of the CFA lamina I NK1R+ neuron (Fig. 3C) that had a small polysynaptic Aβ and clear monosynaptic Aδ-fiber input. In the presence of the GABAA and glycine receptor antagonists bicuculline (10 μm) and strychnine (300 nm), this polysynaptic Aβ-fiber input is clearly enhanced (Fig. 4Bii). Figure 4C shows the fold increase in EPSC area under disinhibition conditions in lamina I NK1R+ neurons and illustrates that there is no significant difference between control and CFA-treated groups. Disinhibition revealed novel or enhanced polysynaptic Aβ-fiber input in ∼60% of both control and also CFA lamina I NK1R neurons, similar to that reported previously in naive spinal cord (Torsney and MacDermott, 2006). These data suggest that there is a low-threshold polysynaptic drive to lamina I NK1R+ neurons that is similarly under strong inhibitory regulation in both control and CFA inflamed spinal slices.
CFA inflammation increases the incidence and magnitude of monosynaptic Aδ-fiber but not monosynaptic C-fiber input to lamina I NKIR+ neurons
It has been suggested that the firing pattern of C-fiber afferents in inflammatory pain conditions will lead to a potentiation of monosynaptic C-fiber but not monosynaptic A-fiber synapses with lamina I projection neurons expressing the NK1R (Ikeda et al., 2006). To address this question, the afferent input to lamina I NK1R+ neurons was characterized, as illustrated in Figure 3, and compared between control and CFA inflammation, as summarized in Figure 5. The pie charts in Figure 5A show that lamina I NK1R+ neurons were dominated by primary afferent monosynaptic inputs with only a small proportion in both control and CFA conditions receiving purely polysynaptic inputs in 13 and 15% of neurons, respectively. Under control conditions, this monosynaptic input is predominantly C-fiber mediated, with 69% of neurons receiving monosynaptic C-fiber input, with a smaller proportion (28%) receiving monosynaptic Aδ-fiber input and with some of these neurons (14%) receiving both monosynaptic C-fiber and monosynaptic Aδ-fiber input (Fig. 5Ai). There were few control neurons (6%) with monosynaptic Aβ-fiber input. After CFA inflammation, the pattern of afferent input is significantly altered (Fig. 5Aii) (χ2 analysis, p < 0.05). The overall incidence of C-fiber monosynaptic input is not significantly altered (Fisher's exact test). However, strikingly, the incidence of monosynaptic Aδ-fiber input is significantly increased by twofold, from 28 to 58% (Fisher's exact test, p < 0.01). Both the number of neurons with only monosynaptic Aδ-fiber input and those with monosynaptic Aδ-fiber and monosynaptic C-fiber inputs are increased by twofold (both increased from 14 to 29%), although these just miss significance (Fisher's exact test, both p = 0.08) when the data is subgrouped in this manner. A significant increase in the latter is certainly supported by a significant decrease in the numbers of neurons with only monosynaptic C-fiber input (Fisher's exact test, p < 0.001) despite a lack of significant change in the overall incidence of C-fiber monosynaptic input. The incidence of purely polysynaptic and monosynaptic Aβ-fiber input was not significantly altered by CFA inflammation (Fisher's exact test).
Figure 5.

CFA inflammation increases the incidence and magnitude of monosynaptic Aδ-fiber but not monosynaptic C-fiber input to lamina I NKIR+ neurons. A, The proportion of lamina I NK1R+ neurons with input from different afferent fiber classes in both control (Ai) and CFA inflammation (Aii) groups is summarized (control neurons, n = 56; CFA neurons, n = 41). The wedges in the pie chart refer to the primary afferent monosynaptic input(s) received apart from the “poly” grouping, which reflects those neurons receiving purely polysynaptic inputs. For example “Aδ only” refers to those neurons receiving only monosynaptic Aδ-fiber input, and “Aδ with C” refers to those neurons receiving both monosynaptic Aδ-fiber and monosynaptic C-fiber input. The overall pattern of afferent input is significantly altered by CFA inflammation (χ2 analysis, p < 0.05). The incidence of monosynaptic Aδ-fiber input is significantly increased by twofold (Fisher's exact test, p < 0.01), whereas the incidence of C-fiber monosynaptic input is not significantly affected (Fisher's exact test). B, Effect of CFA inflammation on the peak amplitude of monosynaptic Aδ-fiber-evoked and monosynaptic C-fiber-evoked EPSCs in lamina I NK1R+ neurons. “C all” refers to the C-fiber-evoked peak amplitude in all neurons receiving monosynaptic C-fiber input, “Aδ only” refers to the Aδ-fiber-evoked peak amplitude of those neurons receiving only monosynaptic Aδ-fiber input, and “Aδ with C” refers to the Aδ-fiber-evoked peak amplitude of those neurons receiving both monosynaptic Aδ-fiber and monosynaptic C-fiber input. Averaged data are represented as mean ± SE, and the numbers of neurons analyzed within each class were shown on each bar. Two-way ANOVA followed by Bonferroni's post hoc tests (*p < 0.05) reveals that CFA inflammation significantly increases the peak amplitude in the “Aδ only” grouping.
The increased incidence of monosynaptic Aδ-fiber input is credible and not likely attributable to, for example, C fibers with lowered thresholds after CFA inflammation being misidentified as monosynaptic Aδ-fiber inputs for the following reasons. Compound action potential recordings from isolated dorsal roots (Fig. 2) clearly established that CFA inflammation did not alter the threshold stimulation intensity required to activate the different afferent components (the 100 μA Aδ stimulation intensity used in the patching studies was below the C-fiber threshold in all control and CFA dorsal roots tested). The criteria used to identify a monosynaptic Aδ-fiber input would likely have been too stringent (lack of failures, latency variability ≤2 ms at 2 Hz stimulation) to misidentify a monosynaptic C-fiber input, which show considerable latency variability and failures at rates above 1 Hz, as an apparent monosynaptic Aδ-fiber input (Nakatsuka et al., 2000). If, however, monosynaptic C fibers with lowered thresholds were observed after CFA inflammation and were misidentified as monosynaptic Aδ-fiber inputs, then the incidence of neurons with monosynaptic C-fiber input would be expected to be correspondingly decreased, which they are not (Fig. 5). Furthermore, the estimated conduction velocity of monosynaptic Aδ-fiber inputs was not significantly different between control and inflamed tissue but was significantly different from the faster conducting monosynaptic C-fiber inputs (see Fig. 7). For these reasons, it is strongly held that CFA inflammation increased the incidence of monosynaptic Aδ-fiber input to lamina I NK1R+ neurons.
Figure 7.

Estimated conduction velocity of silent monosynaptic Aδ and conventional (control and CFA) monosynaptic Aδ- and C-fiber inputs. Averaged data are represented as mean ± SE. Control Aδ (n = 14), CFA Aδ (n = 24), silent Aδ (n = 6), control C (n = 37), and CFA C (n = 21). One-way ANOVA followed by Bonferroni's post hoc tests reveals that all Aδ subgroups are not significantly different from one another, that both C subgroups are not significantly different from one another, but that all Aδ subgroups are significantly different from both C subgroups. Post hoc test significance values range from *p < 0.05 to p < 0.001, so for simplicity the least significance is shown.
The peak amplitude of monosynaptic Aδ-fiber-evoked and monosynaptic C-fiber-evoked EPSCs was also compared between control and CFA conditions (Fig. 5B). CFA inflammation resulted in a significant increase (∼75%) in the magnitude of monosynaptic Aδ-fiber-evoked EPSCs in those neurons receiving only monosynaptic Aδ-fiber input (two-way ANOVA, followed by Bonferroni's post hoc tests, p < 0.05) but not in those neurons receiving both monosynaptic Aδ-fiber and monosynaptic C-fiber input, which have smaller amplitude responses. However, CFA inflammation did not significantly alter the magnitude of monosynaptic C-fiber-evoked EPSCs whether or not they received only monosynaptic C-fiber input or both monosynaptic Aδ-fiber and monosynaptic C-fiber input (data not shown) or when all C-fiber-evoked EPSCs are pooled as shown in Figure 5B.
In summary, CFA inflammation results in a significant expansion and potentiation of monosynaptic Aδ-fiber inputs to lamina I NK1R+ neurons without any significant alteration in monosynaptic C-fiber inputs. Compared with the control situation in which monosynaptic C-fiber input was the predominant input to these neurons, in CFA inflammation this was no longer the case. The additional monosynaptic Aδ-fiber inputs that were unmasked by CFA inflammation increased the incidence of monosynaptic Aδ-fiber inputs to a level that was comparable with that of the monosynaptic C-fiber input. Furthermore, although CFA inflammation did not potentiate responses to monosynaptic C-fiber input, it did potentiate responses to monosynaptic Aδ-fiber input in the subset of neurons that received only monosynaptic Aδ-fiber input, such that these monosynaptic Aδ-fiber inputs were now able to evoke even larger EPSCs (∼2200 pA) than that evoked by monosynaptic C-fiber input (∼1200 pA).
Control lamina I NKIR+ neurons received pure NMDA or “silent” monosynaptic Aδ-fiber input
The presence of pure NMDA receptor or “silent” synapses in the spinal dorsal horn has been well established by a number of groups (Bardoni et al., 1998; Li and Zhuo, 1998; Baba et al., 2000; Jung et al., 2005) after the first suggestion of the existence of silent or “ineffective” spinal synapses (Wall, 1977). It has been proposed that activation of these silent synapses, by activity-dependent mechanisms after injury, could provide a mechanism for the acquisition of novel synaptic inputs important for allodynia and hyperalgesia (Kerchner et al., 1999). To address the hypothesis that the novel monosynaptic Aδ-fiber inputs revealed by CFA inflammation, in the present study, could arise as a result of the activation of silent synapses, control lamina I NK1R+ neurons lacking conventional monosynaptic Aδ-fiber inputs were examined in detail for the presence of silent monosynaptic Aδ-fiber inputs. It should be noted that, in the aforementioned studies of dorsal horn silent synapses, specific presynaptic and postsynaptic partners were not identified.
Control lamina I NK1R+ neurons that lacked conventional monosynaptic Aδ-fiber inputs typically exhibited EPSCs in response to 100 μA Aδ-fiber stimulation that were attributable to polysynaptic Aβ-fiber and/or polysynaptic Aδ-fiber input. Figure 6 shows an example of a control lamina I NK1R+ neuron with polysynaptic Aδ-fiber input that was examined for the presence of a silent monosynaptic Aδ-fiber input. This neuron did not respond to Aβ-fiber stimulation (data not shown), and the response evoked by Aδ-fiber stimulation showed 7 ms latency variability when elicited at 2 Hz, clearly indicating its polysynaptic nature (Fig. 6A). Identification of silent synapses generally uses “minimal stimulation” techniques whereby the stimulus strength is reduced to a level at which AMPA-receptor-mediated EPSCs are no longer observed at −70 mV, allowing the experimenter to then change to a positive holding potential to determine whether there are pure NMDA-receptor-mediated synapses present (Isaac et al., 1995; Liao et al., 1995). This minimal stimulation technique could not be used in the present study because the stimulation strength could not be reduced from the 100 μA Aδ-fiber stimulation intensity because the goal was to determine the existence of silent monosynaptic Aδ-fiber inputs. To circumvent this issue, the AMPA receptor antagonist NBQX (10 μm) was applied to eliminate the polysynaptic activity (Torsney and MacDermott, 2006) in the presence of the GABAA and glycine receptor antagonists bicuculline (10 μm) and strychnine (1 μm) to block inhibition (Fig. 6B). Moving to a positive holding potential (+50 mV) revealed a slow outward current that is presumed to be a pure NMDA or silent monosynaptic Aδ-fiber input for the following reasons (Fig. 6C). The response was evidently NMDA receptor mediated because the slow outward current observed at +50 mV was no longer present on return to −70 mV, as expected given the voltage dependence of the NMDA receptor and magnesium block at hyperpolarized potentials (Fig. 6D). It was also blocked by the NMDA receptor antagonist APV (30 μm) at +50 mV (Fig. 6E), and this was reversible during washout (Fig. 6F). Regarding the monosynaptic/polysynaptic nature of the NMDA-receptor-mediated response, silent synapses show failures at stimulation frequencies even lower than 2 Hz (Isaac et al., 1995, 1997) even when minimal stimulation techniques are not used (Durand et al., 1996), and so the criteria for identifying a conventional monosynaptic Aδ-fiber input (lack of failures and latency variability ≤2 ms at 2 Hz stimulation rates) could not be used here. However, although silent synapses show failures with repetitive stimulation, the response is clearly evident during averaging (Isaac et al., 1995, 1997) as was observed in the current example after high-frequency stimulation at 2 Hz (Fig. 6G) as would be expected for a monosynaptic input. Moreover, the latency of the slow outward current (Fig. 6C) was shorter than that of the polysynaptic Aδ-fiber input observed at −70 mV (Fig. 6A) as would be predicted for a monosynaptic Aδ-fiber input. If the slow outward current was not monosynaptic and was an NMDA-receptor-mediated polysynaptic component of the polysynaptic input observed at −70 mV, it would most likely have the same or a longer latency, but it instead had a shorter latency. In addition, as discussed in detail above, blocking inhibition facilitates polysynaptic A-fiber input to these neurons (Torsney and MacDermott, 2006), but such polysynaptic input generally had a longer latency than the preexisting input to the neuron. Therefore, the shorter-latency slow outward current observed here is not likely attributable to a disinhibition revealed polysynaptic input. Furthermore, it would seem unlikely that the NMDA-receptor-mediated response could be polysynaptic anyway given that the slow outward current was recorded in the presence of the AMPA receptor antagonist NBQX (10 μm), and so the polysynaptic input would need to be solely reliant on NMDA receptor activity, yet only the recorded neuron was held at +50 mV. It should be noted that activation of NMDA receptors alone can drive action potential firing of dorsal horn neurons (Bardoni et al., 2000), but this is only when neurons are stimulated at very high frequency (40 Hz) in order for EPSPs to summate sufficiently to action potential threshold, which could not be observed at the considerably lower frequencies (0.05 and 2 Hz) used in the present study. Therefore, the most plausible interpretation is that this lamina I NK1R+ neuron, which lacked a conventional monosynaptic Aδ-fiber input, received a silent monosynaptic Aδ-fiber input.
Figure 6.
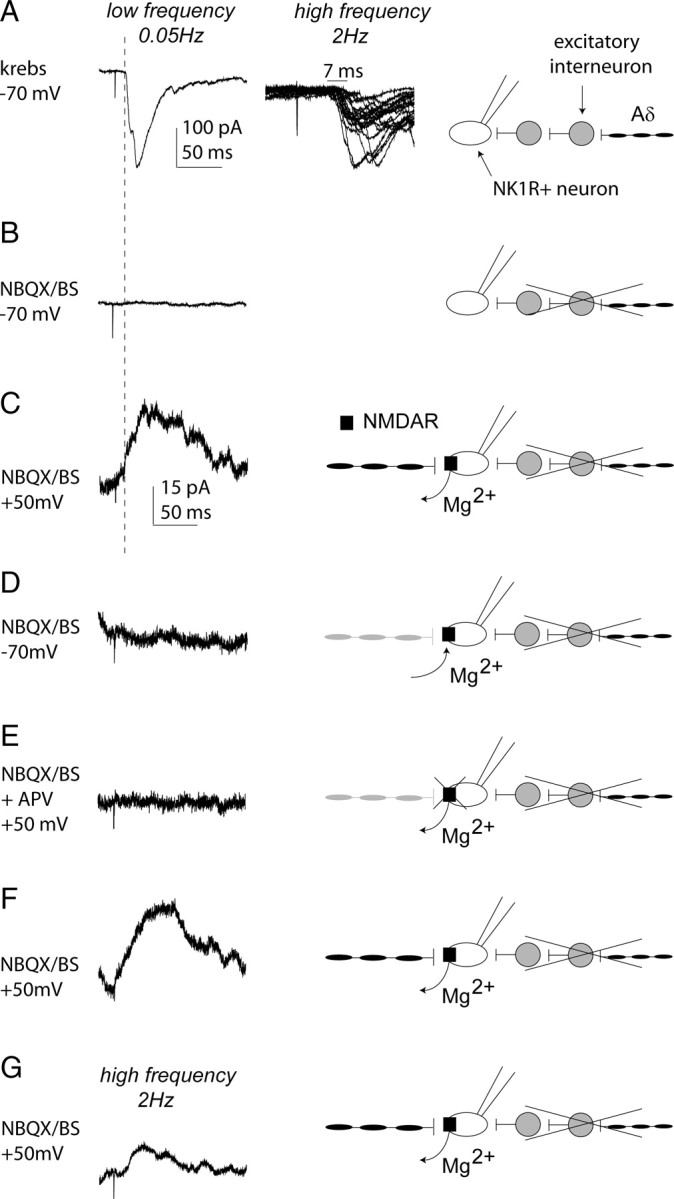
Silent monosynaptic Aδ-fiber input to a control lamina I NK1R+ neuron. A, Characterization of primary afferent synaptic input to a control lamina I NK1R+ neuron receiving polysynaptic Aδ-fiber input. In A–F, the left trace shows examples of EPSCs evoked by stimulation (0.1 ms) using Aδ (100 μA)-fiber stimulation intensities at low frequency. Each trace comprises three averaged traces evoked at 0.05 Hz. The second trace in A shows examples of EPSCs evoked by higher-frequency stimulation (100 μA/2 Hz) and comprises 20 superimposed traces exhibiting a latency variability of 7 ms and is shown at a faster timescale than the left trace. In A–G, the schematic on the right illustrates the input recorded under the different experimental conditions. Scale bars for the left trace in A also apply to B. In B–G, all recordings are performed in the presence of NBQX (10 μm) to block polysynaptic activity and bicuculline (B; 10 μm) and strychnine (S; 1 μm) to block inhibition. B, Stimulation of the neuron, held at −70 mV under these conditions, produced no detectable EPSC, and the block of polysynaptic input is shown schematically. C, When the neuron is held at +50 mV, a slow outward current was observed because of the removal of the voltage-dependent Mg2+ block of the NMDA receptor and unmasking of a silent monosynaptic Aδ-fiber input, as illustrated. Note that the slow outward current has a shorter latency than the polysynaptic input in A (see dotted line). D, The slow outward current was no longer evident on return to −70 mV because of the voltage-dependent Mg2+ block of the NMDA receptor and remasking of the silent monosynaptic Aδ-fiber input, as depicted. E, The outward current was completely blocked by APV (30 μm) as expected for a pure NMDA receptor synapse; this block was reversible (F), and these are shown schematically. In G, the trace comprises 20 averaged traces evoked by 2 Hz Aδ (100 μA)-fiber stimulation intensity (0.1 ms), and the silent monosynaptic Aδ-fiber input is again illustrated. Scale bars in C apply to C–G.
Control lamina I NK1R+ neurons, which lacked conventional monosynaptic Aδ-fiber input, were similarly assessed, and 6 of the 12 neurons examined were shown to receive silent monosynaptic Aδ-fiber input. In summary, these slow outward currents observed at +50 mV were only observed at Aδ-fiber and not Aβ-fiber stimulation intensities, were blocked by 30 μm APV, were no longer evident after return to a holding current of −70 mV, and were present during averaging when evoked by 2 Hz dorsal root stimulation. The average peak amplitude of the outward currents was 52.6 ± 9.0 pA, and these events had slow 10–90% rise times (17.7 ± 3.6 ms), which are very similar to the rise time kinetics of pure NMDA-receptor-mediated but not mixed AMPA/NMDA-receptor-mediated synapses, reported previously in the dorsal horn (Bardoni et al., 1998).
The monosynaptic nature of the primary afferent evoked slow outward current observed at +50 mV can be clearly dissociated from the primary-afferent-evoked polysynaptic activity observed in these neurons at −70 mV. In those neurons with polysynaptic Aδ-fiber input, the latency of the slow outward current observed at +50 mV was always shorter than that of the polysynaptic Aδ-fiber input recorded at −70 mV (n = 3), consistent with a monosynaptic Aδ-fiber input. In those neurons with polysynaptic Aβ-fiber input (n = 3), dorsal root stimulation only evoked slow outward currents when stimulated at Aδ-fiber but not Aβ-fiber strength. Finally, only a proportion of neurons (6 of 10) with polysynaptic activity observed at −70 mV showed outward currents at +50 mV.
To further confirm the monosynaptic Aδ nature of the outward current observed at +50 mV, the estimated conduction velocity of the afferent component evoking the response was calculated and compared with the estimated conduction velocity of conventional monosynaptic Aδ- and C-fiber inputs in control and CFA conditions. Figure 7 shows that the estimated conduction velocity of the silent Aδ-fiber inputs were not significantly different from values obtained for conventional monosynaptic Aδ-fiber inputs but were, as expected, significantly faster than the values for conventional monosynaptic C-fiber inputs, in both control and CFA conditions (one-way ANOVA). These data provide additional confirmation that a proportion (50%) of lamina I NK1R+ neurons that lacked conventional monosynaptic Aδ-fiber inputs received pure NMDA or silent monosynaptic Aδ-fiber input.
In summary, in contrast to previous studies describing pure NMDA receptor expression at dorsal horn synapses with unknown presynaptic and postsynaptic partners (Bardoni et al., 1998; Li and Zhuo, 1998; Baba et al., 2000; Jung et al., 2005), these data provide novel evidence for the presence of silent synapses specifically between monosynaptic Aδ-fiber inputs that are key for mechanical hyperalgesia (Treede and Magerl, 2000) and lamina I NK1R+ neurons that are important for chronic pain (Mantyh et al., 1997; Nichols et al., 1999).
Discussion
CFA inflammation increased the incidence and magnitude of monosynaptic Aδ-fiber but not monosynaptic C-fiber input to lamina I NK1R+ neurons. A proportion of control neurons received silent monosynaptic Aδ-fiber inputs, suggesting that these may provide the substrate for the novel Aδ inputs observed after CFA inflammation. The fact that 50% of lamina I NK1R+ neurons that lacked conventional monosynaptic Aδ-fiber input received silent monosynaptic Aδ-fiber input correlates well with the fact that 40% of these neurons acquired novel monosynaptic Aδ-fiber input during CFA inflammation.
Inflammation induced alterations in afferent input
Spinal disinhibition facilitates polysynaptic Aβ-fiber input to lamina I NK1R+ neurons (Torsney and MacDermott, 2006); therefore, it was predicted that peripheral inflammation, which disrupts spinal inhibition (Zeilhofer and Zeilhofer, 2008), would similarly facilitate this input. However, CFA inflammation did not alter the degree of polysynaptic Aβ-fiber input to lamina I NK1R+ neurons. Nonetheless, it could be that a mediator, such as prostaglandin E2, which plays a pivotal role in inflammatory spinal disinhibition (Zeilhofer and Zeilhofer, 2008), is simply not maintained in spinal slices in vitro. In addition, ongoing peripheral input, which would be eliminated by the slice preparation, is required for the maintenance of allodynia (but interestingly not hyperalgesia) (LaMotte et al., 1991; Koltzenburg et al., 1994), providing another explanation for the unaltered Aβ input observed. However, facilitation of polysynaptic Aβ-fiber input to substantia gelatinosa neurons has been observed in adult slices after CFA inflammation (Baba et al., 1999), suggesting that these factors may not be an issue. The use of juvenile rather than adult rats, in the present study, is not a concern given that robust mechanical hypersensitivity was observed and inflammation induced spinal hyperexcitability, at this age, is comparable with the adult (Torsney and Fitzgerald, 2002). Furthermore, spinal disinhibition similarly facilitated polysynaptic Aβ-fiber input in both control and CFA neurons, suggesting that low-threshold drive to lamina I NK1R+ neurons is similarly under strong inhibitory control in both conditions. Therefore, unmasking of polysynaptic Aβ-fiber input to lamina I NK1R+ neurons may not be important for inflammatory allodynia, although it may have relevance for neuropathic pain (Keller et al., 2007). Inflammatory allodynia may instead be mediated by other spinal circuits driven by Aβ fibers (Miraucourt et al., 2007, 2009) or low-threshold C-fiber mechanoreceptors (Seal et al., 2009).
Lamina I projection neurons, expressing the NK1R, exhibit homosynaptic potentiation of C-fiber monosynaptic inputs when these inputs are electrically stimulated in vitro, at rates that mimic their inflammatory firing pattern (Ikeda et al., 2006). However, in the present study, CFA inflammation did not potentiate responses to monosynaptic C-fiber input in lamina I NK1R+ neurons. Notably, homosynaptic potentiation was only observed in a subpopulation that projected to the periaqueductal gray (PAG) (Ikeda et al., 2006), and so a comparable effect in the current study may have been diminished by studying lamina I NK1R+ neurons that project to a variety of sites, not only the PAG (Marshall et al., 1996; Todd et al., 2000; Spike et al., 2003; Al-Khater et al., 2008). Alternatively, given that this subpopulation exhibit lower NK1R immunoreactivity than other lamina I projection neurons (Spike et al., 2003), they may not have been detected with TMR-SP labeling. However, if this subpopulation was included in the current study, the lack of potentiation may simply reflect the differing timescales, days after an inflammatory insult versus minutes after an artificial induction protocol (Ikeda et al., 2006). In summary, although this study does not provide support for homosynaptic potentiation of C-fiber monosynaptic inputs to lamina I NK1R+ neurons in inflammatory pain, neither does it specifically refute inflammation-induced potentiation of monosynaptic C-fiber-evoked responses in lamina I NK1R+ PAG projecting neurons.
It is well established that Aδ fibers mediate mechanical hyperalgesia via central spinal mechanisms after injury, but to date the circuitry engaged by these afferents is unknown (Treede and Magerl, 2000). The present study has demonstrated unmasking of novel and potentiated monosynaptic Aδ-fiber inputs to lamina I NK1R+ neurons, which are essential for hyperalgesia, after CFA inflammation. These findings therefore reveal spinal circuitry that is only engaged by Aδ fibers after injury and as such may underlie or contribute to the sensation of mechanical hyperalgesia. In support, both noxious mechanical stimulation and electrical stimulation of Aδ fibers, a proportion of which express substance P (Lawson et al., 1997), induce NK1R internalization in a significantly larger proportion of lamina I NK1R+ neurons, after CFA inflammation (Abbadie et al., 1997; Allen et al., 1999; Honor et al., 1999). Furthermore, CFA inflammation enlarges noxious mechanical receptive field sizes of lamina I projection neurons, interestingly without unmasking any low-threshold mechanoreceptive drive (Hylden et al., 1989), which correlates well with the novel Aδ and unaltered Aβ input observed in the current study.
Silent synapses
Pure NMDA receptor or silent synapses have been identified in the dorsal horn by various groups (Bardoni et al., 1998; Li and Zhuo, 1998; Baba et al., 2000; Jung et al., 2005), but their existence has recently been disputed (Yasaka et al., 2009). In the latter study, during the silent synapse identification protocol, paired presynaptic stimulation (0.2 Hz) and postsynaptic depolarization (0 mV) was applied to verify pharmacological blockade of IPSCs. Because this “pairing” protocol induces AMPA receptor expression at previously silent synapses (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996), any silent synapses present may have been inadvertently altered and therefore the lack of evidence for silent synapses reported is inconclusive. Dorsal horn silent synapses have been proposed to be a developmental phenomenon, prominent mainly in the first postnatal week (Li and Zhuo, 1998; Baba et al., 2000), but their expression correlates with susceptibility to plasticity (Isaac et al., 1997). Therefore, their expression in lamina I NK1R+ neurons, implicated in synaptic plasticity (Ikeda et al., 2003, 2006; Géranton et al., 2007, 2009), is not surprising. A postsynaptic model of silent synapses is generally assumed in which NMDA but not AMPA receptors are expressed postsynaptically. Presynaptic models also exist in which low-level glutamate release, from partially functional presynaptic terminals or from adjacent functional synapses that spill over, leads to the activation of only high-affinity NMDA but not low-affinity AMPA receptors present postsynaptically (Kerchner and Nicoll, 2008). Postsynaptically silent synapses are believed to be activated by AMPA receptor insertion postsynaptically. Presynaptically silent synapses become apparent by enhanced presynaptic glutamate release. In the dorsal horn, evidence thus far is for the postsynaptic model (Bardoni et al., 1998; Li and Zhuo, 1998; Li et al., 1999; Baba et al., 2000; Jung et al., 2005). If, as proposed, the silent synapses in the present study are the substrate for the novel Aδ inputs observed in inflammation, it seems likely, therefore, that postsynaptic mechanisms would be involved, especially given the altered postsynaptic AMPA receptor subunit composition in inflammatory pain (Katano et al., 2008; Larsson and Broman, 2008; Vikman et al., 2008). However, presynaptic mechanisms may also play a role, particularly given that the silent synapses were observed at room temperature, conditions under which spillover is enhanced (Asztely et al., 1997). Interestingly, to achieve maximal potentiation, it has been suggested that synapses draw on both presynaptic and postsynaptic resources, with presynaptic potentiation emerging once silent synapses are unsilenced by postsynaptic mechanisms (Ward et al., 2006; Kerchner and Nicoll, 2008). Therefore, the emergence of novel Aδ-fiber inputs after CFA inflammation may well result from a combination of both postsynaptic and presynaptic mechanisms. It will also be important to determine whether this unmasking is driven by activity in particular afferent inputs or in descending serotonergic systems (Li and Zhuo, 1998), which interestingly have been shown to regulate mechanical hypersensitivity in inflammatory pain (Géranton et al., 2008; Wei et al., 2010).
Heterosynaptic facilitation
It has been proposed that a heterosynaptic facilitation model of central sensitization is necessary to explain secondary mechanical hyperalgesia and mechanical allodynia (Treede and Magerl, 2000; Latremoliere and Woolf, 2009, 2010; Sandkühler, 2010) and potentially also primary mechanical hyperalgesia (Klein et al., 2008). This central sensitization is driven by C-fiber nociceptors and facilitates Aβ-fiber and capsaicin-insensitive Aδ-fiber input to distinct spinal circuitries, hitherto undefined, which mediate mechanical allodynia and hyperalgesia, respectively, without facilitation of C-fiber inputs. It is argued that models of homosynaptic potentiation of C-fiber inputs with projection neurons are insufficient to explain these phenomena and instead may contribute, together with peripheral sensitization to primary hyperalgesia. However, the current study provides novel evidence for a spinal circuitry that complies with the heterosynaptic facilitation model and may be relevant for mechanical hyperalgesia. Specifically, inflammatory pain, which is driven by C-fiber nociceptors (Abrahamsen et al., 2008), facilitates monosynaptic Aδ-fiber input to lamina I NK1R+ neurons, without facilitation of monosynaptic C-fiber inputs. If, as proposed, this novel circuitry contributes to mechanical hyperalgesia, elucidating the mechanisms by which this circuitry is unmasked and identifying agents that reduce its activity could aid in the targeting of pathological versus nociceptive pain.
Footnotes
This work was supported by the Caledonian Research Foundation, The Royal Society, and The Carnegie Trust for the Universities of Scotland. I thank Prof. Amy B. MacDermott for helpful discussion and critical reading of this manuscript.
The author declares no competing financial interests.
References
- Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI. Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J Neurosci. 1997;17:8049–8060. doi: 10.1523/JNEUROSCI.17-20-08049.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Al Ghamdi KS, Polgár E, Todd AJ. Soma size distinguishes projection neurons from neurokinin 1 receptor-expressing interneurons in lamina I of the rat lumbar spinal dorsal horn. Neuroscience. 2009;164:1794–1804. doi: 10.1016/j.neuroscience.2009.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khater KM, Kerr R, Todd AJ. A quantitative study of spinothalamic neurons in laminae I, III, and IV in lumbar and cervical segments of the rat spinal cord. J Comp Neurol. 2008;511:1–18. doi: 10.1002/cne.21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BJ, Li J, Menning PM, Rogers SD, Ghilardi J, Mantyh PW, Simone DA. Primary afferent fibers that contribute to increased substance P receptor internalization in the spinal cord after injury. J Neurophysiol. 1999;81:1379–1390. doi: 10.1152/jn.1999.81.3.1379. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Baba H, Doubell TP, Woolf CJ. Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J Neurosci. 1999;19:859–867. doi: 10.1523/JNEUROSCI.19-02-00859.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Doubell TP, Moore KA, Woolf CJ. Silent NMDA receptor-mediated synapses are developmentally regulated in the dorsal horn of the rat spinal cord. J Neurophysiol. 2000;83:955–962. doi: 10.1152/jn.2000.83.2.955. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Magherini PC, MacDermott AB. NMDA EPSCs at glutamatergic synapses in the spinal cord dorsal horn of the postnatal rat. J Neurosci. 1998;18:6558–6567. doi: 10.1523/JNEUROSCI.18-16-06558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Magherini PC, MacDermott AB. Activation of NMDA receptors drives action potentials in superficial dorsal horn from neonatal rats. Neuroreport. 2000;11:1721–1727. doi: 10.1097/00001756-200006050-00025. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett VJ, Simmons MA. Analysis of fluorescently labeled substance P analogs: binding, imaging and receptor activation. BMC Chem Biol. 2001;1:1. doi: 10.1186/1472-6769-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard L, Hill RG, Morris R. The correlation between the distribution of the NK1 receptor and the actions of tachykinin agonists in the dorsal horn of the rat indicates that substance P does not have a functional role on substantia gelatinosa (lamina II) neurons. J Neurosci. 1994;14:7655–7664. doi: 10.1523/JNEUROSCI.14-12-07655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Liu H, Maggio JE, Vigna SR, Mantyh PW, Basbaum AI. Morphological characterization of substance P receptor-immunoreactive neurons in the rat spinal cord and trigeminal nucleus caudalis. J Comp Neurol. 1995;356:327–344. doi: 10.1002/cne.903560302. [DOI] [PubMed] [Google Scholar]
- Cheunsuang O, Morris R. Spinal lamina I neurons that express neurokinin 1 receptors: morphological analysis. Neuroscience. 2000;97:335–345. doi: 10.1016/s0306-4522(00)00035-x. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Fratto V, Tochiki KK, Hunt SP. Descending serotonergic controls regulate inflammation-induced mechanical sensitivity and methyl-CpG-binding protein 2 phosphorylation in the rat superficial dorsal horn. Mol Pain. 2008;4:35. doi: 10.1186/1744-8069-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Jiménez-Díaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29:15017–15027. doi: 10.1523/JNEUROSCI.3451-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honor P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, Mantyh PW. Spinal substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci. 1999;19:7670–7678. doi: 10.1523/JNEUROSCI.19-17-07670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Nahin RL, Traub RJ, Dubner R. Expansion of receptive fields of spinal lamina I projection neurons in rats with unilateral adjuvant-induced inflammation: the contribution of dorsal horn mechanisms. Pain. 1989;37:229–243. doi: 10.1016/0304-3959(89)90135-8. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Jung SJ, Kim YS, Kim DK, Kim J, Kim SJ. Long-term potentiation of silent synapses in substantia gelatinosa neurons. Neuroreport. 2005;16:961–965. doi: 10.1097/00001756-200506210-00016. [DOI] [PubMed] [Google Scholar]
- Katano T, Furue H, Okuda-Ashitaka E, Tagaya M, Watanabe M, Yoshimura M, Ito S. N-ethylmaleimide-sensitive fusion protein (NSF) is involved in central sensitization in the spinal cord through GluR2 subunit composition switch after inflammation. Eur J Neurosci. 2008;27:3161–3170. doi: 10.1111/j.1460-9568.2008.06293.x. [DOI] [PubMed] [Google Scholar]
- Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Li P, Zhuo M. Speaking out of turn: a role for silent synapses in pain. IUBMB Life. 1999;48:251–256. doi: 10.1080/713803505. [DOI] [PubMed] [Google Scholar]
- Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22:9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabov SG, Ghilardi JR, Mantyh PW, Simone DA. Spinal neurons that express NK-1 receptors modulate descending controls that project through the dorsolateral funiculus. J Neurophysiol. 2005;93:998–1006. doi: 10.1152/jn.01160.2003. [DOI] [PubMed] [Google Scholar]
- Klein T, Stahn S, Magerl W, Treede RD. The role of heterosynaptic facilitation in long-term potentiation (LTP) of human pain sensation. Pain. 2008;139:507–519. doi: 10.1016/j.pain.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Torebjörk HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- Labrakakis C, MacDermott AB. Neurokinin receptor 1-expressing spinal cord neurons in lamina I and III/IV of postnatal rats receive inputs from capsaicin sensitive fibers. Neurosci Lett. 2003;352:121–124. doi: 10.1016/j.neulet.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Labrakakis C, Tong CK, Weissman T, Torsney C, MacDermott AB. Localization and function of ATP and GABAA receptors expressed by nociceptors and other postnatal sensory neurons in rat. J Physiol. 2003;549:131–142. doi: 10.1113/jphysiol.2002.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMotte RH, Shain CN, Simone DA, Tsai EF. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. J Neurophysiol. 1991;66:190–211. doi: 10.1152/jn.1991.66.1.190. [DOI] [PubMed] [Google Scholar]
- Larsson M, Broman J. Translocation of GluR1-containing AMPA receptors to a spinal nociceptive synapse during acute noxious stimulation. J Neurosci. 2008;28:7084–7090. doi: 10.1523/JNEUROSCI.5749-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Synaptic plasticity and central sensitization: author reply. J Pain. 2010;11:801–803. doi: 10.1016/j.jpain.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, Crepps BA, Perl ER. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. J Physiol. 1997;505:177–191. doi: 10.1111/j.1469-7793.1997.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Lu Y, Park TJ. A plethora of painful molecules. Curr Opin Neurobiol. 2004;14:443–449. doi: 10.1016/j.conb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Li P, Zhuo M. Silent glutamatergic synapses and nociception in mammalian spinal cord. Nature. 1998;393:695–698. doi: 10.1038/31496. [DOI] [PubMed] [Google Scholar]
- Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- Marshall GE, Shehab SA, Spike RC, Todd AJ. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience. 1996;72:255–263. doi: 10.1016/0306-4522(95)00558-7. [DOI] [PubMed] [Google Scholar]
- Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PLoS One. 2007;2:e1116. doi: 10.1371/journal.pone.0001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraucourt LS, Moisset X, Dallel R, Voisin DL. Glycine inhibitory dysfunction induces a selectively dynamic, morphine-resistant, and neurokinin 1 receptor-independent mechanical allodynia. J Neurosci. 2009;29:2519–2527. doi: 10.1523/JNEUROSCI.3923-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuka T, Park JS, Kumamoto E, Tamaki T, Yoshimura M. Plastic changes in sensory inputs to rat substantia gelatinosa neurons following peripheral inflammation. Pain. 1999;82:39–47. doi: 10.1016/S0304-3959(99)00037-8. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Ataka T, Kumamoto E, Tamaki T, Yoshimura M. Alteration in synaptic inputs through C-afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience. 2000;99:549–556. doi: 10.1016/s0306-4522(00)00224-4. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Rahman W, Suzuki R, Hunt SP, Dickenson AH. Selective ablation of dorsal horn NK1 expressing cells reveals a modulation of spinal alpha2-adrenergic inhibition of dorsal horn neurones. Neuropharmacology. 2008;54:1208–1214. doi: 10.1016/j.neuropharm.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Ruscheweyh R, Ikeda H, Heinke B, Sandkühler J. Distinctive membrane and discharge properties of rat spinal lamina I projection neurones in vitro. J Physiol. 2004;555:527–543. doi: 10.1113/jphysiol.2003.054049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycroft BK, Vikman KS, Christie MJ. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto NK1 receptor-positive lamina I neurons in the rat dorsal horn. J Physiol. 2007;580:883–894. doi: 10.1111/j.1469-7793.2000.t01-1-02117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J. Central sensitization versus synaptic long-term potentiation (LTP): a critical comment. J Pain. 2010;11:798–800. doi: 10.1016/j.jpain.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci [Suppl] 2002;5:1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, Edwards RH. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature. 2009;462:651–655. doi: 10.1038/nature08505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike RC, Puskár Z, Andrew D, Todd AJ. A quantitative and morphological study of projection neurons in lamina I of the rat lumbar spinal cord. Eur J Neurosci. 2003;18:2433–2448. doi: 10.1046/j.1460-9568.2003.02981.x. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Polgár E. A quantitative study of neurons which express neurokinin-1 or somatostatin sst2a receptor in rat spinal dorsal horn. Neuroscience. 1998;85:459–473. doi: 10.1016/s0306-4522(97)00669-6. [DOI] [PubMed] [Google Scholar]
- Todd AJ, McGill MM, Shehab SA. Neurokinin 1 receptor expression by neurons in laminae I, III and IV of the rat spinal dorsal horn that project to the brainstem. Eur J Neurosci. 2000;12:689–700. doi: 10.1046/j.1460-9568.2000.00950.x. [DOI] [PubMed] [Google Scholar]
- Tong CK, MacDermott AB. Both Ca2+-permeable and -impermeable AMPA receptors contribute to primary synaptic drive onto rat dorsal horn neurons. J Physiol. 2006;575:133–144. doi: 10.1113/jphysiol.2006.110072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsney C, Fitzgerald M. Age-dependent effects of peripheral inflammation on the electrophysiological properties of neonatal rat dorsal horn neurons. J Neurophysiol. 2002;87:1311–1317. doi: 10.1152/jn.00462.2001. [DOI] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Magerl W. Multiple mechanisms of secondary hyperalgesia. Prog Brain Res. 2000;129:331–341. doi: 10.1016/S0079-6123(00)29025-0. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Rycroft BK, Christie MJ. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J Physiol. 2008;586:515–527. doi: 10.1113/jphysiol.2007.145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall PD. The presence of ineffective synapses and the circumstances which unmask them. Philos Trans R Soc Lond B Biol Sci. 1977;278:361–372. doi: 10.1098/rstb.1977.0048. [DOI] [PubMed] [Google Scholar]
- Ward B, McGuinness L, Akerman CJ, Fine A, Bliss TV, Emptage NJ. State-dependent mechanisms of LTP expression revealed by optical quantal analysis. Neuron. 2006;52:649–661. doi: 10.1016/j.neuron.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Wei F, Dubner R, Zou S, Ren K, Bai G, Wei D, Guo W. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. J Neurosci. 2010;30:8624–8636. doi: 10.1523/JNEUROSCI.5389-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Yasaka T, Hughes DI, Polgár E, Nagy GG, Watanabe M, Riddell JS, Todd AJ. Evidence against AMPA receptor-lacking glutamatergic synapses in the superficial dorsal horn of the rat spinal cord. J Neurosci. 2009;29:13401–13409. doi: 10.1523/JNEUROSCI.2628-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Zeilhofer UB. Spinal dis-inhibition in inflammatory pain. Neurosci Lett. 2008;437:170–174. doi: 10.1016/j.neulet.2008.03.056. [DOI] [PubMed] [Google Scholar]