

Abstract
Brain dopamine (DA) has been linked to error processing. Because high and low (vs medium) prefrontal cortex (PFC) DA levels may facilitate D2-receptor-related modulations of PFC neural activation patterns, we hypothesized that high and low DA predicts increased error-specific transitions of PFC activity. Male human participants (n = 169) were genotyped for the catechol-O-methyltransferase (COMT) Val158Met polymorphism, associated with low (Val) and medium (Met) PFC DA levels. In addition, DRD2TaqIa and 5-HTTLPR, associated with striatal D2 receptor density and serotonin uptake, respectively, were assessed. Participants received placebo or a selective DA–D2 receptor blocker (sulpiride, 200 mg) and performed a Flanker task. EEG was recorded and decomposed into independent brain components (ICs) using independent component analysis. After errors, participants displayed (1) a negative deflection in ICs source-localized to the proximity of the anterior midcingulate cortex [IC-error-related negativity (IC-ERN)], (2) increased midcingulate cortex IC power in the delta/theta frequency range, and (3) slowing in the subsequent trial [posterror slowing (PES)]. Importantly, all, IC-ERN, delta/theta power, and PES were modulated by COMT × Substance interactions such that the Val allele predicted elevated IC-ERN, delta/theta power, and PES after placebo; this association was reversed under sulpiride. Because low doses of sulpiride presumably increase PFC DA levels, the COMT × Substance interaction supports the hypothesis that low (Val, placebo) and high (Met, sulpiride) versus medium (Val, sulpiride; Met, placebo) DA levels elevate reactivity to errors. Consistent with an influence of serotonin on PFC DA, the COMT × Substance interaction was modulated by 5-HTTLPR.
Introduction
Errors rapidly trigger cascades of behavioral (Rabbitt and Phillips, 1967) and neural (Gehring and Knight, 2000) sequelae associated with monitoring and regulating performance. These include dynamic modulations of medial prefrontal cortex (PFC) and basal ganglia (BG) neural activation patterns in response to the error (Holroyd and Coles, 2002) and/or associated response conflicts (Botvinick et al., 2001). According to the dual-state theory of PFC dopamine (DA) function (Durstewitz and Seamans, 2008), low or high DA levels induce a relative dominance of D2 over D1 receptor activation (D2 state) while medium DA promotes D1 receptor activation (D1 state). While D1 states have been associated with relative network stability and presumably improved working memory, D2 states presumably facilitate modulations of PFC neural activation patterns (Durstewitz and Seamans, 2008). Because (1) error feedback sensitivity of single medial PFC neurons (Shima and Tanji, 1998; Hikosaka and Isoda, 2010) and error-related brain activity in humans (Yasuda et al., 2004; Hester et al., 2008) are linked to increased response and/or neural network variability typically found in D2 states (Durstewitz and Seamans, 2008), and (2) brain and behavioral responses to errors reflect dynamic, presumably DA-sensitive, processes, particularly tied to D2 receptor activation (Zirnheld et al., 2004; Frank and Hutchison, 2009), we hypothesized that error-induced transitions of brain activity and behavior are enhanced at relatively high and low DA levels associated with low-stability D2-dominated regimes.
According to the dual-state theory, D2-dominated regimes likely occur in Val allele carriers (Val+) of the catechol-O-methyltransferase (COMT) Val158Met polymorphism (Durstewitz and Seamans, 2008), which show relatively low PFC DA levels (Lachman et al., 1996), while Met/Met carriers, displaying medium DA levels, are characterized by D1-state networks (Durstewitz and Seamans, 2008). Due to the inverted U-model, increasing PFC DA level, for example through administration of sulpiride (Kuroki et al., 1999), should push Val+ carriers toward medium and Met/Met carriers toward high DA levels, associated with D1 and D2 states, respectively (Fig. 1). If D2 states enhance error reactivity, Val+ versus Met/Met carriers should show increased error responses and sulpiride should reverse this genotype effect.
Figure 1.

Postulated relationship between prefrontal dopamine level, COMT, and relative D1- versus D2-receptor activation as previously described by Durstewitz and Seamans (2008). Due to enhanced relative D2 receptor activation in Val+ versus Met/Met carriers, we hypothesized increased error reactivity in Val+ versus Met/Met carriers. By increasing PFC dopamine activity through presynaptic D2 receptor blockade, sulpiride (200 mg) is predicted to shift Val+ carriers into medium and Met/Met carriers into high dopamine levels (red arrows), resulting in reduction or enhancement of error reactivity, respectively.
We tested these hypotheses using neural and behavioral markers for error reactivity. First, we analyzed the error-related negativity (ERN) event-related potential component, which originates from anterior midcingulate cortex (Debener et al., 2005) and is modulated by DA (Ullsperger, 2010). Second, we assessed periresponse increases in mediocentral delta (1–4 Hz) and theta (4–8 Hz) power, which are elevated for error versus correct responses (Luu et al., 2004; Yordanova et al., 2004; Cavanagh et al., 2009). Finally, we analyzed posterror slowing (PES), which reflects increases of reaction times in trials after error commission (Rabbitt and Phillips, 1967).
In addition, we investigated the effect of the DRD2TaqIa polymorphism, which is linked to presynaptic D2 receptor density in striatum (Zhang et al., 2007) and—because PFC dopaminergic neurotransmission is modulated by serotonin (5-HT) (Fink and Göthert, 2007)—the 5-HT transporter polymorphism (5-HTTLPR), associated with 5-HT transporter expression and ERN amplitude (Fallgatter et al., 2004).
Materials and Methods
Participants
Participants (n = 195) were all nonsmoking, right-handed, and male. Before testing, the absence of any illnesses or DSM-IV diagnoses was confirmed using a standardized clinical interview (Margraf, 1994). All participants reported not having used any prescription or illegal drugs during the past 3 months. Participants were required to refrain from consuming alcohol, nicotine, and caffeine 12 h before the beginning of the study. All volunteers gave written informed consent before participating and received a monetary compensation of 70 EUR (90 USD) for ∼7 h involvement in the project. The study protocol was approved by the Ethics Committee of the German Society for Psychology (Deutsche Gesellschaft für Psychologie). Datasets containing grossly artifact-contaminated EEG (n = 13; 6.7%) or less than three error trials remaining following artifact correction (n = 8; 4.1%) were excluded. Further, datasets for which the clustering algorithm (see Independent component analysis decomposition, below) assigned no independent component dipoles to an anterior midcingulate cortex cluster (n = 5; 2.5%) were excluded from further processing, yielding a final sample of n = 169 with an average age of 23.8 years (SD = 3.1 years; for further description, see Table 1).
Table 1.
Sample characteristics and descriptive statistics
| Placebo |
Sulpiride |
|||
|---|---|---|---|---|
| Val (n = 54) | Met (n = 25) | Val (n = 65) | Met (n = 25) | |
| Number of A1− carriers | 39 | 16 | 47 | 14 |
| Number of S+ and LG+ carriers | 39 | 17 | 49 | 13 |
| Age | 23.8 (0.4) | 23.4 (0.6) | 23.8 (0.4) | 23.5 (0.4) |
| Weight, kg | 77 (2) | 79 (3) | 78 (2) | 81 (3) |
| Height, cm | 183 (8) | 183 (7) | 183 (8) | 181 (7) |
| Number of errors | 19.6 (1.3) | 18.5 (1.9) | 20.0 (1.2) | 20.5 (2.3) |
| Reaction time correct, ms | 366 (4) | 357 (5) | 364 (3) | 358 (4) |
| Reaction time error, ms | 335 (5) | 331 (7) | 332 (4) | 326 (7) |
| Post-error slowing, ms | 77 (4) | 56 (4) | 72 (3) | 70 (5) |
| Post-correct slowing, ms | 34 (3) | 29 (4) | 40 (3) | 36 (2) |
| IC-ERN (error trials) | −3.0 (0.2) | −2.4 (0.2) | −2.6 (0.2) | −3.1 (0.3) |
| IC-ERN (correct trials) | −1.1 (0.1) | −1.2 (0.1) | −1.1 (0.1) | −1.1 (0.1) |
| IC-ERN (error − correct) | −1.9 (0.2) | −1.2 (0.3) | −1.4 (0.2) | −1.9 (0.3) |
| IC-delta/theta power (error) | 4.8 (0.3) | 3.6 (0.5) | 3.9 (0.3) | 4.4 (0.5) |
| IC-delta/theta power (correct) | 2.2 (0.1) | 1.9 (0.2) | 1.8 (0.1) | 1.7 (0.2) |
| Channel ERN (error − correct) | −23.3 (1.7) | −19.5 (2.5) | −19.4 (1.6) | −23.8 (2.5) |
Values are given as means (SEM). Channel ERN reflects the difference score (amplitude error minus amplitude correct) measured at scalp channel Fz.
Procedure
All participants gave informed consent and were administered the standardized clinical interview. If no exclusion criteria applied, participants received a standardized breakfast (water/juice, 1–2 rolls with cheese, marmalade, or sausage) and ingested either a sulpiride (200 mg) or placebo capsule with a glass of water. Both capsules had the same appearance to ensure that the experimenter and the participants were blind to the pharmacological treatment. Thereafter participants performed several tasks (e.g., intelligence tests, personality questionnaires, learning tasks) (results will be reported in a future paper). Approximately 4 h after administration of the pill, participants started the Flanker task.
Flanker task
The Flanker task, delivered using Presentation 12.0 (Neurobehavioral Systems), consisted of three blocks of 140 trials each, plus an initial practice block to determine individual response speed, during which no EEG was recorded. A trial began with central screen presentation of a fixation cross for 1000 ms. Thereafter congruent (SSSSS, HHHHH) or incongruent (SSHSS, HHSHH) stimulus arrays were presented for 600 ms with the central target letter onset coming 100 ms after the four flanker letters. Participants were instructed to respond as quickly as possible by pressing one of two buttons under their right index and middle fingers in response to S and H targets, respectively. If participants reacted slower than their mean reaction time plus 1 SD (both determined from the preceding trial block), the feedback “too slow” appeared for 500 ms beginning 900 ms after the onset of the target stimulus. Otherwise performance feedback was given (“error” or “correct”). If participants did not react within 0–900 ms after presentation of central target letter onset, the feedback read “no button press.”
Behavioral data
For behavioral analyses, reaction times were measured as the latency between central target letter presentation and the ensuing subject button press. All responses 3 SDs faster than the average reaction time were discarded. Posterror slowing (Rabbitt and Phillips, 1967) was computed as the difference in reaction times between an error trial and the subsequent trial. To compute a comparable measure for correct trials, a correct trial with similar reaction time was automatically identified for each error trial, and again the difference was taken between this (relatively fast) response and the response time in the subsequent trial. Error rates were also computed.
EEG
Recording.
EEG was recorded at a sampling rate of 512 Hz using an Active Two (BioSemi) active electrode system with driven right leg and common mode sense as active and passive reference, respectively. EEG data of the first 99 and remaining 96 participants were recorded using 32- and 64-channel configurations, respectively. Because normalizing the ERN amplitudes within these two subsets did not affect the effects presented, we provide results for the two subject subgroups pooled together.
Independent component analysis decomposition.
Data were analyzed by custom Matlab (Mathworks) scripts built on the open source EEGLAB toolbox (Delorme and Makeig, 2004). The manually inspected and artifact-removed continuous EEG data were 1 Hz high-pass filtered and decomposed using an adaptive mixture independent component analysis (ICA) algorithm (Palmer et al., 2006, 2008). As explained in more detail previously (Gramann et al., 2010), for each independent component (IC) scalp topography, an equivalent current dipole was computed using a standard adult boundary element head model implemented in the DIPFIT toolbox (Oostenveld and Oostendorp, 2002). Only ICs with dipoles localized within the brain and whose scalp projection (through a spherical forward head model) explained >90% IC scalp topography variance were used in further analyses. ICs of all participants were clustered using a K-means clustering algorithm applied to the estimated equivalent dipole positions. A total of 15 IC clusters, plus one outlier cluster, were thus identified (Fig. 2). The outlier cluster was defined a priori to consist of all ICs whose estimated dipole positions were >3 SD from any IC cluster centroid. As depicted in Figure 3, IC Cluster 9 had maximum dipole density in and near anterior midcingulate cortex (Vogt, 2005), a region of the brain previously implicated in error processing and ERN generation (Debener et al., 2005); 169 participants had at least one IC component in this cluster. For participants with more than one IC in the cluster, the IC exhibiting the largest negative error-response time-locked ERP deflection within 150 ms was automatically identified as this subject's cluster-component ERN (ERN-IC). Both activation time courses and scalp maps of cluster ICs with inverted topographies (e.g., a negative correlation between the IC scalp map and the IC cluster mean scalp map) were polarity reversed (multiplied by −1) before between-subject comparisons to ensure that all IC activation time courses had comparable polarities. ERN amplitudes for the thus derived ERN-IC activations (for measurement see, Error-related negativity below) showed a substantial variance overlap with ERN amplitudes at frontocentral scalp channels Cz and Fz (or the average of both channels), yet they were not redundant (0.53 < rs < 0.58). To measure and visualize dipole position spread, dipole density was computed using a 3D Gaussian smoothing function with SD = 10 mm. The result is plotted in Figure 3a.
Figure 2.

Cluster-mean IC scalp topographies for outlier (Cluster 1, top left) and other IC clusters. Cluster 9 (red box) was used for analyses of error-related brain activity in the present study.
Figure 3.
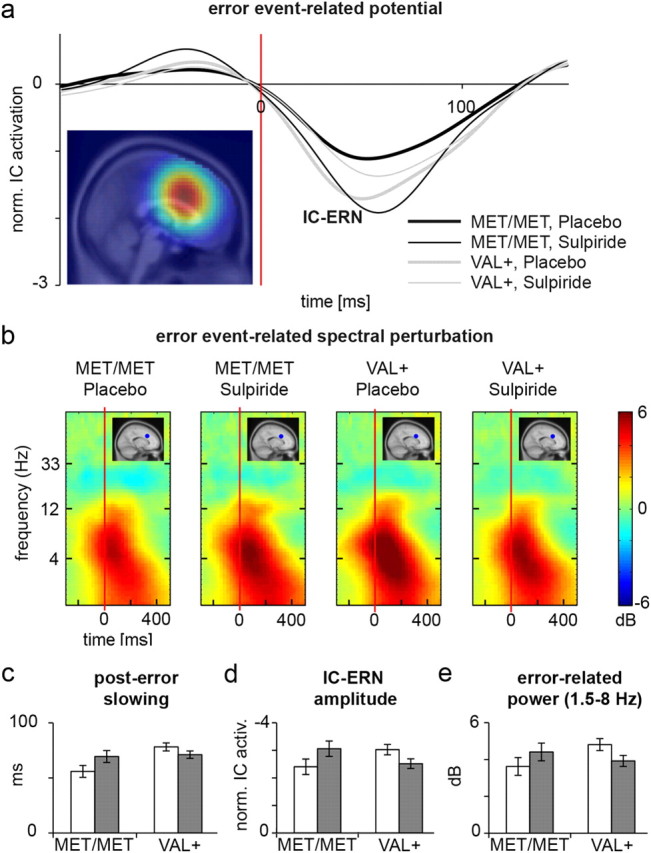
Interactions of Substance and COMT on neural and behavioral error-processing correlates. a, Grand average response-locked event-related potentials for a medial frontal IC-cluster following erroneous button presses for Val+ (gray lines) and Met/Met (black lines) carriers who had received placebo (thick lines) or sulpiride (thin lines). A latency of 0 ms corresponds to the time of the button press. Independent component ERPs were normalized, dividing by the root mean square over the component scalp map projection to all channels before averaging (see Materials and Methods). A standard template brain image (Montreal Neurological Institute) indicates the region of maximum concentration measured as equivalent dipole density for this IC cluster. b, Event-related spectral perturbation plot showing cluster mean differences in log spectral power relative to log spectral power at button press, separately for the two (Met/Met vs Val+) by two (placebo vs sulpiride) matrix of subgroups. A latency of 0 ms corresponds to the time of the button press. For each subgroup, the mean estimated dipole position is also plotted in the upper right corner of the event-related spectral perturbation plot. c–e, Bar plots indicating means (and SEMs) of reaction-time slowing in the subsequent trial following errors (c), peak IC-cluster error-related negativity (d), and average power increase from 1.5 to 8 Hz (e) in the placebo (white) or sulpiride (gray) groups.
Error-related negativity.
For each participant, ERN-IC activations were baseline corrected (using the baseline interval from −800 to −600 ms relative to response onset), then averaged across trials, normalized to make the root-mean square component scalp map projection to all channels (across all time points in the data epochs) 1 μV, and 20 Hz low-pass filtered. Peak-to-peak ERN amplitude was measured as the difference between the most negative-going ERN-IC activation value from 0 to 150 ms following the button press and the most positive-going activation value from −100 to 0 ms before it. In the same way, we also measured the standard scalp-channel peak-to-peak ERN at electrode Fz re-referenced to linked earlobes.
Time-frequency analyses.
Time-frequency analyses were conducted using the EEGLAB function newtimef.m. Event-related spectral perturbations (Makeig, 1993) and intertrial phase coherence (Delorme and Makeig, 2004) were computed for epochs from −1500 to 2000 ms relative to button presses using Hanning-windowed sinusoids as wavelets. The analyzed frequencies increased from 1.5 to 100 Hz in 65 logarithmically spaced steps while the number of cycles in the corresponding wavelets linearly increased from one cycle at 1.5 Hz to eight cycles at 100 Hz. Power was converted to a decibel (dB) scale (log10 [power(t)/log10(0 ms)]) and baseline corrected for −800 to −600 ms relative to the button press. Error reactivity was analyzed for a time window from −100 to 300 ms (Cavanagh et al., 2009).
Genotyping
DNA was extracted from buccal cells, purified, and genotyped for COMT Val158Met as previously described (Reuter and Hennig, 2005). Using the methods reported by Reuter et al. (2006), all participants were also genotyped for DRD2 TaqIA, a polymorphism associated with individual differences in D2 dopamine receptor density (Pohjalainen et al., 1998), presumably via linkage disequilibrium with another functional polymorphism located directly on the DRD2 gene (Zhang et al., 2007). Finally, we genotyped participants for 5-HTTLPR as described before (Osinsky et al., 2008), which occurs in the three variants short (S), long G (LG), and long A (LA) (Nakamura et al., 2000), the former two being associated with low, nearly equivalent 5-HT transporter expression (Hu et al., 2006).
The resulting genotype distributions were as follows. COMT Val158Met: n = 33, 86, and 50 for Val/Val, Val/Met, and Met/Met, respectively; DRD2TaqIa: n = 6, 47, and 116 for A1/A1, A1/A2, and A2/A2, respectively; 5-HTTLPR: n = 27, 10, 70, 0, 11, and 50 for S/S, S/LG, S/LA, LG/LG, LG/LA, and LA/LA, respectively. For all statistical analyses, homozygotes of the major allele were compared with the remainder. This resulted in the following comparisons: Met/Met (n = 50) versus Val+ (n = 119), A2/A2 (n = 116) versus A1+ (n = 53), and LA/LA (n = 50) versus LG+, S+ (n = 119). With regard to 5-HTTLPR, LG+ and S+ were grouped based on the almost identical 5-HT transporter expression (Hu et al., 2006). All genotype distributions were in Hardy-Weinberg equilibrium (all X2s < 0.3).
Sulpiride
Sulpiride is a substituted benzamide that acts as a selective D2-receptor antagonist. A single acute dose of 200 mg of sulpiride results in considerably lower levels of D2 receptor occupancy than considered efficacious in the treatment of schizophrenia and is thought to primarily block presynaptic autoreceptors, thereby leading to increased DA level (Mereu et al., 1983; Kuroki et al., 1999). Sulpiride is generally well tolerated; does not appear to significantly block other types of receptors (e.g., histaminergic, cholinergic, serotonergic, adrenergic, and GABA receptors); is slowly absorbed from the gastrointestinal tract, with peak serum levels occurring within one to 6 h after oral ingestion; and the average elimination half life is in the range of 3 to 10 h (Mauri et al., 1996).
Statistical analyses
To reduce the impact of potential outliers, we first Winsorized the data (both reaction times and IC activations) by replacing the lower and upper 10% of the data with the 10th and 90th percentile values, respectively (Erceg-Hurn and Mirosevich, 2008). To test for main effects or interactions we computed standard parametric ANOVAs with the repeated measures factor Response (error vs correct) and the between-subjects factors COMT (Val+ vs Met/Met) and Substance (sulpiride vs placebo). To analyze time-frequency data, the additional repeated measures factor Band (delta, 1.5–4 Hz vs theta, 4–8 Hz) was included. Post hoc comparisons were performed using unpaired t tests with one-tailed significance thresholds for predicted effects. All statistical tests were conducted using SPSS 15.0.
Results
Behavioral data
As expected, reaction times (RT) following errors were significantly larger than reaction times following RT-matched correct responses, indicating significant PES (main effect Response: F(1,165) = 259.6, p < 0.0001, ηp2 = 0.61). Importantly, the Response × COMT × Substance interaction (F(1,165) = 4.9, p = 0.03, ηp2 = 0.03) was significant (Fig. 3). To follow up on this interaction, we calculated difference scores (slowing after errors minus slowing after RT-matched correct trials) and compared COMT genotypes on this error-specific net effect separately for placebo and sulpiride. As predicted, in the placebo group, Val+ carriers showed significantly more error-specific slowing than Met/Met carriers (t(78) = 2.75, p = 0.004, one-tailed). When analyzing the raw scores in the next step, this effect could be attributed to slowing following errors (t(78) = 4.0, p < 0.001, one-tailed) rather than slowing following correct responses (t(78) = 0.99, p > 0.15: Table 1). Of particular relevance, the genotype effect on the difference score was completely eliminated in participants who received sulpiride (t(87) = 0.22, p > 0.4, one-tailed), primarily due to a significant reduction in Val+ carriers after sulpiride versus placebo intake (t(117) = 2.57, p = 0.006, one-tailed) and a nonsignificant increase in Met/Met carriers (t(48) = 1.02, p = 0.16, one-tailed). There were no main effects or interactions on the total number of errors or on the mean reaction time following correct trials, supporting the specificity of the present findings with regard to posterror processing.
We also tested for conflict adaptation effects (Gratton et al., 1992; Botvinick et al., 1999). Trials in which the Flanker stimuli did not match the target stimulus were considered high conflict trials. Individuals showed increased reaction times in high versus low conflict trials (F(1,165) = 2405.4, p < 0.001, ηp2 = 0.94) and this conflict effect was potentiated in individuals who received sulpiride versus placebo, regardless of genotype (F(1,165) = 6.37, p < 0.015, ηp2 = 0.04). The effect of high versus low conflict was reduced in trials following an error response (F(1,165) = 36.7, p < 0.001, ηp2 = 0.19) (Ridderinkhof et al., 2002) or a high conflict trial (F(1,165) = 209.8, p < 0.001, ηp2 = 0.56) (Gratton et al., 1992). However, Substance and COMT did not further modulate these interactions (ps > 0.3).
EEG data
Independent-component event-related potentials
As shown in Figure 3, errors triggered a clear negative deflection in independent EEG sources localized to anterior midcingulate cortex (i.e., IC-ERN) that was dramatically reduced following correct responses (main effect Response: F(1,165) = 225.3, p < 0.0001, ηp2 = 0.57). Mirroring the behavioral data, there was a significant Response × COMT × Substance interaction (F(1,165) = 6.8, p = 0.01, ηp2 = 0.04; Fig. 3). Analogous to the behavioral data, we followed up on this three-way interaction by first calculating difference scores indicating the error-specific net effect (amplitude error minus amplitude correct) and then testing these difference scores separately for both substance conditions. In the placebo group, Val+ carriers showed significantly higher difference scores than Met/Met carriers (t(78) = 1.98, p = 0.026, one-tailed). Analysis of the raw amplitudes revealed that this effect was driven by IC-amplitudes after error-trials (t(78) = 1.8, p = 0.04, one-tailed; Fig. 3) rather than correct responses t(78) = 0.22, p > 0.8; Table 1). Similar to the behavioral data, sulpiride increased the difference score in Met/Met carriers (t(48) = 1.99, p = 0.03, one-tailed) but decreased the difference score in Val+ carriers (t(117) = 1.84, p = 0.04, one-tailed).
Independent component event-related spectral perturbations
Error responses yielded a significant increase in anterior midcingulate cortex cluster delta and theta power from −100 to 300 ms relative to the button press (main effect Response: F(1,165) = 183.01, p < 0.001, ηp2 = 0.53), which was significantly correlated with the IC-ERN amplitude (r(169)= −0.62, p < 0.001). Consistent with the behavioral and the IC-ERP data, the error-related increase of delta and theta was modulated by a significant Response × COMT × Substance interaction (F(1,165) = 4.91, p = 0.03, ηp2 = 0.03; Fig. 3), indicating that error-related power increases were greater in Val+ versus Met/Met carriers after placebo (t(78) = 2.1, p = 0.02, one-tailed) but not after sulpiride (p > 0.2), and that sulpiride reduced error-related delta/theta in Val+ but not Met/Met carriers (t(117) = 2.0, p = 0.02, one-tailed).
Independent component intertrial coherence
Errors versus correct responses also yielded an increase in intertrial coherence in the delta and theta bands (F(1,165) = 23.18, p < 0.001, ηp2 = 0.12), which was correlated with IC-ERN amplitude (r(169)= −0.57, p < 0.001) but unaffected by COMT and Substance (p > 0.5). In addition, there was a significant Substance × Frequency interaction (F(1,165) = 8.0, p = 0.003, ηp2 = 0.5; Fig. 3), indicating that sulpiride reduced intertrial coherence in the delta but not theta range, regardless of Response.
Scalp-channel event-related potentials
Following the same pattern as PES and IC activation, the Response × COMT × Substance interaction on the ERN-amplitude at channel Fz was marginally significant (F(1,165) = 3.2, p = 0.07, ηp2 = 0.02). With regard to difference scores (amplitude error minus amplitude correct), sulpiride induced a decrease in scalp-channel ERN in Val carriers (t(117) = 1.85, p = 0.03, one-tailed) and a nonsignificant increase of amplitude in Met/Met homozygotes (p > 0.15; Table 1). The reduced effect sizes in scalp-channel versus IC data are consistent with lower signal-to-noise ratio in the former (Makeig et al., 2004).
Behavioral and EEG data
To test whether individual differences in IC-ERN and PES were related, we correlated IC activation peaks and reaction time slowing separately for error and correct trials. IC-activation after errors (i.e., IC-ERN) showed a significant correlation with PES (r(169) = 0.16, p = 0.02, one-tailed) but not with slowing after RT-matched correct responses (p > 0.3). Moreover, IC-activation after correct responses correlated neither with PES nor with slowing after RT-matched correct responses (all ps > 0.15), indicating an association between anterior midcingulate cortex IC-activation and reaction time slowing that is specific to error responses.
Epistasis effects with DRD2TaqIa and 5-HTTLPR
To test for epistasis effects, we separately included DRD2TaqIa and 5-HTTLPR into the previously reported ANOVAs (for the distributions of DRD2TaqIa and 5-HTTLPR over the COMT Val158Met genotypes, see Table 1). There were no significant main effects or interactions on PES, IC-ERN, IC-delta/theta, or scalp-channel ERN that involved the DRD2TaqIa polymorphism. However, when 5-HTTLPR was included, there was a COMT × 5-HTTLPR interaction on reaction time slowing independent of Response and Substance (F(1,160) = 4.52, p = 0.04, ηp2 = 0.03), indicating that, among carriers with the less functional 5-HTTLPR alleles (LG and S), Val carriers slowed more (F(1,114) = 8.01, p < 0.006), while this effect was absent in LA homozygotes (p > 0.5). Importantly, a significant Response × COMT × 5-HTTLPR × Substance interaction on IC-ERN (F(1,160) = 3.18, p = 0.05, ηp2 = 0.02) indicated that the previously reported Response × COMT × Substance interaction on error-related IC activation was potentiated in carriers of the less functional 5-HTTLPR variant (F(1,114) = 8.94, p < 0.005, ηp2 = 0.07) but absent in LA homozygotes (p > 0.8; Fig. 4). No interactions involving 5-HTTLPR emerged in a Response × COMT × 5-HTTLPR × Substance ANOVA on IC delta/theta power.
Figure 4.

Interactions of Substance, COMT, and 5-HTTLPR on peak IC cluster event-related potentials. Bar plots indicating means (and SEMs) of peak IC cluster event-related potentials following errors in the placebo (white) or sulpiride (gray) groups.
Discussion
We have shown that a dopaminergic genotype and a dopaminergic drug interact on human error processing, previously linked to DA. Although prior studies have investigated the effect of DA genotypes (for review, see Ullsperger, 2010) or pharmacological challenges (for review, see Jocham and Ullsperger, 2009) alone, this is the first study, to the best of our knowledge, that combines the two approaches to investigate the involvement of DA in error processing. Based on (1) the dual-state theory of PFC DA functioning, claiming that low and high DA levels promote D2 states associated with modulations of neural activation patterns (Durstewitz and Seamans, 2008), and because (2) it has been suggested that PFC DA plays a substantial role in error processing (Holroyd and Coles, 2002), we hypothesized that the DA-related COMT Val158Met polymorphism and intake of a selective D2 blocker would interact such that individuals with presumably low or high (vs medium) DA levels show elevated indices of error-processing. We tested our hypothesis in 169 healthy male participants, who were genotyped for COMT Val158Met and who received a DA D2 receptor blocker or placebo and then performed a Flanker task while EEG was recorded. Val carriers who had taken placebo and Met homozygotes who had received sulpiride showed relatively high PES and error-related brain activity compared with Val+ carriers under sulpiride and Met/Met carriers under placebo. This pattern of results provides strong support for a role of DA and the COMT Val158Met polymorphism on individual differences in ERN amplitude, error-related delta/theta power, and PES, and suggests a U-shaped relationship between error reactivity and DA level.
In individuals who had received placebo, the IC-ERN amplitude and error-related increases in IC delta/theta power were significantly smaller in Met/Met versus Val+ carriers. This was mirrored by the behavioral data, where—after placebo—Met/Met carriers showed significantly less PES than Val+ carriers. Because Val+ carriers presumably show reduced DA levels and may thus occupy D2 rather than D1 states (Durstewitz and Seamans, 2008), these findings are consistent with our hypothesis that D2 states facilitate modulations of PFC networks driven by error responses.
Because 200 mg of sulpiride presumably increase PFC dopamine availability by blocking presynaptic D2-receptors (Mereu et al., 1983; Kuroki et al., 1999), we hypothesized that sulpiride would increase error reactivity in Met/Met carriers by pushing them from medium toward high DA levels and thereby into D2 states, and decrease error reactivity in Val+ carriers by pushing them from low toward medium DA levels and thereby into D1 states. Sulpiride significantly interacted with COMT on ERN, error-related delta/theta power, and PES in the expected directions. Sulpiride significantly reduced PES, ERN amplitude, and error-related IC delta/theta power in Val+ carriers but increased ERN-amplitude and tended to increase PES in Met/Met carriers. Together, this pattern of findings suggests a U-shaped association between PFC DA availability and error processing. Similar nonlinear associations have been found with regard to DA availability and other brain processes (Goldman-Rakic et al., 2000; Tunbridge et al., 2006; Durstewitz and Seamans, 2008). With regard to the processing of infrequently occurring response errors, a possible mechanism for these effects involves D1- versus D2-dominated states in PFC, as outlined in the Introduction, which may affect reactivity to erroneous events (Fig. 1) by influencing the variability of PFC neural activation patterns and behavior. Val carriers after sulpiride and Met/Met carriers after placebo were less variable and more rigid in their task performance and continued in their mode of behavior despite contrary evidence, which is consistent with reduced set shifting abilities in D1 receptor-dominated regimes (Durstewitz and Seamans, 2008) and reduced neuronal trial to trial variability in unmedicated Met/Met carriers (Winterer and Weinberger, 2004).
While herein we hypothesize that DA level serves to modulate the reactivity of PFC networks to errors, the ERN may also be directly generated by dips in phasic mesocortical DA release, which disinhibit midcingulate cortex pyramidal cell neurons (Holroyd and Coles, 2002). The present findings within the placebo groups are consistent with this latter assumption because elevated DA levels in mPFC associated with the Met allele would presumably increase the threshold for ERN generation and thus reduce overall ERN amplitude. However, due to the apparent contradiction between relatively low DA neuron spontaneous firing rate and the temporal precision needed for ERN generation, it has recently been speculated that the ERN is more directly linked to glutamatergic or GABAergic neurotransmission, whereas DA modulates the background of error-related cortical excitability (Lapish et al., 2007; Ullsperger, 2010). The application of the dual-state model to error reactivity presented here is consistent with this latter view. Among others, the stimulation of D1 receptors increases NMDA-dependent currents—boosting recurrent excitation of active cell assemblies—-and GABAA currents, and may thereby reduce the activation probability of new cell assemblies (Zheng et al., 1999; Seamans et al., 2001; Durstewitz and Seamans, 2008). In contrast, the stimulation of D2 receptors—presumably facilitated at relatively low or high DA levels—increases the activation probability of new cell assemblies by downregulating NMDA and inhibitory GABAA currents (Durstewitz and Seamans, 2008). D2 receptor activation could thereby facilitate the transition to error-related neural activation patterns (Zirnheld et al., 2004; Frank and Hutchison, 2009), characterized by increased mediofrontal theta power (Cavanagh et al., 2009) and error-related negativity predictive of posterror slowing (Debener et al., 2005).
Although we interpret our findings within the dual-state model with a focus on the PFC, COMT and sulpiride may also influence error processing at subcortical structures. For example, striatopallidal neurons, which are disinhibited via D2 receptors upon cessation of DA firing, have been related to error processing (Frank et al., 2007). Increases in PFC DA level associated with the Met allele may indirectly downregulate phasic BG DA release (Bilder et al., 2004). Moreover, presynaptic and postsynaptic effects of sulpiride may modulate striatal error reactivity by elevating DA levels or blocking striatopallidal D2 receptors (Kuroki et al., 1999; Frank, 2005), respectively. To better understand the role of the BG in error processing (Ullsperger and von Cramon, 2006), future studies, including pharmacological challenges in combination with neuroimaging methods, could probe how well presynaptic and/or postsynaptic effects at the BG can explain the present results.
Consistent with recent calls to acknowledge the 5-HT system when investigating the effects of dopaminergic drugs (de Almeida et al., 2008), we further found that the COMT × Substance interaction was potentiated in carriers of the less functional 5-HTTLPR variant, which is linked to reduced 5-HT uptake (Lesch et al., 1996). The 5-HT system is a relevant modulator of dopaminergic processes and influences mPFC DA release induced by D2 receptor antagonists (Kuroki et al., 1999). Glutamatergic pyramidal neurons presumably involved in ERN generation (Ullsperger, 2010) receive inputs not only from dopaminergic but also from serotoninergic fibers and GABAergic interneurons, which receive projections from raphe nuclei serotoninergic neurons (de Almeida et al., 2008). Of relevance, the less functional 5-HTTLPR alleles have previously been linked to increased risk for depression after stressful life events (Caspi et al., 2003), which also affect the DA system (Pruessner et al., 2004), and for schizophrenia when individuals also carry the Val allele of the COMT Val158Met polymorphism (Borroni et al., 2006). Epistasis effects involving 5-HT and DA should therefore be targeted in future studies of negative affect/depression and schizophrenia that have been consistently linked to abnormal error processing (Alain et al., 2002; Holmes and Pizzagalli, 2008; Olvet and Hajcak, 2008).
Finally, the limitations of the present study should be acknowledged. Because dopaminergic polymorphisms have previously been shown to have sexually dimorphic effects (Stein et al., 2005; Wacker et al., 2005), we constrained our sample to male participants. In addition, participants were recruited from a not previously genotyped sample, resulting in heterogeneous cell sizes for some analyses (particularly those involving 5-HTTLPR or DRD2 in addition to COMT). Future studies investigating men and women who were preselected based on genotypes would allow further generalization of the present findings. Finally, for pragmatic reasons (i.e., safety, statistical power, conductibility), we only investigated the effect of 200 mg of sulpiride compared with placebo. Future studies including different doses and/or D2 agonists may help to verify and/or modify the interpretations we derived from our findings.
Despite these limitations, the present study clearly shows a link between dopaminergic genes, a dopaminergic challenge, and error processing, thereby extending prior work using smaller samples and either genetic or pharmacological approaches alone in important ways (for review, see Jocham and Ullsperger, 2009; Ullsperger, 2010). The present findings suggest (1) that a significant effect of COMT Val158Met on error processing can be demonstrated using a sufficiently large sample and ICA-decomposed EEG (Makeig et al., 2002), and (2) that modulations of midcingulate ERN amplitudes and increases in delta/theta power after administration of D2 antagonists (Zirnheld et al., 2004) depend on COMT Val158Met genotype. Moreover, the present study provides initial evidence for a modulating involvement of 5-HT in dopaminergic mechanisms of error processing, opening up novel research perspectives.
Footnotes
The work was supported by Deutsche Forschungsgemeinschaft Grants WA 2593/2-1 and WA 2593/2-2, and a Society for Psychophysiological Research Fellowship Training Award to E.M.M.
The authors declare no financial conflicts of interests.
References
- Alain C, McNeely HE, He Y, Christensen BK, West R. Neurophysiological evidence of error-monitoring deficits in patients with schizophrenia. Cereb Cortex. 2002;12:840–846. doi: 10.1093/cercor/12.8.840. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Volavka J, Lachman HM, Grace AA. The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology. 2004;29:1943–1961. doi: 10.1038/sj.npp.1300542. [DOI] [PubMed] [Google Scholar]
- Borroni B, Grassi M, Agosti C, Archetti S, Costanzi C, Cornali C, Caltagirone C, Caimi L, Di Luca M, Padovani A. Cumulative effect of COMT and 5-HTTLPR polymorphisms and their interaction with disease severity and comorbidities on the risk of psychosis in Alzheimer disease. Am J Geriatr Psychiatry. 2006;14:343–351. doi: 10.1097/01.JGP.0000192491.50802.c3. [DOI] [PubMed] [Google Scholar]
- Botvinick M, Nystrom LE, Fissell K, Carter CS, Cohen JD. Conflict monitoring versus selection-for-action in anterior cingulate cortex. Nature. 1999;402:179–181. doi: 10.1038/46035. [DOI] [PubMed] [Google Scholar]
- Botvinick MM, Braver TS, Barch DM, Carter CS, Cohen JD. Conflict monitoring and cognitive control. Psychol Rev. 2001;108:624–652. doi: 10.1037/0033-295x.108.3.624. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Cavanagh JF, Cohen MX, Allen JJ. Prelude to and resolution of an error: EEG phase synchrony reveals cognitive control dynamics during action monitoring. J Neurosci. 2009;29:98–105. doi: 10.1523/JNEUROSCI.4137-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida J, Palacios JM, Mengod G. Distribution of 5-HT and DA receptors in primate prefrontal cortex: implications for pathophysiology and treatment. Prog Brain Res. 2008;172:101–115. doi: 10.1016/S0079-6123(08)00905-9. [DOI] [PubMed] [Google Scholar]
- Debener S, Ullsperger M, Siegel M, Fiehler K, von Cramon DY, Engel AK. Trial-by-trial coupling of concurrent electroencephalogram and functional magnetic resonance imaging identifies the dynamics of performance monitoring. J Neurosci. 2005;25:11730–11737. doi: 10.1523/JNEUROSCI.3286-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK. The dual-state theory of prefrontal cortex dopamine function with relevance to catechol-o-methyltransferase genotypes and schizophrenia. Biol Psychiatry. 2008;64:739–749. doi: 10.1016/j.biopsych.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Erceg-Hurn DM, Mirosevich VM. Modern robust statistical methods: an easy way to maximize the accuracy and power of your research. Am Psychol. 2008;63:591–601. doi: 10.1037/0003-066X.63.7.591. [DOI] [PubMed] [Google Scholar]
- Fallgatter AJ, Herrmann MJ, Roemmler J, Ehlis AC, Wagener A, Heidrich A, Ortega G, Zeng Y, Lesch KP. Allelic variation of serotonin transporter function modulates the brain electrical response for error processing. Neuropsychopharmacology. 2004;29:1506–1511. doi: 10.1038/sj.npp.1300409. [DOI] [PubMed] [Google Scholar]
- Fink KB, Göthert M. 5-HT receptor regulation of neurotransmitter release. Pharmacol Rev. 2007;59:360–417. doi: 10.1124/pr.107.07103. [DOI] [PubMed] [Google Scholar]
- Frank MJ. Dynamic dopamine modulation in the basal ganglia: a neurocomputational account of cognitive deficits in medicated and nonmedicated Parkinsonism. J Cogn Neurosci. 2005;17:51–72. doi: 10.1162/0898929052880093. [DOI] [PubMed] [Google Scholar]
- Frank MJ, Hutchison K. Genetic contributions to avoidance-based decisions: striatal D2 receptor polymorphisms. Neuroscience. 2009;164:131–140. doi: 10.1016/j.neuroscience.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ, D'Lauro C, Curran T. Cross-task individual differences in error processing: neural, electrophysiological, and genetic components. Cogn Affect Behav Neurosci. 2007;7:297–308. doi: 10.3758/cabn.7.4.297. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Knight RT. Prefrontal-cingulate interactions in action monitoring. Nat Neurosci. 2000;3:516–520. doi: 10.1038/74899. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Muly EC, 3rd, Williams GV. D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev. 2000;31:295–301. doi: 10.1016/s0165-0173(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Gramann K, Gwin JT, Bigdely-Shamlo N, Ferris DP, Makeig S. Visual evoked responses during standing and walking. Front Hum Neurosci. 2010;4:202. doi: 10.3389/fnhum.2010.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton G, Coles MG, Donchin E. Optimizing the use of information: strategic control of activation of responses. J Exp Psychol Gen. 1992;121:480–506. doi: 10.1037//0096-3445.121.4.480. [DOI] [PubMed] [Google Scholar]
- Hester R, Barre N, Murphy K, Silk TJ, Mattingley JB. Human medial frontal cortex activity predicts learning from errors. Cereb Cortex. 2008;18:1933–1940. doi: 10.1093/cercor/bhm219. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Isoda M. Switching from automatic to controlled behavior: cortico-basal ganglia mechanisms. Trends Cogn Sci. 2010;14:154–161. doi: 10.1016/j.tics.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AJ, Pizzagalli DA. Spatiotemporal dynamics of error processing dysfunctions in major depressive disorder. Arch Gen Psychiatry. 2008;65:179–188. doi: 10.1001/archgenpsychiatry.2007.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyd CB, Coles MG. The neural basis of human error processing: reinforcement learning, dopamine, and the error-related negativity. Psychol Rev. 2002;109:679–709. doi: 10.1037/0033-295X.109.4.679. [DOI] [PubMed] [Google Scholar]
- Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD, Xu K, Arnold PD, Richter MA, Kennedy JL, Murphy DL, Goldman D. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006;78:815–826. doi: 10.1086/503850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jocham G, Ullsperger M. Neuropharmacology of performance monitoring. Neurosci Biobehav Rev. 2009;33:48–60. doi: 10.1016/j.neubiorev.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Kuroki T, Meltzer HY, Ichikawa J. Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther. 1999;288:774–781. [PubMed] [Google Scholar]
- Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM. Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. 1996;6:243–250. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- Lapish CC, Kroener S, Durstewitz D, Lavin A, Seamans JK. The ability of the mesocortical dopamine system to operate in distinct temporal modes. Psychopharmacology (Berl) 2007;191:609–625. doi: 10.1007/s00213-006-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Müller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Luu P, Tucker DM, Makeig S. Frontal midline theta and the error-related negativity: neurophysiological mechanisms of action regulation. Clin Neurophysiol. 2004;115:1821–1835. doi: 10.1016/j.clinph.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Makeig S. Auditory event-related dynamics of the EEG spectrum and effects of exposure to tones. Electroencephalogr Clin Neurophysiol. 1993;86:283–293. doi: 10.1016/0013-4694(93)90110-h. [DOI] [PubMed] [Google Scholar]
- Makeig S, Westerfield M, Jung TP, Enghoff S, Townsend J, Courchesne E, Sejnowski TJ. Dynamic brain sources of visual evoked responses. Science. 2002;295:690–694. doi: 10.1126/science.1066168. [DOI] [PubMed] [Google Scholar]
- Makeig S, Debener S, Onton J, Delorme A. Mining event-related brain dynamics. Trends Cogn Sci. 2004;8:204–210. doi: 10.1016/j.tics.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Margraf J. Mini-DIPS: Diagnostisches Kurz-Interview bei Psychischen Störungen. Berlin: Springer; 1994. [Google Scholar]
- Mauri MC, Bravin S, Bitetto A, Rudelli R, Invernizzi G. A risk-benefit assessment of sulpiride in the treatment of schizophrenia. Drug Saf. 1996;14:288–298. doi: 10.2165/00002018-199614050-00003. [DOI] [PubMed] [Google Scholar]
- Mereu G, Casu M, Gessa GL. (-)-Sulpiride activates the firing rate and tyrosine hydroxylase activity of dopaminergic neurons in unanesthetized rats. Brain Res. 1983;264:105–110. doi: 10.1016/0006-8993(83)91125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Ueno S, Sano A, Tanabe H. The human serotonin transporter gene linked polymorphism (5-HTTLPR) shows ten novel allelic variants. Mol Psychiatry. 2000;5:32–38. doi: 10.1038/sj.mp.4000698. [DOI] [PubMed] [Google Scholar]
- Olvet DM, Hajcak G. The error-related negativity (ERN) and psychopathology: toward an endophenotype. Clin Psychol Rev. 2008;28:1343–1354. doi: 10.1016/j.cpr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenveld R, Oostendorp TF. Validating the boundary element method for forward and inverse EEG computations in the presence of a hole in the skull. Hum Brain Mapp. 2002;17:179–192. doi: 10.1002/hbm.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osinsky R, Reuter M, Küpper Y, Schmitz A, Kozyra E, Alexander N, Hennig J. Variation in the serotonin transporter gene modulates selective attention to threat. Emotion. 2008;8:584–588. doi: 10.1037/a0012826. [DOI] [PubMed] [Google Scholar]
- Palmer JA, Kreutz-Delgado K, Makeig S. Super-Gaussian mixture source model for ICA. In: Rosca J, Erdogmus D, Principe JC, Haykin S, editors. Lecture notes in computer science. Heidelberg: Springer; 2006. pp. 854–861. [Google Scholar]
- Palmer JA, Makeig S, Delgado KK, Rao BD. Newton method for the ICA mixture model. IEEE International Conference on Acoustics, Speech and Signal Processing, 2008 ICASSP 2008 IEEE; Las Vegas, Nevada. 2008. pp. 1805–1808. [Google Scholar]
- Pohjalainen T, Rinne JO, Någren K, Lehikoinen P, Anttila K, Syvälahti EK, Hietala J. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry. 1998;3:256–260. doi: 10.1038/sj.mp.4000350. [DOI] [PubMed] [Google Scholar]
- Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci. 2004;24:2825–2831. doi: 10.1523/JNEUROSCI.3422-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbitt PM, Phillips S. Error-detection and correction latencies as a function of S-R compatibility. Q J Exp Psychol. 1967;19:37–42. doi: 10.1080/14640746708400065. [DOI] [PubMed] [Google Scholar]
- Reuter M, Hennig J. Association of the functional catechol-O-methyltransferase VAL158MET polymorphism with the personality trait of extraversion. Neuroreport. 2005;16:1135–1138. doi: 10.1097/00001756-200507130-00020. [DOI] [PubMed] [Google Scholar]
- Reuter M, Schmitz A, Corr P, Hennig J. Molecular genetics support Gray's personality theory: the interaction of COMT and DRD2 polymorphisms predicts the behavioural approach system. Int J Neuropsychopharmacol. 2006;9:155–166. doi: 10.1017/S1461145705005419. [DOI] [PubMed] [Google Scholar]
- Ridderinkhof KR, de Vlugt Y, Bramlage A, Spaan M, Elton M, Snel J, Band GP. Alcohol consumption impairs detection of performance errors in mediofrontal cortex. Science. 2002;298:2209–2211. doi: 10.1126/science.1076929. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima K, Tanji J. Role for cingulate motor area cells in voluntary movement selection based on reward. Science. 1998;282:1335–1338. doi: 10.1126/science.282.5392.1335. [DOI] [PubMed] [Google Scholar]
- Stein MB, Fallin MD, Schork NJ, Gelernter J. COMT polymorphisms and anxiety-related personality traits. Neuropsychopharmacology. 2005;30:2092–2102. doi: 10.1038/sj.npp.1300787. [DOI] [PubMed] [Google Scholar]
- Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60:141–151. doi: 10.1016/j.biopsych.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Ullsperger M. Genetic association studies of performance monitoring and learning from feedback: the role of dopamine and serotonin. Neurosci Biobehav Rev. 2010;34:649–659. doi: 10.1016/j.neubiorev.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Ullsperger M, von Cramon DY. The role of intact frontostriatal circuits in error processing. J Cogn Neurosci. 2006;18:651–664. doi: 10.1162/jocn.2006.18.4.651. [DOI] [PubMed] [Google Scholar]
- Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci. 2005;6:533–544. doi: 10.1038/nrn1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker J, Reuter M, Hennig J, Stemmler G. Sexually dimorphic link between dopamine D2 receptor gene and neuroticism-anxiety. Neuroreport. 2005;16:611–614. doi: 10.1097/00001756-200504250-00019. [DOI] [PubMed] [Google Scholar]
- Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. 2004;27:683–690. doi: 10.1016/j.tins.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Yasuda A, Sato A, Miyawaki K, Kumano H, Kuboki T. Error-related negativity reflects detection of negative reward prediction error. Neuroreport. 2004;15:2561–2565. doi: 10.1097/00001756-200411150-00027. [DOI] [PubMed] [Google Scholar]
- Yordanova J, Falkenstein M, Hohnsbein J, Kolev V. Parallel systems of error processing in the brain. Neuroimage. 2004;22:590–602. doi: 10.1016/j.neuroimage.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee ML, Xiao T, Papp A, Wang D, Sadée W. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci U S A. 2007;104:20552–20557. doi: 10.1073/pnas.0707106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Zhang XX, Bunney BS, Shi WX. Opposite modulation of cortical N-methyl-D-aspartate receptor-mediated responses by low and high concentrations of dopamine. Neuroscience. 1999;91:527–535. doi: 10.1016/s0306-4522(98)00604-6. [DOI] [PubMed] [Google Scholar]
- Zirnheld PJ, Carroll CA, Kieffaber PD, O'Donnell BF, Shekhar A, Hetrick WP. Haloperidol impairs learning and error-related negativity in humans. J Cogn Neurosci. 2004;16:1098–1112. doi: 10.1162/0898929041502779. [DOI] [PubMed] [Google Scholar]