

Abstract
Botulinum neurotoxin type A (BoNT/A) is a metalloprotease that blocks synaptic transmission via the cleavage of SNAP-25 (synaptosomal-associated protein of 25 kDa). BoNT/A is successfully used in clinical neurology for the treatment of several neuromuscular pathologies and pain syndromes. Despite its widespread use, relatively little is known on BoNT/A intracellular trafficking in neurons. Using the visual pathway as a model system, here we show that catalytically active BoNT/A is capable of undergoing anterograde axonal transport and transcytosis. Following BoNT/A injection into the rat eye, significant levels of BoNT/A-cleaved SNAP-25 appeared in the retinorecipient layers of the superior colliculus (SC). Anterograde propagation of BoNT/A effects required axonal transport, ruling out a systemic spread of the toxin. Cleaved SNAP-25 was present in presynaptic structures of the tectum, but retinal terminals were devoid of the immunoreactivity, indicative of transcytosis. Experiments based on sequential administration of BoNT/A and BoNT/E showed a persistent catalytic activity of BoNT/A in tectal cells following its injection into the retina. Our findings demonstrate that catalytically active BoNT/A is anterogradely transported from the eye to the SC and transcytosed to tectal synapses. These data are important for a more complete understanding of the mechanisms of action of BoNT/A.
Introduction
Botulinum neurotoxins (BoNTs, seven serotypes, from A to G) and tetanus neurotoxin (TeNT) form the clostridial neurotoxin family and are the most potent poisons known (Johnson, 1999; Rossetto et al., 2006). They bind with high affinity to neuronal membranes and block neurotransmission via the specific cleavage of synaptic SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment receptor) proteins (Schiavo et al., 2000; Rossetto et al., 2001; Turton et al., 2002; Sudhof, 2004; Binz and Rummel, 2009). These toxins are composed of a 100 kDa heavy chain (H chain) and a 50 kDa light chain (L chain) linked by a single disulfide bond. The L chain is endowed with an endopeptidase activity specific for members of the SNARE family (synaptobrevin, syntaxin and SNAP-25—synaptosomal-associated protein of 25 kDa). In particular, BoNT/A and BoNT/E cleave SNAP-25, removing 9 and 26 residues, respectively (Schiavo et al., 2000). These two serotypes differ markedly in duration of action. Intoxication by BoNT/E is short-lived (days), while the neuroparalytic effects of BoNT/A persist for months in humans and weeks in mice (Eleopra et al., 1998; Adler et al., 2001; Meunier et al., 2003; Morbiato et al., 2007).
In the laboratory setting, clostridial neurotoxins are excellent tools for studying cellular physiology and SNARE-mediated membrane fusion. In the clinic, the long-lasting effects of BoNT/A at peripheral nerve terminals have been exploited for the treatment of several neuromuscular and autonomic disorders, such as dystonia, spasticity, and hyperhidrosis (Johnson, 1999; Davletov et al., 2005; Montecucco and Molgo, 2005; Naumann et al., 2008; Simpson et al., 2008a,b). BoNT/A has proven very effective in the treatment of these conditions, and the range of possible clinical applications of BoNT/A is continuously increasing (Evidente and Adler, 2010). Of particular interest, the action of BoNT/A on nociceptive neurons has prompted its use in the management of different types of pain (Casale and Tugnoli, 2008; Pavone and Luvisetto, 2010), and this is currently an area of very active investigation.
Despite the widespread use of BoNT/A, much remains to be understood on the mechanisms involved in binding, sorting and intracellular trafficking of BoNT/A in neurons. Uptake of BoNT/A at the nerve terminal occurs via synaptic vesicle endocytosis, followed by translocation of the L chain into the neuronal cytosol and cleavage of the target substrates (Binz and Rummel, 2009). While most of BoNT/A remains at the injection site, there is experimental evidence in animal models that this toxin can undergo retrograde axonal transport and transcytosis, particularly when high doses are used (Habermann, 1974; Wiegand et al., 1976; Moreno-López et al., 1997; Antonucci et al., 2008b). Transcytosis refers to the process by which a ligand penetrates the neuron at one side, followed by its movement and release at the opposite end, with possible uptake by second-order neurons. This cell-to-cell trafficking is important because it may allow the toxin to exert its actions at a distance from the injection site. Here, we have examined anterograde trafficking and transcytosis of BoNT/A in the rat retinotectal pathway, as this system offers several advantages. In fact, retinal ganglion cells are amenable to surgical manipulation in vivo, and anterograde transport can be precisely quantified (von Bartheld et al., 2001; Caleo and Cenni, 2004; von Bartheld, 2004). We report that catalytically active BoNT/A does undergo anterograde transport and transcytosis in neurons.
Materials and Methods
Adult Long–Evans rats bred in our animal facility were used in this study. The animals were kept on a 12 h light/dark cycle and had access to food and water ad libitum. All experiments were performed in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC) and were approved by the Ministry of Health. A total of 90 animals were used.
BoNT/A and BoNT/E were prepared and tested as described previously (Schiavo and Montecucco, 1995; Costantin et al., 2005; Caleo et al., 2007; Antonucci et al., 2008b).
Intraocular injections.
BoNT/A was slowly injected into the vitreous (1–3 nm, 0.4 μl) via a glass micropipette inserted at the ora serrata (Caleo et al., 1999). Similarly, CTB (cholera toxin β subunit; 1%, 2.5 μl; Sigma) or the axonal transport blocker colchicine (10 μg; Sigma) were injected in some experiments (Antonucci et al., 2008b). Colchicine was injected 16–18 h before BoNT/A. The optic nerve was sectioned using a retroorbital approach (Antonucci et al., 2008b).
BoNT/E and kainic acid injections.
BoNT/E and kainic acid (KA) injections into the SC were performed by a glass micropipette connected to an injector. Injections were performed at the following coordinates (in mm with respect to lambda): anteroposterior, −0.5; ML, 1; 2.8 below dura (Tropea et al., 2003). BoNT/E (50 nm, 0.5 μl) was injected into the SC 3 d after intravitreal BoNT/A delivery in 10 rats. KA (1 nmol in 100 nl of PBS; Sigma; Antonucci et al., 2008a) was injected 1 d before BoNT/A (6 rats).
Immunoblotting.
Immunoblotting was performed as described previously (Caleo et al., 2007; Antonucci et al., 2008b). Proteins were extracted with lysis buffer (1% Triton X-100, 10% glycerol, 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm EDTA, 0.1 mm Na3VO4, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mm PMSF), and the total concentration of the samples was assessed with a protein assay kit (Bio-Rad) using a bovine serum albumin-based standard curve. Protein extracts were separated by electrophoresis and blotted, and filters were incubated with primary antibodies overnight at 4°C (anti-cleaved SNAP-25, 1:500 dilution, characterized in the work of Antonucci et al., 2008b; anti-vGlut-2, 1:1000, Millipore). Blots were then reacted with HRP-conjugated secondary antibodies (Jackson ImmunoResearch) and developed by ECL (GE Healthcare). Filters were also probed with anti-tubulin antibody (monoclonal, 1:15,000 dilution, Sigma; polyclonal, 1:10,000 dilution, Abcam), which serves as an internal standard for protein quantification. Total proteins loaded were 10 μg for retinal extracts, 10 μg for collicular extracts to detect vGlut-2, 100 μg for collicular extracts to detect cleaved SNAP-25. In Western blot experiments for Figures 6 and 8 (see below), we loaded 20 μg of total proteins per lane. Quantification of Western blot experiments was performed as described previously (Caleo et al., 2007; Antonucci et al., 2008b; Mainardi et al., 2010).
Figure 6.

Persistence of cleaved SNAP-25 in the tectum of animals with optic nerve section. Animals were injected intraocularly with BoNT/A. After 3 d, a group of animals was subjected to optic nerve section (ON cut, n = 5), while the remaining rats were left unoperated (CTR, n = 6). After another 7 d, tissues were dissected and Western blot was performed. A, B, Immunoblotting for cleaved SNAP-25 on protein extracts from the SC contralateral to BoNT/A injection. Quantification demonstrated that expression of cleaved SNAP-25 in the SC does not decrease following RGC fiber degeneration. Levels of BoNT/A-truncated SNAP-25 are normalized to those measured in animals without ON cut. C, D, Immunoblotting for vGlut-2 on protein extracts from the SC contralateral to BoNT/A injection. vGlut-2 is specific for RGC terminals and is used as a control for loss of retinal innervation. Quantification demonstrated that expression of vGlut-2 in the SC consistently decreases following ON section. Levels of vGlut-2 are normalized to those measured in animals without ON cut. α-Tubulin, internal standard. Data are mean ± SE. ON cut, n = 5; CTR, n = 6. *p < 0.05.
Figure 8.

Transcytosis of catalytically active BoNT/A. A, Experimental protocol. BoNT/A was injected intraocularly. After 3 d, tectal samples were dissected from a subset of animals, while the other animals were subjected to optic nerve section (ON cut) and BoNT/E injection into the colliculus. Colliculi were taken either 2 or 23 d following BoNT/E injection. B, Immunoblotting for BoNT/A-truncated SNAP-25 on protein extracts from the SC contralateral to the injected retina. BoNT/A-truncated SNAP-25 is clearly detectable after 3 d, disappears 2 d following BoNT/E injection (3d + 2d BoNT/E), and reemerges when BoNT/E action is off, i.e., ∼23 d following BoNT/E injection (3d + 23d BoNT/E). α-Tubulin, internal standard. C, Quantification of the immunoblotting experiments. BoNT/A-truncated SNAP-25 shows modest levels at 3 d (3d), decreases 2 d after collicular injection of BoNT/E (3d + 2d BoNT/E), and then shows significantly higher levels 23 d after BoNT/E (3d + 23d BoNT/E). Levels of cleaved SNAP-25 are normalized to those measured 3 d after intraocular injection. This experiment demonstrates anterograde transfer and persistence of catalytically active BoNT/A. Data are mean ± SE; 3d, n = 5 rats; 3d + 2d BoNT/E, n = 5; 3d + 23d BoNT/E, n = 5. *p < 0.05; **p < 0.01; ***p = 0.001.
Immunohistochemistry.
Animals were deeply anesthetized with chloral hydrate and perfused through the heart with freshly prepared 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Tissues were dissected and postfixed for 2 h at 4°C. Brain sections (40 μm thick) were cut with a freezing microtome. Eye cups and optic nerves were embedded in OCT compound (Leica Microsystems). Retinal vertical sections (20 μm thick) and longitudinal nerve sections (16 μm thick; Caleo et al., 2000; Mandolesi et al., 2005) were cut with a cryostat. For immunostaining, sections were blocked with 10% normal horse serum in PBS containing 0.25% Triton X-100 and then incubated overnight at room temperature with the anti-cleaved SNAP-25 antibody (1:500 dilution). On the following day, sections were incubated with Alexa Fluor 568-conjugated secondary antibody (1:500; Invitrogen) for 2 h at room temperature. Sections were then washed in PBS and mounted with antifading agent (Vectashield; Vector Laboratories). In double-labeling for cleaved SNAP-25 and presynaptic markers, collicular sections were incubated as described previously (see above), with anti-cleaved SNAP-25 and, respectively, mouse monoclonal anti-synapsin (1:200, Synaptic Systems), mouse monoclonal anti-vGlut-1 (1:100, Synaptic Systems), guinea pig anti-vGAT (anti-vesicular GABA transporter; 1:300, Synaptic Systems), mouse monoclonal anti-vGlut-2 (1:1000, Millipore). In the experiments in which CTB (Sigma) was injected, double-labeling between CTB and, respectively, cleaved SNAP-25 and intact SNAP-25 were made by incubating tectal sections with anti-CTB (1:4000, Merck KGaA) and anti-cleaved SNAP-25 or anti-intact SNAP-25 (1:1000, Sternberger Monoclonals). For anti-intact SNAP-25, sections were postfixed in methanol for 5 min at −20°C before the immunohistochemical process. NeuN immunohistochemistry was performed with a mouse monoclonal antibody (1: 500, Millipore). Glial markers were revealed with mouse monoclonal anti-glial fibrillary acidic protein (1:500, Sigma) and mouse monoclonal OX-42 antibody (1:500; BD Biosciences PharMingen). Bound primary antibodies were revealed by Alexa Fluor-conjugated secondaries (Invitrogen).
Immunohistochemical quantification of cleaved SNAP-25 and vGlut-2.
Confocal images were collected with a Leica confocal microscope using a 63× oil-immersion lens. An initial analysis on stained sections was performed to establish settings for laser intensity, gain, offset and pinhole size. Care was taken to avoid saturation at either end of the pixel intensity range (0–255). Confocal settings were then held constant through the study. For each coronal section, two representative fields were acquired using the same settings in the control and denervated SC. All fields were located in the stratum griseum superficiale. Stacks of images were collected at the top face of the tissue section and the image within each stack with the highest average pixel intensity was selected for the quantitative analysis of immunoreactivity. Quantification was performed with MetaMorph software (Molecular Devices). The area occupied by cleaved SNAP-25- and vGlut-2-positive pixels was calculated for each image by applying a threshold that removes background levels of fluorescence, and calculating the percentage of positive pixels in the field (Tropea et al., 2003; Caleo et al., 2007, 2009).
Colocalization analysis.
Colocalization was performed essentially as described by Tropea et al. (2003) using MetaMorph software (Molecular Devices). Two stacks of optical sections separated by 1 μm were collected on a confocal microscope (Leica). One stack contained images of cleaved SNAP-25, while the other contained images of a synaptic terminal label (synapsin, vGlut-1 and -2, vGAT, CTB). All images were collected through a 63× objective. Image size was 512 × 512 pixels, resulting in a pixel size of 0.08 μm. The optical section with highest pixel intensity was selected for colocalization analysis. Images from both channels were thresholded to remove background levels of fluorescence and retain only the brightest pixels. Thus, each image was binarized so that pixels above threshold were set to 1 and pixels with background fluorescence were set to 0. We then determined the percentage of double-labeled pixels in the neuropil. The analysis was perfomed on 8 different confocal stacks per animal. To estimate the amount of colocalization expected based on random overlap of the two labels, we performed the same colocalization analysis after rotation by 90 degrees of the cleaved SNAP-25 image (“shuffled” condition). A synaptic marker was considered to be colocalized with cleaved SNAP-25 when there was a significant difference (t test) in the percentage of double-positive pixels between the normal and shuffled condition.
Results
Anterograde propagation of BoNT/A effects in the visual system
To examine anterograde propagation of BoNT/A effects in the visual system, BoNT/A (1–3 nm, 0.4 μl) was injected into the vitreous humor of adult rats (Fig. 1A). This dose of toxin was chosen based on previous studies of central and peripheral administration of BoNT/A in rats (Franchi, 2002; Franchi and Veronesi, 2004; Antonucci et al., 2008b). Three days later, retinal and tectal sections were immunostained with an antibody specific for BoNT/A-truncated SNAP-25. Previous experiments have demonstrated that this antibody recognizes specifically the cleaved form of SNAP-25 and not the whole protein (Antonucci et al., 2008b; Matak et al., 2011). In the injected retina, strong staining was found in the inner plexiform layer; cell bodies of bipolar cells in the inner nuclear layer were also stained (Fig. 1B, left). A fainter labeling appeared in cell bodies of RGCs and in the optic nerve fiber layer (Fig. 1B, left). To determine whether BoNT/A-truncated SNAP-25 is detectable in RGC axons, we injected BoNT/A intraocularly and we crushed the optic nerve to allow accumulation of the cleaved product. Labeled fibers were clearly evident on the side proximal to the crush 24 h after surgery, indicating presence of BoNT/A-altered SNAP-25 within retinal axons (Fig. 1C). No specific staining was found in the contralateral, uninjected optic nerves and retinas (Fig. 1B, right).
Figure 1.

SNAP-25 cleavage in the BoNT/A-injected retina and optic nerve. A, Experimental protocol. BoNT/A (1–3 nm, 0.4 μl) was injected intraocularly and proteolysis of SNAP-25 was assayed in the retina and superior colliculus. B, Immunohistochemistry showing cleaved form of SNAP-25 (red) in the injected retina (ret ipsi BoNT/A). No specific signal is detectable in the contralateral retina (ret contra BoNT/A). ONL, Outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar, 25 μm. C, Cleaved SNAP-25 immunoreactivity (red) in a longitudinal section from a crushed optic nerve ipsilateral to BoNT/A injection. Staining was performed 24 h after the crush. Note cleaved SNAP-25-positive profiles on the proximal side (left). The site of crush is indicated by an arrow. Scale bar, 100 μm.
Examination of tectal sections revealed consistent labeling of the retinorecipient layers of the SC contralateral to the injected eye (n = 16 BoNT/A-injected rats; Fig. 2A). Labeling was evident 3 d after injection and became particularly prominent at 15 d. Staining for cleaved SNAP-25 was typically stronger in the medial part of the SC, topographically corresponding to the injection site in the ventral retina. Labeling was virtually undetectable in the ipsilateral SC (Fig. 2A, left). Thus, injection of BoNT/A into the eye leads to the appearance of BoNT/A-truncated SNAP-25 in the contralateral tectum. This is consistent with the fact that in the rat, ∼97% of the RGCs project contralaterally (Cowey and Perry, 1979).
Figure 2.

Anterograde propagation of BoNT/A effects along the visual pathway. A, Immunostaining demonstrating cleaved SNAP-25 labeling in the SC contralateral (Contra) to the injected retina. Note labeling in the superficial, retinorecipient layers of the SC. No signal is detectable in the SC ipsilateral (Ipsi) to the injected retina. Scale bar, 500 μm. B, Immunoblotting for cleaved SNAP-25 (cl. SNAP-25) on protein extracts from the injected retina (ret) and the contralateral (C) and ipsilateral (I) SC. Note SNAP-25 cleavage in the SC contralateral to the injected retina. Ten micrograms of protein were loaded for retinal samples, 100 μg for SC samples. α-Tubulin, internal standard. C–E, Immunostaining for cleaved SNAP-25 (red) in the pretectal area (C), dorsal lateral geniculate nucleus (D) and visual cortex (E) contralateral to the BoNT/A-injected eye, 3 d after operation. Scale bar, 10 μm.
Western blotting experiments on retinal and tectal tissue extracts provided further evidence for anterograde propagation of BoNT/A effects. We found robust SNAP-25 cleavage in the injected retina; a smaller, but significant, level of BoNT/A-cleaved SNAP-25 was detected in the contralateral (but not ipsilateral) SC (Fig. 2B). These data clearly indicate that proteolysis of SNAP-25 appears to propagate anterogradely from the eye in the adult retinotectal system following BoNT/A injection into the eye.
Labeling for BoNT/A-altered SNAP-25 also appeared in other retinorecipient nuclei such as the lateral geniculate nucleus (Fig. 2D) and pretectal area (Fig. 2C) 3–15 d after injection. In the deep layers of primary visual cortex contralateral to the injection, we found stained profiles with varicosities, likely axons en passant (Fig. 2E). This might reflect transsynaptic labeling of thalamocortical, corticogeniculate or corticotectal axons, as previously described for tetanus neurotoxin (Manning et al., 1990).
Anterograde propagation of BoNT/A effects is microtubule dependent
We next examined whether anterograde propagation of BoNT/A effects requires axonal transport. To block microtubule-dependent transport, we used intraocular injections of the depolymerizing agent colchicine (10 μg). This dose of colchicine was previously found to effectively prevent axonal transport along the rat optic nerve (Caleo et al., 2000, 2003; Antonucci et al., 2008b). Injection of colchicine blocked the anterograde propagation of BoNT/A effects from the retina to the contralateral SC (BoNT/A, n = 4, BoNT/A + colchicine, n = 5; Fig. 3). The selective appearance of BoNT/A-truncated SNAP-25 in the SC contralateral to the injected retina, and the blockade of this anterograde transfer by colchicine strongly argue against a systemic spread of toxin effects (via blood or CSF).
Figure 3.

Propagation of BoNT/A effects requires axonal transport. A, Immunoblotting showing cleaved SNAP-25 (cl. SNAP-25) levels in the retina and in the superior colliculi (C, contralateral to the injection; I, ipsilateral to the injection) of one animal injected intraocularly with BoNT/A (left lanes) and one animal subjected to colchicine injection (10 μg) 16 h before BoNT/A (right lanes). Note the loss of signal for cleaved SNAP-25 in the contralateral SC of the animal treated with colchicine (BoNT/A + Colch). α-Tubulin, internal standard. B, Quantification of cleaved SNAP-25 levels. Levels of cleaved SNAP-25 in the superior colliculi are normalized to levels of expression in the corresponding retinal samples. In animals treated with colchicine, expression of cleaved SNAP-25 in the superior colliculus contralateral to injected retina is virtually abolished. Data are mean ± SE. BoNT/A, n = 4, BoNT/A + colchicine, n = 5. *p < 0.05.
Expression of BoNT/A-truncated SNAP-25 in the tectum remains unaltered despite degeneration of RGC fibers
We next investigated the localization of BoNT/A-truncated SNAP-25 in the optic tectum. Staining was prominent in the superficial gray of the SC, but sparse in the underlying optic nerve fiber layer (stratum opticum). Using confocal analysis at high magnification, cleaved SNAP-25 was found associated with puncta and processes in the neuropil surrounding tectal cells (Fig. 4A). To determine whether cleaved SNAP-25-positive profiles correspond to synaptic terminals, we performed double-label immunofluorescence for BoNT/A-altered SNAP-25 and the synaptic vesicle marker synapsin. The quantitative analysis (Silver and Stryker, 2000; Tropea et al., 2003) demonstrated a very significant colocalization of cleaved SNAP-25 and synapsin immunoreactivity (Fig. 4B). On average, 26.5 ± 0.5% (SEM) of the pixels in the neuropil were double-stained for BoNT/A-altered SNAP-25 and synapsin. Confocal analysis showed no colocalization with markers of glial cells such as GFAP (a marker of astrocytes; Fig. 4C) and OX-42 (an antibody that recognizes microglia; Fig. 4D).
Figure 4.
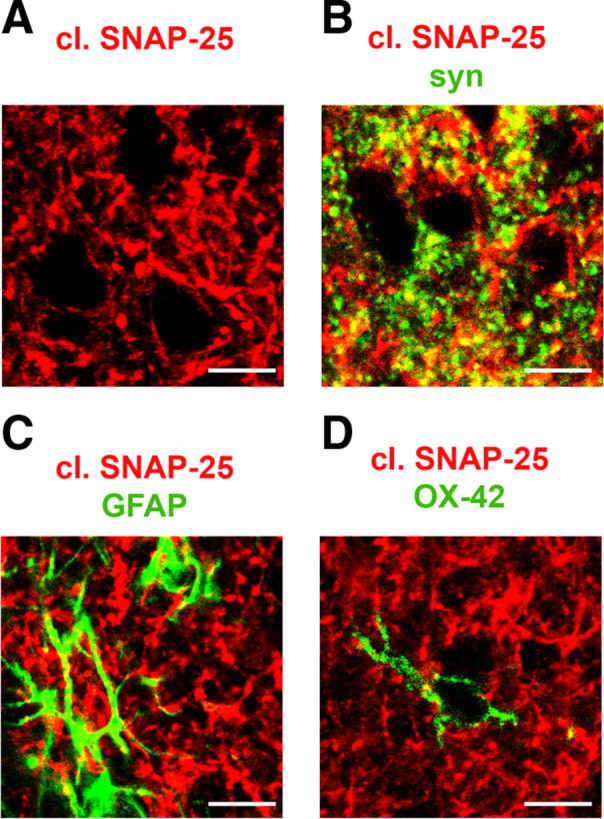
Localization of cleaved SNAP-25 in the tectum. A, B, Animals were injected intraocularly with BoNT/A and killed at 3 d. Immunostaining in SC sections for cleaved SNAP-25 (red) and the synaptic vesicle marker synapsin (green; B). C, D, Animals were injected intraocularly with BoNT/A and killed at 15 d. Immunostaining in SC sections for cleaved SNAP-25 (red) and the glial markers GFAP (green in C) and OX-42 antibody (green in D). Scale bar, 10 μm.
To gain more insight into the type of nerve terminals containing truncated SNAP-25, we performed double-label immunostaining for markers of excitatory (vGlut-1 and -2) and inhibitory synapses (vGAT). In the rat SC, vGlut-2 is specifically expressed by terminals of RGCs (Fujiyama et al., 2003; Caleo et al., 2009). We found that cleaved SNAP-25 displayed significant colocalization with vGlut-1 and vGAT (10.3 ± 0.3% and 14.8 ± 0.6% of double-labeled neuropil pixels, respectively, a percentage significantly different from random colocalization of the two labels, t test, p < 0.001, Fig. 5A,B), while there was no colocalization with the retinal terminal marker vGlut-2 (6.4 ± 0.4% of double-labeled neuropil pixels, a percentage not significantly different from random colocalization of the two labels, t test, p > 0.05; Fig. 5C). Surprised by this initial analysis, we performed a more thorough set of colocalization experiments, in which massive labeling of RGC terminals was achieved by intraocular injection of an anterograde tracer (CTB; n = 4 rats). This resulted in robust staining of retinal fibers in the SC. However, confocal analysis of CTB- and cleaved SNAP-25-positive pixels (Fig. 5D) revealed no significant colocalization of the two markers (5.1 ± 0.7% of pixels in the neuropil, no significantly different from random; t test, p = 0.23; Fig. 5D). Since SNAP-25 is abundantly expressed in RGCs, we used antibodies to intact SNAP-25 as a positive control for our colocalization analysis (Fig. 5E). We found a very significant colocalization when staining for CTB and the intact form of SNAP-25 was combined (30.2 ± 1.3% of double-labeled pixels in the neuropil).
Figure 5.
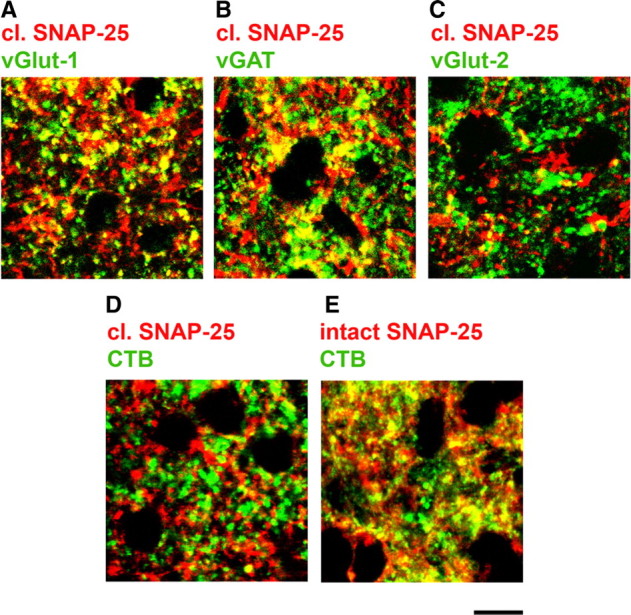
Colocalization of cleaved SNAP-25 with vGlut-1- and vGAT-positive terminals. A–C, Animals were injected intraocularly with BoNT/A and killed at 3 d. Immunostaining in SC sections for cleaved SNAP-25 (red) and presynaptic markers (green: A, vGlut-1; B, vGAT; C, vGlut-2). Note that cleaved SNAP-25 displays an evident colocalization with vGlut-1 and vGAT (A, B), but it does not colocalize with vGlut-2 (C), specific for RGC terminals. D, E, Animals were injected intraocularly with an anterograde tracer (CTB) to label RGC fibers. A subset of animals were injected 1 d later with BoNT/A and killed after another 3 d. Note the lack of colocalization between CTB (green) and cleaved SNAP-25 (red) in D. In contrast, there is clear colocalization between CTB (green) and the intact form of SNAP-25 (red) in E. Scale bar, 10 μm.
These immunostaining data suggest that cleaved SNAP-25 is not present within RGC terminals, but a lack of colocalization can be caused by several technical factors, including incomplete penetration of the antibodies, different subcellular localization of CTB/vGlut-2 and truncated SNAP-25, and poor resolution of synaptic puncta by confocal microscopy. To explore more directly whether cleaved SNAP-25 is present in RGC fibers, we took advantage of the fact that cleaved SNAP-25 associated with retinal terminals would be rapidly lost after section of the optic nerve, due to Wallerian degeneration of transected fibers. Therefore, we injected unilaterally BoNT/A into the vitreous in a group of rats (n = 11), and allowed 3 d for anterograde transport to the SC, and then we cut the optic nerve corresponding to the injected eye in a subset of the animals (n = 5). The remaining animals (n = 6) were left unoperated. We allowed another 7 d for Wallerian degeneration of transected RGC fibers and then performed immunoblotting for cleaved SNAP-25 in tectal protein extracts (contralateral to the injected retina) in control and optic nerve-sectioned rats. We found that BoNT/A-truncated SNAP-25 was not decreased in the denervated tectum (control vs denervated, t test, p = 0.26; Fig. 6A,B). Levels of BoNT/A-truncated SNAP-25 in the injected retina were similar between groups (t test, p = 0.73). Conversely, expression of vGlut-2 (marker of retinal terminals in the tectum) was consistently dampened in the deafferented SC, consistent with the effects of optic nerve section (Fig. 6C,D; t test, p < 0.05).
In a second protocol, we used bilateral, intravitreal injections of BoNT/A, followed by section of the left optic nerve 3 d later and immunohistochemistry for cleaved SNAP-25 at 10 d (n = 6 rats). This protocol allows one to compare levels of BoNT/A-truncated SNAP-25 in the denervated and control SC of the same animal. We found that staining for cleaved SNAP-25 was not abolished in the deafferented tectum (Fig. 7A). Quantitative analysis of the immunostaining revealed no differences between levels of BoNT/A-truncated SNAP-25 in the SC contralateral and ipsilateral to optic nerve section (Fig. 7B; control vs denervated, paired t test, p = 0.38). Importantly, quantification of the retinal fiber marker vGlut-2 indicated a very robust decrease of the immunostaining (Fig. 7C,D; control vs denervated, paired t test, p < 0.001), demonstrating that optic nerve section effectively removes most of retinal terminals from the contralateral SC. The immunohistochemical decrease of vGlut-2 following optic nerve section was quantitatively greater than that observed by Western blot (Fig. 6B), likely because quantification of the immunostaining could be restricted to the retinorecipient superficial gray of the SC, while surgical dissection of the colliculi for immunoblotting inevitably leads to pickup of tissue from nonvisual deep layers of the SC (such as the intermediate gray layer), in which vGlut-2 is not decreased. In summary, these sets of experiments demonstrate that expression of BoNT/A-truncated SNAP-25 in the tectum remains unaltered despite degeneration of RGC fibers, indicating that structures other than retinal terminals contain cleaved SNAP-25.
Figure 7.

Immunohistochemical expression of BoNT/A-truncated SNAP-25 persists following retinal fiber degeneration. BoNT/A was injected into both eyes and after 3 d all animals were subjected to unilateral optic nerve section (ON cut). The other side served as control (CTR). After an additional 7 d, rats were perfused and immunohistochemistry was performed. A, C, Immunostaining for cleaved SNAP-25 (red, A) and vGlut-2 (green, C) on tectal sections. In the zoomed images (boxes) it is possible to note that staining for cleaved SNAP-25 is similar in both sides, while staining for vGlut-2 is strongly reduced in the side corresponding to ON section, indicating the degeneration of RGCs terminals. Scale bars: top, 100 μm; bottom, 10 μm. B, D, Quantification of cleaved SNAP-25 (B) and vGlut-2 levels (D) on tectal sections. There is no statistically significant difference in cleaved SNAP-25 staining between the two colliculi. Note, instead, the clear reduction of vGlut-2 staining in the colliculus contralateral to the ON section, demonstrating degeneration of RGCs terminals. Data are mean ± SE. CTR side, n = 6; ON cut side, n = 6. ***p = 0.001.
Active BoNT/A is anterogradely transported and transcytosed in the retinotectal system
The lack of presence of BoNT/A-truncated SNAP-25 in RGC fibers suggests a process of anterograde transport and transcytosis, by which BoNT/A (and/or its cleaved substrate) is released from RGC terminals and subsequently concentrates in other synaptic terminals in the tectum. The localization of cleaved SNAP-25 with vGlut-1/vGAT (but not vGlut-2)-positive puncta in the tectum is consistent with this scenario (Fig. 5). However, as only the distribution of BoNT/A-cleaved SNAP-25 was assayed, it could be argued that the cleaved substrate is transported and transcytosed, rather than the toxin itself. In addition, it remained undetermined whether BoNT/A can be transported and transcytosed in a catalytically active form, thus capable of long-range anterograde actions. To address these issues, we designed an experiment (Fig. 8A) to test whether the catalytic activity of BoNT/A appears in the SC following toxin injection into the retina. For this experiment, we injected unilaterally BoNT/A into the vitreous of 15 rats, and five of them were killed 3 d after injection to visualize the expected anterograde propagation of BoNT/A effects to the tectum (3 d; Fig. 8B, left lanes). In the remaining animals, we performed section of the optic nerve (to prevent further transport along the visual pathway) and BoNT/E was injected into the tectum contralateral to BoNT/A. BoNT/A removes the last 9 residues from the C terminus of SNAP-25, while BoNT/E cleaves a larger 26 residue fragment from the same region of SNAP-25; in addition, the action of BoNT/E is short-lasting (Meunier et al., 2003; Caleo et al., 2007), while BoNT/A catalytic activity is known to persist for much longer. In the animals killed 2 d after BoNT/E/optic nerve section (3 d + 2 d BoNT/E; n = 5), we found that injection of BoNT/E strongly reduced expression of BoNT/A-truncated SNAP-25 in the tectum (t test, 3 d vs 3 d + 2 d BoNT/E, p < 0.05; Fig. 8B, middle lanes; Fig. 8C). However, the catalytic activity of BoNT/A reappeared in the SC at the completion of BoNT/E effects (23 d; t test, 3 d + 2 d BoNT/E vs 3 d + 23 d BoNT/E, p < 0.001; Fig. 8B, right lanes; Fig. 8C), indicating that active BoNT/A is present within tectal terminals and capable of generating new truncated substrate in the SC. If BoNT/A-cleaved SNAP-25 was transported anterogradely, rather than the toxin itself, the reduction of BoNT/A-truncated SNAP-25 would have been permanent. Conversely, levels of BoNT/A-altered SNAP-25 in the tectum 23 d after the optic nerve cut were greater than that observed 3 d after BoNT/A (t test, p = 0.01; Fig. 8C), indicating the long-term presence of the BoNT/A metalloprotease activity within the SC. These data allow us to conclude that active BoNT/A is anterogradely transported from the eye to the SC and transcytosed to tectal cells, where it maintains catalytic activity for a relatively long duration (23 d).
BoNT/A transfer from retinal to tectal terminals does not necessarily require internalization by postsynaptic cells
We finally investigated in more detail the mechanisms by which BoNT/A moves from the RGC terminals to tectal synapses. This transfer could be direct (release from retinal terminals and uptake by presynaptic sites in the vicinity), or involve a step of internalization by tectal cells followed by further release and uptake. To determine whether internalization by postsynaptic neurons is required for anterograde propagation of BoNT/A effects, we performed an experiment in which tectal cells were ablated in a portion of the superficial gray by injection of a minute amount (100 nl, 1 nmol) of the excitotoxin KA. Injection of KA is known to lead to local neuronal loss while sparing retinocollicular and other afferent axons (Carpenter et al., 1986). A complete loss of immunostaining for the neuronal marker NeuN was found in the injected area (Fig. 9A). One day after the lesion, BoNT/A was injected intraocularly and three further days were allowed for anterograde transport. We found that cleaved SNAP-25-positive profiles were detectable in the tectal areas completely devoid of NeuN immunoreactivity, although their density was lower than that found in adjacent, unlesioned areas (Fig. 9B,C). Thus, tectal cells are dispensable for the anterograde propagation of BoNT/A effects.
Figure 9.

Tectal cells are dispensable for the anterograde propagation of BoNT/A effects. A, NeuN immunostaining (red) in a coronal section through both colliculi, 4 d after unilateral delivery of KA. Borders of the lesion are indicated by arrows. Scale bar, 500 μm. B, C, Double-label immunohistochemistry for NeuN (red) and cleaved SNAP-25 (green). Staining was performed 3 d after intravitreal BoNT/A. Zoomed images are taken from a medial, unlesioned area of the SC (B) and from within the damaged zone (C). Scale bar, 10 μm.
Discussion
The data presented in this article demonstrate anterograde trafficking and transcytosis of catalytically active BoNT/A in neurons. They were obtained with a very relevant model system: the rat retinotectal pathway which allows to assay for transcytosis via optic nerve section and use of RGC-specific markers (von Bartheld et al., 1996; Caleo et al., 2000, 2009; von Bartheld and Butowt, 2000). In the rat, virtually all RGCs send a unidirectional projection to the superior colliculus, with 97% of the axons crossing at the chiasm and innervating the contralateral tectum (Cowey and Perry, 1979). Cell bodies of RGCs can be easily exposed to exogenous ligands by injections into the vitreous humor, and the optic nerve is amenable to surgical manipulation (Manning et al., 1990; von Bartheld et al., 1996; Caleo et al., 2000, 2009; von Bartheld and Butowt, 2000). This model system has been extensively used for studies of anterograde transport and transcytosis (von Bartheld et al., 2001; Caleo and Cenni, 2004; von Bartheld, 2004).
In this article, we have used the cleavage of the BoNT/A substrate SNAP-25 as a marker for toxin action in distant sites. This choice is justified by the fact that detection of BoNT/A itself is technically challenging due to the very tiny amounts of injected toxin; in addition, measuring cleavage is a more sensitive assay for BoNT/A trafficking, as a small number of toxin molecules can cleave massive amounts of the substrate, thus providing a dramatic amplifying effect. We found significant levels of BoNT/A-truncated SNAP-25 in the tectum after BoNT/A delivery into the eye. The blockade of this anterograde propagation of BoNT/A effects by coinjection of colchicine ruled out a systemic spread of the toxin (via blood or CSF). Interestingly, cleaved SNAP-25 in the tectum apparently accumulated in synaptic endings, but was not associated with RGC terminals. This points to a process of transcytosis by which BoNT/A and/or the cleaved substrate are transferred from RGCs to tectal terminals. Here, the question arises as to whether detection of the cleaved substrate can be unequivocally interpreted as presence of the BoNT/A protease. The BoNT/E injection into the tectum coupled with optic nerve section a few days after intravitreal delivery of BoNT/A provides compelling functional evidence that a persistent proteolytic activity of BoNT/A resides in tectal synapses. In particular, tectal delivery of BoNT/E produced a consistent reduction of BoNT/A-altered SNAP-25, that returned at high levels at the completion of BoNT/E effects. The reappearance of BoNT/A-truncated SNAP-25 cannot be explained by ongoing anterograde transport from the eye, as the optic nerve was severed concomitantly with BoNT/E. Rather, the return of BoNT/A-cleaved SNAP-25 indicates persistent BoNT/A catalytic activity in the superior colliculus, since there are no other means to generate new truncated substrate in the tectum. This proves that catalytically active BoNT/A was transferred to tectal terminals during the first 3 d following intraocular administration. The demonstration of cell-to-cell transfer of active BoNT/A is extremely relevant because it provides proof-of-principle that this toxin can affect cellular targets that are distant from the administration site. It will be important in future studies to determine what are the functional consequences of this distant cleavage at the level of individual synaptic boutons. Previous work from our group (Antonucci et al., 2008b) supports long-distance functional changes after BoNT/A delivery, as shown by the fact that trans-hemispheric propagation of BoNT/A effects in the hippocampus results in electrophysiological consequences. Here, we choose not to provide functional measures in the SC, because of the confounding effects of blockade of afferent retinal activity, that likely impacts per se physiological parameters in the tectum.
The mechanisms and cellular compartments involved in the anterograde axonal transport and transcytosis of BoNT/A remain to be elucidated. Molecules that are well known to enter a trancytosis pathway include TeNT, lectins, and trophic factors such as brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and glial cell line-derived neurotrophic factor (GDNF). Following injection into the eye of the rat and rabbit, fragment C of TeNT undergoes anterograde direct transport to all retinorecipient areas. From these areas, TeNT exhibits transcytosis to neurons that project to retinorecipient areas, including neurons in layers V and VI of visual cortex (Manning et al., 1990). Certain neurotrophic factors can also be anterogradely transported and transcytosed. For example, in developing chick brain, NT-3 is transported in the anterograde direction, from cell bodies to the axon terminals, and the intact neurotrophin is released after anterograde transport, taken up and used by second-order visual neurons (von Bartheld et al., 1996; von Bartheld and Butowt, 2000). BDNF travels in an anterograde direction along the rat optic nerve and has rapid effects on retinal target neurons in the superior colliculus and lateral geniculate nucleus of the brain (Caleo et al., 2000, 2003). GDNF is anterogradely transported from the cell body of sensory neurons into the spinal cord (von Bartheld et al., 2001). Neurotrophic factors are packaged in large dense core vesicles during anterograde axonal transport, released via activity-dependent mechanisms, and once secreted they may act at either presynaptic or postsynaptic sites (von Bartheld et al., 2001; von Bartheld, 2004). Whether BoNT/A uses a similar pathway for anterograde trafficking remains to be investigated. The findings that the fibroblast growth factor (FGF) receptor 3 has been identified as a component of the receptor complex for BoNT/A (Fernandez-Salas et al., 2008), and FGF is known to travel anterogradely in the optic nerve (Ferguson and Johnson, 1991), suggest that BoNT/A may use an anterograde trafficking pathway shared with trophic factors. In this context, it is known that TeNT enters a vesicular pathway shared with neurotrophins and their receptors, at least during retrograde transport (Deinhardt et al., 2006; Salinas et al., 2010).
Our colocalization analysis clearly indicates significant colocalization of cleaved SNAP-25 with synapsin-positive terminals (specifically, with a subset of vGlut-1- and vGAT-stained terminals), but no significant colocalization with the retinal markers vGlut-2 and CTB. Importantly, persistence of cleaved SNAP-25 in the tectum after optic nerve lesion confirms expression of BoNT/A-altered SNAP-25 in tectal, but not RGC terminals. Thus, RGCs appear to be mostly involved in uptake, transport and release of the active toxin, while the cleaved product accumulates in other neuronal types in the tectum. One question is how the toxin moves from the RGC terminals to the vGlut-1 and vGAT-positive synapses. By performing an excitotoxic lesion in the SC, we have shown that internalization by tectal cells is not necessarily required for anterograde propagation of BoNT/A effects. Therefore, these data indicate that a direct, presynaptic to presynaptic route is possible (release from RGC afferents and uptake by other synaptic terminals in the vicinity). Such synaptic terminals may include a contingent of corticotectal, vGlut-1-positive synapses. In addition, the extensive colocalization between vGAT and BoNT/A-truncated SNAP-25 hints at BoNT/A effects in local GABAergic interneurons and possibly afferent inhibitory projections from the parabigeminal nucleus. The reduced density of cleaved SNAP-25 profiles in the neuropil after excitotoxic lesions (Fig. 9) is also consistent with uptake of BoNT/A by terminals of local GABAergic neurons, that degenerate after such injury.
Our data demonstrating anterograde transport and transcytosis of BoNT/A may have implications for the understanding of the mechanisms of action of BoNT/A in pain. Several studies have shown that BoNT/A can be effectively used to treat several pathological pain conditions (Aoki, 2005) and this line of research is rapidly growing (Pavone and Luvisetto, 2010). A likely mechanism by which BoNT/A exerts antinociceptive actions is the inhibition of release of neurotransmitter and neuropeptides (substance P, calcitonin gene-related peptide) from the peripheral branch of primary sensory neurons. However, several recent data demonstrate bilateral antinociceptive effects of BoNT/A following unilateral, peripheral administration of the toxin (Bach-Rojecky and Lacković, 2009; Favre-Guilmard et al., 2009; Bach-Rojecky et al., 2010). This bilateral effect suggests the involvement of the CNS in the mechanisms of action of BoNT/A. This could occur either indirectly (through synaptic plasticity), or directly (via axonal transport of the toxin), as suggested by the fact that the bilateral effect can be prevented by peripheral administration of colchicine (Bach-Rojecky and Lacković, 2009). To exert a spinal effect, it is required that the toxin ascends retrogradely along the peripheral branch of nociceptive neurons and then penetrates the spinal cord through anterograde transport and transcytosis (von Bartheld et al., 2001; von Bartheld, 2004). This possibility has never been supported experimentally before, and our data indicate for the first time that BoNT/A is indeed capable of undergoing anterograde trafficking and transcytosis across synapses.
In summary, we have provided proof-of-principle that BoNT/A can undergo anterograde axonal transport and transcytosis in neurons. These data are important for a more complete understanding of the mechanisms of action and trafficking of clostridial toxins, for the future design of drug delivery vehicles capable to be transferred specifically across synapses, and to target specific neural circuits for long-term therapeutic effects on neuronal function.
Footnotes
This work was supported by the Italian Ministry of Health (PRIN—Progetti di Rilevante Interesse Nazionale—2008; M.C. and O.R.), by Tuscany region (Health Program 2009; M.C.), and by FIRB (Fondo per gli Investimenti della Ricerca de Base) project “CHEM-PROFARMA-NET.” We thank Elena Novelli (Fondazione Bietti, Rome) for contributing to data collection for Figure 1. Carlo Gianfranceschi and Eleonora Vannini participated in some experiments. We are grateful to Cesare Montecucco for critical reading of the manuscript.
References
- Adler M, Keller JE, Sheridan RE, Deshpande SS. Persistence of botulinum neurotoxin A demonstrated by sequential administration of serotypes A and E in rat EDL muscle. Toxicon. 2001;39:233–243. doi: 10.1016/s0041-0101(00)00120-3. [DOI] [PubMed] [Google Scholar]
- Antonucci F, Di Garbo A, Novelli E, Manno I, Sartucci F, Bozzi Y, Caleo M. Botulinum neurotoxin E (BoNT/E) reduces CA1 neuron loss and granule cell dispersion, with no effects on chronic seizures, in a mouse model of temporal lobe epilepsy. Exp Neurol. 2008a;210:388–401. doi: 10.1016/j.expneurol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Antonucci F, Rossi C, Gianfranceschi L, Rossetto O, Caleo M. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008b;28:3689–3696. doi: 10.1523/JNEUROSCI.0375-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki KR. Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology. 2005;26:785–793. doi: 10.1016/j.neuro.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Bach-Rojecky L, Lackoviæ Z. Central origin of the antinociceptive action of botulinum toxin type A. Pharmacol Biochem Behav. 2009;94:234–238. doi: 10.1016/j.pbb.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Bach-Rojecky L, Salkoviæ-Petrisiæ M, Lackoviæ Z. Botulinum toxin type A reduces pain supersensitivity in experimental diabetic neuropathy: bilateral effect after unilateral injection. Eur J Pharmacol. 2010;633:10–14. doi: 10.1016/j.ejphar.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Binz T, Rummel A. Cell entry strategy of clostridial neurotoxins. J Neurochem. 2009;109:1584–1595. doi: 10.1111/j.1471-4159.2009.06093.x. [DOI] [PubMed] [Google Scholar]
- Caleo M, Cenni MC. Anterograde transport of neurotrophic factors: possible therapeutic implications. Mol Neurobiol. 2004;29:179–196. doi: 10.1385/mn:29:2:179. [DOI] [PubMed] [Google Scholar]
- Caleo M, Lodovichi C, Maffei L. Effects of nerve growth factor on visual cortical plasticity require afferent electrical activity. Eur J Neurosci. 1999;11:2979–2984. doi: 10.1046/j.1460-9568.1999.00737.x. [DOI] [PubMed] [Google Scholar]
- Caleo M, Menna E, Chierzi S, Cenni MC, Maffei L. Brain-derived neurotrophic factor is an anterograde survival factor in the rat visual system. Curr Biol. 2000;10:1155–1161. doi: 10.1016/s0960-9822(00)00713-2. [DOI] [PubMed] [Google Scholar]
- Caleo M, Medini P, von Bartheld CS, Maffei L. Provision of brain-derived neurotrophic factor via anterograde transport from the eye preserves the physiological responses of axotomized geniculate neurons. J Neurosci. 2003;23:287–296. doi: 10.1523/JNEUROSCI.23-01-00287.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M, Restani L, Gianfranceschi L, Costantin L, Rossi C, Rossetto O, Montecucco C, Maffei L. Transient synaptic silencing of developing striate cortex has persistent effects on visual function and plasticity. J Neurosci. 2007;27:4530–4540. doi: 10.1523/JNEUROSCI.0772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M, Tropea D, Rossi C, Gianfranceschi L, Maffei L. Environmental enrichment promotes fiber sprouting after deafferentation of the superior colliculus in the adult rat brain. Exp Neurol. 2009;216:515–519. doi: 10.1016/j.expneurol.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Carpenter P, Sefton AJ, Dreher B, Lim WL. Role of target tissue in regulating the development of retinal ganglion cells in the albino rat: effects of kainate lesions in the superior colliculus. J Comp Neurol. 1986;251:240–259. doi: 10.1002/cne.902510208. [DOI] [PubMed] [Google Scholar]
- Casale R, Tugnoli V. Botulinum toxin for pain. Drugs R D. 2008;9:11–27. doi: 10.2165/00126839-200809010-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantin L, Bozzi Y, Richichi C, Viegi A, Antonucci F, Funicello M, Gobbi M, Mennini T, Rossetto O, Montecucco C, Maffei L, Vezzani A, Caleo M. Antiepileptic effects of botulinum neurotoxin E. J Neurosci. 2005;25:1943–1951. doi: 10.1523/JNEUROSCI.4402-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowey A, Perry VH. The projection of the temporal retina in rats, studied by retrograde transport of horseradish peroxidase. Exp Brain Res. 1979;35:457–464. doi: 10.1007/BF00236764. [DOI] [PubMed] [Google Scholar]
- Davletov B, Bajohrs M, Binz T. Beyond BOTOX: advantages and limitations of individual botulinum neurotoxins. Trends Neurosci. 2005;28:446–452. doi: 10.1016/j.tins.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Eleopra R, Tugnoli V, Rossetto O, De Grandis D, Montecucco C. Different time courses of recovery after poisoning with botulinum neurotoxin serotypes A and E in humans. Neurosci Lett. 1998;256:135–138. doi: 10.1016/s0304-3940(98)00775-7. [DOI] [PubMed] [Google Scholar]
- Evidente VG, Adler CH. An update on the neurologic applications of botulinum toxins. Curr Neurol Neurosci Rep. 2010;10:338–344. doi: 10.1007/s11910-010-0129-z. [DOI] [PubMed] [Google Scholar]
- Favre-Guilmard C, Auguet M, Chabrier PE. Different antinociceptive effects of botulinum toxin type A in inflammatory and peripheral polyneuropathic rat models. Eur J Pharmacol. 2009;617:48–53. doi: 10.1016/j.ejphar.2009.06.047. [DOI] [PubMed] [Google Scholar]
- Ferguson IA, Johnson EM., Jr Fibroblast growth factor receptor-bearing neurons in the CNS: identification by receptor-mediated retrograde transport. J Comp Neurol. 1991;313:693–706. doi: 10.1002/cne.903130412. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salas E, Garay P, Jacky B, Dupuy J, Wang J, Nelson J, Raymond CS, Aoki KR. Identification of the fibroblast growth factor receptor FGFR3 as a component of the receptor complex for Botulinum Neurotoxin Type A. Toxicon. 2008;5(Suppl 1):3.25. [Google Scholar]
- Franchi G. Time course of motor cortex reorganization following botulinum toxin injection into the vibrissal pad of the adult rat. Eur J Neurosci. 2002;16:1333–1348. doi: 10.1046/j.1460-9568.2002.02195.x. [DOI] [PubMed] [Google Scholar]
- Franchi G, Veronesi C. Time course for the reappearance of vibrissal motor representation following botulinum toxin injection into the vibrissal pad of the adult rat. Eur J Neurosci. 2004;20:1873–1884. doi: 10.1111/j.1460-9568.2004.03653.x. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Hioki H, Tomioka R, Taki K, Tamamaki N, Nomura S, Okamoto K, Kaneko T. Changes of immunocytochemical localization of vesicular glutamate transporters in the rat visual system after the retinofugal denervation. J Comp Neurol. 2003;465:234–249. doi: 10.1002/cne.10848. [DOI] [PubMed] [Google Scholar]
- Habermann E. 125I-labeled neurotoxin from Clostridium botulinum A: preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn Schmiedebergs Arch Pharmacol. 1974;281:47–56. doi: 10.1007/BF00500611. [DOI] [PubMed] [Google Scholar]
- Johnson EA. Clostridial toxins as therapeutic agents: benefits of nature's most toxic proteins. Annu Rev Microbiol. 1999;53:551–575. doi: 10.1146/annurev.micro.53.1.551. [DOI] [PubMed] [Google Scholar]
- Mainardi M, Landi S, Gianfranceschi L, Baldini S, De Pasquale R, Berardi N, Maffei L, Caleo M. Environmental enrichment potentiates thalamocortical transmission and plasticity in the adult rat visual cortex. J Neurosci Res. 2010;88:3048–3059. doi: 10.1002/jnr.22461. [DOI] [PubMed] [Google Scholar]
- Mandolesi G, Menna E, Harauzov A, von Bartheld CS, Caleo M, Maffei L. A role for retinal brain-derived neurotrophic factor in ocular dominance plasticity. Curr Biol. 2005;15:2119–2124. doi: 10.1016/j.cub.2005.10.045. [DOI] [PubMed] [Google Scholar]
- Manning KA, Erichsen JT, Evinger C. Retrograde transneuronal transport properties of fragment C of tetanus toxin. Neuroscience. 1990;34:251–263. doi: 10.1016/0306-4522(90)90319-y. [DOI] [PubMed] [Google Scholar]
- Matak I, Bach-Rojecky L, Filipoviæ B, Lackoviæ Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience. 2011;186:201–207. doi: 10.1016/j.neuroscience.2011.04.026. [DOI] [PubMed] [Google Scholar]
- Meunier FA, Lisk G, Sesardic D, Dolly JO. Dynamics of motor nerve terminal remodeling unveiled using SNARE-cleaving botulinum toxins: the extent and duration are dictated by the sites of SNAP-25 truncation. Mol Cell Neurosci. 2003;22:454–466. doi: 10.1016/s1044-7431(02)00016-7. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Molgó J. Botulinal neurotoxins: revival of an old killer. Curr Opin Pharmacol. 2005;5:274–279. doi: 10.1016/j.coph.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Morbiato L, Carli L, Johnson EA, Montecucco C, Molgó J, Rossetto O. Neuromuscular paralysis and recovery in mice injected with botulinum neurotoxins A and C. Eur J Neurosci. 2007;25:2697–2704. doi: 10.1111/j.1460-9568.2007.05529.x. [DOI] [PubMed] [Google Scholar]
- Moreno-López B, Pastor AM, de la Cruz RR, Delgado-García JM. Dose-dependent, central effects of botulinum neurotoxin type A: a pilot study in the alert behaving cat. Neurology. 1997;48:456–464. doi: 10.1212/wnl.48.2.456. [DOI] [PubMed] [Google Scholar]
- Naumann M, So Y, Argoff CE, Childers MK, Dykstra DD, Gronseth GS, Jabbari B, Kaufmann HC, Schurch B, Silberstein SD, Simpson DM. Assessment: Botulinum neurotoxin in the treatment of autonomic disorders and pain (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70:1707–1714. doi: 10.1212/01.wnl.0000311390.87642.d8. [DOI] [PubMed] [Google Scholar]
- Pavone F, Luvisetto S. Botulinum neurotoxin for pain management: insights from animals models. Toxins. 2010;12:2890–2913. doi: 10.3390/toxins2122890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto O, Seveso M, Caccin P, Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: turning bad guys into good by research. Toxicon. 2001;39:27–41. doi: 10.1016/s0041-0101(00)00163-x. [DOI] [PubMed] [Google Scholar]
- Rossetto O, Morbiato L, Caccin P, Rigoni M, Montecucco C. Presynaptic enzymatic neurotoxins. J Neurochem. 2006;97:1534–1545. doi: 10.1111/j.1471-4159.2006.03965.x. [DOI] [PubMed] [Google Scholar]
- Salinas S, Schiavo G, Kremer EJ. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol. 2010;8:645–655. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Montecucco C. Tetanus and botulism neurotoxins: isolation and assay. Methods Enzymol. 1995;248:643–652. doi: 10.1016/0076-6879(95)48041-2. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- Silver MA, Stryker MP. Distributions of synaptic vesicle proteins and GAD65 in deprived and nondeprived ocular dominance columns in layer IV of kitten primary visual cortex are unaffected by monocular deprivation. J Comp Neurol. 2000;422:652–664. doi: 10.1002/1096-9861(20000710)422:4<652::aid-cne11>3.0.co;2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DM, Blitzer A, Brashear A, Comella C, Dubinsky R, Hallett M, Jankovic J, Karp B, Ludlow CL, Miyasaki JM, Naumann M, So Y. Assessment: Botulinum neurotoxin for the treatment of movement disorders (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008a;70:1699–1706. doi: 10.1212/01.wnl.0000311389.26145.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DM, Gracies JM, Graham HK, Miyasaki JM, Naumann M, Russman B, Simpson LL, So Y. Assessment: Botulinum neurotoxin for the treatment of spasticity (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008b;70:1691–1698. doi: 10.1212/01.wnl.0000311391.00944.c4. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tropea D, Caleo M, Maffei L. Synergistic effects of brain-derived neurotrophic factor and chondroitinase ABC on retinal fiber sprouting after denervation of the superior colliculus in adult rats. J Neurosci. 2003;23:7034–7044. doi: 10.1523/JNEUROSCI.23-18-07034.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turton K, Chaddock JA, Acharya KR. Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem Sci. 2002;27:552–558. doi: 10.1016/s0968-0004(02)02177-1. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS. Axonal transport and neuronal transcytosis of trophic factors, tracers, and pathogens. J Neurobiol. 2004;58:295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Butowt R. Expression of neurotrophin-3 (NT-3) and anterograde axonal transport of endogenous NT-3 by retinal ganglion cells in chick embryos. J Neurosci. 2000;20:736–748. doi: 10.1523/JNEUROSCI.20-02-00736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Wang X, Butowt R. Anterograde axonal transport, transcytosis, and recycling of neurotrophic factors: the concept of trophic currencies in neural networks. Mol Neurobiol. 2001;24:1–28. doi: 10.1385/MN:24:1-3:001. [DOI] [PubMed] [Google Scholar]
- Wiegand H, Erdmann G, Wellhöner HH. 125I-labelled botulinum A neurotoxin: pharmacokinetics in cats after intramuscular injection. Naunyn Schmiedebergs Arch Pharmacol. 1976;292:161–165. doi: 10.1007/BF00498587. [DOI] [PubMed] [Google Scholar]