

Abstract
Peripheral nerve lesion triggers alterations in the spinal microenvironment that contribute to the pathogenesis of neuropathic pain. While neurons and glia have been implicated in these functional changes, it remains largely underexplored whether the blood–spinal cord barrier (BSCB) is also involved. The BSCB is an important component in the CNS homeostasis, and compromised BSCB has been associated with different pathologies affecting the spinal cord. Here, we demonstrated that a remote injury on the peripheral nerve in rats triggered a leakage of the BSCB, which was independent of spinal microglial activation. The increase of BSCB permeability to different size tracers, such as Evans Blue and sodium fluorescein, was restricted to the lumbar spinal cord and prominent for at least 4 weeks after injury. The spinal inflammatory reaction triggered by nerve injury was a key player in modulating BSCB permeability. We identified MCP-1 as an endogenous trigger for the BSCB leakage. BSCB permeability can also be impaired by circulating IL-1β. In contrast, antiinflammatory cytokines TGF-β1 and IL-10 were able to shut down the openings of the BSCB following nerve injury. Peripheral nerve injury caused a decrease in tight junction and caveolae-associated proteins. Interestingly, ZO-1 and occludin, but not caveolin-1, were rescued by TGF-β1. Furthermore, our data provide direct evidence that disrupted BSCB following nerve injury contributed to the influx of inflammatory mediators and the recruitment of spinal blood borne monocytes/macrophages, which played a major role in the development of neuropathic pain. These findings highlight the importance of inflammation in BSCB integrity and in spinal cord homeostasis.
Introduction
As an interface between the spinal cord and the periphery, the blood–spinal cord barrier (BSCB) constitutes a physical/biochemical barrier between the CNS and the systemic circulation, which serves to protect the microenvironment of the spinal cord. Similar to the blood–brain barrier (BBB), the BSCB is composed of a monolayer of nonfenestrated endothelial cells, interconnected by tight junctions (TJs) and surrounded by end feet of astrocytes (Pardridge, 1999). Once thought to be static, the BSCB/BBB is actually a dynamic structure capable of rapid modulation. Breakdown of the BBB/BSCB has been reported in different pathologies affecting the nervous system, including Alzheimer's disease, stroke, multiple sclerosis, and traumatic brain/spinal cord injury; and it is mostly associated with in situ damages in the CNS (Carvey et al., 2009).
Injury to the peripheral nervous system leading to neuropathic pain triggers multidimensional changes along the pain signaling pathway (Basbaum et al., 2009). One of the most salient observations is the inflammatory reaction. After nerve injury, proinflammatory mediators are released from injured nerve fibers and adjacent immune cells (Scholz and Woolf, 2007). Within the spinal cords, remote injury induces substantial changes in microglia and astrocytes, which are important players in the central inflammatory response (Zhang and De Koninck, 2009). In addition, peripheral nerve injury (PNI) evoked trafficking of immune cells from circulation into the spinal cord parenchyma (Zhang et al., 2007; Cao and DeLeo, 2008; Costigan et al., 2009). These neuroimmune interactions participate in the pathogenesis of chronic pain states (McMahon and Malcangio, 2009; White et al., 2009). As a key component of the spinal cord microenvironment, the BSCB could play critical roles in both separating and conjoining the immune system and the CNS. However, the functional states of the BSCB following PNI have not been thoroughly explored. Many questions remain unanswered: whether a remote injury can affect the integrity of the BSCB, and if so, what are the underlying mechanisms regulating such disturbance and what are the consequences of a BSCB breakdown in the spinal cord homeostasis and development of pathological pain.
Two main structural features that contribute to the impermeable characteristics of BBB/BSCB are the presence of TJs and fewer caveolae in CNS endothelium. TJs form a continuous wall that limits paracellular flux of solutes and cells across the endothelial barriers (Huber et al., 2001). Caveolae, flask-shaped invaginations of the plasma membrane, participate in vesicle transport (Couet et al., 2001). How these structures are regulated in relation to BBB/BSCB function in the context of PNI is again not known.
In the present study, we analyzed the permeability of the BSCB following a partial ligation on the sciatic nerve. Our results revealed that PNI disrupted the integrity of the BSCB and inflammatory mediators are key regulators of BSCB function. Disruption of the BSCB following nerve injury causes the influx of inflammatory mediators and the recruitment of circulating immune cells into the spinal parenchyma, which play a critical role in neuroimmune interactions and the development of neuropathic pain.
Materials and Methods
Animals
Male Sprague Dawley rats weighing 250–275 g and male/female C57BL/6 mice weighing ∼25 g (Charles River) were housed under standard 12 h light/dark conditions and received food ad libitum. All protocols were performed in accordance with guidelines from the Canadian Council on Animal Care and were approved by the McGill University and the Laval University Animal Care Committees.
Generation of GFP+ chimeric mice
GFP+ chimeric mice were generated as described previously (Zhang et al., 2007). Briefly, recipient C57BL/6 mice were exposed to irradiation using a cobalt-60 source (Nordion) and then injected via tail vein with 5 × 106 bone marrow cells freshly collected from GFP+ transgenic mice.
Induction of peripheral nerve injury and pain behavioral testing
Partial sciatic nerve ligation was performed according to the method of Seltzer et al. (1990). Rats were anesthetized with isoflurane, and under aseptic conditions the left sciatic nerve was exposed at high-thigh level. The dorsal one-third to one-half of the nerve thickness was tightly ligated with 6-0 silk sutures. Sham-operated rats underwent the same surgical procedure, but the nerve was exposed and left intact. Survival times were 3, 7, 14, 60, and 95 d after surgery. A group of naive rats was included in the protocol as a control. Mechanical and thermal sensitivities were monitored using well calibrated von Frey hair (Stoelting) and Hargreaves apparatus, respectively, with the methods described previously (Hargreaves et al., 1988; Chaplan et al., 1994). Investigators were blinded to the assignment of the groups. GFP+ chimeric mice received a partial ligation on the left sciatic nerve 10 weeks after irradiation and bone marrow transplantation and were killed 14 d after surgery to collect spinal cords.
Induction of experimental autoimmune encephalomyelitis
To induce experimental autoimmune encephalomyelitis (EAE), a group of female C57BL/6 mice were immunized with 200 μg/mouse MOG (myelin oligodendrocyte glycoprotein) peptide 35–55 (custom-synthesized by CS Bio) emulsified in 100 μl of complete Freund's adjuvant (CFA). Control group was inoculated with equal volumes of CFA. Mice were administrated intraperitoneally 200 ng of pertussis toxin dissolved in 200 μl of PBS on the day of immunization and 48 h later. Mice were monitored daily for clinical signs of EAE and scored as follows: 0, no abnormality; 1, limp tail; 2, mild hindlimb weakness; 3, severe hindlimb weakness; 4, complete hindlimb weakness; 5, quadriplegia or premoribund state; and 6, death. At 10–14 d following inoculation, mice with clinical scores of 2 were used for BBB/BSCB integrity evaluation.
Evaluation of BSCB permeability
Evans Blue leakage.
Evans Blue (EB) leakage was assessed with a well established protocol adapted from Xu et al. (2001). EB dye (3% w/v in saline) [molecular weight (MW), 961 Da; Sigma-Aldrich] was injected intravenously under anesthesia. Thirty minutes after the injection, rats were perfused transcardially with saline and rinsed thoroughly until no more blue dye flew out of the right atrium. The choroid plexuses and meninges were removed. Spinal cords and brains were dissected into different regions: lumbar and thoracic/cervical segments for the spinal cord, left and right cerebral cortex, and midbrain/brainstem. Each region was weighed for quantitative measurement of EB extravasation. EB content within the parenchyma was extracted by incubating the tissue in formamide for 48 h at room temperature. The diluents were filtered and subject to spectrophotometric measurements at 620 nm. All measurements were within the range of detection established by the standard curve. R2 of the standard curve was 0.9–1.0. The dye concentration was calculated as the ratio of absorbance relative to the amount of tissue (n = 3–10).
Sodium fluorescein leakage.
BBB and BSCB permeability was also assessed using a micromolecular tracer dye sodium fluorescein (NaFlu) (MW, 376; Sigma-Aldrich) according to a modified procedure by Kaya et al. (2008). Briefly, NaFlu was administered intravenously (10%; 2 ml/kg) and allowed to circulate for 30 min. Then, rats were transcardially perfused with cold saline, for ∼12 min to remove intravascular NaFlu. The choroid plexuses and meninges were removed. Spinal cords and brains were dissected into different regions: lumbar spinal cord, left and right cerebral cortex, and midbrain/brainstem. Each region was weighed for quantitative measurement of NaFlu extravasation. Samples were homogenized in 1 ml of PBS and mixed with a vortex for 2 min after the addition of equal volumes of 60% trichloroacetic acid to precipitate protein. Samples were later cooled for 30 min and centrifuged at 14,000 × g for 10 min. The concentration of NaFlu in supernatant was measured at excitation wavelength of 440 nm and emission wavelength of 525 nm using a spectrophotofluorometer. A standard curve of different amounts of NaFlu was drawn under identical conditions of the assay for calculating dye concentrations in brain and spinal cord. All measurements were within the range of detection established by the standard curve. R2 of the standard curve was 0.85–0.9. NaFlu was expressed as micrograms per gram of tissue.
Plasma protein extravasations.
Plasma protein extravasations were additionally evaluated using immunohistochemical labeling with antibodies against the plasma protein IgG (MW, 53 kDa) and fibronectin (MW, 400 kDa). Free-floating lumbar spinal cord sections were immunolabeled with either rabbit anti-fibronectin (1:500; Millipore Bioscience Research Reagents) or goat anti-rat IgG (1:200; Invitrogen). Vascular markers CD31 and CD34 were used to delineate blood vessels.
Intrathecal infusion of minocycline
The microglial inhibitor, minocycline hydrochloride (Sigma-Aldrich), was freshly dissolved in 0.9% sterile, isotonic saline and delivered intrathecally at a dose of 150 μg per day for 7 d (day 0 to day 7) using an osmotic pump (Alzet) with a flow rate of 1 μl/h connected to an intrathecal catheter (n = 5–8 in each group). The dose for intrathecal minocycline was selected according to previous studies (Ledeboer et al., 2005; Lin et al., 2007).
Immune modulation of BSCB function
Recombinant proinflammatory or antiinflammatory molecules were dissolved in sterile 0.9% saline and delivered either intravenously or intrathecally into naive or nerve-injured animals. Detailed protocols are listed in Table 1.
Table 1.
Experimental protocols for immune modulation of BSCB integrity
| Molecules | Animals | Duration | Administration route | Doses (source) |
|---|---|---|---|---|
| Rat recombinant MCP-1 | Naive rats | 3 d | Intrathecal (osmotic pumps) | 2.5 μg (PeproTech) |
| Rat MCP-1 neutralizing antibody | Nerve-injured rats | 3 d (day 0 to day 3) | Intrathecal (osmotic pumps) | 3 μg (PeproTech) |
| Recombinant TGF-β1 (mammalian derived) | Nerve-injured rats | 3 d (day 0 to day 3) | Intrathecal (osmotic pumps) | 2 μg (PeproTech) |
| Recombinant IL-10 (mammalian derived) | Nerve-injured rats | 3 d (day 0 to day 3) | Intrathecal (osmotic pumps) | 3 μg (PeproTech) |
| Recombinant TGF-β1 (mammalian derived) | Naive rats | 3 d | Intrathecal (osmotic pumps) | 2 μg (PeproTech) |
| Recombinant TGF-β1 (mammalian derived) | Nerve-injured GFP+ chimeric mice | 14 d (day 0 to day 14) | Intrathecal, punction (every 3 d) | 1 μg/injection (PeproTech) |
| 125I-recombinant human IL-1β | Naive rats | 10 min | Intravenous | 20–5000 immune units (PerkinElmer) |
Permeation to circulating IL-1β
BSCB permeation to circulating IL-1β was assessed using 125I-labeled IL-1β according to a previously described method (Pan et al., 2003). Briefly, 125I-IL-1β was dissolved in 0.9% saline and a bolus of 150 μl of the radiolabeled IL-1β (0.7 μCi) was injected into the left ventricle. The blood sample was collected 10 min later from the right ventricle. The animals were then perfused with saline and lumbar spinal cords were collected. The 125I radioactivity in serum and in lumbar spinal cord was measured separately using a gamma counter (PerkinElmer). The ratio of radioactivity in the spinal cord to that of serum was plotted against exposure time, the latter being the theoretical value of steady-state serum radioactivity. Ten minute exposure to 125I-IL-1β was chosen based on the fact that, during this short period, the radioactivity in the tissue parenchyma represents most likely intact 125I-cytokine rather than degraded products (Pardridge, 1999). A control group was included in which 125I-IL-1β was dissolved in 3% EB. After measurement of the radioactivity, the spinal cord samples were incubated in formamide for 48 h in the dark to extract and assess EB content in the parenchyma as described above.
Microvessel isolation
Spinal microvessels from naive and injured rats (day 3) were isolated according to the method described by Hoehn et al. (2002). Rats were decapitated under anesthesia, and spinal cords were removed quickly. The meninges were excised and the lumbar segments were homogenized in microvessel isolation buffer containing protease inhibitor (Sigma-Aldrich). Dextran (26% m/v) was added to the homogenate afterward at 4°C. Samples were then gently vortexed and centrifuged (5600 × g; 4°C) for 10 min, and the supernatant was discarded. Pellets were resuspended in fresh microvessel isolation buffer and passed through a 70 μm filter (BD Biosciences). Filtered homogenates were pelleted by centrifugation at 3000 × g for 10 min. The supernatant was removed, and the pellet, which was enriched in spinal microvessels, was collected for protein extraction. The purity of collected microvessels was verified with immunostaining using two endothelial markers von Willebrand factor (VWF) (1:1000; Abcam) and Glut-1 (1:500; Millipore).
Western blot
The pellet containing spinal microvessels was resuspended in 500 μl of CellLytic (Sigma-Aldrich) with protease inhibitor. Thirty micrograms of proteins extracted from the above preparation were loaded and separated on 10% SDS-PAGE gel. After the transfer, blots were incubated overnight at 4°C with the following antibodies: zonula occludens-1 (ZO-1) (1:200; Cell Signaling), occludin (1:100; Cell Signaling), caveolin-1 (1:250; Cell Signaling), TGF-β receptor I (TGF-β-RI) (1:200; Santa Cruz), and p-Smad2/3 (1:500; Santa Cruz). For loading control, blots were probed with β-actin antibody (1:10,000; Sigma-Aldrich). Density of specific bands from Western blotting was quantified with a computer-assisted imaging analysis system (Image Pro Plus). Three separate experiments where each treatment group consists of pooled microvessels from two animals were included.
Immunohistochemistry
To identify the expression of TGF-β receptors on endothelial cells, immunofluorescent staining was performed on spinal microvessels extracted as explained above. Microvessels were fixed with 4% PFA for 30 min and incubated overnight at 4°C with rabbit anti-TGF-β-RI polyclonal antibody (1:250; Santa Cruz Biotechnology) and rat anti-CD31 (for endothelial cells; 1:250; BD Biosciences), followed by a 60 min incubation at room temperature in fluorochrome-conjugated goat secondary antibody. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was also used as a nuclear counterstain (1:10,000; Sigma-Aldrich).
The following antibodies were also used to examine the spinal microglial activation and immune cell trafficking following nerve injury. Free-floating sections from lumbar spinal cords were incubated overnight at 4°C with rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba-1) polyclonal antibody (for microglia, 1:1000; Wako Chemicals) and anti-CD3 monoclonal antibody (for T-lymphocytes, 1:200; AbD Serotec), followed by a 60 min incubation at room temperature in fluorochrome-conjugated goat secondary antibody. Anti-CD2 monoclonal antibody (1:100; AbD Serotec) was incubated for 72 h, at 4°C, and followed by tyramide signal amplification (PerkinElmer).
Image acquisition
Images were acquired using an Olympus BX51 microscope equipped with a color digital camera (Olympus DP71) or Olympus confocal laser-scanning microscope (Fluoview 1000). GFP+ cells were counted by a blind investigator in four different regions of interest [ipsilateral dorsal horn (DHi), contralateral dorsal horn (DHc), ipsilateral ventral horn (VHi), and contralateral ventral horn (VHc)], in mice treated with saline or with recombinant TGF-β1. Only ramified GFP+ cells within parenchymal gray matter were included. Colocalization of Iba-1 with GFP and the distribution of GFP+ cells outside of Glut-1-labeled vessels were ensured with confocal Z stacks at 0.8 μm intervals and visualization in three-dimensional orthogonal planes. Similar approaches were applied to the quantification of CD2+ and CD3+ cells in the dorsal horns of the spinal cords.
Statistical analysis
To determine statistical significance, unpaired t test was used for the difference between groups (TGF-β1/MCP-1/IL-1β/IL-10 treated vs saline treated at each time point); paired t test was used for the difference between ipsilateral and contralateral sides within the same group; two-way ANOVA followed by Bonferroni's post tests were used for the analysis of behavioral data. A value of p < 0.05 was accepted as statistically significant. Values are presented as means ± SEM.
Results
Development of neuropathic pain following peripheral nerve injury
Hypersensitivity to mechanical and thermal stimuli are characteristic behaviors developed following nerve injury. In this study, shortly after the lesion on the sciatic nerve, rats exhibited mechanical allodynia (Fig. 1A,B) and thermal hyperalgesia (Fig. 1C,D), which lasted for at least 2 months, the end of the testing period.
Figure 1.

Development of neuropathic pain following injury on sciatic nerve. Shortly after the partial ligation on the sciatic nerve, rats develop mechanical allodynia and thermal hyperalgesia in the ipsilateral hindpaws. Withdrawal threshold of both ipsilateral (A) and contralateral (B) paws to calibrated von Frey hair stimulation decreased significantly, starting from day 3 until at least day 63 after injury. Withdrawal latency to noxious heat stimuli was also decreased at ipsilateral side (C), and to a less extent at the contralateral paw (D). N = 6/group. Values are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 from baseline.
Peripheral nerve injury leads to leakage of different size molecules into the lumbar spinal cord parenchyma
BSCB permeability following sciatic nerve injury was assessed by different approaches. Intravenous administration of dyes with molecular weight >180 Da precludes passage across an intact barrier. As one of the largest tracers, EB (MW, 961 Da) has a high affinity for plasma protein albumin in blood circulation, which gives rise to a high-molecular complex (EB-albumin, 68,500 Da) with limited penetration of the BSCB and BBB in normal conditions. Although the injury occurred on the sciatic nerve and the spinal cord was physically intact, the EB content within the lumbar spinal cord (1 cm segment around the L4–L5 level) was significantly increased when compared with naive rats (Fig. 2A). The leakage was most prominent at early time points, day 3 to day 7 after injury (p < 0.01), and remained significant at least for 1 month (p < 0.05). Two months after the nerve ligation, BSCB permeability to EB returned to normal levels as seen in naive rats. No significant increase of EB was detected in the thoracic spinal cord and in the brain of rats with sciatic nerve injury (Fig. 2B). To confirm whether there is any leakage of small molecules at chronic stages (>2 months) or diffused propagation into other CNS regions, we applied intravenously one of the smallest tracers, NaFlu (MW, 376 Da) in rats having nerve injury. The content of NaFlu within lumbar spinal cords at 2 and 3 months after nerve injury was similar to that of naive and sham-operated animals, whereas the increase of NaFlu in lumbar spinal cords at 3 d after injury was dramatic (Fig. 2C). The extravasation of NaFlu was again restricted locally to the lumbar spinal cord (Fig. 2D). To further localize anatomical distribution of the leakage, additional assessment was performed by using immunolabeling with antibodies against rat IgG and fibronectin, two plasma proteins that should not be found in normal spinal cord parenchyma. As shown in Figure 2E, while no positive IgG or fibronectin staining was detected in naive spinal cords, IgG- and fibronectin-immunopositive deposits were found within the spinal cord of rats following sciatic nerve injury. The signal (extracellular aggregates) was essentially located in the ipsilateral side. Extravasation of IgG and fibronectin was confirmed by costaining with the vascular markers CD34 and CD31 (Fig. 2E). To better understand the scope of the nerve injury-triggered BSCB disruption, we used the same protocol to compare the EB leakage in EAE mice, an animal model of multiple sclerosis where the BBB/BSCB impairment has been well established (Hawkins and Davis, 2005; Fabis et al., 2007). As illustrated in Figure 2F, EAE-associated EB leakage is much more pronounced and diffused in different segments of the spinal cords and in the brain. In contrast, peripheral nerve injury-triggered BSCB compromise is most likely restricted in the corresponding lumbar spinal cord.
Figure 2.
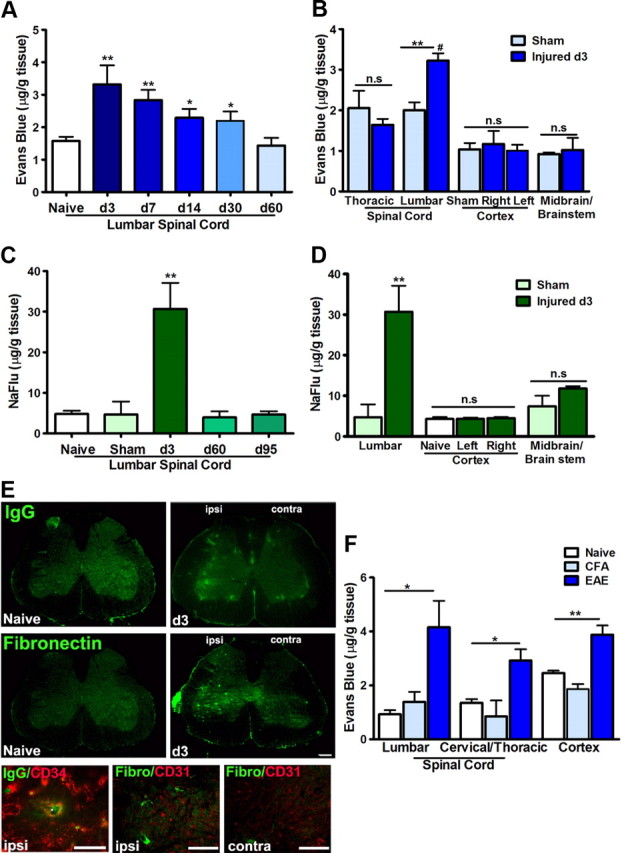
Peripheral nerve injury impairs BSCB permeability. A, Evans Blue (MW, 961 Da; EB-albumin, MW, 68,500 Da) extravasation within lumbar spinal cords at different time points after nerve injury. Note there is a prominent increase of EB content within lumbar spinal cords during the first 4 weeks after nerve injury, which returns to normal levels at 2 months. B, Quantification of EB leakage in different regions of the spinal cord and brain at day 3 after injury; the increase of EB is only significant in the lumbar spinal cord. C, NaFlu (MW, 376 Da) content at different time points after nerve injury. At late time points (>2 months), while BSCB permeability to EB is back to normal levels, the passage of small molecular (NaFlu) through BSCB is at the similar levels of naive animals as well; however, the increase of NaFlu at day 3 after injury is dramatic. D, NaFlu content in lumbar and brain regions at day 3 after injury. A significant extravasation is only observed in the lumbar segment. E, Leakage of plasma protein IgG and fibronectin in the spinal parenchyma 3 d after nerve injury with preference in the ipsilateral side. Endothelium markers CD31 and CD34 delineated blood vessels and confirmed the presence of plasma protein in the parenchyma. F, Evans Blue extravasation is generalized throughout the spinal cord and brain in EAE mice. Values are presented as means ± SEM. **p < 0.01, *p < 0.05 versus naive or sham; #p < 0.05 versus thoracic segment. N = 6–10/group. Scale bar, 50 μm.
Microglial activation is not required for BSCB disruption
To understand whether nerve injury modulates the BSCB permeability directly or such effect depends on the activation of resident microglial cells in the spinal cord, we infused intrathecally a microglial inhibitor minocycline from day 0 to day 7. As reported previously (Lin et al., 2007), such a treatment significantly prevented the increase of Iba-1 immunoreactivity (IR) (Fig. 3A,B) and partially reduced pain development (Fig. 3C). However, minocycline did not affect the extravasation of EB (Fig. 3D), when compared with saline (intrathecally)-treated animals, which suggest that BSCB disruption is not dependent on microglial activation in the spinal cord.
Figure 3.

BSCB disruption is not affected by the suppression of spinal microglial activation. A, Photomicrographs depicting Iba-1 staining in the lumbar spinal cord. Partial sciatic nerve ligation induced a remarkable increase of Iba-1 IR at the ipsilateral side in the dorsal and ventral horns. Intrathecal minocycline (150 μg/d for 7 d) successfully reduced this Iba-1+ IR. B, Quantification of the area occupied by the Iba-1+ IR reveals a striking reduction of microglial activation in the minocycline-treated animals compared with saline-treated animals. C, Mechanical allodynia (left panel) and thermal hyperalgesia (right panel) were partially attenuated in minocycline-treated animals. D, Despite inhibition of microglia, EB leakage into the spinal cord remained elevated in minocycline-treated animals at day 7. Data are shown as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, minocycline-treated versus saline-treated groups. N = 6/group. Scale bar, 250 μm.
Inflammatory mediators modulate BSCB function
MCP-1 is critical in nerve injury-induced BSCB disruption
The chemokine MCP-1 is released by damaged neurons following nerve injury (Zhang and De Koninck, 2006). To assess the role of endogenous MCP-1 in BSCB integrity, we intrathecally infused an antibody against MCP-1 (3 μg) in injured animals. Compared with injured animals infused with saline, MCP-1 antibody significantly reduced EB extravasation in the spinal cord at day 3 after injury (Fig. 4A) (p < 0.05 vs saline). Next, we intrathecally infused naive animals with recombinant MCP-1 (2.5 μg) for 3 d and then assessed permeability of BSCB to EB. Exogenous MCP-1 evoked an increase of EB content in the spinal parenchyma in naive animals similar to that seen in nerve-injured animals at day 3 (Fig. 4B) (p < 0.01 vs naive).
Figure 4.

Inflammatory mediators modulate BSCB integrity. A, Neutralizing endogenous MCP-1 with a MCP-1 antibody (3 μg for 3 d) significantly reduced the leakage of EB in the spinal cord observed at day 3 after injury. B, Intrathecal infusion of recombinant murine MCP-1 (2.5 μg for 3 d) produced an increase of EB content in the spinal cord in naive animals, similar to that seen in nerve-injured animals. Intrathecal catheterization provoked a slight, but nonsignificant increase of EB content in the spinal cord. C, IL-1β injected intravenously caused a dose-dependent impairment of BSCB permeability to EB. D, Intrathecal administration of antiinflammatory cytokines TGF-β1 (2 μg) and IL-10 (3 μg) for 3 d significantly reduced the EB content in the spinal cord increased by peripheral nerve injury. TGF-β1 did not affect the BSCB permeability in naive rats. N = 3–6/group. Values are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus naive rats without any treatment; #p < 0.05 versus injured saline-treated.
Systemic IL-1β induces EB extravasation in the spinal cord
Low doses of IL-1β, 10–50 immune units (IU) injected intravenously did not provoke any significant increase of EB content in the spinal cord (Fig. 3C). However, higher concentrations of IL-1β, 1000–5000 IU did dose-dependently increase the EB permeation in the spinal cord, which is significant at 5000 IU (p < 0.001 vs saline) (Fig. 4C).
TGF-β1 and IL-10 block EB extravasation in the spinal cord following peripheral nerve injury
To evaluate whether TGF-β1, as an antiinflammatory cytokine, is effective in preventing nerve injury-induced BSCB disruption, we administrated TGF-β1 (2 μg) in rats having ligation on the sciatic nerve. Three day intrathecal infusion (from day 0 to day 3) of TGF-β1 successfully prevented the EB extravasation into the spinal cord following PNI (p < 0.01 vs saline) (Fig. 4D). Similar blocking effects were observed using another antiinflammatory cytokine IL-10 (Fig. 4D). TGF-β1 did not alter the normal physiological properties of the BSCB in naive animals (Fig. 4D).
Peripheral nerve injury alters tight junction complex protein expression
In view of the critical roles of tight junctions in maintaining BSCB integrity, we examined the effect of PNI on the expression of some proteins important to tight junction structure and function, including plasma protein ZO-1 and membrane protein occludin. As caveolae complexes play an important role in transcytosis of numerous substrates (Tuma and Hubbard, 2003; Kirkham and Parton, 2005), including immune mediators (Middleton et al., 1997), we also sought to determine whether the expression of caveolin-1, the major structural protein of caveolae (Cohen et al., 2004; Stan, 2005) was altered in the spinal cord by nerve injury. Western blot analysis was performed using microvessels isolated from lumbar spinal cord segments (Fig. 5A). In comparison with naive or sham-operated rats, all three examined proteins, ZO-1 (Fig. 5B), occludin (Fig. 5C), and caveolin-1 (Fig. 5D), were downregulated 3 d after a partial ligation on the sciatic nerve, when maximal disruption of the BSCB was observed. Relative levels of targeted protein expression were determined by densitometric analysis and shown in Figure 5E.
Figure 5.

Downregulation of tight junction proteins following peripheral nerve injury. Microvessels isolated from the spinal cord were positive for endothelium markers, VWF and Glut-1 (A). The expression of tight junction-associated protein ZO-1 (B), occludin-1 (C), and caveolin-1, a major player in the formation of caveolae (D), was significantly lower in microvessels of animals day 3 after nerve lesion than controls (naive and sham). Quantitative assessment of protein levels is illustrated in E. Three separate experiments in which each treatment group consists of pooled microvessels from two animals were included. Values are presented as means ± SEM. *p < 0.05, **p < 0.01. Scale bar, 20 μm.
TGF-β1 prevents the downregulation of tight junction proteins following peripheral nerve injury
To determine how TGF-β1 prevents the BSCB dysfunction associated with nerve injury, we examined the effects of TGF-β1 on the expression of tight junction proteins. Intrathecal infusion of TGF-β1 was able to prevent the downregulation of tight junction proteins ZO-1 and occludin following PNI (Fig. 6A,B,D). However, TGF-β1 failed to rescue the nerve injury-associated downregulation of caveolin-1 (Fig. 6C,D). We have reported previously that, as a potent antiinflammatory cytokine, TGF-β1 inhibited the neuronal MCP-1 expression and reduced the spinal cytokine level in the same nerve injury model through its pleiotropic effects on neurons and glia (Echeverry et al., 2009). To further explore the mechanism of TGF-β1 in modulating BSCB integrity, we examined the direct effects of TGF-β1 on endothelial cells. TGF-β-RI was detected in isolated endothelial cells (Fig. 6E). Neither nerve injury nor exogenous ligand modified significantly the quantity of the receptor on microvessels (Fig. 6F). However, intrathecal infusion of TGF-β1 did activate the Smad2/3 signaling pathway in endothelial cells, evidenced by an increase of phosphorylated Smad2/3 (Fig. 6G). Therefore, we think that TGF-β1 regulates BSCB integrity through its direct effects on endothelial cells and its indirect effects in inhibiting local inflammation.
Figure 6.
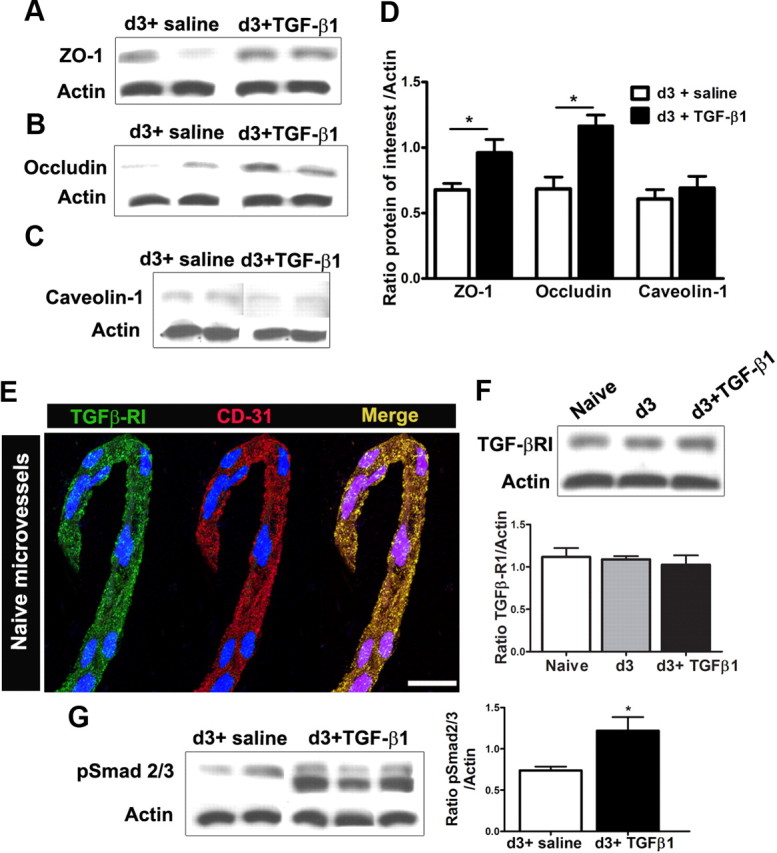
TGF-β1 alters tight junction protein levels in spinal cord microvessels. Intrathecal administration of TGF-β1 (2.5 μg for day 0 to day 3) successfully prevents the decrease in tight junction protein levels ZO-1 (A, D) and occludin (B, D) observed after peripheral nerve injury. Levels of caveolae structural component caveolin-1, however, remained unchanged (C, D). The receptor for TGF-β1 (TGF-β-RI) is found in isolated spinal cord endothelial cells, confirmed by the colocalization with vascular marker CD31. DAPI staining (blue) was used for cellular identification (E). The protein levels of the TGF-β-RI remain unchanged after peripheral nerve injury, with or without TGF-β1 infusion (F). TGF-β1 treatment induced the phosphorylation of signaling pathway proteins Smad2/3 (pSmad2/3) in spinal cord microvessels (G). Three separate experiments in which each treatment group consists of pooled microvessels from two animals were included. Values are presented as means ± SEM. *p < 0.05 versus d3 + saline. Scale bar, 10 μm.
Peripheral nerve injury promotes cytokine penetration and immune cell infiltration into the spinal cord parenchyma
Permeation to circulating cytokine IL-1β
125I-labeled recombinant human IL-1β was used as a tracer to test whether the disruption of the BSCB caused by PNI could be accompanied by a passive influx of proinflammatory cytokines into the CNS parenchyma. We first coadministrated 125I-IL-1β (0.7 μCi/150 μl) with EB, to identify a minimal dose for the detection of radioactivity and to ensure the increased entrance of IL-1β occurred through preexisting compromised BSCB provoked by PNI, not due to the interference of injected IL-1β as seen in Figure 4C. As shown in Figure 7A, the EB content was similar in 125I-IL-1β-injected and in saline-injected rats, either naive or nerve-injured animals, which confirms that the dose of IL-1β used in this experiment, with 10 min exposure, did not alter the BSCB permeability. However, 125I radioactivity detected in the spinal parenchyma was significantly higher in rats 3 d after PNI than those in naive rats (Fig. 7B).
Figure 7.

Peripheral nerve injury induced disruption of BSCB provided access to circulating immune mediators into the spinal cord parenchyma. A, Intracardiac injection of 125I-IL-1β (0.7 μCi/150 μl) did not affect BSCB permeability to EB in either naive or nerve-injured rats. B, There was a significant increase in the uptake of 125I-IL-1β in the lumbar spinal cord 3 d after sciatic nerve injury compared with control groups. Values are presented as means ± SEM. *p < 0.05, **p < 0.01 versus naive.
Infiltration of GFP+ bone marrow-derived monocytes/macrophages and CD2+/CD3+ lymphocytes into the spinal cord
We previously reported a remarkable and selective infiltration of circulating monocytes into the spinal cord parenchyma after PNI (Zhang et al., 2007). However, whether a compromised BSCB is required and plays a critical role in such cell recruitment is unknown. As shown in Figure 4D, TGF-β1 has the ability to restore BSCB function. Here, we injected intrathecally recombinant TGF-β1 in GFP+ chimeric mice, starting from the day of the injury (day 0) and until day 14, to evaluate whether a compromised BSCB is necessary for GFP+ cell infiltration in the spinal cord parenchyma. Consistent with our previous findings, a striking number of GFP+ cells, derived from bloodborne monocytes/macrophages were detected in the ipsilateral side of the L4–L5 spinal cord in injured animals (Fig. 8A). These recruited GFP+ cells were differentiated into ramified microglia and located in the parenchyma, beyond blood vessels (Fig. 8A). The immune cell trafficking was almost completely prevented in injured animals following recombinant TGF-β1 administration (Fig. 8A). The number of ramified GFP+ cells in TGF-β1-injected mice (5.42 ± 0.67 in DHi and 4.91 ± 0.94 in VHi) was significantly lower than that injected with saline (29.08 ± 1.17 in DHi and 30.25 ± 8.92) (Fig. 8A). Although the most predominant phenotype of cells infiltrating into the spinal cord is monocyte/macrophages, we also found few CD2+ and CD3+ lymphocytes recruited into the CNS. This T-cell trafficking was prevented at the presence of TGF-β1 (Fig. 8C,D).
Figure 8.

Peripheral nerve injury-induced disruption of BSCB provided access to circulating immune cells into the spinal cord parenchyma. A, GFP+ bone marrow-derived cells infiltrated into the spinal cords after PNI, which differentiated into Iba-1+ microglia. Intrathecal injection of TGF-β1 prevented the entrance of circulating immune cells at 14 d after nerve injury. Evidence on localization of infiltrated GFP+ bone marrow-derived cells in the parenchyma is shown by coimmunostaining with vessel marker Glut-1. Scale bars: left, 250 μm; middle, 10 μm; right, 20 μm. B, Quantitative analysis of GFP+ cells in the spinal cords of GFP chimeric mice after PNI and TGF-β1 treatment. C, To compare with naive animals, there was a marked infiltration of CD2+ and CD3+ lymphocytes into the ipsilateral side of spinal cords after peripheral nerve injury (white arrows). Intrathecal injection of TGF-β1 prevented the entrance of circulating lymphocytes at 7 d after nerve injury. Scale bar, 50 μm. D, Quantitative analysis of CD2+ and CD3+ cells in the dorsal horns of spinal cords of rats after nerve injury and TGF-β1 treatment. TGF-β1 successfully reduced the number of cells invading the spinal parenchyma. N = 6/group. Values are expressed as means ± SEM. *p < 0.05, **p < 0.01 versus controls (naive and sham); #p < 0.05, ##p < 0.01, ###p < 0.001 versus saline.
Discussion
The present study provides evidence that PNI can impair BSCB permeability in a transient and restricted manner, which is mediated by the chemokine MCP-1 released from damaged neurons. The compromised BSCB allows penetration of both inflammatory molecules and immune cells into the spinal cord, participating in the central inflammatory response, a critical process for the development of neuropathic pain.
BBB/BSCB disruption has been observed in many CNS pathologies as a consequence of direct trauma or inflammation in the CNS parenchyma (de Vries et al., 1997; Hawkins and Davis, 2005; Banks and Erickson, 2010). Recent work has shown that insults in the periphery can also open the BBB/BSCB. Injection of complete Freund's adjuvant into the hindpaw increased BBB permeability (Brooks et al., 2005). Injury on peripheral nerves altered BSCB function (Gordh et al., 2006; Beggs et al., 2010). Here, we describe a detailed spatial and temporal kinetics of BSCB alteration associated with partial ligation on the sciatic nerve (Seltzer et al., 1990). The BSCB displayed an early, but transient (up to 1 month), extravasation of large plasma proteins (EB-albumin complex; MW, 68,500 Da), as well as small molecules (NaFlu; MW, 376 Da) into the spinal cord parenchyma. This was restricted to the corresponding lumbar segment, without any significant impact in other regions of the spinal cord or the brain. The BSCB compromise was consistent, both temporally and spatially, with the spinal inflammatory reaction following PNI (Basbaum et al., 2009; Zhang and De Koninck, 2009). However, it is in contrast with the extensive and diffused BBB/BSCB disruption in EAE mice observed by us and others (Bennett et al., 2010), which is accompanied by a generalized inflammatory reaction in the disease pathology.
Much progress has been made toward illustrating the mechanisms underlying the spinal inflammatory reaction in response to PNI. Among various inflammatory mediators released in the spinal cord, the chemokine MCP-1 stands out as an important trigger of microglial activation (Zhang et al., 2007). Some in vitro and in vivo studies have shown that MCP-1 may also regulate endothelial function (Stamatovic et al., 2005; Ge et al., 2008). We demonstrated here that MCP-1 is necessary for BSCB disruption following peripheral nerve injury, because MCP-1 antibody treatment prevented the increase of permeability to EB, and recombinant MCP-1 delivered in naive animals mimicked the effects seen in nerve-injured rats. The expression of CCR2 on brain endothelium (Dzenko et al., 2005), especially on the abluminal surface (Andjelkovic et al., 1999), supports the findings that parenchymally deposited MCP-1 has the ability to open the BSCB and to stimulate immune cell trafficking across the BSCB. Together with our previous report (Zhang et al., 2007), we implicate MCP-1, released by damaged neurons, not only as a necessary chemoattractant for microglial activation but also as an important player for regulating BSCB function. The key role of neuronal-derived MCP-1 in altering BSCB integrity is further supported by the fact that activated microglia, where MCP-1 expression is absent (Zhang and De Koninck, 2006), is not required for opening the BSCB.
Beggs et al. (2010) have demonstrated that electrical stimulation of the sciatic nerve at intensity sufficient to activate C-fibers can trigger a delayed increase in BSCB permeability. The phenomenon can be explained by a neurogenic inflammation following electrical stimulation. Activation of C-fibers stimulates the release of substance P and CGRP (calcitonin gene-related peptide) from small peptidergic neurons (Zieglgänsberger et al., 2005), and subsequent release of other inflammatory mediators including cytokines (Sahbaie et al., 2009). All these molecules can be good candidates to affect BSCB permeability. This is probably why there was a delay in the increase of BSCB permeability following C-fiber stimulation, and moreover stimulation of A-fibers alone was not able to modify BSCB integrity. It should be noted that, in addition to act as a local anesthetic, lidocaine also has powerful antiinflammatory properties (Sinclair et al., 1993; Craner et al., 2005; Gu et al., 2008). Therefore, the observed effects using lidocaine may not be entirely attributed to the blockade of electrical activities. Further investigation is needed to clarify whether central release of MCP-1 depends on electrical firing.
Cytokines are important molecules of the immune system involved in the alteration of the BBB/BSCB integrity (McColl et al., 2007), and their roles in neuropathic pain have been extensively studied (Ren and Torres, 2009). Elevated serum levels of proinflammatory cytokines have been reported in neuropathic pain patients (Davies et al., 2007; Yu et al., 2009). A dose-dependent effect of systemic IL-1β on EB extravasation suggests that circulating inflammatory molecules also have the capability to modulate the BBB/BSCB permeability. Systemic IL-1β-induced alterations in BSCB can be mediated through a direct interaction with the IL-1β receptor located in the endothelium (Konsman et al., 2004) or indirectly, through IL-1β-stimulated release of other chemokines having their corresponding receptors on blood vessels (Shaw and Greig, 1999; Ubogu et al., 2006).
Further support for such an inflammation-mediated regulation of BSCB/BBB function is that, while proinflammatory molecules promoted the openings of BSCB, antiinflammatory cytokines, TGF-β1 or IL-10, which have been shown to be able to inhibit spinal microglial activation and to reduce the expression of proinflammatory molecules (Moore et al., 2001; Echeverry et al., 2009; Soderquist et al., 2010), prevented the disruption of BSCB triggered by nerve injury. These antiinflammatory cytokines were effective in relieving experimental neuropathic pain (Echeverry et al., 2009; Soderquist et al., 2010).
The molecular and cellular mechanisms underlying the detrimental effects of nerve injury on BSCB permeability remain unknown. Regulation and redistribution of TJ-associated proteins have been postulated as key players. Accompanied by a significant increase of BSCB permeability, a rapid decrease of cytoplasmic ZO-1 and transmembrane occludin have been found in spinal microvessels 3 d after injury. In addition, the loss of TJ proteins can be rescued by exogenous TGF-β1, in parallel with the recovery of the BSCB integrity. We assume that observed effects of TGF-β1 in modulating TJ protein expression and BBB/BSCB function might yield from its antiinflammatory effects, although they may be attributed to a direct activation of endothelial cells. TGF-β1 receptors are present on the surface of these cells and the Smad2/3 signaling pathway is activated in response to the ligand (Ronaldson et al., 2009). Another structural feature that can contribute to the relative impermeability of BBB/BSCB is caveolae-associated changes. Endothelial cells allow transport of substances from the blood to the CNS via several routes among which transcytosis is highly mediated by caveolae complexes. Caveolae functions rely on caveolin-1, a scaffolding protein that drives the formation of plasma membrane caveolae and anchors them to the actin cytoskeleton. Caveolin-1 is increased in different pathological conditions associated with alterations of BBB permeability through enhanced transcytosis (Nag et al., 2009). However, we have detected a significant decrease of cavelolin-1 protein expression in microvessels isolated from lumbar spinal cords of rats having PNI. These results might indicate a lack of major involvement of the transcytosis route in PNI-triggered BSCB compromise. Recent work, however, has also highlighted the role of caveolin-1 in the organization of TJ protein (Nusrat et al., 2000). Interestingly, reduced expression of caveolin-1 has been shown to cause TJ-associated protein disruption in a mechanism dependent on endothelial cell exposure to MCP-1 (Song et al., 2007). Whether changes of caveolin-1 are relevant to BSCB alterations and whether such changes can affect the spatial reorganization of TJ in our context are not clear and warrant more experiments for clarification.
Loss of BSCB integrity may have manifold consequences (Banks and Erickson, 2010), through which circulating immune mediators could reach the spinal cord. Our results provided clear evidence that systemic IL-1β has access to the CNS via a disrupted BSCB. Although the active transport was not tested in this study, the passage of IL-1β most likely corresponded to a passive diffusion into the spinal cord parenchyma since the human IL-1β used in this study cannot cross the BSCB through active mechanisms in the rat (Plotkin et al., 2000). Increasing evidence suggests that proinflammatory cytokines enhance pain via central mechanisms (DeLeo and Yezierski, 2001; Kawasaki et al., 2008). We demonstrated here that, in addition to being produced in situ by activated glial cells and/or damaged neurons, cytokines, such as IL-1β, can also be available by permeation from blood to the spinal cord. The breakdown of BSCB allowed the entrance of circulating immune cells. PNI-induced recruitment of GFP+ cells was also significantly reduced in the spinal cord parenchyma of chimeric mice treated with TGF-β1. The ability of circulating immune cells to populate the CNS is further supported by T-lymphocyte infiltration observed in rats having spared nerve injury (Costigan et al., 2009) or partial sciatic nerve injury (current study) and in mice having spinal nerve transection (Cao and DeLeo, 2008). The cross talk between the immune and nervous systems through impaired BBB/BSCB may have significant impact on long-term molecular and cellular changes in pain pathways and persistent pain behavior. Another significant clinical consequence of these findings that deserves to be mentioned is that the period in which BSCB remains more permeable may directly influence the pharmacokinetics of drugs that are administered systemically.
Footnotes
This work was supported by Canadian Institutes for Health Research (CIHR) Grant MOP-77624 (J.Z.), the Fonds de la Recherche en Santé du Québec/Quebec Pain Research Network/Pfizer partnership (J.Z.), The Louise and Alan Edwards Foundation (J.Z.), and The Alan Edwards Center for Research on Pain/AstraZeneca Alliance (J.Z.), and by a Canadian Pain Society Trainee Grant (S.E.). We acknowledge the support from the CIHR Neuroinflammation Training Program. S.E. holds a CIHR doctoral studentship. J.Z. is a CIHR new investigator, and S.R. is a Canadian Research Chair in Neuroimmunology.
References
- Andjelkovic AV, Spencer DD, Pachter JS. Visualization of chemokine binding sites on human brain microvessels. J Cell Biol. 1999;145:403–412. doi: 10.1083/jcb.145.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs S, Liu XJ, Kwan C, Salter MW. Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol Pain. 2010;6:74. doi: 10.1186/1744-8069-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, McQuaid S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. 2010;229:180–191. doi: 10.1016/j.jneuroim.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. Am J Physiol Heart Circ Physiol. 2005;289:H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. 2008;38:448–458. doi: 10.1002/eji.200737485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvey PM, Hendey B, Monahan AJ. The blood-brain barrier in neurodegenerative disease: a rhetorical perspective. J Neurochem. 2009;111:291–314. doi: 10.1111/j.1471-4159.2009.06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couet J, Belanger MM, Roussel E, Drolet MC. Cell biology of caveolae and caveolin. Adv Drug Deliv Rev. 2001;49:223–235. doi: 10.1016/s0169-409x(01)00139-9. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia. 2005;49:220–229. doi: 10.1002/glia.20112. [DOI] [PubMed] [Google Scholar]
- Davies AL, Hayes KC, Dekaban GA. Clinical correlates of elevated serum concentrations of cytokines and autoantibodies in patients with spinal cord injury. Arch Phys Med Rehabil. 2007;88:1384–1393. doi: 10.1016/j.apmr.2007.08.004. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- de Vries HE, Kuiper J, de Boer AG, Van Berkel TJ, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev. 1997;49:143–155. [PubMed] [Google Scholar]
- Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc Res. 2005;70:53–64. doi: 10.1016/j.mvr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Haw A, Liu H, Zhang ZW, Zhang J. Transforming growth factor-beta1 impairs neuropathic pain through pleiotropic effects. Mol Pain. 2009;5:16. doi: 10.1186/1744-8069-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabis MJ, Scott GS, Kean RB, Koprowski H, Hooper DC. Loss of blood-brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc Natl Acad Sci U S A. 2007;104:5656–5661. doi: 10.1073/pnas.0701252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Song L, Serwanski DR, Kuziel WA, Pachter JS. Transcellular transport of CCL2 across brain microvascular endothelial cells. J Neurochem. 2008;104:1219–1232. doi: 10.1111/j.1471-4159.2007.05056.x. [DOI] [PubMed] [Google Scholar]
- Gordh T, Chu H, Sharma HS. Spinal nerve lesion alters blood-spinal cord barrier function and activates astrocytes in the rat. Pain. 2006;124:211–221. doi: 10.1016/j.pain.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Gu YW, Su DS, Tian J, Wang XR. Attenuating phosphorylation of p38 MAPK in the activated microglia: a new mechanism for intrathecal lidocaine reversing tactile allodynia following chronic constriction injury in rats. Neurosci Lett. 2008;431:129–134. doi: 10.1016/j.neulet.2007.11.065. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hoehn BD, Harik SI, Hudetz AG. VEGF mRNA expressed in microvessels of neonatal and adult rat cerebral cortex. Brain Res Mol Brain Res. 2002;101:103–108. doi: 10.1016/s0169-328x(02)00175-4. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya M, Gurses C, Kalayci R, Ekizoglu O, Ahishali B, Orhan N, Oku B, Arican N, Ustek D, Bilgic B, Elmas I, Kucuk M, Kemikler G. Morphological and functional changes of blood-brain barrier in kindled rats with cortical dysplasia. Brain Res. 2008;1208:181–191. doi: 10.1016/j.brainres.2008.02.101. [DOI] [PubMed] [Google Scholar]
- Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1745:273–286. doi: 10.1016/j.bbamcr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol. 2004;472:113–129. doi: 10.1002/cne.20052. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lin CS, Tsaur ML, Chen CC, Wang TY, Lin CF, Lai YL, Hsu TC, Pan YY, Yang CH, Cheng JK. Chronic intrathecal infusion of minocycline prevents the development of spinal-nerve ligation-induced pain in rats. Reg Anesth Pain Med. 2007;32:209–216. doi: 10.1016/j.rapm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Nag S, Manias JL, Stewart DJ. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol. 2009;35:417–426. doi: 10.1111/j.1365-2990.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- Pan W, Zhang L, Liao J, Csernus B, Kastin AJ. Selective increase in TNF alpha permeation across the blood-spinal cord barrier after SCI. J Neuroimmunol. 2003;134:111–117. doi: 10.1016/s0165-5728(02)00426-5. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- Plotkin SR, Banks WA, Maness LM, Kastin AJ. Differential transport of rat and human interleukin-1alpha across the blood-brain barrier and blood-testis barrier in rats. Brain Res. 2000;881:57–61. doi: 10.1016/s0006-8993(00)02772-4. [DOI] [PubMed] [Google Scholar]
- Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev. 2009;60:57–64. doi: 10.1016/j.brainresrev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29:1084–1098. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahbaie P, Shi X, Guo TZ, Qiao Y, Yeomans DC, Kingery WS, Clark JD. Role of substance P signaling in enhanced nociceptive sensitization and local cytokine production after incision. Pain. 2009;145:341–349. doi: 10.1016/j.pain.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Shaw KT, Greig NH. Chemokine receptor mRNA expression at the in vitro blood-brain barrier during HIV infection. Neuroreport. 1999;10:53–56. doi: 10.1097/00001756-199901180-00010. [DOI] [PubMed] [Google Scholar]
- Sinclair R, Eriksson AS, Gretzer C, Cassuto J, Thomsen P. Inhibitory effects of amide local anaesthetics on stimulus-induced human leukocyte metabolic activation, LTB4 release and IL-1 secretion in vitro. Acta Anaesthesiol Scand. 1993;37:159–165. doi: 10.1111/j.1399-6576.1993.tb03693.x. [DOI] [PubMed] [Google Scholar]
- Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, Amer LD, Young CS, Lewis MT, Poole S, Frank MG, Watkins LR, Milligan ED, Mahoney MJ. Release of plasmid DNA-encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm Res. 2010;27:841–854. doi: 10.1007/s11095-010-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25:593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- Stan RV. Structure of caveolae. Biochim Biophys Acta. 2005;1746:334–348. doi: 10.1016/j.bbamcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, Callahan MK, Tucky BH, Ransohoff RM. Determinants of CCL5-driven mononuclear cell migration across the blood-brain barrier. Implications for therapeutically modulating neuroinflammation. J Neuroimmunol. 2006;179:132–144. doi: 10.1016/j.jneuroim.2006.06.004. [DOI] [PubMed] [Google Scholar]
- White FA, Feldman P, Miller RJ. Chemokine signaling and the management of neuropathic pain. Mol Interv. 2009;9:188–195. doi: 10.1124/mi.9.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Qaum T, Adamis AP. Sensitive blood-retinal barrier breakdown quantitation using Evans Blue. Invest Ophthalmol Vis Sci. 2001;42:789–794. [PubMed] [Google Scholar]
- Yu LN, Yang XS, Hua Z, Xie W. Serum levels of pro-inflammatory cytokines in diabetic patients with peripheral neuropathic pain and the correlation among them (in Chinese) Zhonghua Yi Xue Za Zhi. 2009;89:469–471. [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Central neuroglial interactions in the pathophysiology of neuropathic pain. In: Mayer EA, Bushnell MC, editors. Functional pain syndromes: presentation and pathophysiology. Seattle: IASP; 2009. pp. 319–336. [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieglgänsberger W, Berthele A, Tölle TR. Understanding neuropathic pain. CNS Spectr. 2005;10:298–308. doi: 10.1017/s1092852900022628. [DOI] [PubMed] [Google Scholar]