

Abstract
Focal adhesion kinase (FAK) is an intracellular kinase and scaffold protein that regulates migration in many different cellular contexts but whose function in neuronal migration remains controversial. Here, we have analyzed the function of FAK in two populations of neurons with very distinct migratory behaviors: cortical interneurons, which migrate tangentially and independently of radial glia; and pyramidal cells, which undergo glial-dependent migration. We found that FAK is dispensable for glial-independent migration but is cell-autonomously required for the normal interaction of pyramidal cells with radial glial fibers. Loss of FAK function disrupts the normal morphology of migrating pyramidal cells, delays migration, and increases the tangential dispersion of neurons arising from the same radial unit. FAK mediates this process by regulating the assembly of Connexin-26 contact points in the membrane of migrating pyramidal cells. These results indicate that FAK plays a fundamental role in the dynamic regulation of Gap-mediated adhesions during glial-guided neuronal migration in the mouse.
Introduction
Newborn neurons reach their final destination through two different mechanisms, radial and tangential migration. Radial migration is the primary mechanism by which developing neurons reach their final position, and it courses in parallel to the radial glia scaffold that spans the neural tube (Rakic, 2007). Tangential migration allows newborn neurons to disperse orthogonally to the radial glia axis, and it is involved in increasing neuronal diversity in specific brain circuits (Marín and Rubenstein, 2001). Although there are cellular and molecular elements that are common to both types of migration (Marín et al., 2006), a major difference between both mechanisms is their different dependency on migratory substrates. Thus, radial migration is largely dependent on the interaction of migrating neurons with radial glial fibers, whereas tangential migration is not (Marín and Rubenstein, 2003; Rakic, 2006).
The developing cerebral cortex is perhaps the best model to investigate the molecular underpinnings of radial and tangential migration, because its two main neuronal constituents—pyramidal cells and interneurons—distinctly use these different modes of migration. In the neocortex, for example, the pallial ventricular zone (VZ) generates several consecutive cohorts of pyramidal cells that migrate radially and settle into progressively more superficial positions within the cortical plate (CP) (Gupta et al., 2002). This “inside-out” pattern of cortical lamination is then refined during the differentiation of pyramidal neurons to conform the adult architecture of the six-layered neocortex (Caviness et al., 2008). Concurrently, progenitor cells located in several regions of the subpallium generate cortical interneurons that migrate tangentially to colonize the entire cerebral cortex before changing their mode of migration to adopt specific laminar positions (Batista-Brito and Fishell, 2009).
One of the main characteristics that distinguish radial and tangential migration is their differential dependency on radial glial cells (RGCs). RGCs reside in the VZ and have a long basal process that extends to the pial surface, where it anchors into the basement membrane. Of note, the first cohorts of newborn pyramidal cells reach the CP through a mechanism known as somal translocation, which is independent of their interaction with the processes of radial glial cells (Miyata et al., 2001; Nadarajah et al., 2001; Kriegstein and Noctor, 2004). As the cortex grows, however, pyramidal cells migrate progressively longer distances to reach the CP, and this requires the support of RGCs. This process of glial-guided migration involves a dynamic interaction between migrating neurons and the basal process of RGCs, which serves as the main substrate to support their movement (Rakic, 1972, 1974; Kriegstein and Noctor, 2004).
Molecules controlling the adhesion of migrating neurons to RGCs are thought to play a prominent role in glial-guided migration. Recent work has shown that the adhesive properties of connexins are essential for glial-guided radial migration, while dispensable for tangential migration (Elias et al., 2007, 2010; Cina et al., 2009). Two different connexins have been shown to form part of these cell–cell junctions: Connexin-26 (Cx26) puncta seem to preferentially mediate the interaction of the soma and proximal leading process with RGCs, while Cx43 is enriched along the entire leading process. Considering the dynamic behavior of migrating neurons, it is expected that the assembly and disassembly of connexin-mediated adhesions would be a very dynamic process, but the mechanisms underlying this process have not been explored yet.
The intracellular kinase and scaffold protein focal adhesion kinase (FAK) have been shown to regulate the formation and disassembly of adhesions during the migration of multiple cell types (Avizienyte and Frame, 2005; Mitra et al., 2005). FAK is strongly expressed in the brain (Grant et al., 1995), in particular during embryonic development (Xie et al., 2003), but its function in migrating neurons is somehow controversial (Beggs et al., 2003; Xie et al., 2003). Here, we have investigated the function of this protein in the tangential migration of cortical interneurons and in the radial migration of pyramidal cells. We have found that FAK cell-autonomously modulates glial-guided radial migration, but it is dispensable for the migration of cells whose movement is independent of RGCs (interneurons and early-born pyramidal cells undergoing somal translocation). FAK modulates glial-guided migration by regulating the interaction between migrating neurons and RGCs, a process that depends on the function of FAK in the assembly of Cx26-mediated adhesions in the membrane of migrating pyramidal cells.
Materials and Methods
Mouse lines.
Fakflox/flox (Beggs et al., 2003), NEX-Cre (Goebbels et al., 2006), and NEX-Cre;Fakflox/flox were maintained in a CD1 background. All control animals used in our experiments were obtained from the identical familial genetic background than the corresponding mutant mice. The day of vaginal plug was considered to be embryonic day 0.5 (E0.5). Mice were kept at the Instituto de Neurociencias in accordance with Spanish and European Union regulations.
DNA constructs.
Fak chicken cDNA (kindly provided by J. T. Parsons, University of Virginia, Charlottesville, VA) was subcloned into a pCAGIG vector (11159; Addgene), and all point mutations were based on this construct (GenScript). All short hairpin RNA (shRNA) were expressed from U6 promoter-containing vectors. shLuc was obtained from Ambion, shFak was kindly provided by J. L. Guan (University of Michigan, Ann Arbor, MI), and shFak(2) was cloned in the pSilencer vector (Ambion) using the sequence described by Tilghman et al. (2005). shCx26, based on the same sequence tested by Elias et al. (2007), was also cloned in the pSilencer vector. A second shRNA against Cx26 [shCx26(2); target sequence, AACGAGTTTAAGGACATCGAA] was also tested and gave similar results as the original one (data not shown). DsRed, Cre-i-Gfp, and ND1 plasmids were kindly provided by F. Guillemot (National Institute for Medical Research, London, UK) and F. Polleux (Scripps Research Institute, La Jolla, CA); the mRfp plasmid was provided by J. Sanes and J. Livet (both Harvard University, Cambridge, MA). Gfp was cloned into the β-actin promoter expression vector (pCAGGS). Cx26-Gfp, provided by M. Falk (Lehigh University, Bethlehem, PA) (Falk, 2000), was also cloned into pCAGGS.
Cell lines.
N2A, COS, and C6 cell lines were cotransfected (Fugene; Roche) with the indicated plasmids at 0.9 μg/μl and incubated for 72 h.
Immunoprecipitation and immunoblot analysis.
Cells or tissue were homogenized in TBS-T (50 mm Tris, pH 7.4, 150 mm NaCl, 10% glycerol, 5 mm EDTA, 1% Triton X-100, and a mixture of proteases inhibitors). Lysates were clarified by centrifugation at 15,000 × g for 10 min and precleared by incubation with recombinant protein G-agarose (rProtein G Agarose; Sigma-Aldrich) for 1 h at 4°C. After removal of the protein G-agarose by brief centrifugation, the supernatants were transferred into a fresh tube, and proteins were immunoprecipitated overnight at 4°C on an orbital shaker with the recommended volume of the immunoprecipitating antibody (rabbit anti-FAK; 1:100; 06-543; Millipore). The immunocomplexes were captured by adding 30 μl of protein G-agarose bead slurry (15 μl of packed beads). Then they were gently rocked on an orbital shaker 2 h at 4°C. Immunoprecipitates were washed three times with appropriated buffer, reconstituted in 30–60 μl of 2× SDS sample buffer [50 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 100 mm DTT (dithiothreitol), and bromophenol blue], boiled for 5 min to dissociate the immunocomplexes from the beads, and further analyzed by SDS-PAGE and immunoblot analysis.
Proteins were resolved by SDS-PAGE on 10 or 12.5% gels and blotted to 0.2 mm nitrocellulose in transfer buffer, pH 8.3 (25 mm Tris, 192 mm glycine, and 20% v/v methanol). Immunoblots were blocked for 1 h in TBS-T (20 mm Tris, pH 7.4, 150 mm NaCl, and 0.2% Tween 20) containing 5% BSA and incubated overnight at 4°C with primary antibodies diluted in TBS-T containing 1% BSA powder. The following primary antibodies were used: rabbit anti-FAK (1:3000; 06-543; Millipore), rabbit anti-Cx26 (1:1000; 71-0500; Zymed), mouse anti-actin (1:3000; ab11003; Abcam), mouse anti-βIII-tubulin (1:1000; MMS-410-P; Covance), and anti-green fluorescent protein (GFP) (1:1000; A11122; Invitrogen). Membranes were then washed in TBS-T, incubated for 1 h with secondary antibodies (goat anti-mouse or anti-rabbit horseradish peroxidase-conjugated IgG; 1:25,000; 31444 and 31460; Pierce) diluted in TBS-T, washed, and incubated for 1 min with ECL chemiluminescence reagents (Immobilon Western; Millipore).
Immunohistochemistry.
Embryos and postnatal mice were perfused with 4% PFA, postfixed overnight at 4°C, equilibrated in 15 and 30% sucrose in PBS, and cut on a freezing microtome (HM450; Microm) into 40-μm-thick coronal sections. Alternatively, brains were postfixed 30 min at room temperature plus 90 min at 4°C, and then 40–80 μm thickness coronal sections were cut on a vibratome (VT1000S; Leica). Immunohistochemistry was also performed in medial ganglionic eminence (MGE) explants dissected out from E13.5 coronal slices of the telencephalon, as described previously (Martini et al., 2009). Free-floating sections were processed for immunohistochemistry by blocking with 0.3% Triton X-100 and 1% BSA in PBS for 1 h, followed by overnight incubation with primary antibody diluted in 0.1% Triton X-100 and 0.5% BSA in PBS. The following primary antibodies were used: rabbit anti-GFP (1:1000; A11122; Invitrogen), chicken anti-GFP (1:1000; GFP-1020; Aves Labs), rabbit anti-FAK (1:300; 06-543; Millipore), rabbit anti-Cx26 (1:25; 51-2800; Zymed), rabbit anti-Cx43 (1:50; 71-0700; Zymed), rabbit anti-laminin (1:200; AB2034; Millipore), rabbit anti-Cux1 (sc-13024; Santa Cruz), rat anti-BrdU (1:200; AB6326; Abcam), and rabbit anti-RFP (1:150; AB62341; Abcam). The specificity of FAK and Cx26 antibodies has been tested previously in genetic deletion and knockdown experiments (Elias et al., 2007; Chacón et al., 2010). Secondary antibodies diluted in 0.1% X-100 and 0.5% BSA in PBS were incubated for 90 min. The following secondary antibodies were used: donkey anti-rabbit 488 (1:500; A21206; Invitrogen), goat anti-chicken 488 (1:500; A11039; Invitrogen), donkey anti-rabbit 555 (1:400; A31572; Invitrogen), and donkey anti-mouse 555 (1:200; A31570; Invitrogen). Cell nuclei were stained using bis-benzamide (1:1000; Sigma-Aldrich), and sections were mounted with Mowiol (Sigma-Aldrich) with NPG (Calbiochem). Cx26 and Cx43 immunohistochemistry was performed as described before (Elias et al., 2007).
Focal electroporation.
Organotypic slice cultures of the embryonic mouse telencephalon were prepared as previously described (Anderson et al., 1997). pCAGGS-based Gfp and DsRed expression vectors were pressure injected focally into the MGE of coronal slice cultures and focally electroporated as described previously (Flames et al., 2004).
In utero electroporation and retroviral infection.
Pregnant females were anesthetized with isoflurane, their uterine horns were exposed, and the lateral ventricles of embryos were injected through the uterus wall using pulled capillaries (1B120F-4; World Precision Instruments) filled with either DNA (0.8–2.25 μg/μl) or retrovirus stock (107 cfu/ml) diluted in PBS and colored with 0.5% Fast Green (Sigma-Aldrich). DNA-injected embryos were electroporated using a 35–45 V/50 ms/950 ms/five pulses program (CUY21/CUY650P3/CUY650P5; Nepa Gene). Previous experiments in the laboratory have determined that the ratio of coexpression of two different plasmids electroporated at similar concentrations is nearly 95%. Retroviruses were prepared as described previously (Tashiro et al., 2006). After appropriate survival times, mice were deeply anesthetized and killed using cervical dislocation. In some cases, pregnant females were injected with a 40 mg/kg BrdU solution (Sigma-Aldrich).
Imaging.
Images were acquired using fluorescence microscopes (DM5000B/CTR5000 and DMIRB; Leica) coupled to digital cameras (DC500 or DFC350FX; Leica) or a confocal microscope (DMIRE2/CTRMIC/TCS SP2; Leica). Neurolucida software (MBF Bioscience) was used for the reconstruction of cell morphologies using a fluorescence microscope with a 63× oil objective (DM4000B; Leica) coupled to a digital camera (QICAM Fast 1394; QImaging).
Quantification.
Quantification of images was done using Canvas (ACD Systems), NIH ImageJ (http://rsb.info.nih.gov/ij/), or Neurolucida Explorer (MBF Bioscience) software. For the quantification of tangential migration, we used methods described previously (Cobos et al., 2007; Nóbrega-Pereira et al., 2008). For quantification of radial migration, layers were drawn following nuclear staining and GFP-expressing cells were quantified in a common boxed region through the somatosensory cortex at the same rostrocaudal level for each brain. Swelling quantification was performed acquiring photos every 3 μm from electroporated cortices and identifying cytoplasmic dilatations in the region of the leading process preceding the nucleus. Neurolucida Explorer was used to quantify the number of branches (>5 μm in length) in drawn cells. In retroviral experiments, GFP+ clones were acquired using an inverted confocal microscope. For each clone, the radial glia process was initially localized under the microscope, and then the shortest distance from the centroid of every clonally related migrating neuron to the apical process of their mother glia cell was calculated using ImageJ. In postnatal brains, layers were drawn according to nuclear staining, and the total numbers of cells located below layer IV were quantified. Images of FAK, Cx26, and Cx43 puncta were acquired using an inverted confocal microscope. x- and y-axis sectioning of the acquired images was performed using Leica confocal software. Cx26 and Cx43 puncta were quantified using ImageJ, and their number normalized with the total area of the cell. Western blots were quantified using Quantity One (Bio-Rad). A two-tailed t test was used for single pairwise comparisons to examine differences in number of swellings, number of processes, adhesion assays, distance to radial glial process following retrovirus injection, and number of Cx26 or Cx43 puncta. A χ2 test was used to examine differences between subpopulations of neurons following in utero electroporation.
Results
FAK is not required for the tangential migration of interneurons
FAK is strongly expressed in the developing brain (Xie et al., 2003), including migrating interneurons derived from the MGE (Fig. 1A). To unravel the function of FAK in the tangential migration of cortical interneurons, we knocked down FAK protein levels by using RNA interference (RNAi). In brief, plasmids encoding control or shRNA constructs that produced significant knockdown of mouse FAK (Fig. 1B), along with a plasmid encoding the enhanced GFP, were cotransfected in progenitor cells in the MGE by focal electroporation in organotypic slice cultures at E13.5, and the distribution of migrating neurons was analyzed 48 h later (Fig. 1C). Consistent with previous reports (Marín et al., 2001; Cobos et al., 2007), analysis of slices electroporated with control plasmids (shLuc) revealed that many MGE interneurons had reached the cortex after 48 h (Fig. 1D). FAK knockdown using two different plasmids encoding Fak shRNA did not perturb the movement of MGE-derived interneurons toward the cortex (Fig. 1E) (percentage of slices with >70 cells in cortex, mean ± SEM, shLuc: 60.85 ± 11.03, n = 28 slices; shFak: 59.05 ± 5.85, n = 23 slices; t test, p = 0.90200), which suggested that FAK function might be dispensable for tangential migration.
Figure 1.

FAK knockdown does not perturb the tangential migration of cortical interneurons. A, Migrating interneurons leaving the MGE at E13.5 express FAK. B, Representative immunoblots of FAK protein levels in N2A cells 72 h after transfection with a control plasmid (empty vector pCAGIG) plus a plasmid encoding Fak, shFak plus Fak, or shFak(2) plus Fak. The graph shows the quantification of FAK protein levels in each case. Histograms show mean ± SEM from three experiments. shLuc, 94.90 ± 13.85; shFak, 41.59 ± 6.35; shFak(2), 49.59 ± 7.67. shLuc and shFak comparison: t test, *p = 0.0128. shLuc and shFak(2) comparison: t test, *p = 0.0287. C, Schematic diagram of experimental design. D, E, Coronal slices after electroporation of embryonic MGE at E13.5 with plasmids encoding Gfp and shLuc (B) or shFak (C) incubated 48 h in culture.
To strengthen this conclusion, we used alternative approaches to disrupt FAK function in tangentially migrating interneurons. We generated interneuron-specific conditional Fak mutant mice by breeding Dlx5/6-Cre-i-Gfp mice (Stenman et al., 2003) with mice homozygous for loxP-flanked Fak alleles (Beggs et al., 2003). Consistent with the RNAi experiments, analysis of the distribution of GFP+ interneurons in the cortex of control and Fak mutant E13.5 embryos revealed no significant differences in the number, route, or extent of interneuron migration between both genotypes (Fig. 2A–D), despite the effective removal of FAK from these cells (Fig. 2E). We next wondered whether FAK function might only be required for a population of MGE-derived interneurons, and thus the analysis of the entire complement of MGE-derived interneurons could mask a possible phenotype. To solve this question, we performed focal electroporation experiments with a plasmid encoding a red fluorescent protein (DsRed) in organotypic slices obtained from E13.5 control (Dlx5/6-Cre-i-Gfp;Fak+/+) and interneuron-specific Fak mutants (Dlx5/6-Cre-i-Gfp;Fakflox/flox) (Fig. 2F). Analysis of these experiments revealed no significant differences in the number, route, or extent of interneuron migration between control and interneuron-specific Fak mutant slices (Fig. 2G,H) (number of interneurons reaching the cortex, mean ± SEM, Dlx5/6-Cre-i-Gfp;Fak+/+: 105.17 ± 11.10, n = 6 slices; Dlx5/6-Cre-i-Gfp;Fakflox/flox: 85.25 ± 17.84, n = 8 slices; t test, p = 0.40094).
Figure 2.
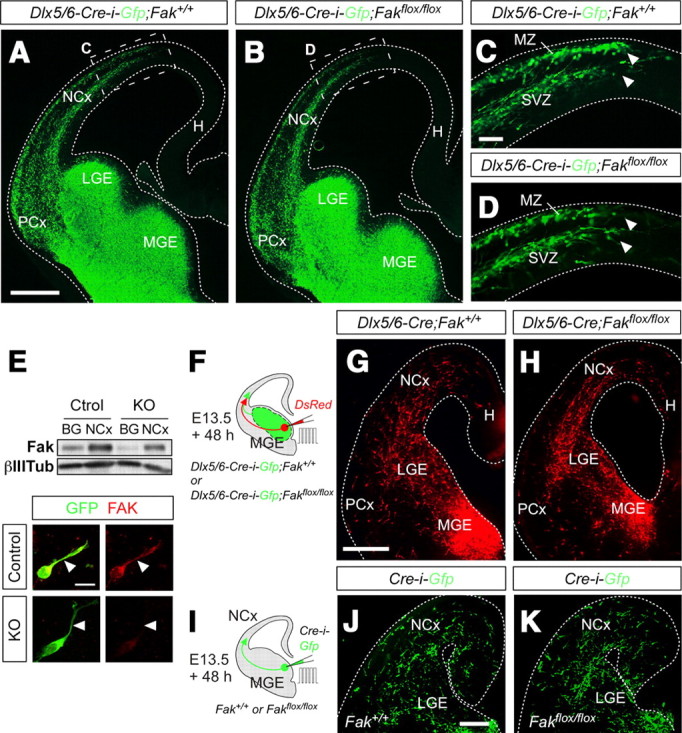
FAK function is not required for the tangential migration of cortical interneurons. A–D, Coronal sections through the telencephalon of E13.5 control (A, C) and interneuron-specific conditional Fak mutant (B, D) embryos. G and H are high-magnification images of the boxed areas shown in E and F, respectively. Note that GFP+ interneurons migrate through the marginal zone (MZ) and SVZ in control and Fak mutant embryos, and they reach the same mediolateral position at this stage. E, Western blot analysis revealed that FAK levels are decreased in the BG and NCx of interneuron-specific Fak mutants (KO) compared with controls (Ctrol). Arbitrary units normalized for control NCx ± SEM from two animals per condition: NCx Ctrol, 100.00 ± 1.80; BG Ctrol, 45.70 ± 7.75; NCx KO, 76.58 ± 6.85; BG KO, 21.97 ± 0.84. Similarly, immunohistochemistry experiments in primary cultures of MGE revealed that FAK protein levels are efficiently reduced in interneurons obtained from interneuron-specific Fak mutants (KO) compared with controls. Mean fluorescence arbitrary units ± SEM from at least three animals per condition: control, 0.89 ± 0.16 (n = 18 cells); KO, 0.05 ± 0.03 (n = 9 cells). F, Schematic diagram of experimental design. G, H, Coronal slices after electroporation of embryonic MGE of control (G) and interneuron-specific conditional Fak mutant embryos (H) at E13.5 with a plasmid encoding DsRed and 48 h in culture. I, Schematic diagram of experimental design. J, K, Coronal slices after electroporation of embryonic MGE from Fak+/+ (J) or Fakflox/flox (K) embryos at E13.5 with a plasmid encoding Cre-i-Gfp. Scale bars: C, D, 50 μm; A, B, G, H, J, K, 100 μm.
Dlx5 and Dlx6 are expressed by progenitor cells in the subventricular zone (SVZ) of the subpallium, but it is largely absent from VZ progenitors (Eisenstat et al., 1999). Thus, although Dlx5/6-Cre-i-Gfp mice seem to efficiently eliminate FAK from cortical interneurons (Fig. 2E), we speculated that early deletion of Fak from VZ progenitors might unravel a possible role for FAK in the tangential migration of interneurons. To test this idea, we performed focal electroporation experiments with a plasmid encoding the recombinase Cre [Cre-i-Gfp, where i stands for IRES (internal ribosome entry site)] in organotypic slices obtained from E13.5 embryos harboring wild-type (Fak+/+) or loxP-flanked Fak alleles (Fakflox/flox) (Fig. 2I). As in the other cases, analysis of these experiments revealed no significant differences in the tangential migration of normal or FAK-deficient interneurons (Fig. 2J,K) (percentage of slices with >70 cells in cortex, mean ± SEM, Fak+/+: 68.12 ± 10.20, n = 12 slices; Fakflox/flox: 70.20 ± 8.14, n = 12 slices; t test, p = 0.804). Together, these experiments demonstrated that FAK function is not required for the tangential migration of cortical interneurons.
FAK is required for the radial migration of pyramidal cells
To unravel the function of FAK in the migration of pyramidal neurons, we first performed in utero electroporation experiments targeting progenitor cells in the dorsal pallium with plasmids encoding control (shLuc) or shFak constructs along with Gfp at E14.5, and the distribution of migrating pyramidal cells was analyzed at E18.5 (Fig. 3A). Consistent with previous reports (Bai et al., 2003; Kawauchi et al., 2003; Shu et al., 2004; Hand et al., 2005), analysis of mouse embryos electroporated with shLuc revealed that most cortical neurons had reached the CP at this stage (40.76 ± 2.66%; n = 11) (Fig. 3C,F). By contrast, FAK knockdown led to a pronounced migration defect, with a marked reduction in the proportion of neurons reaching the CP compared with controls [shFak: 20.47 ± 3.76%, n = 6; shFak(2): 30.73 ± 2.17%, n = 11] (Fig. 3D,F). To verify the target specificity of the shRNA effect and demonstrate that the observed phenotype was specifically due to loss of FAK, we took advantage of the fact that one of the shRNA constructs used in our assays targets a sequence that is not conserved in chicken Fak (4 bp mismatches) (see Materials and Methods). In utero electroporation of E14.5 mouse embryos with plasmids encoding Gfp, shFak, and chicken Fak resulted in a complete recovery of the migration phenotype by E18.5, with a similar proportion of transfected neurons invading the CP than in controls (43.63 ± 4.28%; n = 10) (Fig. 3E,F).
Figure 3.

Removal of FAK disrupts radial migration of projection neurons. A, B, Schematic diagrams of experimental designs. C–E, Coronal sections through the somatosensory cortex of E18.5 wild-type embryos after E14.5 in utero electroporation with plasmids encoding Gfp and shLuc (C), shFak (D), or shFak plus Fak (E). Schemas with dots depict representative distributions of GFP-expressing neurons in each condition. F, Quantification of the relative distribution (percentage) of GFP-expressing cells in the CP and the layers below it for each condition. Histograms show mean ± SEM from at least three different brains. shLuc: CP, 40.76 ± 2.66; layers IV–VI, 59.24 ± 2.66. shFak: CP, 20.47 ± 3.76; layer IV–VI, 79.53 ± 3.76. χ2 test for shLuc and shFak comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. shFak plus Fak: CP, 43.63 ± 4.28; layers IV–VI, 56.37 ± 4.28. χ2 test for shFak and shFak plus Fak comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. G, H, Coronal sections through the somatosensory cortex of E18.5 Fak+/+ (G) and Fakflox/flox (H) embryos after E14.5 in utero electroporation with a plasmid encoding Cre-i-Gfp. Schemas with dots depict representative distributions of GFP-expressing neurons in each condition. I, Quantification of the relative distribution (percentage) of GFP-expressing cells in the CP and the layers below it for each condition. Histograms show mean ± SEM from at least three different brains. Control (Cre-i-Gfp in Fak+/+ embryos, Gfp in Fakflox/flox embryos, or Gfp in Fak+/+ embryos): CP, 52.66 ± 0.83; layer IV–VI, 47.34 ± 0.83. Cre-i-Gfp in Fakflox/flox embryos: CP, 35.41 ± 1.22; layer IV–VI, 64.59 ± 1.22. χ2 test for the comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. Scale bar, 100 μm.
To further ensure that the phenotype observed in FAK knockdown experiments was exclusively due to loss of FAK, we next deleted Fak from cortical progenitors by electroporating a plasmid encoding Cre-i-Gfp in the dorsal pallium of E14.5 Fakflox/flox embryos. As in RNAi experiments, Cre-mediated deletion of Fak reduced the proportion of neurons reaching the CP compared with control embryos (Gfp: 52.66 ± 0.83%, n = 6; Cre-i-Gfp: 35.41 ± 1.22%, n = 4) (Fig. 3B,G–I). We observed that genetic deletion of Fak caused a milder migratory phenotype than Fak knockdown. This disparity is likely due to the different time required for FAK removal in both types of experiments, since conditional deletion of Fak must be preceded by expression of Cre and the subsequent recombination of the targeted locus. Consistent with this idea, when the analysis of the migratory phenotype was restricted to the population of neurons born 1 d after Cre electroporation (as assayed by BrdU incorporation), the migratory phenotype was very similar to that found in Fak knockdown experiments (data not shown). Together, these experiments demonstrated that FAK function is required for the efficient migration of pyramidal neurons.
FAK is cell-autonomously required for the migration of pyramidal cells
In utero electroporation primarily targets progenitor cells contacting the ventricle (Noctor et al., 2001), so loss of FAK function could impair cortical migration by affecting radial glia, migrating neurons, or both. Previous work has suggested that targeted deletion of Fak in the dorsal telencephalon perturbs neuronal migration by disrupting radial glia and the cortical basement membrane, but not by directly affecting migrating neurons (Beggs et al., 2003). In our experiments, however, loss of FAK (via shFak in wild-type embryos or Cre in Fakflox/flox embryos) did not perturb the morphology of the radial glia cells or the organization of the cortical basal lamina overlaying the electroporated region (data not shown). These results suggested that loss of FAK in migrating neurons might indeed perturb migration. To test this hypothesis, we generated conditional mutant embryos in which Fak was deleted using NEX-Cre mice (Goebbels et al., 2006). In this strain, the promoter region of the transcription factor Math2 drives the expression of Cre, which is thereby confined to most migrating cortical pyramidal neurons and a small subset of SVZ progenitors, but excluded from RGCs (Wu et al., 2005). To analyze the migration of pyramidal neurons, we electroporated in utero a plasmid encoding Gfp in either Fakflox/flox (control) or NEX-Cre;Fakflox/flox (mutant) embryos (Fig. 4A). We found that loss of Fak function in postmitotic neurons perturb their migration, with relatively fewer cells reaching the CP in mutant embryos than in controls (controls: 46.67 ± 2.61%, n = 4; mutants: 37.38 ± 2.79%, n = 4) (Fig. 4B–D). We noticed that this approach led to milder defects than those observed in Fak knockdown experiments (Fig. 3), which could be attributed to the late removal of FAK in postmitotic neurons. Alternatively, this result could indicate that FAK is primarily required in RGCs, with a minor contribution in migrating neurons. To distinguish between both possibilities, we performed additional experiments in which we deleted Fak from cortical progenitors (Cre-i-Gfp in Fakflox/flox embryos) while simultaneously expressing a plasmid in which the promoter region of the transcription factor NeuroD1 drives the expression of Fak (ND1-Fak) (Fig. 4E). Using this approach, Fak is removed from RGCs and their progeny, but only newborn cortical neurons are able to reexpress Fak because the NeuroD1 promoter is only active in these cells (Heng et al., 2008). We found that reexpression of Fak in migrating cortical neurons is able to rescue the migratory phenotype caused by genetic ablation of Fak in cortical progenitors (Cre-i-Gfp: 35.41 ± 1.22%, n = 4; Cre-i-Gfp plus ND1-Fak: 49.74 ± 2.36%, n = 3) (Fig. 4F–H). Together, our experiments demonstrated that FAK function is cell-autonomously required in pyramidal neurons for radial migration.
Figure 4.

FAK removal from neurons disrupts radial migration in a cell-autonomous manner. A, E, I, Schematic diagrams of experimental designs. B, C, Coronal sections through the somatosensory cortex of E18.5 Fakflox/flox (B) and NEX-Cre;Fakflox/flox (C) embryos after E14.5 in utero electroporation with a plasmid encoding Gfp. D, Quantification of the relative distribution (percentage) of GFP-expressing in CP and the layers below it for each condition. Histograms show mean ± SEM from at least four different brains. Fakflox/flox: CP, 46.67 ± 2.61; layer IV–VI, 53.33 ± 2.61. NEX-Cre;Fakflox/flox: CP, 37.38 ± 2.79; layer IV–VI, 62.62 ± 2.79. χ2 test for comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. F, G, Coronal sections through the somatosensory cortex of E18.5 Fakflox/flox embryos after E14.5 in utero electroporation with a plasmid encoding Cre-i-Gfp (F) or Cre-i-Gfp plus ND1-Fak (G). H, Quantification of the relative distribution (percentage) of GFP-expressing in CP and the layers below it for each condition. Histograms show mean ± SEM from at least three different brains. Cre-i-Gfp in Fakflox/flox embryos: CP, 35.41 ± 1.22; layer IV–VI, 64.59 ± 1.22. Cre-i-Gfp plus ND1-Fak in Fakflox/flox embryos: CP, 49.74 ± 2.36; layer IV–VI, 50.26 ± 2.36. χ2 test for comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. J, K, Coronal sections through the somatosensory cortex of P7 wild-type mice after E14.5 in utero electroporation with plasmids encoding Gfp and shLuc (J) or shFak (K). At P7, most cells were found in a similar position in both conditions, although ectopic cells with highly disorganized dendrite orientation (high magnification in K) were also present in deep layers of the cortex (below layer IV) in Gfp plus shFak electroporated brains. L, Quantification of the number of ectopic cells per field below layer IV for both conditions. Histograms show mean ± SEM from seven different brains in every condition. shLuc: 14.57 ± 9.33. shFak: 80.22 ± 15.11. t test, **p = 0.0040. M, N, Coronal sections through the somatosensory cortex of P7 Fakflox/flox (M) and NEX-Cre;Fakflox/flox (N) embryos stained with Cux1 antibody. O, Quantification of the number of Cux1-expressing cells below layer IV for each condition. Histograms show mean ± SEM from two different brains. Fakflox/flox: 12.67 ± 5.49. NEX-Cre;Fakflox/flox: 35.80 ± 4.50. t test, *p = 0.0458. Scale bars: B, C, F, G, J, K, 100 μm; M, N, 50 μm.
Fak deletion leads to subtle lamination defects in the postnatal cortex
Our previous experiments suggested that disruption of FAK function in newborn neurons impairs radial migration, which in the embryo leads to a significant delay in the movement of these cells. To evaluate the postnatal consequences of this defect, we analyzed the distribution of neurons in postnatal day 7 (P7) mice that were electroporated with control and Fak shRNA plasmids at E14.5. We found that most electroporated neurons managed to reach the superficial layers of the cortex even after Fak knockdown (Fig. 4I–K), reinforcing the notion that FAK deficiency delays rather than blocks the migration of pyramidal neurons. However, we also found a significant number of FAK-deficient neurons in ectopic locations throughout the white matter and layers V and VI (Fig. 4K,L), many of which displayed abnormal dendritic morphology (Fig. 4K, inset). Similarly, analysis of the distribution of Cux1-expressing pyramidal neurons, which are normally restricted to layers II–IV in wild-type mice, revealed the existence of misplaced neurons in P7 NEX-Cre;Fakflox/flox mutant mice (Fig. 4M–O). In sum, this analysis indicated that loss of FAK function causes a delay the migration of pyramidal cells, which in some case results in defective laminar acquisition.
Loss of FAK impairs the morphology of cortical migrating neurons
To get insight into the mechanisms through which FAK may contribute to the migration of pyramidal neurons, we examined the morphology of migrating neurons before their entrance in the CP, where most FAK-deficient neurons accumulated. As part of their migratory cycle, migrating neurons form a cytoplasmic dilatation in the leading process that contains the centrosome and the Golgi apparatus, toward which the nucleus translocates forward in a subsequent stage (Bellion et al., 2005). Consistent with previous reports (Solecki et al., 2004), control transfected neurons were found to contain either one or none dilatation in the leading process, depending on the stage of their migratory cycle at which they were fixed (Fig. 5A–C). In contrast, we observed that migrating neurons transfected with shFak frequently contained several of such dilatations (Fig. 5A,B,D) (percentage of cells with two or more dilatations in the leading process, mean ± SEM, shLuc: 10.83 ± 0.36, n = 176 cells; shFak: 45.33 ± 5.21, n = 173 cells; t test, **p = 0.0027). In addition, migrating neurons transfected with shFak were found to harbor elaborated leading processes (Fig. 5F,H), whereas control transfected neurons typically had a single or bifurcated leading process (Fig. 5E,G) (number of leading processes, mean ± SEM, shLuc: 2.2 ± 0.13, n = 30 cells; shFak: 4.13 ± 0.37, n = 30 cells; t test, ***p = 7.46432E-06). Of note, these phenotypes were not unique to RNAi experiments, as similar morphological defects were found in migrating neurons after conditional deletion of Fak from cortical progenitors (Cre-i-Gfp in Fakflox/flox embryos) or from cells that have already left the VZ (NEX-Cre;Fakflox/flox mutant embryos) (data not shown). Thus, loss of FAK leads to profound modifications in the morphology of migrating pyramidal neurons.
Figure 5.

Morphological defects in migrating FAK-deficient projection neurons. A, Schematic diagram of experimental design. B, Representative example of a coronal section through the somatosensory cortex of an E18.5 wild-type embryo after E14.5 in utero electroporation showing the approximate region containing the neurons analyzed. Cell morphology was analyzed in layer VI, where most cells accumulated upon Fak knockdown. C, D, Representative examples of GFP-expressing migrating cells after in utero electroporation with plasmids encoding Gfp and shLuc (C) or shFak (D). Note the increased number of cytoplasmic dilatations in FAK-deficient neurons. E, F, Drawings illustrate the morphology of migrating cells expressing Gfp and shLuc (E) or shFak (F). Note the increased number of leading process branches in Fak-deficient neurons (F). G, H, Representative examples of GFP-expressing migrating cells after in utero electroporation with plasmids encoding Gfp and shLuc (G) or shFak (H). Note the increased number of leading process branches in Fak-deficient neurons (H). Scale bars: C, D, G, H, 15 μm; E, F, 25 μm.
Disrupted radial glia–neuron interaction in the absence of FAK
The abnormal morphology of FAK-deficient pyramidal cells is highly reminiscent of other cases in which the interaction between migrating neurons and radial glial fibers has been disrupted (Gupta et al., 2003; Sanada et al., 2004; Elias et al., 2007). Considering also that FAK is dispensable in tangentially migrating neurons (Figs. 1, 2), this observation prompted us to test the hypothesis that FAK might be primarily required in migrating neurons to regulate their interaction with radial glial fibers. We reasoned that, if this were the case, then very early-born pyramidal cells, which reach the CP without interacting with the radial glia scaffold (Miyata et al., 2001; Nadarajah et al., 2001; Hatanaka et al., 2004), should also be unaffected by the loss of FAK function. To test this, we electroporated the dorsal pallium of wild-type embryos at E12.5 with plasmids encoding Gfp and either shLuc or shFak, and the distribution of transfected neurons was analyzed at E16.5 (Fig. 6A). We found no differences in the percentage of neurons that reached the CP between control and shFak-electroporated embryos (shLuc: 71.16 ± 2.69%, n = 3; shFak: 67.72 ± 0.84%, n = 3) (Fig. 6B–D), suggesting that FAK function is dispensable for radial migration that is independent of radial glial fibers. In addition, we did not detect important morphological differences between control and shFak-transfected neurons at this stage (Fig. 6E,F), reinforcing the view that the disruption of cell morphology found at later ages (Fig. 5) might be secondary to a defect in the interaction between migrating neurons and radial glia.
Figure 6.
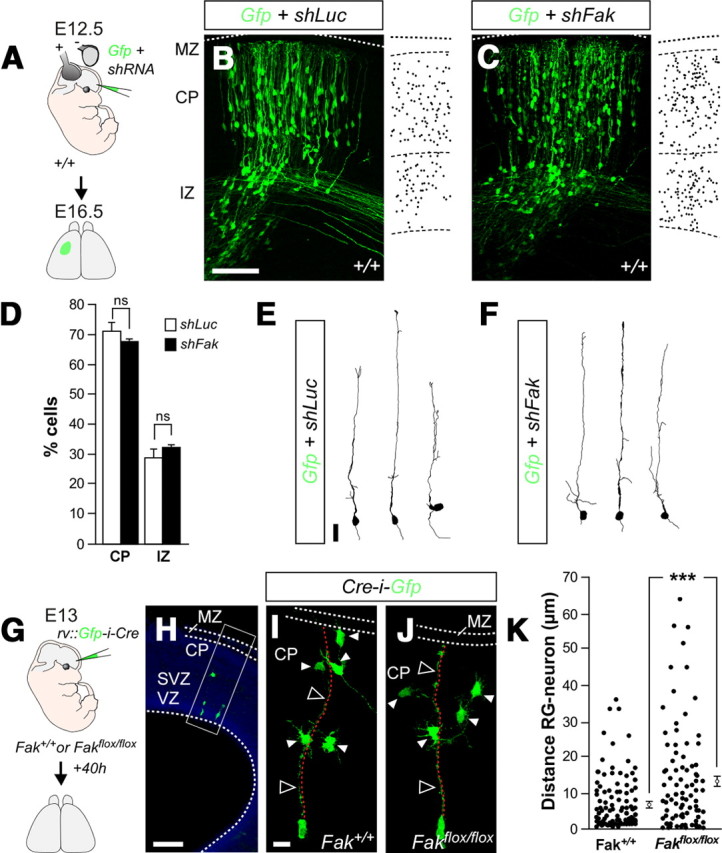
FAK regulates the interaction of migrating neurons with radial glia fibers. A, G, Schematic diagram of experimental designs. B, C, Coronal sections through the somatosensory cortex of E16.5 wild-type embryos after E12.5 in utero electroporation with plasmids encoding Gfp and shLuc (B) or shFak (C). D, Quantification of the relative distribution (percentage) of GFP-expressing in different cortical compartments for each condition. Histograms show mean ± SEM from at least three different brains. shLuc: CP, 71.16 ± 2.69; IZ, 28.84 ± 2.69. shFak: CP, 67.72 ± 0.84; IZ, 32.10 ± 0.89. χ2 test for shLuc and shFak comparison: CP, ns; IZ, ns. E, F, Drawings illustrate the morphology of migrating cells expressing Gfp and shLuc (E) or shFak (F). H, Representative example of a coronal section through the somatosensory cortex of an ∼E14.5 wild-type embryo after E13 infection with retroviruses expressing Gfp-i-Cre, showing the approximate region containing the clones analyzed in I and J. I, J, Representative examples of single clones containing GFP-expressing cells after infection of Fak+/+ (I) and Fakflox/flox (J) embryos with retroviruses encoding Gfp-i-Cre. Note that FAK-deficient cells are often located at greater distance from the basal process (dotted red lines) of the parental RGC than wild-type cells. K, Quantification of the distance between migrating neurons and the corresponding parental radial glia fiber. The dots represent individual cells from multiple experiments. Histograms show mean ± SEM from three different brains. Fak+/+, 7.14 ± 0.76; Fakflox/flox, 13.46 ± 1.52. t test, ***p = 1.4483E-04. Scale bars: B, C, 100 μm; E, F, 20 μm; H, 50 μm; I, J, 15 μm.
We next examined whether wild-type and FAK-deficient neurons that migrate at later stages of corticogenesis have differential properties of adhesion to radial glia. To this end, we examined the spatial relationship between individual radial glial processes and their progeny of migrating neurons by performing clonal analyses. Low-titer Gfp-i-Cre-expressing retroviruses were injected in the lateral ventricles of E13 wild-type or Fakflox/flox mouse embryos to sporadically label RGCs throughout the telencephalon (Fig. 6G). We chose this stage because retroviruses only infect progenitor cells, and so their integration and subsequent expression of GFP would take ∼16–24 h, labeling pyramidal cells that start to migrate around E14. Forty hours after infection, labeled radial glial units typically comprised a radial glial cell in the VZ, a few multipolar cells located between the SVZ and the intermediate zone (IZ), and some cells near or within the CP (Fig. 6H,I). We chose to examine our experiments at this relatively early stage (around E15) because pyramidal cells would normally detach from RGCs once they reach their final position. We found that wild-type migrating neurons were located in a range between 0.29 and 35.67 μm from the basal process of their parental RGCs (Fig. 6I,K). In contrast, migrating FAK-deficient neurons were dispersed across a wide territory, with distances from the basal process of their parental RGCs ranging from 0.29 to 63.68 μm (Fig. 6J,K). Consequently, the average distance between neurons and their parental radial glial processes was significantly higher in FAK-deficient clones (13.46 ± 1.52 μm; n = 115 cells from 3 embryos) (Fig. 6K) than in wild-type clones (7.14 ± 0.76 μm; n = 146 cells from 3 embryos) (Fig. 6K). These experiments suggested that either loss of FAK promote the tangential migration of pyramidal cells or FAK is required for the normal interaction between migrating neurons and RGCs.
Disruption of Connexin-26 puncta in FAK-deficient migrating neurons
The abnormally loose contact of FAK-deficient migrating neurons with their parental RGCs suggested a possible defect in some of the cell adhesion molecules that mediate these interactions. Recent studies have shown that the adhesive properties of some connexins are required during glial-guided migration (Elias et al., 2007; Cina et al., 2009), while they are dispensable for the tangential migration of cortical interneurons (Elias et al., 2010). Interestingly, Cx26 puncta are abundant in the perinuclear region and in the cytoplasmic dilatation formed in the proximal leading process before nucleokinesis (Fig. 7A–B′), a region that has been suggested to play a key role in the adhesion of migrating neurons (Schaar and McConnell, 2005). Considering that FAK is known to regulate cell–cell adhesions in other cellular contexts (Schaller, 2004; Yano et al., 2004), we hypothesized that FAK could regulate the interactions between migrating neurons and radial glia by controlling the assembly of Cx26 puncta. Consistent with this idea, we found that FAK is highly abundant in large points distributed throughout the cytoplasmic dilatation of migrating neurons (Fig. 7C,C′), in a pattern that resembled the distribution of Cx26 puncta. To test whether FAK and Cx26 cooperate in vivo, we prepared protein lysates from the embryonic cortex and performed coimmunoprecipitation experiments. We found that FAK and Cx26 indeed interact in the embryonic cortex (Fig. 7D), which reinforced the view that FAK might be required for Cx26 function.
Figure 7.

FAK interacts with Cx26 and is required for the establishment of Cx26 puncta. A, Schematic drawing showing the location of images displayed in B–C′ and F–G′. B–C′, Distribution of Cx26 (B, B′) and FAK (C, C′) puncta (arrows) in the cytoplasmic dilatation or swelling (s) preceding the nucleus (n) in migrating GFP-expressing pyramidal neurons. D, Coimmunoprecipitation of Cx26 with FAK from the neocortex of E16 wild-type embryos. E, Schema of experimental design. F–G′, Distribution of Cx26 puncta in the cytoplasmic dilatation (white arrowheads) preceding the nucleus in migrating control (F, F′) and FAK-deficient (G, G′) pyramidal neurons. The open arrowheads point to Cx26 puncta found in non-electroporated cells. Scale bar, 5 μm.
We next examined whether FAK is needed for the formation of Cx26 puncta in migrating neurons. To this end, we electroporated progenitor cells in the dorsal pallium with control or Fak shRNA at E14.5 and analyzed Cx26 protein in migrating pyramidal cells at E18.5 (Fig. 7E). We found that the number of Cx26-containing puncta in migrating neurons transfected with shFak was dramatically reduced compared with control neurons (Fig. 7F–G′) (puncta per cell, mean ± SEM, shLuc: 100 ± 18.43, n = 20 cells; shFak: 33.89 ± 11.70, n = 21 cells; t test, **p = 0.0040). In contrast, we did not observe changes in the number puncta containing Cx43 (mean ± SEM, shLuc: 100 ± 12.58, n = 19 cells; shFak: 102.65 ± 23.11, n = 12 cells; t test, p = 0.9133), a related connexin that is also found in radially migrating neurons (Elias et al., 2007; Cina et al., 2009). These results indicated that FAK mediate its function during neuronal migration at least in part by regulating neuron–glia interactions specifically through Cx26.
We next performed genetic experiments to confirm that Fak and Cx26 function in the same molecular pathway during glial-guided migration (Fig. 8A). We reasoned that if both genes were in the same genetic pathway in this context, then simultaneous loss of function for Cx26 and Fak should not cause a stronger migratory phenotype than loss of function for any of the genes alone. Analysis of mouse embryos electroporated with Cx26 shRNA confirmed that loss of this connexin disrupts glial-guided migration (Elias et al., 2007) (Fig. 8B,L). Remarkably, coelectroporation of shCx26 and shFak caused a phenotype that was almost identical with that produced by shCx26 and shFak alone (Fig. 8B–D,L). So, these experiments reinforced the view that FAK and Cx26 function in the same genetic pathway during glial-guided migration.
Figure 8.
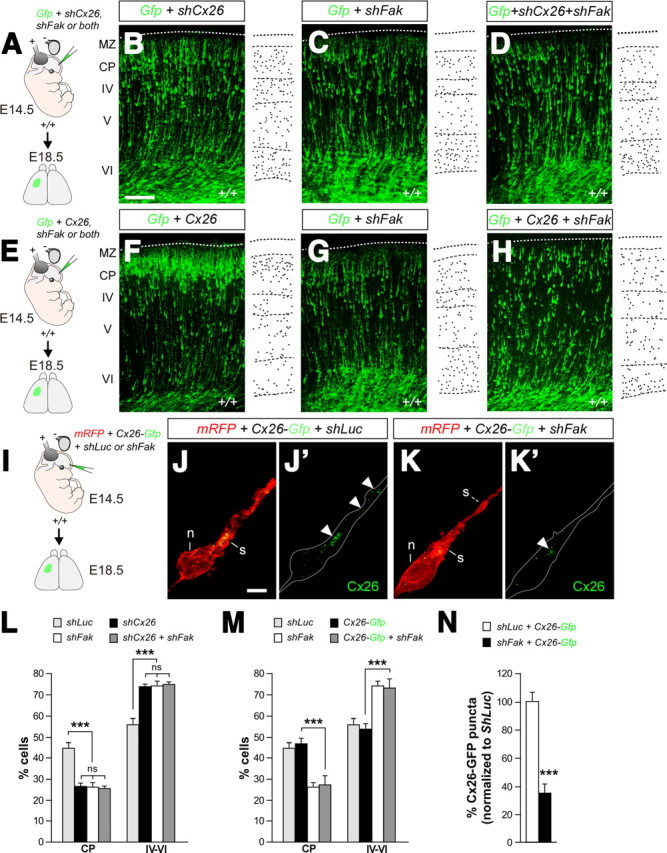
Genetic analysis of FAK–Cx26 interactions. A, Schematic diagram of experimental design. B–D, Coronal sections through the somatosensory cortex of E18.5 wild-type embryos after E14.5 in utero electroporation with plasmids encoding Gfp and shCx26 (B), shFak (C), or shCx26 plus shFak (D). Schemas with dots depict representative distributions of GFP-expressing neurons in each condition. E, Schematic diagram of experimental design. F–H, Coronal sections through the somatosensory cortex of E18.5 wild-type embryos after E14.5 in utero electroporation with plasmids encoding Gfp and Cx26 (F), shFak (G), or Cx26 plus shFak (H). Schemas with dots depict representative distributions of GFP-expressing neurons in each condition. I, Schematic diagram of experimental design. J–K′, Distribution of exogenous Cx26-GFP puncta in the cytoplasmic dilatation (white arrowheads) preceding the nucleus in migrating control (J, J′) and FAK-deficient (K, K′) pyramidal neurons. L, Quantification of the relative distribution (percentage) of GFP-expressing cells in the CP and the layers below it for each condition. Histograms show mean ± SEM from at least three different brains. shLuc: CP, 44.27 ± 2.66; layers IV–VI, 55.73 ± 2.66. shCx26: CP, 26.44 ± 1.21; layers IV–VI, 73.56 ± 1.21. shFak: CP, 25.96 ± 2.09; layers IV–VI, 73.99 ± 2.08. shCx26 plus shFak: CP, 25.28 ± 1.20; layers IV–VI, 74.72 ± 1.20. χ2 test for shLuc and shCx26, shFak or shCx26 plus shFak comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. χ2 test for shCx26 and shFak comparison: CP, ns; IV–VI, ns. χ2 test for shCx26 and shCx26 plus shFak comparison: CP, ns; IV–VI, ns. χ2 test for shFak and shCx26 plus shFak comparison: CP, ns; IV–VI, ns. M, Quantification of the relative distribution (percentage) of GFP-expressing cells in the CP and the layers below it for each condition. Histograms show mean ± SEM from at least three different brains. shLuc: CP, 44.27 ± 2.66; layers IV–VI, 55.73 ± 2.66. Cx26-Gfp: CP, 46.55 ± 2.54; layers IV–VI, 53.45 ± 2.54. shFak: CP, 25.96 ± 2.09; layers IV–VI, 73.99 ± 2.08. Cx26-Gfp plus shFak: CP, 26.95 ± 4.16; layers IV–VI, 73.05 ± 4.16. χ2 test for Cx26-Gfp and Cx26-Gfp plus shFak comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. N, Quantification of the relative (percentage) number of Cx26-GFP puncta in migrating neurons normalized to shLuc. Histograms show mean ± SEM from three different brains. shLuc plus Cx26-Gfp: 100 ± 6.53, n = 20 cells; shFak plus Cx26-Gfp: 34.93 ± 6.05, n = 19 cells. t test, ***p = 1.1612E-08. Scale bars: B–D, F–H, 100 μm; J–K′, 5 μm.
The reduced number of Cx26 puncta found in neurons lacking FAK suggested that this protein might be required to sustain normal levels of Cx26, either by promoting its synthesis or preventing its degradation. To test this idea, we attempted to rescue the migratory phenotype observed in mouse embryos electroporated with shFak by simultaneously transfecting cortical cells with a plasmid encoding full-length Cx26 (Fig. 8E). Somehow surprisingly, we found that expression of Cx26 did not rescue the phenotype caused by loss of FAK function (Fig. 8F–H,M). This result suggested that loss of FAK function might not interfere with the levels of Cx26, but rather with its ability to aggregate at adhesion points. To evaluate this possibility, we repeated the previous experiments using a variant of Cx26 that is fused to GFP (Cx26-Gfp) and a plasmid encoding a modified version of the red fluorescent protein that target this molecule to the membrane (mRFP), and analyzed the number of GFP puncta that aggregate in the membrane of migrating neurons electroporated with shLuc or shFak (Fig. 8I). Similar to endogenous Cx26 (Fig. 7F,F′), we found that exogenous Cx26-GFP aggregated in puncta preferentially located in the cytoplasmic swelling ahead of the nucleus in control cells (Fig. 8J,J′). By contrast, the number of Cx26-GFP puncta found in cells expressing shFak was significantly smaller than controls (Fig. 8K,K′,N). These experiments reinforced the view that FAK might be required for Cx26 to aggregate at adhesion points in radially migrating cells.
FAK is required for the recruitment of Cx26 to adhesion points
Our previous in vivo experiments indicated that the absence of Cx26 puncta in migrating neurons lacking FAK is probably due to a failure in the transport or aggregation of Cx26 at adhesion points rather than a deficit in the levels of this protein. Given the difficulty of measuring Cx26 levels in migrating cells in vivo, we performed a series of in vitro experiments using a C6 cell line that contains FAK but lacks endogenous expression of connexins (Naus et al., 1991; Hwang et al., 2006). We first performed coimmunoprecipitation experiments after transfection with a plasmid encoding Cx26-GFP. As in migrating neurons, we found that FAK and Cx26 interact in C6 cells (Fig. 9A). In contrast, we did not detect any interaction with Cx43 (Fig. 9A), reinforcing the view that FAK–Cx26 interaction is rather specific. We next examined whether perturbing FAK function leads to a decrease in the total levels of Cx26. We found that the level of Cx26 protein found in transfected C6 cells was not altered by the loss of FAK (Fig. 9B,C). These experiments confirmed that FAK is not required to regulate the levels of Cx26.
Figure 9.
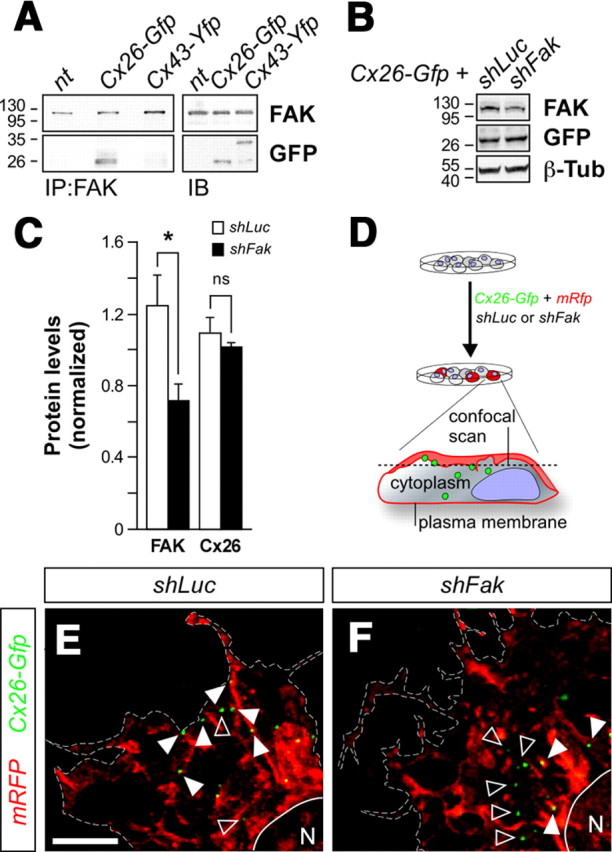
Decreased Cx26 in the membrane of FAK deficient cells. A, Coimmunoprecipitation of Cx26 but not Cx43 with FAK in C6 cells. B, Western blot of COS cells transfected with shLuc or shFak and Cx26-Gfp. Total levels of Cx26 were analyzed using GFP tag. C, Quantification of FAK and Cx26 protein levels following shLuc or shFak transfection. FAK: t test, *p = 0.04787; Cx26: t test, p = 0.4060. D, Schema of experimental design. E, F, Distribution of Cx26-GFP molecules associated with the membrane (white arrowheads) or the cytoplasm (open arrowheads) in C6 cells transfected with either shLuc (E) or shFak (F), membrane-directed RFP (mRFP), and Cx26-Gfp. Scale bar, 5 μm.
The previous experiments, along with our in vivo observations, suggested that the most plausible hypothesis to explain the absence of Cx26 puncta in FAK-deficient neurons is that FAK enables the formation of Cx26 puncta in the membrane. To directly test this hypothesis, we used high-resolution imaging to examine the distribution of Cx26 puncta in C6 cells transfected with either control or shFak. To distinguish between those connexins that were preassembled in the cytoplasm from those that have already reached the plasma membrane, we also transfected the cells with mRFP (Fig. 9D). Consistent with our previous biochemical observations, quantification of total number of Cx26 puncta revealed no differences between cells transfected with shLuc or shFak (Fig. 8E,F) (mean ± SEM, shLuc: 67.00 ± 5.63, n = 15 cells; shFak: 74.94 ± 9.23, n = 16 cells; t test, p = 0.4758). In contrast, we found that the ratio between membrane-bound and cytoplasmic Cx26 puncta was significantly lower in cells in which FAK function was perturbed (Fig. 8E,F) (membrane/cytoplasm ratio, mean ± SEM, shLuc: 3.15 ± 0.54, n = 15 cells; shFak: 1.51 ± 0.18, n = 16 cells; t test, **p = 0.0011). These experiments demonstrated that FAK function is required for the assembly or stability of Cx26 puncta in the plasma membrane.
FAK recruitment to the membrane is needed for Cx26 assembly
The previous experiments strongly suggested that FAK mediates the recruitment of Cx26 plaques to the membrane of migrating neurons, where they mediate glial-guided migration. We reasoned that FAK might be directly required to anchor Cx26 to adhesion sites, a function that has been described for FAK in relation to other proteins that are recruited to adhesions (Mitra et al., 2005). To gain support for this idea, we performed additional experiments in which we perturbed the recruitment of FAK to the plasma membrane. If FAK is required for the distribution of Cx26 in membrane of migrating neurons, then preventing FAK recruitment to adhesion points should be enough to perturb radial migration, and this process should be also associated with a decrease in the number of Cx26 puncta found in migrating neurons. To this end, we performed a series of rescue experiments using FakI937E/I999E, which encodes for a mutant form of FAK that cannot bind to Paxillin (Fig. 10A). Since Paxillin is required for the recruitment of FAK to adhesion points, expression of FakI937E/I999E prevents the targeting of FAK to the membrane (Hayashi et al., 2002). Expression of wild-type Fak rescued the migration phenotype observed in Cre-electroporated Fakflox/flox pyramidal neurons (Fig. 10B,C,H). In contrast, disrupting those residues required for its interaction with Paxillin prevented FAK from restoring pyramidal neuron migration (Fig. 10D,H). These results demonstrated that the recruitment of FAK to the membrane is essential for neuronal migration.
Figure 10.

FAK targeting to adhesion points is required for Cx26 puncta recruitment. A, Schematic diagram of experimental design. B–D, Coronal sections through the somatosensory cortex of E18.5 Fakflox/flox embryos after E14.5 in utero electroporation with plasmids encoding Cre-i-Gfp (B), Cre-i-Gfp plus Fak (C), Cre-i-Gfp plus FakI937E/I999E (D). E–G′, Distribution of Cx26 puncta in the cytoplasmic dilatation (white arrowheads) preceding the nucleus in migrating Fak-deficient (E, E′), Fak-rescued (F, F′), and FakI937E/I999E-rescued (G, G′) pyramidal neurons. The open arrowheads show Cx26 puncta in nonelectroporated cells. H, Quantification of the relative distribution (percentage) of GFP-expressing cells in the CP and the layers below it for each condition shown in B–D. Histograms show mean ± SEM from at least three different brains for each condition. Cre-i-Gfp: CP, 35.41 ± 1.22; layers IV–VI, 64.59 ± 1.22. Cre-i-Gfp plus Fak: CP, 47.08 ± 1.84; layers IV–VI, 52.92 ± 1.84. χ2 test for Cre-i-Gfp and Cre-i-Gfp plus Fak comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. Cre-i-Gfp plus FakI937E/I999E: CP, 36.00 ± 0.53; layers IV–VI, 64.00 ± 0.53. χ2 test for Cre-i-Gfp and Cre-i-Gfp plus FakI937E/I999E comparison: CP, ns; IV–VI, ns. χ2 test for Cre-i-Gfp plus Fak and Cre-i-Gfp plus FakI937E/I999E comparison: CP, ***p < 0.001; IV–VI, ***p < 0.001. I, Quantification of the relative number (percentage) of Cx26 puncta in migrating neurons normalized to Fak-rescued cells. Histograms show mean ± SEM from at least three different brains. Cre-i-Gfp: 46.75 ± 4.15, n = 20 cells; Cre-i-Gfp plus Fak: 100.00 ± 12.45, n = 16 cells; Cre-i-Gfp plus FakI937E/I999E: 55.15 ± 7.33, n = 17 cells. Cre-i-Gfp and Cre-i-Gfp plus Fak comparison: t test, ***p = 9.3371E-05. Cre-i-Gfp plus Fak and Cre-i-Gfp plus FakI937E/I999E comparison: t test, **p = 0.0036. Cre-i-Gfp and Cre-i-Gfp plus FakI937E/I999E comparison: t test, p = 0.3078. Scale bars: B–D, 100 μm; E–G′, 5 μm.
We next wondered whether abnormal recruitment of FAK to adhesion points would alter the pattern of Cx26 puncta in migrating cells. To this end, we quantified the number of Cx26 puncta in the cytoplasmic swelling of migrating pyramidal neurons in each of the three previous experimental conditions. As expected, we found that expression of Cre in Fakflox/flox pyramidal neurons reduced the normal levels of Cx26-containing puncta, as previously shown after expression of shFak in wild-type neurons (compare Figs. 7G,G′, 10E,E′,I). In addition, we observed that reexpression of Fak in Cre-electroporated Fakflox/flox pyramidal neurons was enough to rescue the number of Cx26 puncta in migrating neurons (Fig. 10F,F′,I), while expression of FakI937E/I999E did not (Fig. 10G,G′,I). These experiments reinforced the view that FAK is required for the recruitment of Cx26 complexes to adhesion points, thereby regulating the association of migrating neurons with radial glia processes during glial-guided migration.
Discussion
In this study, we demonstrate that the intracellular kinase FAK plays a critical role in glial-guided locomotion, while it is dispensable for the movement of neurons that migrate independently of radial glia, such as cortical interneurons or early-born pyramidal cells. In the absence of FAK, the interaction between migrating pyramidal cells and RGCs is perturbed (Fig. 11), which leads to impaired migration. Our results suggest that this defective interaction is likely mediated by connexins, which either fail to reach the membrane or are unable to stabilize in migrating neurons in which FAK function is compromised.
Figure 11.
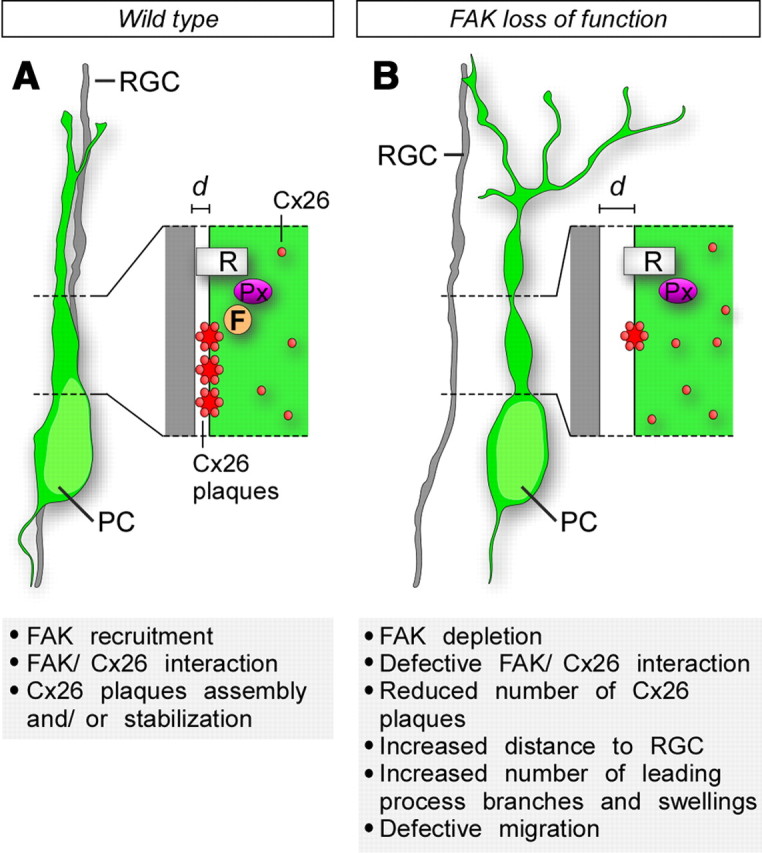
Cx26-dependent glial-guided migration is regulated by FAK. A, Recruitment of FAK (F) to adhesion points through its interaction with Paxillin (Px) is necessary for the recruitment of Cx26 to the membrane and/or to stabilize its aggregation in discrete plaques. Note that the schema only represents FAK and connexins present in migrating neurons, as the present study has not investigated their role in RGCs. B, FAK ablation leads to a failure in the formation/stabilization of Cx26 plaques at the membrane and disruption of radial migration. Pyramidal cells (PC) continue to migrate in the absence of FAK, but they are no longer able to maintain its attachment to the radial glia and the distance (d) between PCs and their parental RGC increases. Detachment from RGC also leads to important morphological modifications (multiple swelling, increased number of processes) in migrating neurons. R, Receptor.
FAK function in neuronal migration
Two previous studies have explored the function of FAK in radial migration (Beggs et al., 2003; Xie et al., 2003), with conflicting results. Beggs et al. (2003) used a genetic loss-of-function approach to study the consequences of conditionally ablating Fak from cortical progenitors (with an Emx1-Cre mouse strain) or migrating neurons (with the same NEX-Cre mouse strain used in this study). They reported that loss of Fak in RGCs produces cortical ectopias, caused by a disruption of the integrity of the pial basement membrane (Beggs et al., 2003). Beggs et al. (2003) did not observe any obvious migratory alteration after conditional deletion of Fak in migrating pyramidal neurons, which led them to propose that FAK function in neuronal migration was primarily non-cell-autonomous. Our results suggest that FAK is indeed required in cortical neurons for their normal migration. Several factors may account for this apparent discrepancy, including the time of analysis (adult vs E18.5 and P7) or the analytical methods (Nissl staining vs Gfp electroporation and Cux1 expression). We found that loss of FAK in NEX-Cre;Fakflox/flox embryos causes a smaller migratory phenotype than the early deletion of FAK by Cre electroporation or, even more so, than knockdown of Fak using shRNA. Nevertheless, reexpression of FAK in migrating neurons is enough to rescue the migratory phenotype caused by loss of Fak in progenitor cells (Fig. 4), which demonstrate that FAK function in neurons is necessary for neuronal migration. The differences observed using the three different manipulations to interfere with FAK function are likely explained by the time required for FAK elimination in each case (NEX-Cre > Cre in VZ > Fak shRNA in VZ).
The analysis of the distribution of migrating pyramidal neurons in E18.5 embryos revealed that FAK-deficient delayed neurons accumulated primarily in the upper intermediate zone and lower cortical layers. In addition, although FAK-deficient neurons displayed an abnormal morphology with multiple branches in the leading process, cells remained highly polarized. These observations suggest that the transition from the multipolar to bipolar stage, a critical step during pyramidal neuronal migration (LoTurco and Bai, 2006), is not dramatically affected by the loss of FAK (data not shown). Instead, loss of FAK function seems to primarily affect pyramidal neurons as they engage in the main phase of glial-guided migration. We also observed that most FAK-deficient pyramidal cells manage to reach the superficial layers of the cortex in postnatal mice, which suggest that FAK deficiency delays rather than blocks the migration of pyramidal neurons. Nevertheless, we consistently found some FAK-deficient neurons accumulating in ectopic locations of the postnatal cortex. Interestingly, these neurons displayed abnormal dendritic morphology, as described by Beggs et al. (2003) in NEX-Cre;Fakflox/flox adult mice.
It is worth mentioning that our experiments do not rule out a role for FAK in regulating the interaction between the basal processes of RGCs and the basal lamina, as proposed by Beggs et al. (2003), because there are important technical differences between both approaches. First, the reported disruptions of the basal lamina found in Emx1-Cre;Fakflox/flox embryos were primarily confined to the medial cortical wall (Beggs et al., 2003), while our analysis has been performed in the somatosensory cortex. It is conceivable that only the alteration of RGCs close to the midline may cause a disruption of the basal lamina. Moreover, the electroporation approach used in our study only affects a restricted number of RGCs, and so every electroporated region contains many wild-type cells that have not been targeted. This is in sharp contrast with the genetic approach used by Beggs et al. (2003), in which FAK function was compromised in all progenitors. Thus, we believe that our findings are not incompatible with a model in which FAK also regulate the interaction between radial glial fibers and the basal lamina (Beggs et al., 2003).
In this study, we have also analyzed the function of FAK during glial-independent neuronal migration. Our experiments demonstrate that FAK function is dispensable in this process. This conclusion is based not only on the absence of migratory defects in the tangential migration of cortical interneurons (Fig. 1) but also on the observation that early-born pyramidal cells, which move independently of radial glia (Miyata et al., 2001; Nadarajah et al., 2001; Kriegstein and Noctor, 2004), migrate normally in the absence of FAK (Fig. 5). It is conceivable that another protein may compensate for the loss of FAK in tangentially migrating cells. For example, the related kinase Pyk2 has been shown to cooperate with FAK in regulating the migration of endothelial cells (Weis et al., 2008). However, our preliminary experiments indicate that cortical interneurons lacking both FAK and Pyk2 function also migrate to the cortex (our unpublished observations), which reinforces the notion that these proteins are not required for glial-independent migration. It remains to be determined whether FAK is required for the final allocation of interneurons into specific cortical layers, a process that may involve glial-guided migration (Yokota et al., 2007). Unfortunately, Dlx5/6-Cre-i-Gfp;Fakflox/flox mice die at birth due to cleft palate (our unpublished observations), thus preventing the analysis of the role of FAK during this phase of interneuron migration.
Regulation of connexin adhesions by FAK
It is well established that FAK plays a prominent role in regulating the dynamic behavior of adhesion contacts that are formed upon activation of integrins and other growth factor receptors. However, FAK has also been involved in the regulation of cell–cell junctions (Avizienyte and Frame, 2005; Mitra et al., 2005), as suggested here for migrating neurons. In HeLa cells, for example, FAK activity has been associated with the formation or turnover of contacts in N-cadherin-based cell–cell junctions (Schaller, 2004; Yano et al., 2004). Recent studies suggest that N-cadherin plays a role in glial-independent radial migration, but it is not required for the interaction of pyramidal cells with the basal processes of radial glial fibers (Franco et al., 2011; Jossin and Cooper, 2011). Instead, this process seems to rely on the dynamic regulation of Gap junction adhesions (Elias et al., 2007). Gap junctions are best known for their role in intercellular communication by electrically and chemically coupling cells or by forming hemichannels for the extracellular release of substrates such as ATP (Söhl et al., 2005). However, recent work has shown that connexin-mediated adhesion, and not channel function, is required for pyramidal neuron migration (Elias et al., 2007; Cina et al., 2009). Our study suggest that FAK is involved in the dynamic regulation of this novel type of cell–cell adhesions, since loss of FAK in migrating neurons prevents Cx26 complexes to either reach or stabilize at the membrane, and impairs migration.
In neurons, adhesion contacts have only been previously analyzed in growth cones, where they take the shape of small, discrete point contacts with a high turnover rate (Gomez et al., 1996; Robles and Gomez, 2006; Bechara et al., 2008; Chacón et al., 2010). In migrating pyramidal neurons, point contacts are enriched in the perinuclear region, in particular in the cytoplasmic dilatation of the leading process that precedes the nucleus. The existence of discrete sites of adhesion in this later location has been hypothesized as a mechanism to regulate the saltatory movement of migrating neurons (Schaar and McConnell, 2005; Shieh et al., 2011). Our biochemical and genetic experiments suggest that adhesion points in migrating neurons contain not only classical adhesion proteins such as FAK but also Cx26. Interestingly, while FAK activity is primarily involved in the disassembly of adhesion contacts in most cell types, FAK signaling seems to be particularly important for the assembly of adhesion points in migrating neurons (this study), as it has been previously shown for growth cones (Robles and Gomez, 2006). This suggests that fundamental differences exist in the dynamic regulation of adhesion complexes between neuronal versus non-neuronal cells.
We found that FAK interacts with Cx26, as described in prostate cells (Tate et al., 2006), which suggests that this connexin might be a direct substrate of FAK activity. Alternatively, FAK and Cx26 may form part of a larger protein complex in which FAK regulates the stability of a protein that contribute to the assembly of Cx26 in the membrane. For example, FAK has been shown to stabilize occludin and zonula occludens-1 (ZO-1) in tight junctions (Siu et al., 2009a,b), and ZO-1 has also been described as part of the architecture of gap junctions (Giepmans, 2004). This mechanism is compatible with a possible function of FAK as a scaffold protein that brings together all these components to the adhesion point, and is consistent with the observation that preventing FAK recruitment to adhesion points is sufficient to impair the assembly of Cx26 puncta in the membrane. Finally, we cannot completely rule out the possibility that FAK contributes to the transport of Cx26 puncta to the membrane. Indeed, the intracellular transport of Cx26 depends on intact actin filaments (Thomas et al., 2001), and loss of FAK function destabilizes the actin cytoskeleton assembled around adhesions (Mitra et al., 2005). Thus, FAK may also contribute to the recruitment of Cx26 puncta by directly regulating the assembly of actin fibers near the adhesion point. Future studies should address clarifying the detailed molecular mechanisms through which FAK regulates this process.
Footnotes
This work was supported by Spanish Ministry of Science and Innovation (MICINN) Grants SAF2008-00770 and SAF 2009-08049-E, and CONSOLIDER Grant CSD2007-00023 (O.M.). M.V. was supported by a fellowship from the Generalitat Valenciana (CTBPRA/2005/021). G.C. is the recipient of a FPI fellowship from MICINN. We thank T. Gil and M. Pérez for excellent technical assistance; H. E. Beggs and L. F. Reichardt (Fakflox), and S. Goebbels and K.-A. Nave (NEX-Cre) for mouse strains; F. H. Gage for retroviruses; and M. Falk, J. L. Guan, F. Guillemot, J. Livet, T. Parsons, F. Polleux, J. Sanes, and S. S. Yum for plasmids and reagents. We are also thankful to members of the Marín, Rico, and Borrell Laboratories for helpful discussions and comments.
References
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005;17:542–547. doi: 10.1016/j.ceb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Batista-Brito R, Fishell G. The developmental integration of cortical interneurons into a functional network. Curr Top Dev Biol. 2009;87:81–118. doi: 10.1016/S0070-2153(09)01203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Nawabi H, Moret F, Yaron A, Weaver E, Bozon M, Abouzid K, Guan JL, Tessier-Lavigne M, Lemmon V, Castellani V. FAK-MAPK-dependent adhesion disassembly downstream of L1 contributes to semaphorin3A-induced collapse. EMBO J. 2008;27:1549–1562. doi: 10.1038/emboj.2008.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellion A, Baudoin JP, Alvarez C, Bornens M, Métin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J Neurosci. 2005;25:5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS, Bhide PG, Nowakowski RS. Histogenetic processes leading to the laminated neocortex: migration is only a part of the story. Dev Neurosci. 2008;30:82–95. doi: 10.1159/000109854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón MR, Fernández G, Rico B. Focal adhesion kinase functions downstream of Sema3A signaling during axonal remodeling. Mol Cell Neurosci. 2010;44:30–42. doi: 10.1016/j.mcn.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Cina C, Maass K, Theis M, Willecke K, Bechberger JF, Naus CC. Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J Neurosci. 2009;29:2009–2021. doi: 10.1523/JNEUROSCI.5025-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Borello U, Rubenstein JL. Dlx transcription factors promote migration through repression of axon and dendrite growth. Neuron. 2007;54:873–888. doi: 10.1016/j.neuron.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstat DD, Liu JK, Mione M, Zhong W, Yu G, Anderson SA, Ghattas I, Puelles L, Rubenstein JL. DLX-1, DLX-2, and DLX-5 expression define distinct stages of basal forebrain differentiation. J Comp Neurol. 1999;414:217–237. doi: 10.1002/(sici)1096-9861(19991115)414:2<217::aid-cne6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Elias LA, Turmaine M, Parnavelas JG, Kriegstein AR. Connexin 43 mediates the tangential to radial migratory switch in ventrally derived cortical interneurons. J Neurosci. 2010;30:7072–7077. doi: 10.1523/JNEUROSCI.5728-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM. Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci. 2000;113:4109–4120. doi: 10.1242/jcs.113.22.4109. [DOI] [PubMed] [Google Scholar]
- Flames N, Long JE, Garratt AN, Fischer TM, Gassmann M, Birchmeier C, Lai C, Rubenstein JL, Marín O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004;44:251–261. doi: 10.1016/j.neuron.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Müller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–497. doi: 10.1016/j.neuron.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Roche FK, Letourneau PC. Chick sensory neuronal growth cones distinguish fibronectin from laminin by making substratum contacts that resemble focal contacts. J Neurobiol. 1996;29:18–34. doi: 10.1002/(SICI)1097-4695(199601)29:1<18::AID-NEU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Grant SG, Karl KA, Kiebler MA, Kandel ER. Focal adhesion kinase in the brain: novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev. 1995;9:1909–1921. doi: 10.1101/gad.9.15.1909. [DOI] [PubMed] [Google Scholar]
- Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sanada K, Miyamoto DT, Rovelstad S, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH. Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat Neurosci. 2003;6:1284–1291. doi: 10.1038/nn1151. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Hisanaga S, Heizmann CW, Murakami F. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J Comp Neurol. 2004;479:1–14. doi: 10.1002/cne.20256. [DOI] [PubMed] [Google Scholar]
- Hayashi I, Vuori K, Liddington RC. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol. 2002;9:101–106. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, Skowronska-Krawczyk D, Bedogni F, Matter JM, Hevner R, Guillemot F. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature. 2008;455:114–118. doi: 10.1038/nature07198. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Jung JW, Jeong JS, Kim YJ, Oh ES, Kim TH, Kim JY, Cho KH, Han IO. Dominant-negative Rac increases both inherent and ionizing radiation-induced cell migration in C6 rat glioma cells. Int J Cancer. 2006;118:2056–2063. doi: 10.1002/ijc.21574. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Cooper JA. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci. 2011;14:697–703. doi: 10.1038/nn.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Marín O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin/neuropilin interactions. Science. 2001;293:872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- Marín O, Valdeolmillos M, Moya F. Neurons in motion: same principles for different shapes? Trends Neurosci. 2006;29:655–661. doi: 10.1016/j.tins.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Martini FJ, Valiente M, López Bendito G, Szabó G, Moya F, Valdeolmillos M, Marín O. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development. 2009;136:41–50. doi: 10.1242/dev.025502. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 2001;31:727–741. doi: 10.1016/s0896-6273(01)00420-2. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Naus CC, Bechberger JF, Caveney S, Wilson JX. Expression of gap junction genes in astrocytes and C6 glioma cells. Neurosci Lett. 1991;126:33–36. doi: 10.1016/0304-3940(91)90364-y. [DOI] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Kessaris N, Du T, Kimura S, Anderson SA, Marín O. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59:733–745. doi: 10.1016/j.neuron.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Rakic P. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science. 1974;183:425–427. doi: 10.1126/science.183.4123.425. [DOI] [PubMed] [Google Scholar]
- Rakic P. A century of progress in corticoneurogenesis: from silver impregnation to genetic engineering. Cereb Cortex. 2006;16(Suppl 1):i3–i17. doi: 10.1093/cercor/bhk036. [DOI] [PubMed] [Google Scholar]
- Rakic P. The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res Rev. 2007;55:204–219. doi: 10.1016/j.brainresrev.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles E, Gomez TM. Focal adhesion kinase signaling at sites of integrin-mediated adhesion controls axon pathfinding. Nat Neurosci. 2006;9:1274–1283. doi: 10.1038/nn1762. [DOI] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci U S A. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD. FAK and paxillin: regulators of N-cadherin adhesion and inhibitors of cell migration? J Cell Biol. 2004;166:157–159. doi: 10.1083/jcb.200406151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JC, Schaar BT, Srinivasan K, Brodsky FM, McConnell SK. Endocytosis regulates cell soma translocation and the distribution of adhesion proteins in migrating neurons. PLoS One. 2011;6:e17802. doi: 10.1371/journal.pone.0017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Siu ER, Wong EW, Mruk DD, Porto CS, Cheng CY. Focal adhesion kinase is a blood-testis barrier regulator. Proc Natl Acad Sci U S A. 2009a;106:9298–9303. doi: 10.1073/pnas.0813113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu ER, Wong EW, Mruk DD, Sze KL, Porto CS, Cheng CY. An occludin-focal adhesion kinase protein complex at the blood-testis barrier: a study using the cadmium model. Endocrinology. 2009b;150:3336–3344. doi: 10.1210/en.2008-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6:191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, Hatten ME. Par6alpha signaling controls glial-guided neuronal migration. Nat Neurosci. 2004;7:1195–1203. doi: 10.1038/nn1332. [DOI] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. 2006;442:929–933. doi: 10.1038/nature05028. [DOI] [PubMed] [Google Scholar]
- Tate AW, Lung T, Radhakrishnan A, Lim SD, Lin X, Edlund M. Changes in gap junctional connexin isoforms during prostate cancer progression. Prostate. 2006;66:19–31. doi: 10.1002/pros.20317. [DOI] [PubMed] [Google Scholar]
- Thomas T, Jordan K, Laird DW. Role of cytoskeletal elements in the recruitment of Cx43-GFP and Cx26-YFP into gap junctions. Cell Commun Adhes. 2001;8:231–236. doi: 10.3109/15419060109080729. [DOI] [PubMed] [Google Scholar]
- Tilghman RW, Slack-Davis JK, Sergina N, Martin KH, Iwanicki M, Hershey ED, Beggs HE, Reichardt LF, Parsons JT. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J Cell Sci. 2005;118:2613–2623. doi: 10.1242/jcs.02380. [DOI] [PubMed] [Google Scholar]
- Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA, Chen XL, Göthert JR, Shen TL, Guan JL, Schlaepfer DD, Cheresh DA. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SX, Goebbels S, Nakamura K, Kometani K, Minato N, Kaneko T, Nave KA, Tamamaki N. Pyramidal neurons of upper cortical layers generated by NEX-positive progenitor cells in the subventricular zone. Proc Natl Acad Sci U S A. 2005;102:17172–17177. doi: 10.1073/pnas.0508560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell. 2003;114:469–482. doi: 10.1016/s0092-8674(03)00605-6. [DOI] [PubMed] [Google Scholar]
- Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. J Cell Biol. 2004;166:283–295. doi: 10.1083/jcb.200312013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Gashghaei HT, Han C, Watson H, Campbell KJ, Anton ES. Radial glial dependent and independent dynamics of interneuronal migration in the developing cerebral cortex. PLoS One. 2007;2:e794. doi: 10.1371/journal.pone.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]