

Abstract
The mitochondrial DNA (mtDNA) of neural stem cells (NSCs) is vulnerable to oxidation damage. Subtle manipulations of the cellular redox state affect mtDNA integrity in addition to regulating the NSC differentiation lineage, suggesting a molecular link between mtDNA integrity and regulation of differentiation. Here we show that 8-oxoguanine DNA glycosylase (OGG1) is essential for repair of mtDNA damage and NSC viability during mitochondrial oxidative stress. Differentiating neural cells from ogg1−/− knock-out mice spontaneously accumulate mtDNA damage and concomitantly shift their differentiation direction toward an astrocytic lineage, similar to wt NSCs subjected to mtDNA damaging insults. Antioxidant treatments reversed mtDNA damage accumulation and separately increased neurogenesis in ogg1−/− cells. NSCs from a transgenic ogg1−/− mouse expressing mitochondrially targeted human OGG1 were protected from mtDNA damage during differentiation, and displayed elevated neurogenesis. The underlying mechanisms for this shift in differentiation direction involve the astrogenesis promoting Sirt1 via an increased NAD/NADH ratio in ogg1−/− cells. Redox manipulations to alter mtDNA damage level correspondingly activated Sirt1 in both cell types. Our results demonstrate for the first time the interdependence between mtDNA integrity and NSC differentiation fate, suggesting that mtDNA damage is the primary signal for the elevated astrogliosis and lack of neurogenesis seen during repair of neuronal injury.
Introduction
Neural stem cells (NSCs) with multipotent and self-renewing abilities are critical in maintaining brain functions and in brain repair process after acute or chronic injuries (Reynolds and Weiss, 1992; Björklund and Lindvall, 2000; Gage, 2000; Ming and Song, 2005). In the adult mammalian brain, there are two distinct germinal areas containing the multipotent NSCs: the subventricular zone of the lateral ventricles and the subgranular layer of the hippocampal dentate gyrus (Gage et al., 1998; Doetsch et al., 1999; Gage, 2000). Generation and expansion of in vitro free-floating neurospheres containing NSCs becomes a promising therapeutic potential for a variety of neurodegenerative disorders, but is hampered by excessive astrocyte formation in neurodegeneration and brain injuries characterized by oxidative stress and inflammation. It becomes evident that the NSC differentiation fate is determined by interplay between extracellular signal modification and intrinsic intracellular machinery, leading to the activation of lineage-specific pathways determining the differentiation fate being neurons or astrocytes (Guillemot, 2007). Interestingly, reactive oxygen species (ROS) levels can alter differentiation fate of mouse progenitor cells toward either neuronal or astroglial lineages by a mechanism involving Sirt1 and Mash1 (Prozorovski et al., 2008).
Mitochondria can regulate cellular ROS levels, and we have recently shown that the increased aerobic activity during differentiation generates oxidative damage to mtDNA (Wang et al., 2010). The potential risk of DNA damage in NSCs is envisioned by the enhanced expression of the DNA repair protein 8-oxoguanine DNA glycosylase (OGG1) during early embryonic development (Hildrestrand et al., 2007). OGG1 removes oxidized purine lesions, such as 8-oxoguanine and formamidopyrimidines from both nuclear and mitochondrial DNA. The OGG1 incision represents the first step in a multienzyme DNA repair cascade known as the base excision repair. This repair pathway is held as the major pathway for maintaining mtDNA. The ogg1−/− mouse accumulates 8-oxoguanine particularly in mtDNA but is otherwise normal under normal breeding conditions (de Souza-Pinto et al., 2001; Klungland et al., 1999). However, OGG1 has been shown to protect cells during mitochondrial oxidative stress (Oka et al., 2008), and we recently discovered that OGG1 was essential for protecting mtDNA from differentiation-mediated oxidation damage (Wang et al., 2010). Hence, OGG1 deficiency appears to be manifested as reduced mtDNA integrity under conditions with elevated ROS, such as increased respiration activity.
The functional importance of mitochondria and mtDNA in the NSC biology has been largely neglected. However, we have recently found evidence that mtDNA is readily modified by subtle changes in intracellular redox ratios that independently alter differentiation direction of NSCs. We tested the hypothesis that differentiation fate depends on the mtDNA integrity, using an established in vitro differentiation system with primary NSCs from dentate gyrus of hippocampus from wt and ogg1−/− mouse. NSCs from the latter progressively accumulate spontaneous mtDNA damage during differentiation.
Materials and Methods
NSCs and differentiation.
Primary NSCs were prepared from hippocampal dentate gyrus of C57BL/6 and C57BL/6 ogg1−/− knock-out mice (of either sex, not littermates) at postnatal day 5 and propagated as previously described (Wang et al., 2010). Briefly, initial passages of cells were cultured for 7 d as floating neurospheres in proliferation medium consisting of serum-free Neurobasal-A medium with 2% B27 supplement, 20 ng/ml basic fibroblast growth factor, 10 ng/ml epidermal growth factor, 2 mm l-glutamine, and penicillin/streptomycin. Subsequently neurospheres were plated onto poly-l-lysine-coated plates or flasks in differentiation medium (proliferation medium without epidermal growth factor) until further experiments.
To generate transgenic mouse expressing isoform 1a of human OGG1 (mtOGG1), the hOGG1 gene was fused to a mitochondrial translocation signal similarly as described previously (Dobson et al., 2000) and provided with a kozak sequence and set under the control of a CMV/AG promoter. Positive carriers from pronuclei injection (performed by Norsk Transgen Senter) were crossed with ogg1−/− knock-out mice to provide homozygote C57BL/6 ogg1−/−/mtOGG1. mtOGG1 carriers were identified by PCR amplification of transgenic fragment using the 5′-TGGCGTGTGACCGGCGGCTCTA and 5′-GCAGAAGCGCGCGGGCAGGCAT primers.
To evaluate pro-oxidant/antioxidant effect on differentiation parameters, buthionine sulfoxide (BSO; 2.5 μm), lipoic acid (LA; 5 μm), and N-acetylcysteine (NAC; 0.75 mm) were added to the differentiation medium. 2-Methyl-1,4-naphthoquinone (menadione; 25 μm) exposure was limited to 1 h, after NSCs have adhered to the plate for 4 h.
Immunocytochemistry.
Immunocytochemistry was performed as described previously (Wang et al., 2010). Following blocking with 5% BSA, 5% goat serum, and 0.1 Triton X-100 in PBS for 30 min, the cells were incubated with monoclonal anti-neuron-specific β-III tubulin (Tuj-1, 1:200, clone Tuj-1, R&D Systems), rabbit polyclonal astrocyte-specific anti-glial fibrillary acidic protein (GFAP, 1:500, clone G-A-5, Sigma), or monoclonal anti-nestin (1:200, clone rat-401, Millipore Bioscience Research Reagents) in PBS containing 0.5% BSA, 0.5% goat serum, and 0.1% Tween 20 at 4°C overnight. Anti-mouse secondary antibody was Alexa 594 (1:500, Invitrogen). To obtain the percentage of each cell type, 1300–1500 cells that were morphologically identified in 10 random fields of each treatment from three different cultures were counted under a 10× objective. Percentage of positive cells was calculated in relation to total number of cells, visualized by DAPI nuclear staining (1 μg/ml: Invitrogen).
Quantitative real-time PCR.
Total RNA from cultured cells was isolated using the RNeasy Kit from Qiagen and cDNA was prepared using Omniscript RT Kit (Qiagen). Quantitative real-time PCR was performed with a 7900HT Fast Real-Time PCR System (Applied Biosystems) using the Power SYBR green PCR Master mix (AB Applied Biosystems). Oligonucleotides used were as follows: Tuj-1, forward 5′-ccaagacaagcagcatctgt, reverse 5′-cagagccaagtggactcaca (Kamnasaran et al., 2008); GFAP, forward 5′-tcctggaacagcaaaacaag, reverse 5′-cagcctcaggttggtttcat (Sandhir et al., 2008); Sirt1, forward 5′-gatgacgatgacagaacgtcaca, reverse 5′-ggatcggtgccaatcatgag (Prozorovski et al., 2008); GAPDH, forward 5′-tcgtcccgtagacaaaatggt, reverse 5′-cgcccaatacggccaaa (Hildrestrand et al., 2007).
mtDNA damage detection.
mtDNA damage in NSCs was detected by the following: (1) quantitative PCR-based amplification of a large mtDNA fragment (Santos et al., 2002) and (2) mtDNA damage-mediated inhibition of restriction enzyme cleavage (Vermulst et al., 2007; Wang et al., 2010).
mtOGG1 DNA glycosylase assay.
Mitochondrial OGG1 measurement in brain mitochondrial extract was measured as described previously (de Souza-Pinto et al., 2001). Briefly, crude brain mitochondrial extracts were incubated with 1 fmol of 32P-end-labeled duplex DNA containing 8-oxoguanine in a defined position for 3 h at 37°C. The incised product was separated by denaturing PAGE and subjected to phosphoimaging by a Typhoon scanner. Recombinant hOGG1 was from our collection.
MTT viability assay.
Cell viability was determined with Cell Proliferation kit (Roche Diagnostics) and absorbance was measured on a Wallac 1420 Multilabel Counter at a wavelength of 570 nm and reference wavelength of 630 nm. The cell viability was calculated as percentage relative to control cells set as 100%.
NAD/NADH assessment.
The NAD+ and NADH levels were determined using a NAD/NADH assay kit from Abcam according to the manufacturer's instructions. Absorbance was measured within a linear range on a Wallac 1420 Multilabel Counter at a wavelength of 450 nm. A standard curve was established and protein concentrations of cell lysates were determined using Bio-Rad protein assay kit.
Statistical analysis.
All data are presented as the mean ± SD. The mean values between two groups were compared by two-tailed paired Student's t test. Differences were considered to be statistically significant at a p value of <0.05.
Results
To address the role of OGG1 protein in genome maintenance and NSC survival, we exposed NSCs derived from hippocampus of wt and ogg1−/− mice to two different types of pro-oxidants: BSO, which is an inhibitor of glutathione synthesis, and menadione, which is a superoxide anion inducer. While BSO generally heightens cellular peroxyl species via glutathione (millimolar range) depletion, menadione more specifically targets mitochondrial compartments. These two agents exert distinct effects on wt and ogg1−/− cells. BSO killed both types of NSCs equally efficient, whereas ogg1−/− NSCs were significantly more sensitive to menadione than to wt NSCs (Fig. 1A). Thus, OGG1 is essential to restore NSC resistance to mitochondrial oxidation. The mtDNA damaging potential of menadione was evaluated by two distinct methods. The mtDNA damage level was similar in the nontreated wt and ogg1−/− NSCs. Both methods demonstrate significant mtDNA damage formation in ogg1−/− NSCs that were exposed to menadione at concentrations ≥25 μm (Fig. 1B,C). Interestingly, increased mtDNA damage was not observed in wt NSC, implying that the repair capacity of OGG1 is sufficient to continuously remove menadione-induced mtDNA damage.
Figure 1.

DNA glycosylase OGG1 is essential for protection against mtDNA damage in NSCs. A, Sensitivity of wt and ogg1−/− NSCs to pro-oxidants. Single NSCs were plated in differentiation medium for 4 h before exposure. B, mtDNA damage detection by the relative quantitative amplification of a small (117 bp) and large (10 kb) of mtDNA. Top, A representative result. Bottom, The mean with SD from more than three independent experiments. C, mtDNA damage detection by the ability to inhibit restriction cleavage, quantified by real-time PCR. Data are mean with SD from more than three independent experiments. *p < 0.05; **p < 0.01.
We have recently shown that mtDNA suffers from damaging insults after the first day in differentiation medium, and that OGG1 is essential for removal of these spontaneously formed mtDNA damages (Wang et al., 2010). Given the protective role of OGG1 against ROS-induced mtDNA damage and NSC sensitivity as well as the reported impact of ROS on NSC differentiation fate, we were prompted to analyze differentiation capacity in ogg1−/− NSCs cells that fail to protect mtDNA from spontaneously formed oxidation damage. The differentiation in vitro can be assessed by ability to express nestin, a precursor filament protein expressed in NSC and neural progenitor cells. The fraction of wt cells expressing nestin dropped from 95% to <10% in 4 d (data not shown) upon initiation of differentiation as reported (Reynolds and Weiss, 1992; Kunke et al., 2009). The decrease in NSCs with the corresponding generation of neurons and astrocytes was evaluated by gene expression analyses of nestin, Tuj-1, and GFAP, respectively (Fig. 2A). Expression of Tuj-1 was relatively more active than GFAP for the first 24 h, indicating that neurogenesis precedes the development of astrocytes.
Figure 2.
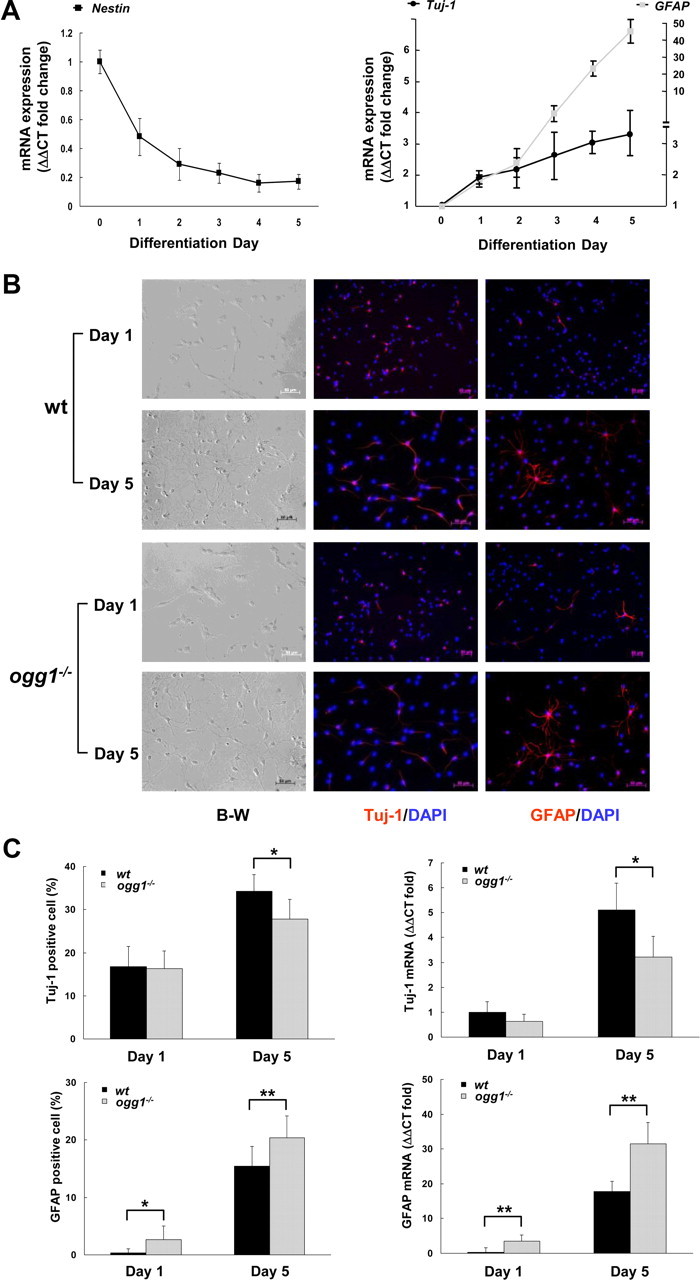
Differentiation of wt and ogg1−/− NSCs. A, Growth factor removal-induced differentiation of NSCs is manifested as reduced levels of nestin and corresponding elevations in the neuronal Tuj-1 and astrocytic GFAP markers. B, Progressive reduction of neurogenesis and increased astrogenesis from ogg1−/− NSCs. Left, Differentiating cells from wt and ogg1−/− NSCs are morphological similar (B-W, black and white phase-contrast images). Neurons and astrocytes at differentiation day 1 and 5 were identified by immunocytochemistry using antibodies against Tuj-1 (middle) and GFAP (right). C, Quantitative analysis of neurons and astrocytes at day 1 and day 5 in differentiation. Gene expression analyses, presented relative to wt at day 1. Data are mean with SD from more than three independent experiments. *p < 0.05; **p < 0.01.
To determine whether the mtDNA damage accumulation in ogg1−/− cells (Fig. 1B,C) affects NSCs, differentiation patterns of wt and ogg1−/− NSCs were compared using Tuj-1 and GFAP antibodies to identify neurons and astrocytes. Differentiating NSCs were morphological similar in both cell types (Fig. 2B). At the first day in differentiation medium, at the time when mtDNA was intact in both cells, the number of neurons was equal in wt and ogg1−/− (16.3 ± 4.1% vs 16.8 ± 4.7%) (Fig. 2C). Further formation of ogg1−/− neurons, however, was significantly attenuated compared to wt neurons (27.5 ± 4.6% in ogg1−/− vs 34.6 ± 3.9% in wt cells at day 5). It can be inferred from Figure 2B that neurogenesis from ogg1−/− NSCs is only 60% [(27.5 − 16.5)/(34.6 − 16.5)] of that in wt NSC after the first day. In contrast, astrocytic development was promoted during ogg1−/− differentiation, and ogg1−/− astrocytes (GFAP-positive cells) were evident already on the first day (2.7 ± 2.3% vs 0.4 ± 0.7% in wt). After additional 4 d in differentiation medium, ogg1−/− astrocytes yielded 20.4 ± 3.8% compared with 15.4 ± 3.5% in wt (Fig. 2B). The altered differentiation lineage identified by immunocytochemistry was confirmed by gene expression analyses (Fig. 2C). The total differentiation capacity (total frequency of neurons and astrocytes) was similar, suggesting that the OGG1 deficiency involves mtDNA damage accumulation that results in a shifted differentiation lineage, with more astrocytes being formed.
Sirt1 plays a critical role in the regulation of neural stem/progenitor cell differentiation (Prozorovski et al., 2008) and is sensitive to changes of intracellular NAD+ levels (Imai et al., 2000; Landry et al., 2000; Fulco et al., 2003). Possible Sirt1 activation in ogg1−/− cells was supported by elevated NAD during differentiation along with increased expression of Sirt1 (Fig. 3A,B), which previously has been shown to support Mash1-mediated astrogliosis (Prozorovski et al., 2008). Importantly, there was no difference in Sirt1 expression in wt and ogg1−/− NSCs before differentiation (Fig. 3B, D0), at the time when mtDNA integrity is similar in the two cells (Wang et al., 2010). In coherence with the shifted differentiation, Sirt1 expression was reduced by reducing agents in the ogg1−/− cells, whereas oxidants increased expression (Fig. 3C). Together, this implies that mtDNA damage accumulation alters cellular redox levels that are sufficient to shift differentiation lineage via Sirt1 signaling.
Figure 3.
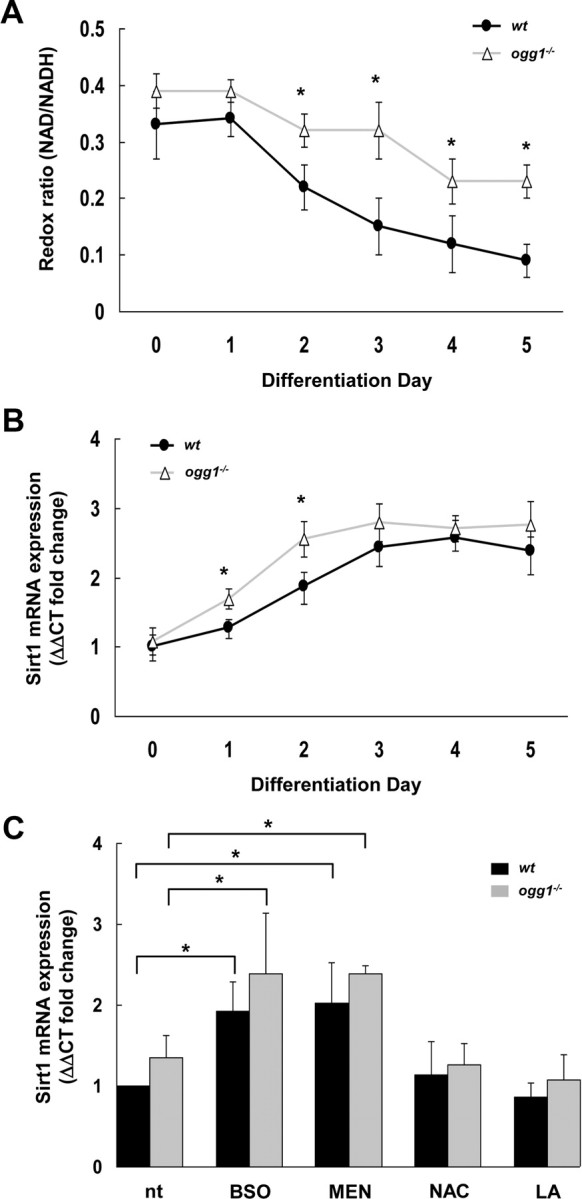
Altered redox levels and Sirt1 activation in ogg1−/− cells. A, Cellular redox levels were analyzed as NAD/NADH ratio in differentiation cells, as described in Materials and Methods. B, Sirt1 expression in cell cultures from wt and ogg1−/− NSC during differentiation. C, Redox manipulations correspondingly involve Sirt1. Sirt1 expression levels after treatment with oxidants [BSO and menadione (MEN)] and antioxidants (NAC and LA) were assessed by RT-PCR. All data are mean with SD from more than three independent experiments *p < 0.05.
Since mtDNA is readily damaged by both endogenous and exogenous factors, we aimed to investigate whether manipulation of NSC mtDNA integrity exogenously by exposure to either pro-oxidants or antioxidants would correspondingly influence on differentiation. As demonstrated in Figure 1, menadione, like BSO, induces mtDNA damage in NSCs, while reducing agents like N-acetylcysteine and lipoic acid protect NSC mtDNA as shown in our previous report (Wang et al., 2010). Cells in differentiation medium containing BSO formed apparent normal neurons and astrocytes, but the proportion of neurons was shifted toward astrocytes (Fig. 4A, top), confirming previous reports on other types of NSCs (Prozorovski et al., 2008). Menadione induced a similar shifting of differentiation pattern but exerted a stronger suppressive effect on neuronal differentiation than BSO, and resulted in a lower proportion of total recovered differentiated cells (neurons + astrocytes) (43.9% vs 50.0% of nontreated wt cells) (Fig. 4A, top). In the ogg1−/− NSC, an even more prominent inhibitory effect on neuronal differentiation was observed following both BSO and menadione treatments. In these cells, BSO treatment reduced neuronal differentiation without significant concomitant increase in astrocytic lineage, while menadione deteriorated the differentiation capacity even further, resulting in 39.5% and 29.1% recovered cells (Fig. 4A, bottom). The reversible phenomenon was demonstrated as exposure to reducing agents reduced astrogliosis and simultaneous increased neurogenesis in ogg1−/− NSCs (Fig. 4B). LA or NAC, which reduce mtDNA damage accumulation (Wang et al., 2010), increased the fraction of neurons by 12.6% and 16.8%, respectively, and in parallel reduced astrocytic fraction by 18.4% and 25.8%, respectively (numbers estimated with neurons/astrocytes in nontreated cultures cells set as 100%).
Figure 4.

NSC mtDNA integrity determines differentiation capacity. A, Pro-oxidants shift differentiation toward astrogenesis in wt cells, but suppress total differentiation capacity in ogg1−/− NSCs. Cells were treated with BSO or menadione and astrocytes/neurons determined by immunocytochemistry at day 5. B, Antioxidants suppress generation of astrocytes in ogg1−/− NSCs with corresponding increase in neurogenesis. Cell were treated with NAC, LA and astrocytes/neurons identified by immunocytochemistry at day 5. C, Strong 8-oxoguanine DNA glycosylase activity in brain mitochondria from transgenic ogg1−/− mice overexpressing mitochondrial hOGG1 (mtOGG1). Brain mitochondrial extracts (6 μg) from six different P5 littermates of mtOGG1 (1–6), adult wt (wt), and adult ogg1−/− (ogg1−/−) mice were assayed for OGG1 DNA glycosylase activity as described. Recombinant human OGG1 (25 ng) is used as positive control. D, Reversal of spontaneous mtDNA damage by mitochondrial hOGG1 results in increased neurogenesis. Top, mtDNA integrity in cells from differentiation day 1 and 5 (D1 and D5, respectively) was determined as in Figure 1B. Numbers are average with SD from three independent experiments. Bottom, Spontaneous differentiation pattern in wt and ogg1−/− cells and ogg1−/− cells expressing mtOGG1, determined as in Figure 2D. E, Induced mtDNA damage in NSCs increases astrogliosis. Top, NSCs were treated with menadione and mtDNA integrity was examined as in D, after (1 h) and 4 h after treatment. Bottom, Different NSCs treated with menadione and recovered for 4 h as above, were cultivated in differentiation medium and the differentiation pattern determined after 1 d, as in Figure 2D. Results shown are experiments from two independent cultures. *p < 0.05, **p < 0.01.
To confirm the important role of mtDNA integrity in NSC differentiation, we generated a transgenic ogg1−/− mouse that expresses a mitochondrially targeted human OGG1 (mtOGG1). Brain mitochondria from mtOGG1 displayed strong OGG1 activity, compared to wt (Fig. 4C). Differentiating NSCs from mtOGG1 were protected from spontaneous mtDNA damage accumulation, and this resulted in increased neurogenesis (Fig. 4D). As expected, increased astrogenesis in ogg1−/− coincided with increased mtDNA damage (Fig. 4D). The apparent enhanced astrogenesis in mtOGG1 compared to wt between day 1 and day 5 (Fig. 4E and Fig. 4D, respectively) could be due to increased astrocytic proliferation promoted by elevated mtDNA repair capacity. Finally, we wanted to evaluate the impact of mtDNA damage in NSC while still in the proliferation state. mtDNA damage was induced by menadione in proliferation medium (Fig. 4E). The mtDNA was repaired in wt and mtOGG1 cells after 4 h, but not in ogg1−/− cells. When differentiation was initiated at this time, astrocyte generation was significantly increased in ogg1−/− cells, thereby demonstrating the interrelation between mtDNA damage and preferential astrocytic development. Although not statistically significant, neurogenesis potential was elevated in nontreated cells and especially mtOGG1 even at day 1. At this point (day 1), neurogenesis was higher in nontreated than in menadione-treated cells and highest in mtOGG1, although the differences were not statistically significant. Thus, increased astrogenesis can result from spontaneously formed mtDNA damage in NSCs during differentiation or from accumulated mtDNA damage in the NSCs population before differentiation initiation.
Discussion
This report demonstrates a novel link between mtDNA damage and differentiation fate of NSC. In the in vitro model, the initial differentiation from NSCs to neurons and astrocytes is independent of OGG1. However along with differentiation progression, differentiation shifts toward astrocytic lineage in ogg1−/− cells that accumulate mtDNA damage, compared to wt cells. Antioxidant treatment that reduced damage accumulation correspondingly shifted differentiation direction toward neurons on the expense of astrocytes in ogg1−/− cells, thereby manifesting the link between mtDNA integrity and differentiation direction. The parallel alterations in redox levels and mtDNA damage accumulation in wt and ogg1−/− cells were mirrored by corresponding responses on Sirt1 expression levels, unraveling underlying mechanisms for how mtDNA integrity influences differentiation fate.
We have previously demonstrated that mtDNA damage forms along with differentiation and that OGG1 is important for maintaining the integrity of mtDNA. The accumulated mtDNA damage secondarily suppressed normal mitochondrial maturation in ogg1−/− cells, resulting in reduced mitochondrial activity in these cells (Wang et al., 2010). Mitochondrial activity is important for cellular NAD/NADH ratio, which in turn is a key regulator of Sirt1. Thus, the underlying mechanism for how mtDNA damage influences on differentiation lineage might originate from mitochondrial alterations producing redox-altered regulation of Sirt1. It is not likely that OGG1's role in repair of nuclear DNA is responsible for the shifted differentiation in ogg1−/− NSCs, since there are several backup functions for removal of oxidative base damage in nuclear DNA (Robertson et al., 2009). In contrast, OGG1 was found to be essential for repair of mtDNA (Fig. 1). Like menadione, BSO is capable of inducing NSC mtDNA damage (Wang et al., 2010), which may seem contradictory to the fact that ogg1−/− cells are not hypersensitive to BSO as to menadione. The explanation probably resides in the distinct oxidation mechanism by the two agents, and that BSO via its glutathione depletion kills cells by mtDNA damage-independent mechanisms.
Oxidative stress and mitochondrial dysfunction are hallmarks of aging and a number of neurodegenerative disorders (Wallace, 2002). Emerging evidence demonstrates the impact of mitochondria-affected DNA damage and neurodegenerative disease, including accumulation of damage in promoter regions of nuclear genes related to mitochondrial function as well as damage to mtDNA (Lu et al., 2004; Lee and Wei, 2007). Damaged mtDNA is repaired by specific repair systems in the neurons and astrocytes, and excessive damage has been hypothesized to give neuronal death (Harrison et al., 2005). However, our results hereby provide an alternative explanation for the connection between oxidative stress, mtDNA damage formation and neurodegeneration. The presence of NSC even in adult animals (Ahlenius et al., 2009) implies that continuous replenishment of neurons is a normal property of the brain. In the extension of this, neurodegeneration may therefore not necessarily be the consequence of increased apoptosis but rather insufficient neuronal renewal because of mtDNA damage accumulation in the NSC population hampering differentiation fate. The results also suggest that antioxidant treatment could reverse the biased differentiation. It remains to evaluate the potential of Sirt1 inhibitors to circumvent the effect of accumulated mtDNA damage in the aging brain.
Footnotes
This work was supported by grants from Norwegian Research Council (NevroNOR and Norwegian stem cell program) to L.E. and M.B.
The authors declare no competing financial interests.
References
- Ahlenius H, Visan V, Kokaia M, Lindvall O, Kokaia Z. Neural stem and progenitor cells retain their potential for proliferation and differentiation into functional neurons despite lower number in aged brain. J Neurosci. 2009;29:4408–4419. doi: 10.1523/JNEUROSCI.6003-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund A, Lindvall O. Self-repair in the brain. Nature. 2000;405:892–893. 895. doi: 10.1038/35016175. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem. 2000;275:37518–37523. doi: 10.1074/jbc.M000831200. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Res. 2005;33:4660–4671. doi: 10.1093/nar/gki759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildrestrand GA, Diep DB, Kunke D, Bolstad N, Bjørås M, Krauss S, Luna L. The capacity to remove 8-oxoG is enhanced in newborn neural stem/progenitor cells and decreases in juvenile mice and upon cell differentiation. DNA Repair (Amst) 2007;6:723–732. doi: 10.1016/j.dnarep.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kamnasaran D, Hawkins C, Guha A. Characterization and transformation potential of “synthetic” astrocytes differentiated from murine embryonic stem cells. Glia. 2008;56:457–470. doi: 10.1002/glia.20631. [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunke D, Bryja V, Mygland L, Arenas E, Krauss S. Inhibition of canonical Wnt signaling promotes gliogenesis in P0-NSCs. Biochem Biophys Res Commun. 2009;386:628–633. doi: 10.1016/j.bbrc.2009.06.084. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 2007;232:592–606. [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008;27:421–432. doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schröter F, Ninnemann O, Siegert E, Bendix I, Brüstle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008;213:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Animal models for mitochondrial disease. Methods Mol Biol. 2002;197:3–54. doi: 10.1385/1-59259-284-8:003. [DOI] [PubMed] [Google Scholar]
- Wang W, Osenbroch P, Skinnes R, Esbensen Y, Bjørås M, Eide L. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells. 2010;28:2195–2204. doi: 10.1002/stem.542. [DOI] [PubMed] [Google Scholar]