

Abstract
As in other diseases associated with mental retardation, dendrite morphology and synaptic plasticity are impaired in Down's syndrome (DS). Both these features of neurons are critically influenced by BDNF, which regulates local dendritic translation through phosphatidylinositol 3-kinase-Akt-mammalian target of rapamycin (mTOR) and Ras–ERK signaling cascades. Here we show that the levels of BDNF and phosphorylated Akt–mTOR (but not Ras–ERK) pathway proteins are augmented in hippocampal dendrites of Ts1Cje mice, a DS model. Consequently, the rate of local dendritic translation is abnormally high and the modulatory effect of exogenous BDNF is lost. Interestingly, rapamycin (a Food and Drug Administration-approved drug) restores normal levels of phosphorylated Akt-mTOR proteins and normal rates of local translation in Ts1Cje neurons, opening new therapeutic perspectives for DS. The NMDAR inhibitors APV, MK-801, and memantine also restore the normal levels of phospho-mTOR in dendrites of Ts1Cje hippocampal neurons. We propose a model to explain how BDNF-mediated regulation of local translation is lost in the Ts1Cje hippocampus through the establishment of a glutamatergic positive-feedback loop. Together, these findings help elucidate the mechanisms underlying altered synaptic plasticity in DS.
Introduction
Down's syndrome (DS) is produced by a trisomy of human chromosome 21 (HSA21), and it is the most frequent genetic cause of mental retardation (Antonarakis et al., 2004). As in other mental retardation diseases, synaptic plasticity and dendritic morphology are impaired in DS (Kaufmann and Moser, 2000). The Ts1Cje mouse is a DS model in which ∼80 orthologous HSA21 genes are trisomic, including the DS critical region responsible for memory and learning impairments (Delabar et al., 1993; Belichenko et al., 2009a). Indeed, these mice exhibit altered synaptic plasticity and morphological dendritic abnormalities (Siarey et al., 2005; Belichenko et al., 2007).
In neurons, the mammalian target of rapamycin (mTOR) signaling pathway regulates dendritic morphogenesis and synaptic plasticity, and it represents an interesting candidate that could be involved in the neuronal pathology of DS. The mTOR pathway controls the growth and branching of dendrites in cultured hippocampal neurons (Jaworski et al., 2005; Kumar et al., 2005), and it is implicated in BDNF-mediated synaptic plasticity in the adult brain, regulating local dendritic translation (Tang et al., 2002; Santos et al., 2010). In this case, the interaction of BDNF with its TrkB receptor activates phosphatidylinositol 3-kinase (PI3K), leading to phosphatidylinositol 3,4,5-trisphosphate (PIP3) synthesis. This PIP3 binds to Akt and promotes its association with the membrane, where it is phosphorylated (i.e., activated). Accordingly, Akt can then activate mTOR, which induces the following: (1) phosphorylation of the inhibitory protein 4EBP1 that provokes its dissociation from eIF4E, enabling the latter to interact with eIF4G, a mandatory step to initiate translation; (2) phosphorylation of p70S6 kinase (p70S6K), which in turn phosphorylates ribosomal protein S6, enhancing the synthesis of translational machinery proteins. Thus, BDNF regulates the local translation of a series of mRNAs via the mTOR pathway, including that of α-CaMKII, Arc, Homer2, Limk1, NMDAR1, and GluR1 (Schratt et al., 2004). Interestingly, hyperactivation of the mTOR signaling cascade has been reported in mouse models of two mental retardation diseases, tuberous sclerosis (Ehninger et al., 2008) and fragile X syndrome (Sharma et al., 2010).
Here we show that the Akt–mTOR signaling pathway is hyperactivated in hippocampal dendrites of trisomic Ts1Cje mice. In these neurons, exposure to exogenous BDNF failed to further increase the levels of phosphorylation of Akt, mTOR, p70S6K, S6, and 4EBP1 proteins, probably due to the high basal levels of BDNF expression and release in the Ts1Cje hippocampus. Accordingly, the rates of local dendritic translation were higher in the hippocampus of Ts1Cje mice than in wild-type (WT) mice, and exogenous BDNF failed to augment local mRNA translation in these trisomic mice. Remarkably, rapamycin restored normal levels of both phosphorylated mTOR protein and local dendritic translation in the trisomic hippocampus. Our findings reveal that BDNF signaling through the Akt–mTOR pathway is impaired in Ts1Cje hippocampal neurons, and they shed light on the mechanisms underlying altered synaptic plasticity in DS. Furthermore, our results provide a new therapeutic perspective in DS, and suggest that the use of rapamycin, a Food and Drug Administration (FDA)-approved drug, may improve the cognitive functions affected in this disease.
Materials and Methods
Animals.
Animals were maintained, handled, and killed in accordance with national and international laws and policies, and all the protocols used were approved by the University of Seville Animal Care and Use Committee. Partially trisomic Ts1Cje mice were obtained from Jackson Laboratories and maintained on a mixed genetic background as recommended, backcrossing Ts1Cje males to C57BL/6JOlaHsd × C3H/HeNHsd F1 females (B6C3F1 hybrid females, Harlan). Littermates were used in all experiments to ensure that the genetic background did not influence the results.
Hippocampal cultures and cell treatments.
The hippocampus from postnatal day 0 (P0) wild-type or trisomic mouse littermates was dissected out in HBSS medium (Invitrogen) and mechanically dissociated after a 10 min treatment with trypsin (0.2%) at 37°C in DB1 culture medium (DMEM high glucose with l-glutamine, without sodium pyruvate; 10% fetal bovine serum; GlutaMAX; 0.8% glucose; penicillin/streptomycin). The cells were seeded on poly-l-lysine-coated (0.5 mg/ml) coverslips, and cultured in 24 well plates in Neurobasal A medium (Neurobasal A; B27 supplement; GlutaMAX; penicillin/streptomycin). After 48 h in culture, 5-fluoro-2′-deoxyuridine (0.3 mm, Sigma-Aldrich) and uridine (0.8 mm, Sigma-Aldrich) were added to the culture medium to inhibit glial growth. At day in vitro (DIV) 12 (or 14 when indicated), cultures were treated with BDNF (Sigma-Aldrich), rapamycin (Cell Signaling Technology or Sigma-Aldrich), recombinant human TrkB/Fc chimera (TrkB-Fc, R&D Systems), recombinant mouse NGF R /p75 NTR/TNFRSF16/Fc chimera (p75NTR-Fc, R&D Systems), APV (Sigma-Aldrich), memantine (Sigma-Aldrich), or MK-801 (Sigma-Aldrich) as indicated. Lactate dehydrogenase (LDH) activity in the culture medium was measured in some experiments, using the Cytotoxicity Detection Kit (Roche).
Immunochemistry and image analysis.
For immunohistochemical analyses, adult mice were first perfused with PBS and then with 4% paraformaldehyde in PBS. The brains were dissected out and postfixed overnight at 4°C with 4% paraformaldehyde in PBS. Coronal vibratome sections (50 μm) were permeabilized for 10 min at room temperature with 1% Triton X-100 in PBS, washed in PBS, and blocked for 1 h at room temperature in the same solution supplemented with 2% bovine serum albumin (fraction V). Primary antibodies were diluted in the same buffer, and the brain slices were incubated overnight at 4°C.
For immunocytochemistry, neurons were fixed in 4% paraformaldehyde in PBS for 20 min and after fixation, washed three times with PBS at room temperature, and blocked for 30 min at 37°C in PBS, 0.1% Triton X-100, and 10% fetal bovine serum. The corresponding primary antibodies were diluted in blocking buffer and the cells were incubated overnight at 4°C. The cells were then washed three times in PBS for 10 min at room temperature.
Primary polyclonal antisera raised against phospho-mTOR (Ser2448), phospho-mTOR (Ser2481), phospho-Akt (Ser473), phospho-70S6K (Thr389), phospho-S6 (Ser235/236), phospho-4EBP1 (Ser65), phospho-Mnk1 (Thr197/202), phospho-eIF4E (Ser209), or mTOR (all from Cell Signaling Technology) were used at a concentration of 1:100; antibodies raised against BDNF and TrkB.T1 [BDNF antibody (N-20):sc-546, and Trk antibody (C-13):sc-119, Santa Cruz Biotechnology] were used at concentrations of 1:100 for immunohistochemistry and 1:200 for immunocytochemistry. Monoclonal anti-GluR1 antibody [GluR-1 antibody (E-6):sc-13152, Santa Cruz Biotechnology] was used at a concentration of 1:50. Monoclonal anti-MAP2 (Millipore Bioscience Research Reagents) or polyclonal anti-MAP2 (Synaptic Systems) antibodies were used at a concentration of 1:1000.
Secondary antibodies conjugated to Alexa488 (Invitrogen), Cy3, or Cy5 (Jackson Laboratories) were diluted at a concentration of 1:1000 and applied for 1 h at room temperature in blocking buffer with 3% fetal bovine serum but without Triton X-100. The cells or slices were then washed three times in PBS for 10 min. ProLong Gold antifade reagent (Invitrogen) was used on cells or slices for preserving immunofluorescence.
Images were acquired on an Olympus Fluoview FV1000 confocal microscope (objective: PLAPON 60×; numerical aperture 1.42 for cultures; objective 10× for tissue slices). For each tested antibody, two to four independent hippocampal neuron cultures from wild-type and Ts1Cje littermates were performed, and the results from a representative experiment (i.e., culture) is shown in each case. For each culture, at least two coverslips containing wild-type or Ts1Cje neurons, treated or not with the appropriate reagent, were subjected to the corresponding immunocytochemistry, and 4–11 confocal images were taken per condition (n represents the number of images, which is specified in the text or in the corresponding figure legend; as can be observed in the figures, each image usually contained one neuron). Image resolution and size were 16 bits and 1024 × 1024 pixels, respectively.
Special precautions were taken for ensuring accurate quantification of immunofluorescent signals to be compared in each experiment. Thus, the confocal settings were adjusted for each particular experiment to maximize the dynamic range of pixel intensity for dendritic labeling of the protein of interest, avoiding saturation of the signal. Saturation was monitored with the confocal software (Fluoview, Olympus). For each experiment, a set of coverslips was especially devoted to confocal setting adjustments, and they were not used for quantification to avoid errors due to photobleaching effects. Matlab software (Mathworks) was used to determine the mean pixel intensity for the corresponding immunofluorescent signal in dendrites that were identified with MAP2 as a marker. To do this, the Matlab routine we developed reads two images that are converted into matrices: one corresponding to the immunodetection of the protein of interest (red channel) and the other corresponding to the immunodetection of MAP2 (green channel). Images were then normalized to show pixel intensity values ranging from 0 to 1 (where 1 represents the maximal pixel intensity among the images to be compared). On the MAP2 image, a mask containing the somatic region was manually delineated. Values outside the mask were converted to 1; inside the mask, values were converted to 0. The mask matrix was then multiplied by the normalized MAP2 matrix, element by element, to eliminate the somatic region. The resulting matrix was then converted to binary, and multiplied by the red channel matrix, element by element; the elements of this matrix different from 0 represent dendritic pixels, the values being the corresponding pixel intensity in the red channel. In this way, the intensity of each pixel in the red channel that coincided with a MAP2-positive pixel was determined, and the mean pixel intensity was calculated and shown in arbitrary units. Note that, as confocal settings are different for each experiment, arbitrary units cannot be compared among different experiments.
Metabolic labeling of synaptoneurosomes.
Hippocampal synaptoneurosomes were isolated and resuspended in synaptoneurosome buffer as described previously (Troca-Marín et al., 2010). After preincubation at 37°C for 5 min, they were incubated for 30 min at 37°C in the presence of EasyTag Express Protein Labeling Mix 35S (PerkinElmer), with or without BDNF (100 ng/ml, Sigma-Aldrich). Where indicated, synaptoneurosomes were preincubated with 20 nm rapamycin (Cell Signaling Technology or Sigma-Aldrich) for 10–30 min before radioactive labeling of proteins.
Samples were pelleted, washed with synaptoneurosome buffer, subjected to SDS-PAGE, and transferred to PVDF membranes. The radiolabeled proteins were detected by exposing the membrane to a BioMax MS film (Kodak) using a BioMax TranScreen-LE intensifying screen (Kodak), or using a Cyclone Plus Phosphor Imager system (PerkinElmer) for quantification.
As a control, the level of phospho-S6 (Ser235/236) protein in synaptoneurosome preparations was quantified in Western blots.
Statistical analysis.
The quantitative data are presented as the mean ± SEM. The significance for comparisons was calculated using the Student's t test (SigmaStat software), and statistical significance was accepted where the p value was <0.05. The Mann–Whitney rank sum test was used where tests of normality or equal variance failed.
Results
mTOR is hyperactivated in the hippocampus of adult Ts1Cje mice
The phosphorylation of mTOR at Ser2448 is a biochemical indicator of its activation (Hay and Sonenberg, 2004) and, hence, we first examined the phosphorylation state of mTOR in the hippocampus of adult WT and Ts1Cje littermates by immunohistochemistry. Increased phospho-mTOR (Ser2448) labeling was evident in the dendritic hippocampal layers of Ts1Cje mice when compared with the WT mice (Fig. 1A). When we quantified the dendritic (i.e., MAP2-positive) phospho-mTOR (Ser2448) staining in primary cultures of hippocampal neurons by fluorescent immunocytochemistry, it was higher in Ts1Cje than in WT neurons after DIV12 (WT: 0.0713 ± 0.00747 a.u.; Ts1Cje: 0.2390 ± 0.01740 a.u.; n = 4; p < 0.001, t test) (Fig. 1B). By contrast, there were no significant differences in the amount of phospho-mTOR (Ser2481) (WT: 0.254 ± 0.0352 a.u.; Ts1Cje: 0.278 ± 0.0378 a.u.; n = 7–8; p = 0.654, t test) (Fig. 1C) or total mTOR protein (i.e., phosphorylated and nonphosphorylated) (WT: 0.139 ± 0.0286 a.u.; Ts1Cje: 0.111 ± 0.0136 a.u.; n = 6; p = 0.402, t test) (Fig. 1D). The PI3K–Akt signaling pathway is responsible for mTOR phosphorylation at Ser2448 (Hay and Sonenberg, 2004), whereas Ser2481 is autophosphorylated under conditions of translational repression (Peterson et al., 2000). Thus, these results suggest that the PI3K–Akt–mTOR pathway is hyperactivated in dendrites of Ts1Cje hippocampal neurons.
Figure 1.
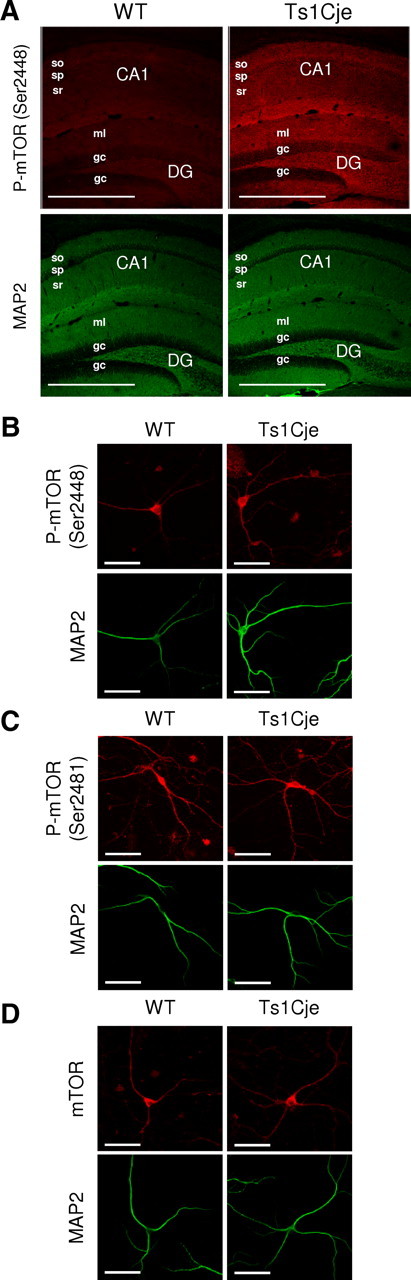
mTOR is hyperphosphorylated in Ts1Cje hippocampus. A, Immunohistochemical detection of phospho-mTOR (Ser2448) protein showing an increased signal in the dendritic layers (identified as MAP2 positive) of the Ts1Cje when compared with the WT adult hippocampus. The maximal projection of a stacking of confocal images is shown in each case. Scale bars, 600 μm. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; DG, dentate gyrus; ml, molecular layer; gc, granule cell layer. B–D, Representative immunocytochemistry images of phospho-mTOR (Ser2448), phospho-mTOR (Ser2481), and mTOR (total protein), respectively, in WT and Ts1Cje hippocampal neurons at DIV12. MAP2 was used as a dendritic marker. Scale bars, 60 μm.
Impaired BDNF signaling via the Akt–mTOR cascade in dendrites of Ts1Cje hippocampal neurons
In neurons, BDNF is a key upstream activator of the PI3K–Akt–mTOR and the Ras–ERK (extracellular signal-regulated kinase) signaling pathways (Bramham and Wells, 2007; Santos et al., 2010). BDNF acts via these pathways to promote dendritic branching, and to regulate dendrite size, as well as the number and morphology of dendritic spines (Kumar et al., 2005). Moreover, BDNF signaling through the PI3K–Akt–mTOR and Ras–ERK cascades plays a pivotal role in synaptic plasticity within the adult nervous system (Santos et al., 2010).
To further analyze the activity of the Akt–mTOR pathway in Ts1Cje hippocampus, phospho-Akt (Ser473), phospho-mTOR (Ser2448), phospho-p70S6K (Thr389), phospho-S6 (Ser235/236), and phospho-4EBP1 (Ser65) levels in dendrites were quantified in primary DIV12 cultures of WT and trisomic neurons in the presence or absence of BDNF for 10 min. As previously reported (Takei et al., 2004), BDNF increased the phosphorylation levels of these proteins in the dendrites of WT neurons (Fig. 2). Interestingly, the basal phosphorylation of all proteins was higher in the dendrites of Ts1Cje neurons, and their levels were not further augmented by exposure to exogenous BDNF (Fig. 2).
Figure 2.
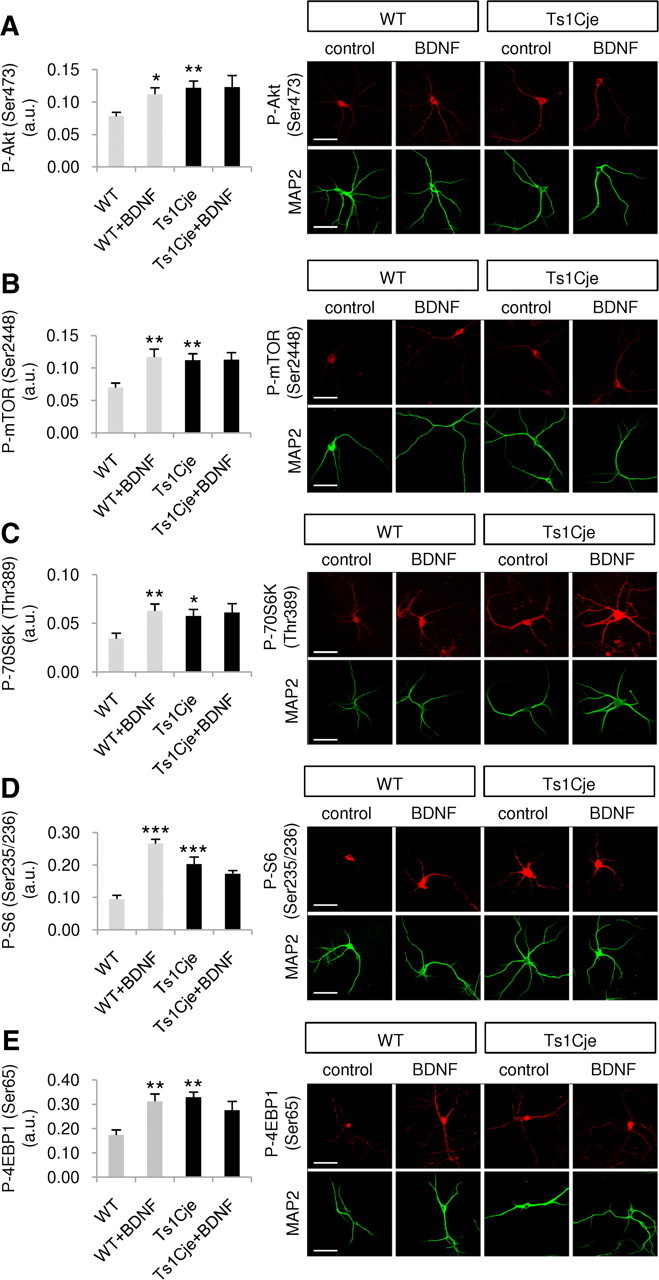
BDNF signaling through the Akt–mTOR cascade is impaired in dendrites of Ts1Cje hippocampal neurons. The fluorescence intensity of the indicated immunolabeled proteins was quantified in the dendrites of WT and Ts1Cje hippocampal neurons, in the presence or absence of BDNF (100 ng/ml for 10 min). A, Significant increases were observed in phospho-Akt (Ser473) in BDNF-treated versus untreated wild-type cells (*p = 0.019, t test), and in nontreated Ts1Cje versus wild-type cells (**p = 0.005, t test); n = 6. B, Significant increases in phospho-mTOR (Ser2448) were observed in BDNF-treated versus untreated wild-type cells (**p = 0.010, t test) and in untreated Ts1Cje versus wild-type cells (**p = 0.010, t test); n = 5–6. C, Significant increases in phospho-p70S6K (Thr389) were observed in BDNF-treated versus untreated wild-type cells (**p = 0.008, t test) and in untreated Ts1Cje versus wild-type cells (*p = 0.023, t test); n = 7–8. D, Significant increases in phospho-S6 (Ser235/236) were observed in BDNF-treated versus untreated wild-type cells (***p < 0.001, t test) and in untreated Ts1Cje versus wild-type cells (***p < 0.001, t test); n = 7–9. E, Significant increases in phospho-4EBP1 (Ser65) were observed in BDNF-treated versus untreated wild-type cells (**p = 0.004, t test) and in untreated Ts1Cje versus wild-type cells (**p = 0.002, Mann–Whitney test); n = 6. Data are expressed as the mean ± SEM. Representative immunocytochemistry images are shown in each case, and MAP2 was used as a dendritic marker. Scale bars, 60 μm.
BDNF signaling through the Ras–ERK cascade activates Mnk1 (mitogen-activated protein kinase integrating kinase 1), which in turn phosphorylates eIF4E. In contrast to the elements of the Akt–mTOR pathway, basal levels of phospho-Mnk1 (Thr197/202) and phospho-eIF4E (Ser209) were similar in the dendrites of WT and Ts1Cje neurons. Moreover, exposure to BDNF produced a similar increase in both these phosphoproteins in WT and Ts1Cje neurons (Fig. 3). Together, these results clearly demonstrate that in dendrites of Ts1Cje hippocampal neurons the Akt–mTOR pathway is constitutively hyperactivated, while the Ras–ERK pathway remains unchanged.
Figure 3.

BDNF signaling through the Ras–ERK cascade is not altered in dendrites of Ts1Cje hippocampal neurons. The fluorescence intensity of the indicated immunolabeled protein was quantified in the dendrites of WT and Ts1Cje hippocampal neurons, in the presence or absence of BDNF (100 ng/ml for 10 min). A, Significant increases in phospho-Mnk1 (Thr197/202) were observed in BDNF-treated versus untreated wild-type cells (**p = 0.008, t test) and in BDNF-treated versus untreated Ts1Cje cells (**p = 0.010, t test). No significant difference was detected between nontreated Ts1Cje and wild-type neurons (p = 0.618, t test); n = 6. B, Significant increases in phospho-eIF4E (Ser209) were observed in BDNF-treated versus untreated wild-type cells (*p = 0.014, t test) and in BDNF-treated versus untreated Ts1Cje cells (**p = 0.005, t test). No significant difference was detected between untreated Ts1Cje and wild-type neurons (p = 0.992, t test); n = 4. Data are expressed as the mean ± SEM. Representative immunocytochemistry images are shown in each case, and MAP2 was used as a dendritic marker. Scale bars, 60 μm.
Intracellular and secreted BDNF are abnormally elevated in the Ts1Cje hippocampus
The basal hyperactivation of the Akt–mTOR pathway observed in Ts1Cje neurons suggests that BDNF signaling may be exacerbated, reflecting an increase in the level of either BDNF or its cognate TrkB receptor. Mouse neurons express a 145 kDa, full-length TrkB isoform (TrkB.FL) and a 95 kDa truncated TrkB isoform (TrkB.T1), the latter lacking intrinsic tyrosine kinase activity. This truncated TrkB is the predominant isoform in the adult hippocampus (Silhol et al., 2005), and interestingly, it has been shown to affect BDNF signaling via specific downstream pathways. Thus, overexpression or loss of TrkB.T1 selectively affects BDNF-mediated Akt activation, but the phosphorylation of ERK is unaffected (Dorsey et al., 2006). As BDNF signaling via Akt but not ERK was altered in Ts1Cje neurons, we studied the expression of the TrkB.T1 isoform using a specific antibody. We failed to detect differences in the hippocampal distribution of TrkB.T1 in adult WT and Ts1Cje mice or in its dendritic levels in cultured hippocampal Ts1Cje and WT neurons (WT: 0.0854 ± 0.00775 a.u.; Ts1Cje: 0.0883 ± 0.01260 a.u.; n = 8; p = 0.848, t test) (Fig. 4). By contrast, BDNF immunohistochemistry using an antibody that recognizes both mature BDNF and its precursor pro-BDNF clearly demonstrated higher levels of BDNF proteins in the trisomic hippocampus, particularly in the CA1 strata pyramidale and radiatum, and in the granule cell layer of the dentate gyrus (Fig. 5A). Measurement of BDNF immunofluorescence in dendrites of DIV12 hippocampal cultures revealed a 1.7-fold increase in Ts1Cje neurons when compared with their WT counterparts (WT: 0.0474 ± 0.00723 a.u.; Ts1Cje: 0.0808 ± 0.01210 a.u.; n = 8; p = 0.033, t test).
Figure 4.
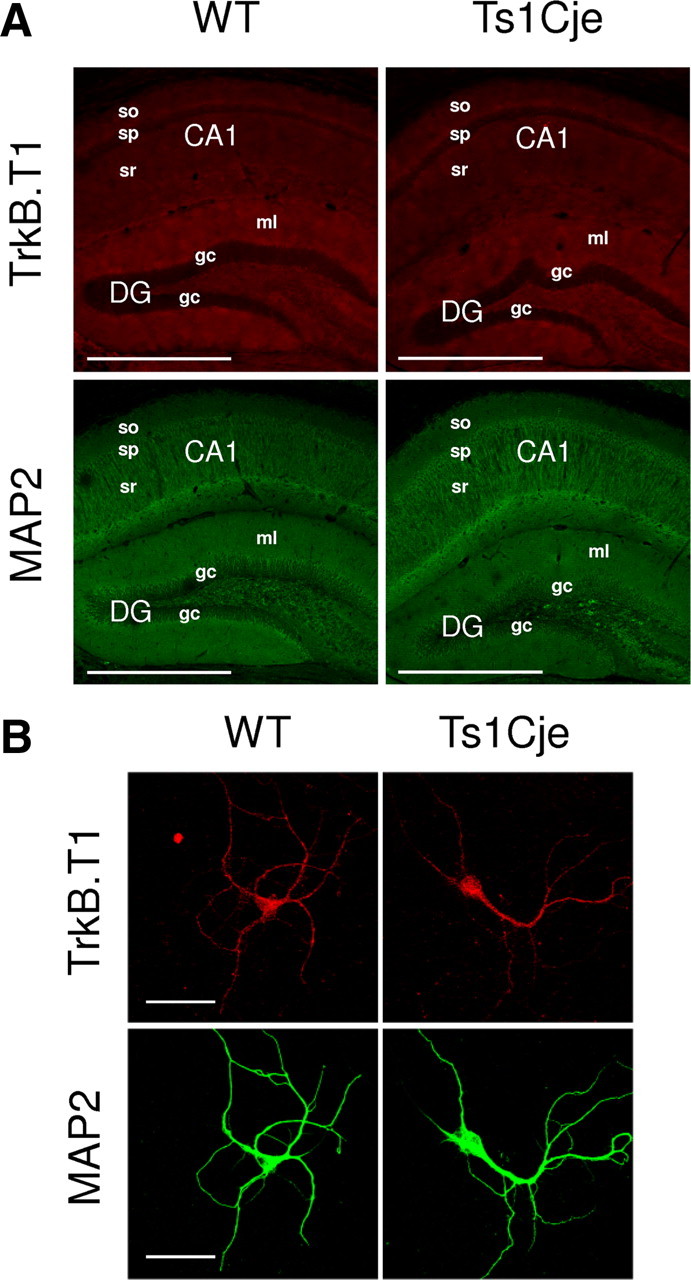
TrkB.T1 expression is unchanged in the Ts1Cje hippocampus. A, Immunohistochemical detection of TrkB.T1 protein showing similar levels of expression in the Ts1Cje and WT adult hippocampus. The maximal projection of a stacking of confocal images is shown in each case. Scale bars, 600 μm. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; DG, dentate gyrus; ml, molecular layer; gc, granule cell layer. B, Representative immunocytochemistry images of TrkB.T1 in WT and Ts1Cje hippocampal neurons at DIV12. MAP2 was used as a dendritic marker. Scale bars, 60 μm.
Figure 5.
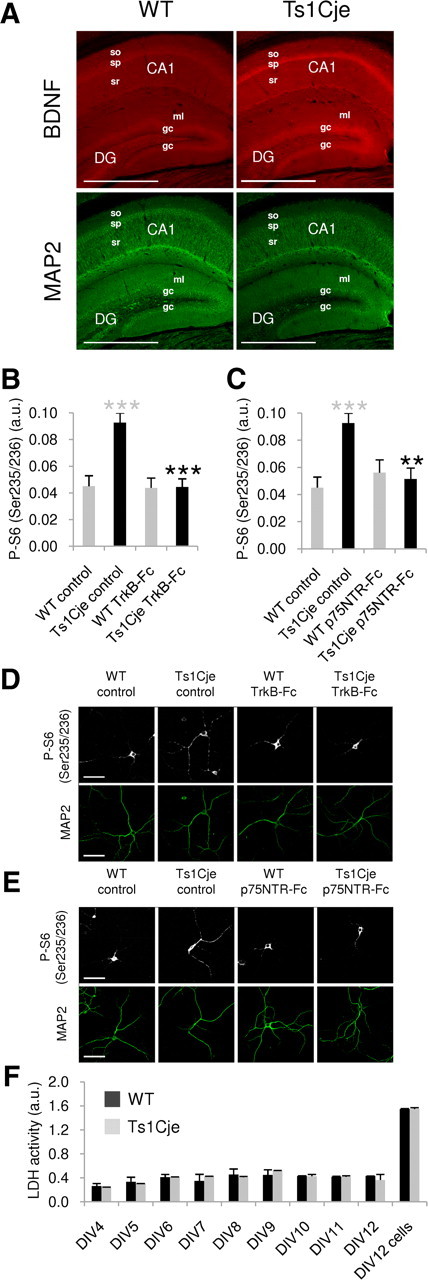
Basal BDNF levels are increased in the Ts1Cje hippocampus. A, Immunohistochemical detection of BDNF proteins (mature and pro-BDNF) showing an increase in the dendritic layers (identified as MAP2 positive) of Ts1Cje versus WT adult hippocampus. Stronger BDNF expression in somatic layers (stratum pyramidale and granule cells) is also appreciable. The maximal projection of a stacking of confocal images is shown in each case. Scale bars, 600 μm. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; DG, dentate gyrus; ml, molecular layer; gc, granule cell layer. B, Quantification of phospho-S6 (Ser235/236) fluorescence intensity in dendrites of WT and Ts1Cje hippocampal neurons at DIV12, in the presence or absence of the BDNF scavenger TrkB-Fc (1 μg/ml for 24 h), as indicated. A significant decrease was observed in TrkB-Fc-treated versus untreated Ts1Cje cells (n = 9; ***p < 0.001, t test). Ts1Cje neurons exhibited increased dendritic labeling when compared with wild-type neurons in basal conditions (n = 9–11; ***p < 0.001, t test). The data are expressed as the mean ± SEM. C, Quantification of phospho-S6 (Ser235/236) fluorescence intensity in dendrites of WT and Ts1Cje hippocampal neurons at DIV12, in the presence or absence of the pro-BDNF scavenger p75NTR-Fc (1 μg/ml for 24 h), as indicated. A significant decrease was observed in Ts1Cje neurons incubated with p75NTR-Fc versus untreated Ts1Cje cells (n = 9–11; **p = 0.001, t test). Data for wild-type and Ts1Cje neurons in basal conditions are the same as in B, as both experiments were done in parallel on neurons coming from the same culture experiment. Data are expressed as the mean ± SEM. D, E, Representative phospho-S6 (Ser235/236) immunocytochemistry images of experiments summarized in B and C, respectively. MAP2 was used as a dendritic marker. Scale bars, 60 μm. F, Time course of LDH activity in the culture media of WT and Ts1Cje hippocampal neurons. LDH activity was measured at the indicated day in vitro. For the sake of comparison, LDH was also measured in cell lysates at DIV12 (DIV12 cells). No significant differences were detected between cultures of WT and Ts1Cje hippocampal neurons, indicating that cell lysis was similar in both cases. Data are expressed as the mean activity in two different culture plates ± SD.
BDNF and pro-BDNF are secreted by the dendrites of hippocampal neurons in an activity-dependent manner, exerting modulatory effects on synaptic plasticity (Waterhouse and Xu, 2009). As intracellular dendritic levels of BDNF proteins were higher in the trisomic hippocampus, the hyperactivation of the Akt–mTOR cascade observed could reflect greater dendritic release of BDNF proteins in basal conditions. Hence, we hypothesized that extracellular application of cell membrane-impermeable scavengers specifically targeting mature BDNF (TrkB–Fc) or pro-BDNF (p75NTR–Fc) could attenuate the observed mTOR hyperactivation. Accordingly, incubation of Ts1Cje neurons with TrkB-Fc for 24 h restored normal dendritic levels of phospho-S6 (Ser235/236) protein, taken as a readout of mTOR activity (Fig. 5B,D). Similar results were obtained when cells were incubated with the p75NTR-Fc (Fig. 5C,E).
To rule out the possibility that the increase in BDNF proteins in the Ts1Cje culture medium was due to increased cell lysis, and the subsequent release of BDNF and pro-BDNF from dead cells, we measured the time course of LDH activity in the culture media. No differences were detected between WT and Ts1Cje cultures (Fig. 5F).
Together, the above results suggests that the hyperphosphorylation of proteins of the Akt–mTOR signaling pathway in Ts1Cje hippocampal dendrites is indeed sustained by the exacerbated release of BDNF and pro-BDNF in basal conditions.
Rapamycin restores normal levels of mTOR (Ser2448) phosphorylation in dendrites of Ts1Cje hippocampal neurons
Rapamycin is a specific inhibitor of mTOR that is used as an antirejection drug in kidney transplants and is approved for the treatment of cardiovascular diseases (Tsang et al., 2007). Rapamycin was capable of counteracting the hyperphosphorylation of dendritic mTOR in Ts1Cje hippocampal neurons, and both short-term (10 min) and long-term (24 h) exposure to rapamycin significantly reduced the levels of phosphorylated dendritic mTOR (Ser2448) to values similar to those observed in WT neurons (Fig. 6).
Figure 6.

Rapamycin restores mTOR activity in hippocampal Ts1Cje neurons. A, B, Phospho-mTOR (Ser2448) fluorescence intensity was quantified in dendrites of WT and Ts1Cje hippocampal neurons at DIV12 in the presence or absence of rapamycin (rapa, 20 nm) for 10 min (A) or 24 h (B), as indicated. Rapamycin produced a significant decrease in the Ts1Cje neurons (10 min: ***p < 0.001; 24 h: **p = 0.002, t test), while only a slight decrease was observed in wild-type cells treated with rapamycin for 24 h (*p = 0.022, Mann–Whitney test). As expected, Ts1Cje neurons exhibited stronger dendritic labeling than wild-type neurons in basal conditions (10 min: **p = 0.002, t test; 24 h: ***p < 0.001, Mann–Whitney test). Data are expressed as the mean ± SEM (n = 7–11). Representative immunocytochemistry images are shown in each case, and MAP2 was used as a dendritic marker. Scale bars, 60 μm.
Regulation of local dendritic mRNA translation by BDNF is lost in Ts1Cje hippocampus, and rapamycin treatment restores normal levels of local translation
As indicated, BDNF signaling through the Akt–mTOR pathway modulates synaptic plasticity by activating local dendritic mRNA translation. Metabolic labeling of synaptoneurosomes is a widely used approach to analyze local translation (Rao and Steward, 1991; Bagni et al., 2000; Schratt et al., 2004; Troca-Marín et al., 2010), and, thus, we isolated hippocampal synaptoneurosomes from WT and Ts1Cje adult littermates to quantify the effect of Akt-mTOR hyperactivation on local Ts1Cje dendritic translation. In WT synaptoneurosomes, dendritic protein synthesis was induced by BDNF, as evident by analyzing the metabolic 35S-Met/Cys labeling, and this effect was attenuated in the presence of rapamycin (Fig. 7A,B). A higher basal level of protein synthesis was detected in Ts1Cje synaptoneurosomes, which was not affected by the addition of BDNF. Nevertheless, rapamycin reduced the rate of local translation in Ts1Cje synaptoneurosomes to a level similar to that seen in WT controls (Fig. 7A,B).
Figure 7.
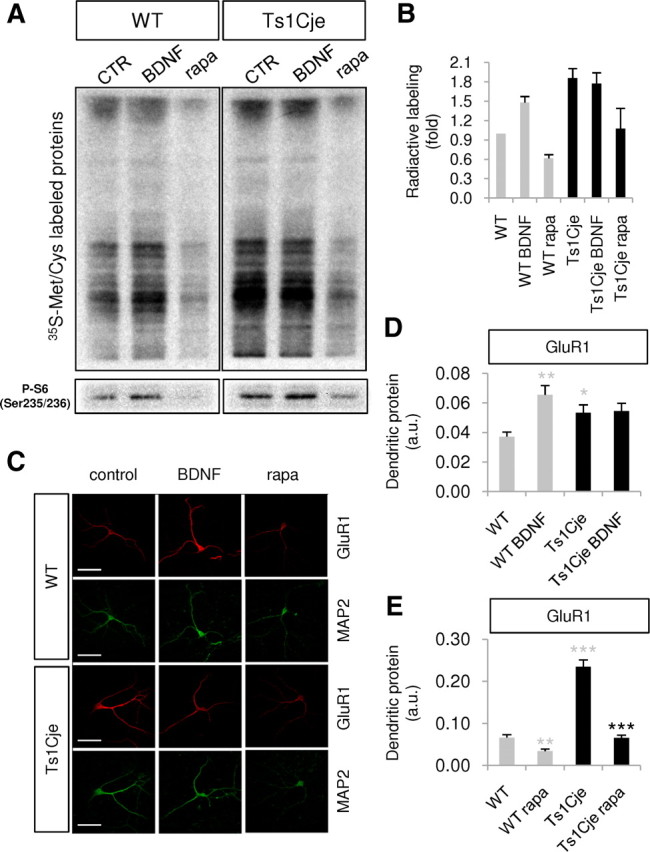
Local dendritic translation increases in the Ts1Cje hippocampus. A, Radioactive, neosynthesized proteins in WT and Ts1Cje hippocampal synaptoneurosomes incubated with 100 ng/ml BDNF or 20 nm rapamycin (rapa) for 30 min, or in control conditions (CTR), as indicated. As a control, the level of phospho-S6 (Ser235/236) was determined in Western blots of each condition. Images correspond to a representative experiment; some lanes between WT and Ts1Cje samples were removed. B, The radioactivity incorporated into neosynthesized proteins was quantified in WT and Ts1Cje hippocampal synaptoneurosomes incubated with 100 ng/ml BDNF or 20 nm rapamycin (rapa), or in control conditions, as shown in A. The data are expressed as the mean ± SEM (n = 2). C, Representative GluR1 immunocytochemistry of an experiment similar to those summarized in D and E. MAP2 was used as a dendritic marker. Scale bars, 60 μm. D, The intensity of GluR1 fluorescence was quantified in dendrites of WT and Ts1Cje hippocampal neurons at DIV14 in the presence or absence of BDNF (100 ng/ml for 10 min), as indicated. Significant increases were observed in BDNF-treated versus untreated wild-type cells (n = 6; **p = 0.004, Mann–Whitney test) and in untreated Ts1Cje versus wild-type cells (n = 6; *p = 0.026, t test). The data are expressed as the mean ± SEM. E, The intensity of GluR1 fluorescence was quantified in dendrites of WT and Ts1Cje hippocampal neurons at DIV12 in the presence or absence of rapamycin (20 nm for 10 min), as indicated. A decrease was observed both in wild-type and Ts1Cje neurons treated with rapamycin (**p = 0.002 for wild-type neurons, t test; ***p < 0.001 for Ts1Cje neurons, Mann–Whitney test; n = 9–10). Similarly to results shown in D, untreated Ts1Cje neurons exhibited stronger dendritic labeling than untreated wild-type neurons (***p < 0.001, t test). The data are expressed as the mean ± SEM.
To corroborate these results, we analyzed the local translation of the AMPA receptor subunit GluR1, a well described dendritic mRNA locally translated and regulated by BDNF through the Akt–mTOR pathway (Schratt et al., 2004; Slipczuk et al., 2009). The amount of GluR1 protein in dendrites of hippocampal neurons was estimated by quantifying the immunocytochemical fluorescent signal, and as expected, exposure to BDNF (10 min) increased the amount of dendritic GluR1 protein in WT hippocampal neurons (Fig. 7C,D). By contrast, Ts1Cje neurons exhibited a higher basal level of dendritic GluR1 protein, which was not further augmented by exposure to exogenous BDNF. Remarkably, the dendritic levels of GluR1 protein diminished in the presence of rapamycin to reach values similar to those measured in WT neurons (Fig. 7C,E).
Together, these results demonstrate that Ts1Cje hippocampal neurons have lost the capacity to detect and respond to variations in the levels of BDNF, probably due to their high basal extracellular levels, which provokes increased and deregulated local translation of dendritic mRNAs. Moreover, the restorative effects of rapamycin on local dendritic translation in the Ts1Cje hippocampus shed light on an important new therapeutic possibility in DS.
The NMDAR inhibitors APV, MK-801, and memantine restore normal levels of mTOR (Ser2448) phosphorylation in dendrites of Ts1Cje hippocampal neurons
Increased release of BDNF proteins from Ts1Cje hippocampal dendrites could result in augmented glutamatergic activity, due to enhanced presynaptic glutamate release (Lessmann et al., 1994; Jovanovic et al., 2000) and/or increased NMDAR signaling (Jarvis et al., 1997; Suen et al., 1997; Levine et al., 1998; Lin et al., 1998). In turn, an increased glutamatergic activity could potentiate the dendritic BDNF secretion (Korte, 2008; Tanaka et al., 2008). This loop could explain the Akt-mTOR hyperactivation observed in Ts1Cje hippocampal dendrites.
To test this idea, the effect of APV, MK-801, and memantine on mTOR phosphorylation was investigated. APV is a competitive antagonist of NMDAR, whereas MK-801 and memantine are high- and moderate-affinity NMDAR channel blockers, respectively. Treatment of Ts1Cje hippocampal neurons with APV, MK-801, or memantine for 10 min or 2 h significantly reduced the levels of phosphorylated dendritic mTOR (Ser2448) to values similar to those observed in WT neurons (Fig. 8), suggesting that enhanced glutamatergic activity through NMDAR is necessary to sustain Akt-mTOR hyperactivation in Ts1Cje hippocampus.
Figure 8.

The NMDAR inhibitors APV, memantine, and MK-801 decrease mTOR activity in hippocampal Ts1Cje neurons. A, Phospho-mTOR (Ser2448) fluorescence intensity was quantified in dendrites of WT and Ts1Cje hippocampal neurons at DIV12 in control conditions or in the presence of APV (120 μm), MK-801 (10 μm), or memantine (30 μm) for 10 min or 2 h, as indicated. The three compounds significantly decreased mTOR (Ser2448) phosphorylation in Ts1Cje neurons both at 10 min (***p < 0.001, Mann–Whitney test for APV and MK-801, t test for memantine) and 2 h (***p < 0.001, t test for APV and memantine, Mann–Whitney test for MK-801). As expected, Ts1Cje neurons exhibited stronger dendritic labeling than wild-type neurons in basal conditions (***p < 0.001, t test). Data are expressed as the mean ± SEM (n = 9–10). B, Representative immunocytochemistry images of experiments summarized in A (10 min treatments) are shown; MAP2 was used as a dendritic marker. Scale bars, 60 μm.
Discussion
In neurons, BDNF signaling through the mTOR pathway is involved in dendritic branching and synaptic plasticity, two key processes that are impaired in DS (Kaufmann and Moser, 2000; Siarey et al., 2005; Belichenko et al., 2007). We found that the PI3K–Akt–mTOR pathway is hyperactivated in the dendrites of hippocampal neurons from trisomic Ts1Cje mice, in agreement with previous reports of increased total hippocampal phospho-Akt protein in Ts65Dn mice (Siarey et al., 2006), another widely used DS model. Most importantly, we found that trisomic neurons lose the capacity to respond to exogenous BDNF and that this trophic factor is overexpressed in the Ts1Cje hippocampus. Interestingly, a recent study of BDNF plasma levels in Down's syndrome patients reported a fivefold increase when compared with control individuals (Dogliotti et al., 2010). As BDNF readily crosses the blood–brain barrier (Pan et al., 1998), this probably reflects an increased level of BDNF in the DS brain.
BDNF is secreted from dendrites in an activity-dependent manner. In glutamatergic synapses, postsynaptically released BDNF appears to act in synergy with presynaptically released glutamate in the context of memory consolidation, inducing local dendritic mRNA translation that in turn modifies synaptic efficacy (Korte, 2008; Tanaka et al., 2008). Both pro-BDNF and mature BDNF are secreted by neurons, pro-BDNF being the primary form released from dendrites (Yang et al., 2009). This pro-BDNF is cleaved extracellularly by tPA/plasmin and matrixmetalloprotease-7 (Waterhouse and Xu, 2009). Indeed, incubation of cultured Ts1Cje hippocampal neurons with the BDNF scavenger TrkB-Fc restored the phosphorylation levels of proteins in the dendritic Akt–mTOR pathway to those observed in WT controls, suggesting that extracellular levels of active BDNF were abnormally high. Interestingly, the extracellular application of p75NTR-Fc, a cell membrane-impermeable scavenger of pro-BDNF, also reduces the phosphorylation of proteins in the dendritic Akt–mTOR pathway in Ts1Cje neurons to levels similar to that found in WT controls. Sequestering pro-BDNF with p75NTR-Fc probably inhibits its cleavage by extracellular proteases, thereby decreasing the availability of mature BDNF to maintain the Akt–mTOR cascade hyperactive in Ts1Cje hippocampal neurons. These observations may explain why exogenous BDNF failed to further increase the phosphorylation of proteins in the Akt–mTOR pathway in dendrites of Ts1Cje neurons.
Surprisingly, the Ras–ERK signaling pathway, also under the control of BDNF through TrkB receptors, was not hyperactivated in Ts1Cje hippocampus. Although the basis for this observation remains to be established, some hypotheses can be anticipated. Thus, it is well known that activated TrkB receptors generate attachment sites for Shc, which binds to the intermediary protein Grb2. This protein can recruit the guanine nucleotide exchange factor Sos to the membrane, resulting in Ras–ERK activation, or can bind Gab proteins to activate the PI3K-Akt–mTOR cascade (Segal, 2003). Because >80 genes are in trisomy in Ts1Cje, overexpression of these genes could affect many different signaling pathways and, for example, modify the relative levels of different TrkB partners (e.g., Sos or Gab proteins), leading to a differential perturbation of the TrkB-downstream cascades.
Other possible explanation is related to signaling through lipid rafts. It has been shown that BDNF rapidly recruits TrkB receptor from nonraft regions to cholesterol-rich lipid rafts (Suzuki et al., 2004). Interestingly, differential activation of ERK and Akt has been found in lipid rafts upon BDNF-TrkB interaction (Suzuki et al., 2004). Taking into account that BDNF elicits cholesterol synthesis in neurons, increasing the cholesterol amount in rafts, but not in nonraft membrane domains (Suzuki et al., 2007), and that BDNF is overexpressed in Ts1Cje hippocampal neurons as shown in this work, it is possible that raft and nonraft domains are altered in Ts1Cje neuronal membranes, leading to a differential activation of Akt–mTOR and Ras–ERK cascades.
In agreement with the fact that BDNF activates the mTOR pathway to induce the local translation of dendritic mRNAs (Bramham and Wells, 2007; Santos et al., 2010), local dendritic translation is elevated in the Ts1Cje hippocampus when compared with WT controls. The addition of exogenous BDNF does not further enhance local translation, as corroborated by analyzing the dendritic expression of the GluR1 AMPA receptor subunit, encoded by a dendritic mRNA regulated by BDNF via the Akt–mTOR cascade (Schratt et al., 2004). As seen with proteins in the mTOR pathway, basal GluR1 protein is more abundant in dendrites of hippocampal Ts1Cje neurons than in WT controls. Moreover, GluR1 protein levels remain unchanged upon addition of exogenous BDNF. Together, these findings support the hypothesis that neurons of the Ts1Cje hippocampus lose their ability to respond to exogenously added BDNF. Hence, in combination with existing data, we propose a model to explain the loss of BDNF-mediated regulation of local translation in the Ts1Cje hippocampus, shedding light on the potential mechanisms underlying altered synaptic plasticity in DS (Fig. 9).
Figure 9.

Model proposed to explain why BDNF fails to regulate local translation in the hippocampus of Ts1Cje mice. Dendritic release of pro-BDNF and BDNF is increased in Ts1Cje hippocampal neurons (1). pro-BDNF is converted to mature BDNF extracellularly by tPA/plasmin, MMP-7 and other extracellular proteases (Waterhouse and Xu, 2009) (2). BDNF acts presynaptically at TrkB receptors (3) to enhance glutamate release (4) (Lessmann et al., 1994; Jovanovic et al., 2000). BDNF acts postsynaptically at TrkB receptors (5), promoting the phosphorylation of NMDAR subunits (6) (Suen et al., 1997; Lin et al., 1998), increasing the probability of NMDA channel opening (Jarvis et al., 1997; Levine et al., 1998) and, hence, increasing excitatory activity. Exacerbated glutamatergic activity provokes increased dendritic release of pro-BDNF and BDNF (7) (Korte, 2008; Tanaka et al., 2008). BDNF binding to postsynaptic TrkB receptors (5) also results in hyperactivation of the PI3K–Akt–mTOR signaling cascade (8), leading to increased local translation of dendritic mRNAs (9) like GluR1(which contributes to the increase in postsynaptic excitatory activity) and NMDAR1 (Schratt et al., 2004). Increased NMDAR-mediated neuronal activity in Ts1Cje hippocampal neurons also enhances CPEB1-mediated local translation of dendritic mRNAs such as DSCAM (10) (Alves-Sampaio et al., 2010). As BDNF mRNA is localized to dendrites (Tongiorgi et al., 1997; An et al., 2008), and its expression is regulated by CPEB1 (Oe and Yoneda, 2010) and a PI3K-dependent pathway (Righi et al., 2000), its local dendritic translation may also increase in trisomic mice, contributing to the overexpression of BDNF in dendrites of the Ts1Cje hippocampus (11). This glutamatergic positive-feedback loop may explain why hippocampal Ts1Cje synapses lose their sensitivity to further increases in BDNF-mediated activity (Alves-Sampaio et al., 2010; data herein) and attain a saturated state that impairs the modulation of synaptic plasticity.
In this model, a positive-feedback loop is established between presynaptic and postsynaptic elements in which exacerbated secretion of pro-BDNF and BDNF from Ts1Cje dendrites increases NMDAR-dependent glutamatergic activity through enhanced presynaptic glutamate release (Lessmann et al., 1994; Jovanovic et al., 2000) and increased phosphorylation of postsynaptic NMDAR subunits (Suen et al., 1997; Lin et al., 1998). This in turn increases the probability of NMDA receptor channel opening (Jarvis et al., 1997; Levine et al., 1998). Increased BDNF release from Ts1Cje dendrites potentiates local mTOR-mediated translation of dendritic mRNAs, including that encoding the AMPA receptor subunit, GluR1 (Schratt et al., 2004; data herein). This modification in expression contributes to the increase in excitatory activity and probably increases dendritic levels of BDNF in Ts1Cje hippocampal neurons (Tongiorgi et al., 1997; Righi et al., 2000; Oe and Yoneda, 2010). The enhanced glutamatergic activity at Ts1Cje dendrites potentiates BDNF secretion (Korte, 2008; Tanaka et al., 2008). In these basal conditions of hyperactivation, the hippocampal Ts1Cje synapses reach a state of “saturation,” losing the capacity to respond to further increases in BDNF activity. We recently demonstrated that the local dendritic translation of DSCAM, a gene in the DS critical region, is regulated by NMDAR-mediated synaptic activity, and that basal levels of dendritic DSCAM in Ts1Cje hippocampal neurons are similar to those found in NMDA-activated wild-type neurons. Although addition of NMDA to Ts1Cje neurons does not further augment dendritic DSCAM, NMDAR signaling is required to maintain DSCAM protein levels above those seen in wild-type neurons under basal conditions, indicating that glutamatergic activity is higher under basal conditions (Alves-Sampaio et al., 2010). We found that APV, MK-801, and memantine, three different inhibitors of NMDARs, were able to restore the normal levels of dendritic phospho-mTOR (Ser2448) in Ts1Cje neurons. APV is a competitive antagonist for glutamate binding to the NR2 subunit of NMDAR; MK-801 and memantine are uncompetitive channel blockers that need prior activation of the NMDAR by glutamate to mediate the blockade action (Chen and Lipton, 2006). Thus, these results support the glutamatergic positive-feedback loop hypothesis we propose (Fig. 9).
Interestingly, this scenario is in agreement with previous studies suggesting increased glutamatergic activity in Ts65Dn mice. Indeed, in this DS model some hippocampal-dependent cognitive deficits are rescued by memantine (Costa et al., 2008; Rueda et al., 2010; Lockrow et al., 2011). Enhanced glutamatergic activity could also explain the reported increases in inhibitory GABAergic synapses in the dentate gyrus of the hippocampus in Ts65Dn, Ts1Cje, and Ts1Rhr DS mice (Belichenko et al., 2007, 2009b). Indeed, these increases are thought to represent a compensatory response to undefined cellular events provoking a persistent increase in excitatory neurotransmission in these mice (Belichenko et al., 2009b). Remarkably, although BDNF enhances excitatory neurotransmission, it potentiates GABAergic inhibition in the epileptic brain (Koyama and Ikegaya, 2005). Furthermore, DS patients have a greater propensity to develop epilepsy (Pueschel et al., 1991), and, thus, the glutamatergic positive-feedback loop model (Fig. 9) may also be relevant to the development of epilepsy in DS patients.
It is important to stress that the effects of BDNF on synaptic physiology are extremely complex, as BDNF influences the formation, maturation, and plasticity of both glutamatergic and GABAergic synapses (Gottmann et al., 2009), and it is a key regulator of homeostatic plasticity (Turrigiano, 2008).
Finally, we found that rapamycin, an FDA-approved drug, restores the levels of phosphorylated proteins in the Akt–mTOR pathway in dendrites of hippocampal Ts1Cje neurons, as well as normal levels of local dendritic translation. Interestingly, the mTOR pathway is hyperactivated in murine models of at least two other mental retardation diseases, tuberous sclerosis (Ehninger et al., 2008) and fragile X (Sharma et al., 2010), and it is emerging as a potential drug target to restore cognitive impairment in mental retardation. In fact, rapamycin rescues the behavioral deficits and those associated with synaptic plasticity in the tuberous sclerosis mouse model Tsc2+/− (Ehninger et al., 2008). Studies into the capacity of rapamycin to reverse learning deficits in Ts1Cje mice are currently underway.
Footnotes
This work was supported by the Fundación Ramón Areces (Spain), the Ministerio de Ciencia e Innovación (Instituto de Salud Carlos III, Spain; Grant PI081401), the Fondation Jérôme Lejeune (France), and the Junta de Andalucía (Grant P09-CTS-4610). We thank Dr. Antonio Leal for very stimulating and helpful discussions on image processing and quantification.
References
- Alves-Sampaio A, Troca-Marín JA, Montesinos ML. NMDA-mediated regulation of DSCAM dendritic local translation is lost in a mouse model of Down's syndrome. J Neurosci. 2010;30:13537–13548. doi: 10.1523/JNEUROSCI.3457-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- Bagni C, Mannucci L, Dotti CG, Amaldi F. Chemical stimulation of synaptosomes modulates α-Ca2+/calmodulin-dependent protein kinase II mRNA association to polysomes. J Neurosci. 2000;20:RC76. doi: 10.1523/JNEUROSCI.20-10-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504:329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci. 2009a;29:5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Masliah E, Wu C, Takimoto-Kimura R, Salehi A, Mobley WC. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J Comp Neurol. 2009b;512:453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- Costa AC, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33:1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, Noel B, Sinet PM. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet. 1993;1:114–124. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- Dogliotti G, Galliera E, Licastro F, Corsi MM. Age-related changes in plasma levels of BDNF in Down syndrome patients. Immun Ageing. 2010;7:2. doi: 10.1186/1742-4933-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey SG, Renn CL, Carim-Todd L, Barrick CA, Bambrick L, Krueger BK, Ward CW, Tessarollo L. In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron. 2006;51:21–28. doi: 10.1016/j.neuron.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199:203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase–Akt–mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Korte M. Neuroscience. A protoplasmic kiss to remember. Science. 2008;319:1627–1628. doi: 10.1126/science.1155748. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist. 2005;11:282–287. doi: 10.1177/1073858405278266. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras–PI3K–Akt–mTOR and Ras–MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav Brain Res. 2011;221:610–622. doi: 10.1016/j.bbr.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oe S, Yoneda Y. Cytoplasmic polyadenylation element-like sequences are involved in dendritic targeting of BDNF mRNA in hippocampal neurons. FEBS Lett. 2010;584:3424–3430. doi: 10.1016/j.febslet.2010.06.040. [DOI] [PubMed] [Google Scholar]
- Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology. 1998;37:1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–7423. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- Pueschel SM, Louis S, McKnight P. Seizure disorders in Down syndrome. Arch Neurol. 1991;48:318–320. doi: 10.1001/archneur.1991.00530150088024. [DOI] [PubMed] [Google Scholar]
- Rao A, Steward O. Evidence that protein constituents of postsynaptic membrane specializations are locally synthesized: analysis of proteins synthesized within synaptosomes. J Neurosci. 1991;11:2881–2895. doi: 10.1523/JNEUROSCI.11-09-02881.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righi M, Tongiorgi E, Cattaneo A. Brain-derived neurotrophic factor (BDNF) induces dendritic targeting of BDNF and tyrosine kinase B mRNAs in hippocampal neurons through a phosphatidylinositol-3 kinase-dependent pathway. J Neurosci. 2000;20:3165–3174. doi: 10.1523/JNEUROSCI.20-09-03165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda N, Llorens-Martín M, Flórez J, Valdizán E, Banerjee P, Trejo JL, Martínez-Cué C. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J Alzheimers Dis. 2010;21:277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- Santos AR, Comprido D, Duarte CB. Regulation of local translation at the synapse by BDNF. Prog Neurobiol. 2010;92:505–516. doi: 10.1016/j.pneurobio.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarey RJ, Villar AJ, Epstein CJ, Galdzicki Z. Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology. 2005;49:122–128. doi: 10.1016/j.neuropharm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Kline-Burgess A, Cho M, Balbo A, Best TK, Harashima C, Klann E, Galdzicki Z. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. J Neurochem. 2006;98:1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x. [DOI] [PubMed] [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience. 2005;132:613–624. doi: 10.1016/j.neuroscience.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci U S A. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Kiyosue K, Hazama S, Ogura A, Kashihara M, Hara T, Koshimizu H, Kojima M. Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J Neurosci. 2007;27:6417–6427. doi: 10.1523/JNEUROSCI.0690-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A. Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci. 1997;17:9492–9505. doi: 10.1523/JNEUROSCI.17-24-09492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troca-Marín JA, Alves-Sampaio A, Tejedor FJ, Montesinos ML. Local translation of dendritic RhoA revealed by an improved synaptoneurosome preparation. Mol Cell Neurosci. 2010;43:308–314. doi: 10.1016/j.mcn.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]