

Abstract
Signaling through extracellular signal-regulated kinase (ERK) is important in multiple signal transduction networks in the CNS. However, the specific role of ERK2 in in vivo brain functions is not fully understood. Here we show that ERK2 play a critical role in regulating social behaviors as well as cognitive and emotional behaviors in mice. To study the brain function of ERK2, we used a conditional, region-specific, genetic approach to target Erk2 using the Cre/loxP strategy with a nestin promoter-driven cre transgenic mouse line to induce recombination in the CNS. The resulting Erk2 conditional knock-out (CKO) mice, in which Erk2 was abrogated specifically in the CNS, were viable and fertile with a normal appearance. These mice, however, exhibited marked anomalies in multiple aspects of social behaviors related to facets of autism-spectrum disorders: elevated aggressive behaviors, deficits in maternal nurturing, poor nest-building, and lower levels of social familiarity and social interaction. Erk2 CKO mice also exhibited decreased anxiety-related behaviors and impaired long-term memory. Pharmacological inhibition of ERK1 phosphorylation in Erk2 CKO mice did not affect the impairments in social behaviors and learning disabilities, indicating that ERK2, but not ERK1 plays a critical role in these behaviors. Our findings suggest that ERK2 has complex and multiple roles in the CNS, with important implications for human psychiatric disorders characterized by deficits in social behaviors.
Introduction
The extracellular signal-regulated kinase (ERK) cascade links transmembrane receptors to downstream effector mechanisms. In neurons, the ERK cascade is activated by stimuli associated with synaptic activity, and in turn activated ERK phosphorylates numerous proteins involved in a diverse array of cellular processes including long-term potentiation, long-term depression, synaptogenesis, and transcriptional and translational regulation (Kelleher et al., 2004; Thomas and Huganir, 2004). Although numerous studies have investigated the role of ERKs in behavioral plasticity, it is controversial whether the ERK isoforms, ERK1 and ERK2, redundantly share their many brain functions and compensate for each other or whether they play distinctive roles. In the analysis of ERK signaling, most experiments use inhibitors of the upstream kinase, MEK. ERK1 and 2 are solely activated by MEK, and thus, it is difficult to examine the specific contribution of each isoform to physiological functions.
Recently, however, accumulating evidence has suggested that the role of each isoform in long-term memory may not be functionally redundant. It was demonstrated that Erk1 knock-out mice did not show a significant impairment in learning ability (Selcher et al., 2001), although the treatment of rodents with a MEK inhibitor impaired memory formation (Kelleher et al., 2004; Thomas and Huganir, 2004). On the other hand, we reported that Erk2 knockdown mice, in which Erk2 expression was partially (20–40%) reduced, showed deficits in long-term memory (Satoh et al., 2007).
Although these results suggest a central contribution of the ERK2 isoform to learning and memory, a specific role of ERK2 for other behavioral profile has not been fully revealed in vivo. However, accumulating evidence has suggested that the ERK pathway is also involved in regulating emotional/affective behaviors (Ailing et al., 2008; Engel et al., 2009) potentially as an integrator at the nexus of multiple neuronal signaling cascades. Moreover, the relevance of ERK to human psychiatric disorder has been speculated (Kumar et al., 2008; Engel et al., 2009). Considering the complex and pleiotropic involvement of ERK in neuronal functions, it is important to examine the behavioral profile of Erk2 conditional knock-out (CKO) mice in detail and dissect the roles of ERK1 and ERK2 to understand the clinical relevance.
Because Erk2 knock-out is embryonically lethal (Satoh et al., 2007), the conditional loss of Erk2 in the nervous system is of great interest to gain a better understanding of the specific functions of ERK2 in vivo. Here, we used a conditional, region-specific, genetic approach to target Erk2 using the Cre/loxP strategy with a nestin promoter-driven cre transgenic mouse line, in which cre activity is confined to the CNS.
We found that Erk2 CKO mice exhibited marked anomalies in social behaviors as well as decreased anxiety-related behaviors and deficits in long-term memory. These anomalies have great relevance to autism-spectrum disorders (ASDs). Our findings suggest pleiotropic roles for ERK2 in neurological and behavioral functions, and that this protein might be a factor underlying human psychiatric disorders.
Materials and Methods
Mice.
All experiments were conducted according to the institutional ethical guidelines for animal experiments and the safety guidelines for gene manipulation experiments of the National Defense Medical College and were approved by the Committee for Animal Research at the National Defense Medical College (Tokorozawa, Saitama, Japan). Mice were maintained on a 12 h light-dark cycle (lights on from 7:00 A.M. to 7:00 P.M.) with room temperature at 21 ± 1°C. Mice had ad libitum access to water and food.
Generation of floxed Erk2 and Erk2 CKO mice.
The Erk2 gene was isolated from a 129X1/SvJ mouse genomic library. To generate the Erk2(floxN) allele, we constructed a targeting vector from 16.8 kb of Erk2 DNA (from the Apa1 site in intron 1 to the Taq1 site in intron 6) (Fig. 1A). For positive selection, a floxed (flanked by loxP sites) Pgk-neo cassette was inserted in the opposite direction into the EcoRI site in intron 1. A third loxP site, KpnI, and SapI sites were inserted into the BglII site in intron 3 (Fig. 1A). Targeted insertion of the plasmid by homologous recombination was performed in 129 derived embryonic stem cells (E14), and derived germline targeted offspring (Erk2+/flox(Neo+) mice) were obtained. While the expression of ERK2 in Erk2flox(Neo+)/flox(Neo+) mice was reduced owing to the presence of the Pgk-neo cassette (Satoh et al., 2007), it resumed normal levels after excision of the neo cassette by in vivo crossing with transgenic mice expressing cre recombinase (EIIA-cre mice) to obtain mice carrying the Erk (flox(Neo−)) allele (Erk2+/flox(Neo−) mice) or the Erk2 (Δflox) allele (Erk2+/Δflox mice). The disruption of the Erk2 gene locus was confirmed by Southern blot analysis of genomic DNA from adult mice (Fig. 1B,C). In the genotyping PCR, the primers used for detection of the Erk2(flox) and Erk2 wild-type alleles were mE2-F3 (5′-GATCTGATGCTTGCCAAAGCC-3′) and mE2-R4 (5′-TGTAAAGTAGCAGCAGATGC-3′) (Fig. 1D). The primers used for detection of the Erk2(Δflox) allele were mE2-F3 and mE2-R1 (5′-CAGAGTTTCATTATGGAGTCCTCGC-3′) (Fig. 1D).
Figure 1.

Generation of Erk2 CKO mice. A, Schematic diagram of targeted knock-out of the mouse Erk2 gene. The Erk2(flox(Neo+)) allele is converted to the Erk2(flox(Neo−)) allele by in vivo cre-mediated partial recombination using EIIA-cre mice. White boxes represent Erk2 exons and black boxes represent the Pgk-neo cassette. The 5′ and 3′ outer probes used for Southern blotting are shown as gray boxes. The locations of the primers used for genotyping are indicated underneath each scheme. B, C, Southern blot analysis of wild-type and mutant mouse genomic DNA. DNA samples are digested with KpnI and hybridized with the 5′ (B) or 3′ (C) outer probe. The positions and sizes of the wild-type and mutant fragments are indicated. D, PCR genotyping of wild-type and mutant mice. The positions and sizes of PCR fragments for wild-type and mutant mice are indicated. E–G, Expression profile in adult Erk2 CKO mice. E, Expression of ERK1 and 2 in extracts of the hippocampus, cortex and cerebellum. F, Phosphorylation status of ERK1 and 2 in basal conditions. In E and F, β-actin serves as the control for protein loading. G, Loss of ERK2 protein in the nervous system in Erk2 CKO mice. Immunohistochemical analysis of the hippocampus (bottom) and hippocampal pyramidal cells (top) show the distribution of ERK2 in neuronal cells from nestin-cre−; Erk2flox/flox (control) mice, and that ERK2 is absent in nestin-cre+; Erk2flox/flox (Erk2 CKO) mice. Slides are counterstained with hematoxylin. Scale bars: top, 10 μm; bottom, 500 μm.
Erk2+/flox(Neo−) mice were backcrossed with C57BL/6J mice for >10 generations. We crossed these mice with nestin promoter-driven cre transgenic mice (Vernay et al., 2005), which were maintained on the same background (C57BL/6J).
Electron microscopy.
Electron microscopy was performed as previously described (Nakata and Yorifuji, 1999). Briefly, under deep anesthesia by intraperitoneal injection of pentobarbital (50 mg/kg), mice (7-weeks of age) were perfused transcardially with 0.1% heparin-PBS followed by a fixative with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were immediately removed from the skull and cut into coronal slices (1 mm thick). Under a stereomicroscope, small pieces (1 × 1 mm) of the slices were extracted from the hippocampal CA1 or layer II/III of the temporal association cortex (at the levels 2.0 mm caudal to bregma). Then, semithin (200 nm) sections were prepared and stained with 1% toluidine blue. Ultrathin (60–70 nm) sections were cut using an ultramicrotome (Ultracut-N; Reichert-Jung), contrasted with uranyl acetate and lead citrate, examined using a JEM-1010 electron microscope (Jeol) and photographed. At least 20 photomicrographs were analyzed for each mouse to quantify the postsynaptic density (PSD) length, spine number and the percentage of perforated spines.
DiI staining.
Lipophilic dye 1,1′-(dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen) staining was performed as described previously (Satoh et al., 2007).
Immunohistochemistry.
Immunohistochemical studies were performed as previously described (Satoh et al., 2011). Briefly, paraffin sections (5 μm thick) were deparaffinized and immersed in unmasking solution (Vector H3300; Vector Laboratories) for antigenic retrieval and heated in an autoclave (121°C) for 5 min. Then, sections were incubated with a nonspecific blocking reagent (Dako) for 1 h to reduce background staining.
For bright-field dye staining (Fig. 1G), sections were then incubated with a primary antibody (anti-ERK2; mouse monoclonal, 1:1000; BD Transduction Laboratories) overnight in a humidified chamber at 4°C, followed by application of a biotinylated horse anti-mouse IgG secondary antibody (Vector Laboratories). For signal amplification, sections were incubated with an avidin-biotin-horseradish peroxidase complex (ABC Elite kit; Vector Laboratories) and visualized by a 3,3′-diaminobenzidine tetrachloride (Vector Laboratories) reaction according to the manufacturer's instructions. Finally, the sections were counterstained with hematoxylin. PBS was used for rinses.
For fluorescent staining, the primary antibodies used were anti-ERK2 (Fig. 2; rabbit polyclonal, 1:400; Cell Signaling Technology), anti-glial fibrillary acidic protein (GFAP) (see Fig. 4; mouse monoclonal, 1:50; Sigma), or anti-NeuN (Fig. 3; mouse monoclonal, 1:100; Millipore). After application of primary antibody overnight in a humidified chamber at 4°C, sections were incubated with secondary antibodies. The secondary antibodies used were Alexa-Fluor 488-conjugated goat anti-mouse IgG (1:400; Invitrogen) or Cy3-conjugated goat anti-mouse IgG (1:400; Jackson Immunoresearch) for primary antibodies derived from mouse. For primary antibody derived from rabbit, Cy3-conjugated goat anti-rabbit IgG antibody (1:400; Jackson Immunoresearch) was used. Sections were examined with a wide-field or confocal fluorescence microscopy using Nikon C1 system (Nikon). Samples from at least four mice per genotype were examined in each experiment.
Figure 2.
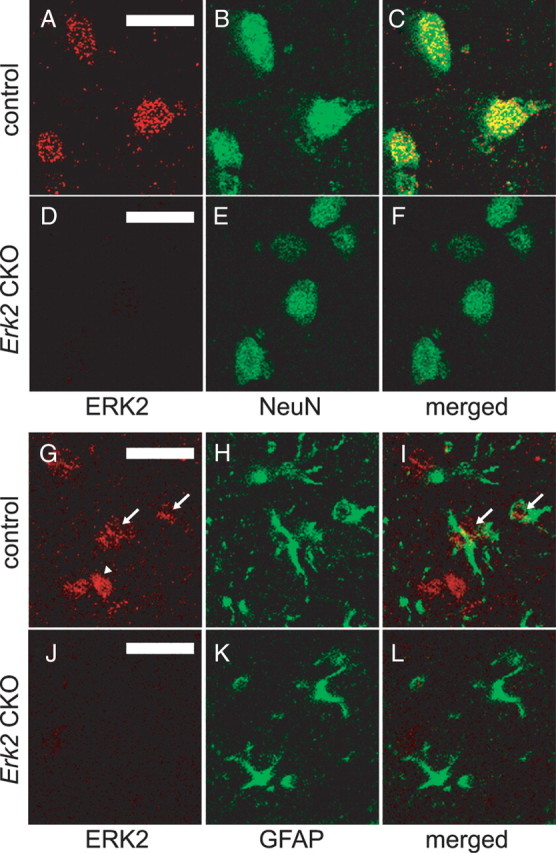
ERK2 protein is abrogated in neuronal and glial cells in Erk2 CKO mice. A–C, ERK2 is expressed in neuronal cells in control mice at 12 weeks of age. Double staining for ERK2 (A) and the postmitotic neuronal marker NeuN (B) in the neocortex show that ERK2 is expressed in neurons, as indicated by colocalization (C). D–F, ERK2 is abrogated in neuronal cells in Erk2 CKO mice. Double staining for ERK2 (D) and NeuN (E) with a merged image (F) show that ERK2 is not detectable in neuronal cells in Erk2 CKO mice. G–I, ERK2 is expressed weakly in astrocytes in control mice. Double staining for ERK2 (G) and the astrocyte marker GFAP (H) in the neocortex with a merged image (I) show partial colocalization of ERK2 and GFAP, indicating that ERK2 is expressed weakly in some astrocytes (arrows) although not in other astrocytes. Arrowhead indicates probable expression of ERK2 in neurons. J–L, ERK2 is abrogated in astrocytes in Erk2 CKO mice. Double staining for ERK2 (J) and GFAP (K) with a merged image (L) show that ERK2 is not detectable in astrocytes in Erk2 CKO mice. Scale bars, 20 μm.
Figure 4.

Inactivation of ERK2 results in more astrocytes within the cerebral cortex. Immunohistochemistry with GFAP, a marker for astrocyte, reveals that GFAP immunoreactivity is increased in the cortices of Erk2 CKO mice compared with controls at 13 weeks of age. Note that GFAP+ cells seem to be more abundant in outer layers than inner layers of cortices in Erk2 CKO mice. Scale bar, 100 μm.
Figure 3.
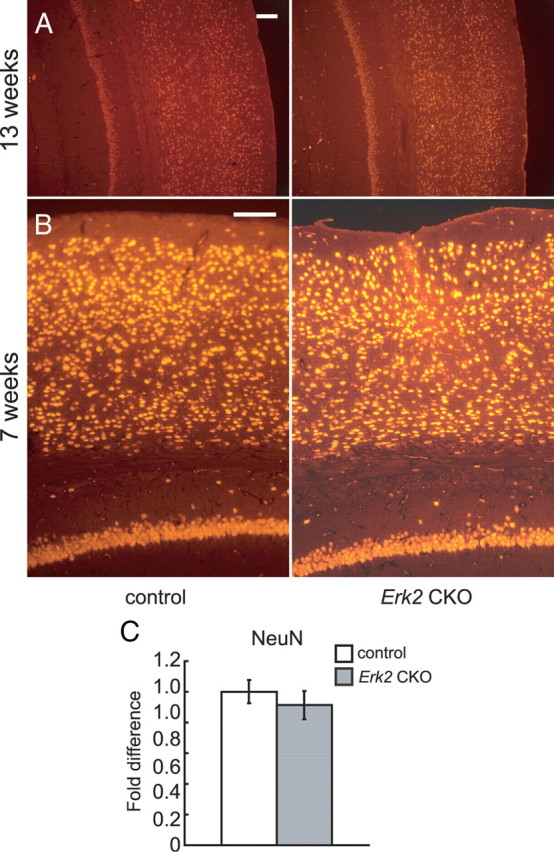
Neuronal number is not altered in Erk2 CKO mice. A, B, Cortical coronal sections are immunostained for the neuronal marker NeuN. No significant difference is detected in the number of NeuN+ cells between controls and Erk2 CKO mice at 13 (A) and 7 (B) weeks of age. C, The fold differences in the numbers of NeuN+ cells at 13 weeks of age are calculated (CKO vs control, n = 5 mice for each, t = 0.33, t test, p > 0.05). Scale bars, 100 μm.
Cell counting.
Cell number was assessed using the StereoInvestigator system (MicroBrightField). The boundaries were drawn using StereoInvestigator and stained cells were counted within sampling frames chosen in a systematically random manner within the areas of interest. The number of cortical cells stained for NeuN was counted in the dorsolateral portion of the cerebral cortex, from the retrosplenial cortex medially up to the rhinal fissure ventrolaterally.
Western blot analysis.
Preparation of protein extracts was performed as previously described (Satoh et al., 2007). Briefly, the amount of protein in each sample was measured using a BCA assay kit (Pierce). Samples were subjected to SDS-PAGE. The proteins were transferred onto an Immobilon-P membrane (Millipore). The blots were then immunoreacted with primary antibodies. The primary antibodies used were anti-ERK1/2 (rabbit polyclonal, 1:1000; Cell Signaling Technology), anti-phospho-ERK1/2 (rabbit polyclonal, 1:1000; Cell Signaling Technology), or anti-β-actin (mouse monoclonal, 1:2500; Sigma). Then, the primary antibodies were recognized using horseradish peroxidase (HRP)-conjugated secondary antibodies. Detection was performed with chemiluminescent substrates for HRP (Super Signal West Pico; Pierce or ECL plus; GE Healthcare). The signals were analyzed using an LAS3000 digital imaging system (Fujifilm). Samples from at least five mice per genotype were examined in each experiment.
Behavioral tests.
Mice used for behavioral tests were age-matched male littermates except for the maternal behavior test and pup exchange test. For the maternal behavior and pup exchange test, age-matched female littermates were used. Since the Erk2 CKO mother does not care well for their pups, all mice used in the behavioral tests were borne and reared by control mothers, except for those used in the pup exchange test. The apparatuses used in this study were made by O'Hara & Co., Ltd. except where described.
Open field test.
The open field test was performed as described previously (Satoh et al., 2007). Briefly, activity was measured as the total distance traveled (meters) in 10 min in the open field chamber (50 cm long × 50 cm wide × 40 cm high). The center square of the open field, comprising 50% of the total area, was defined as the “central area” of the open field. Mice used for the test were 9–11 weeks old.
Elevated plus-maze test.
The elevated plus-maze (EPM) test was performed as described previously (Satoh et al., 2007). Briefly, mouse behavior was recorded during a 10 min test period. The percentage of time spent in the open arms was used as an index of anxiety-like behavior. Mice used for the test were 9–11 weeks old (the same set of mice as in the open field test).
Maternal behavior test.
The maternal behavior test was conducted as described previously (Jin et al., 2007). Briefly, pregnant females were individually housed for a few days before parturition and examined for maternal behavior on the morning of parturition. After a 10 min separation of the mother from her pups, the dam was put in one corner of a cage and three of her pups were placed in different corners of the same cage. Then, she was observed for 20 min with minimal disturbance, and the time spent in crouching over the three pups was recorded. The percentage of newborns scattered was recorded at the end of the test. Mice used for the test were 13–15 weeks old.
Pup exchange test.
The pup exchange test was conducted using six Erk2 CKO and control mother couples. Pups that were born on the same day were exchanged during the first day after birth. The number of surviving pups was followed until weaning. Mice used for the test were 11–15 weeks old.
Resident-intruder test.
The resident-intruder test was performed as previously described (Takayanagi et al., 2005). Twenty-week-old resident males were individually housed for 2 weeks before testing. Eight-week-old wild-type mice, housed in groups, were used as intruders. New intruder mice were used in each test. The attack duration, frequency and latency to first attack were recorded for 10 min.
Social recognition test.
The social recognition test was conducted as described previously (Satomoto et al., 2009). We transferred 16-week-old mice from group to individual housing for 7 d before testing to permit establishment of a home-cage territory. Testing began when an ovariectomized female mouse was introduced into the home cage of each male mouse for a 1 min confrontation. At the end of the 1 min trial, the stimulus animal was removed and returned to an individual cage. This sequence was repeated for four trials with 10 min intertrial intervals, and each stimulus mouse was introduced to the same male resident in all four trials. In a fifth trial, another ovariectomized stimulus female was introduced to a resident male mouse. The stimulus females were all wild-type mice. Investigation was defined as direct, active and olfactory exploration of the female by the subject male mice. In general, this consisted of nosing and sniffing of the perioral and anogenital regions, as well as close following and pursuit. Aggressive posturing and sexual behaviors including mounting were not included in the measures of investigation. Females were exposed to only one male per day to reduce male odor contamination.
Sociability test in the open field chamber.
The preference for interaction with animate (caged adult mouse) versus inanimate (caged dummy mouse) targets (sociability) was examined in the open field chamber according to a slightly modified method of Kwon et al. (2006). Animate or inanimate targets were put into cylindrical cages allowing olfactory but minimal tactile interaction. The cylindrical cage was 10 cm in height, with a diameter of 9 cm and bars spaced 7 mm apart. Sniffing directed at the cage was scored for 10 min under 70 lux lighting conditions. We used 16-week-old mice in this test and all animate targets were wild-type male mice.
Sociability and social novelty test in a three-room chamber.
Social preference for novelty was performed in a three-room chamber as previously described (Moy et al., 2004). Each chamber was 20 cm long × 40.5 cm wide × 30 cm high. Dividing walls were made from clear Plexiglas, with small openings allowing access into each chamber. In the test, mice were initially allowed to explore the chambers for 10 min. After the habituation period, a caged wild-type male mouse, which had no prior contact with the subject mice, was placed in one of the side chambers. The cylindrical cages used were the same as those used in the sociability test in the open field. The subject mouse was placed in the middle chamber, and then the mouse was allowed to interact with an empty cage in one room versus a caged social target in another room for 10 min (sociability test). At the end of the first 10 min session, each mouse was tested in a second 10 min session to quantify social preference for a new stranger (social novelty test). The unfamiliar new stranger mouse was placed in an identical cage in the chamber that had been empty during the first 10 min session. The test mouse had a choice between the first, already-investigated mouse (familiar) and the novel unfamiliar mouse (novel). The time spent in each chamber was measured. Ten-week-old mice were used for the study.
Olfactory test.
The olfactory test was conducted as described previously (Satomoto et al., 2009). Briefly, mice were habituated to the flavor of a novel food (blueberry cheese) for 3 d before testing. On the fourth day, following 24 h food deprivation, a piece of blueberry cheese was buried under 2 cm of bedding in a clean cage. The mice were placed in the cage, and the time required to find the food was measured. The same set of mice was used as in the sociability and social novelty test in the three-room chamber.
Novelty test.
The novelty test was performed as previously described (Satomoto et al., 2009). Mice were housed individually and activity was measured as the total time spent interacting with an inanimate novel object (a small red tube) in 10 min. The same set of mice was used as in the sociability and social novelty test in the three-room chamber.
Nest formation test.
Nest formation was examined as described previously (Lijam et al., 1997) with minor modification. Six cages of male controls and six of male Erk2 CKO mice (n = 4 mice per cage) were used to evaluate nesting patterns. Small pieces of cotton nesting materials were placed in each cage. After 45 min, nest depth was measured. Mice used for the test were 9–11 weeks old.
Fear conditioning test.
The fear conditioning test was performed as previously described (Satoh et al., 2007), with some modifications. Briefly, the conditioning (acquisition) trial for contextual and cued fear conditioning consisted of a 3 min exploration period followed by one or three conditioned stimulus-unconditioned stimulus (CS-US) pairings (US: foot-shock intensity 0.15 mA, duration 1 s; CS: 80 dB white noise, 20 s duration; the US was delivered during the last second of the CS presentation). In the three pairing protocol, each stimulus was separated by 1 min. A context test was performed in the conditioning chamber for 5 min in the absence of white noise at 24 h after conditioning. The level of nonspecific freezing for the context test was monitored for 5 min before the conditioning in the same context. A cued test (for the same set of mice) was performed by presentation of the cue (80 dB white noise, 3 min duration) in an alternative context with distinct visual and tactile cues. The level of nonspecific freezing provoked by the new context was monitored for 3 min before the presentation of the cue in that new context. Mice used for the test were 9–11 weeks old.
MEK inhibitor administration.
To examine the effect of blockade of ERK1 activation in Erk2 CKO mice, MEK was systemically inhibited by intraperitoneal injection of 30 mg/kg SL327 (α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile) (Calbiochem) dissolved in dimethyl sulfoxide, 1 h before the behavioral tests.
Oxytocin assay.
We examined the plasma concentration of oxytocin (Oxt) in 9- to 11-week-old mice. The Oxt assay was performed according to the manufacturer's protocol (Oxytocin Enzyme Immunoassay kit, Assay Designs).
Oxt administration.
Mice were treated with acute subcutaneous injection of Oxt dissolved in saline (10 ng per kg body weight) as described previously (Jin et al., 2007). Mouse brain samples were removed 10 min after Oxt treatment for Western blot analyses. Mice used for the test were 9–11 weeks old.
Statistics.
Data were analyzed using the Student's t test, Welch's t test, two-way ANOVA with Fisher's post hoc test, and two-way repeated measures (RM) ANOVA with Fisher's post hoc test. Values are presented as the mean ± SEM.
Results
Generation of Erk2 CKO mice
To study the function of ERK2 in the CNS, we used a conditional, region-specific, genetic approach to target Erk2 (Fig. 1A). In this study, we used a nestin promoter-driven cre transgenic mouse line, in which cre activity is confined to the CNS (Vernay et al., 2005). The resulting Erk2 CKO mice (nestin-cre+; Erk2flox/flox mice or Erk2ΔCNS/ΔCNS mice) were viable and fertile with normal appearance. As anticipated, Erk2 CKO mice exhibited significantly reduced expression of ERK2 in the cortex, hippocampus and cerebellum compared with littermate controls (Fig. 1E,G), although a little residual expression of ERK2 was observed in the cerebellum (Fig. 1E; cerebellum, long exposure). At the moment, we have no clear explanation for this residual expression, although it is possible that a larger number of nestin-cre-unexpressing cells might exist in the cerebellum. The littermate controls used for this study had one of the following genotypes: nestin-cre−;Erk2+/+, nestin-cre−;Erk2flox/flox, or nestin-cre+; Erk2+/+ (hereafter termed control mice). We did not detect any differences among the different genotypes of the control group and we assumed that any phenotypic differences between them would be minimal.
We found no differences in ERK1 expression levels between the brains of Erk2 CKO and control mice, indicating that there were no compensatory changes in ERK1 expression (Fig. 1E). We also evaluated the in vivo basal activation state of ERK1 and 2 in brain extracts with an antibody against phospho-ERK1/2 (Fig. 1F). In Erk2 CKO mice, the density of the phospho-ERK1 band was greater than that in the control, suggesting the existence of compensatory upregulation in phosphorylation levels as previously reported (Lefloch et al., 2008). Although nestin-cre−; Erk2flox/flox (control) mice were completely normal and showed the expected ERK2 protein expression profile (Fig. 1G, upper left and lower left), Erk2 CKO mice lacked ERK2 immunoreactivity (Fig. 1G, upper right and lower right).
We further confirmed that ERK2 protein was abrogated both in neuronal and glial cells in Erk2 CKO mice by double staining (Fig. 2). ERK2 was expressed in neuronal cells in control mice at 12 weeks of age, as indicated by double staining for ERK2 and NeuN (Fig. 2A–C), but ERK2 was abrogated in neurons of Erk2 CKO mice (Fig. 2D–F). Similarly, double staining for ERK2 and the astrocyte marker GFAP in control mice demonstrated slight expression of ERK2 in astrocytes; this expression was absent in Erk2 CKO mice (Fig. 2G–L).
Normal neuronal number with cortical astrogliosis in Erk2 CKO mice
No significant difference was observed in the neuronal architecture at 7 and 13 weeks of age (Fig. 3A,B). Then, we examined whether the total number of NeuN-positive (NeuN+) cells in cortices of Erk2 CKO mice. Numbers of NeuN+ cells were not significantly different between control and Erk2 CKO mice at 13 weeks of age (Fig. 3C; t test, t = 0.51, p > 0.05), indicating that the effect of ERK2 abrogation on the neuronal population was minimal at this age. On the other hand, immunohistochemical analysis using antibody for GFAP revealed that Erk2 CKO mice contained more GFAP+ cells in their cortices compared with controls at 13 weeks of age (Fig. 4), indicating that ERK2 may be required for regulation of astrogliosis. This is consistent with previous reports, which demonstrated that inhibition of the MEK-ERK pathway resulted in enhanced generation of astrocytes (Paquin et al., 2005; Samuels et al., 2008; Heffron et al., 2009). GFAP+ cells seem to be more abundant in outer layers than inner layers of cortices (Fig. 4), although we do not know the reason.
Spine morphology is normal in Erk2 CKO mice
It has been reported that the ERK pathway might regulate synaptic remodeling (Wu et al., 2001; Goldin and Segal, 2003). Furthermore, the formation and stabilization of dendritic spines via ERK is believed to be involved in long-term storage of information in the CNS (Sweatt, 2004). For instance, the MEK inhibitors PD98059 and U0126 completely prevented a brain-derived neurotrophic factor (BDNF)-induced increase in dendritic spine density (Alonso et al., 2004). Thus we set out to examine the effect of ERK2 abrogation on spine morphology. To examine the number of dendritic spines at the basal level, we used the lipophilic tracer DiI. DiI staining showed that the number of spines per 10 μm dendritic segment in cortical layer II/III was not altered between Erk2 CKO and control mice (Fig. 5A; control, 6.1 ± 0.4, n = 12 neurons from four mice; Erk2 CKO, 6.4 ± 0.5, n = 12 neurons from four mice; t test, t = 0.51, p > 0.05). To compare the structure of individual spines between Erk2 CKO and control mice, we examined spine size using EM. The length of the PSD was used to detect changes in spine size (Meng et al., 2002). In apical dendrites of layer II/III in the temporal cortex, we detected no major changes in the mean PSD length in Erk2 CKO mice compared with controls (Fig. 5B,C; control, 255.0 ± 11.5 nm, n = 5 mice, total 604 synapses; Erk2 CKO, 249.9 ± 8.9 nm, n = 6 mice, total 846 synapses; t test, t = 0.39, p > 0.05). In the same area, the number of spines per 100 μm2 segment was the same in Erk2 CKO mice and controls (Fig. 5D; control, 3.43 ± 0.37, n = 5 mice, total 106 areas; Erk2 CKO, 3.95 ± 0.38, n = 6 mice, total 129 areas; t test, t = 1.0, p > 0.05). Larger spines often have a discontinuous PSD and are classified as complex or perforated spines, whereas smaller spines always have a continuous PSD and are classified as simple spines (Calverley and Jones, 1990). In this area, the proportion of perforated spines was not altered in the Erk2 CKO mice compared with controls (Fig. 5E; control, 9.7 ± 3.4%, n = 5 mice; Erk2 CKO, 8.0 ± 1.4%, n = 6 mice; t test, t = 0.55, p > 0.05).
Figure 5.
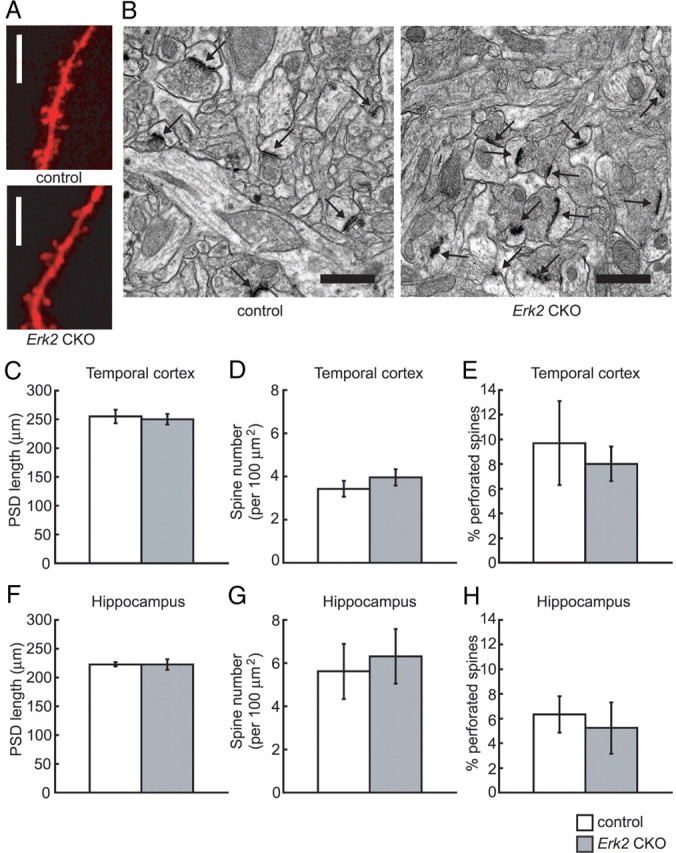
Synaptic density is not altered in Erk2 CKO mice. A, Representative confocal images of DiI-impregnated dendritic segments of layer II/III neurons from control and Erk2 CKO mice. No prominent difference is observed in dendritic spines between the genotypes. B, Representative electron micrographs of layer II/III spines in the temporal cortex from control and Erk2 CKO mice. The postsynaptic density is clearly visible as a dark band located right beneath the postsynaptic membrane in the spine head (arrows). Scale bars: A, 10 μm; B, 1 μm. C, The length of postsynaptic density from layer II/III in the temporal cortex is not significantly different in Erk2 CKO mice compared with controls. D, The number of spines per 100 μm2 segments of layer II/III in the temporal cortex is not significantly different between the groups. E, The percentage of perforated spines in apical dendrites of layer II/III in the temporal cortex is not different between the groups. F, The length of postsynaptic density from the hippocampus is not significantly different in Erk2 CKO mice compared with controls. G, The number of spines per 100 μm2 segments of the hippocampus is not significantly different between the groups. H, The percentage of perforated spines in apical dendrites of the hippocampus is not different between the groups.
Similar to the cortex, we detected no prominent differences in the mean PSD length in pyramidal neurons in stratum radiatum of the hippocampal CA1 area between Erk2 CKO mice and controls (Fig. 5F; control, 222.9 ± 4.0 nm, n = 5 mice, total 2432 synapses; Erk2 CKO, 222.5 ± 9.3 nm, n = 5 mice, total 2589 synapses; t test, t = 0.04, p > 0.05). In the same area, the number of spines per 100 μm2 segment was the same in Erk2 CKO mice and controls (Fig. 5G; control, 5.62 ± 1.28, n = 5 mice, total 217 areas; Erk2 CKO, 6.31 ± 1.26, n = 5 mice, total 213 areas, t test, t = 0.39, p > 0.05). In this area, the proportion of perforated spines was not altered in the Erk2 CKO mice compared with controls (Fig. 5H; control, 6.4 ± 1.5%, n = 5 mice; Erk2 CKO, 5.3 ± 2.1%, n = 5 mice, t test, t = 0.43, p > 0.05).
These results indicated that spine morphology in the basal state is normal in the temporal cortex and hippocampus of Erk2 CKO mice.
Maternal nurturing is impaired in Erk2 CKO mice
We found that whereas Erk2 CKO mice could bear pups, the majority of pups from Erk2 CKO dams died neonatally by postnatal day (P) 3, regardless of the genotype of the pups. On the other hand, pups from control mice were viable regardless of the genotype of the pups (Table 1). Because milk was not observed in the digestive tracts of pups from Erk2 CKO dams, the excess mortality was likely due to defects in lactation. Thus, we investigated the defects in maternal nurturing in Erk2 CKO mice. In pup exchange test, pups (n = 40) born to Erk2 CKO dams (n = 6) were successfully fostered (n = 39) when nursed by control dams. On the other hand, pups (n = 43) born to control dams (n = 6) died when nursed by Erk2 CKO dams. This indicates that the survival defect in the pups lie entirely with the Erk2 CKO dams. Erk2 CKO dams showed a significantly shorter duration of crouching to keep the pups warm and to nurse them (Fig. 6A,B; t test, t = 5.21, p < 0.001). Significantly more pups from Erk2 CKO dams were scattered in their home cages (Fig. 6A,C; t test, t = 8.67, p < 0.001). These results clearly showed abnormal maternal nurturing behavior in postpartum Erk2 CKO mice.
Table 1.
Effect of genotype on reproductive function
| Pairing |
No. of pregnant females observed | No. of pups born | No. of postnatal survivors | Postnatal survivors per total births, % | |
|---|---|---|---|---|---|
| Male | Female | ||||
| Erk2f/f; nestin-cre+ | Erk2f/f; nestin-cre− | 67 | 487 | 449 | 92.2 |
| Erk2+/f; nestin-cre+ | Erk2+/f; nestin-cre− | 16 | 115 | 108 | 93.9 |
| Erk2+/f; nestin-cre− | Erk2+/f; nestin-cre+ | 18 | 125 | 80 | 64.0 |
| Erk2f/f; nestin-cre− | Erk2f/f; nestin-cre+ | 24 | 121 | 3 | 2.5 |
Figure 6.

Erk2 CKO mice are aggressive and impaired in maternal behaviors. A–C, Maternal nurturing in newly postpartum Erk2 CKO mice. A, In nurturing analysis of postpartum females, a mother is deprived of her first litter of pups for 10 min and then rechallenged with three pups placed individually in the corners of her cage. The typical behavior of a control mother is shown, approaching one pup immediately and collecting all three pups into one corner within a short latency (left), then crouching over them. The Erk2 CKO mother typically ignores the pups and does not retrieve them immediately (right). B, Time spent crouching over the pups in the nest of control (n = 10) and Erk2 CKO (n = 10) mothers. C, The percentage of scattered pups from the same set of mothers as in B. D–F, Aggressive behaviors of male Erk2 CKO mice as measured by the resident-intruder test (control, n = 7; Erk2 CKO, n = 7). **p < 0.01, ***p < 0.001.
Erk2 CKO mice display high levels of aggression
When male mice were put together with their siblings after 2 weeks of isolation, some mice were wounded severely. We observed more wounded mice in cages containing Erk2 CKO mice than those containing only control mice (Table 2), so we assessed the aggressive behavior of Erk2 CKO mice using the resident-intruder test. Erk2 CKO resident males attacked the intruder with a shorter latency (Fig. 6D), for a longer duration (Fig. 6E; t test, t = 3.56, p < 0.01) and with higher frequency (Fig. 6F; t test, t = 4.41, p < 0.001) than control residents. These results clearly showed elevated aggressive behavior in Erk2 CKO mice.
Table 2.
The number of wounded mice in siblings when reentered into a cage after isolation for 2 weeks
| Genotypes of members in the same cage | No. of observed mice | No. of wounded mice | % wounded | No. of mice per cage | |
|---|---|---|---|---|---|
| Erk2f/f; nestin-cre− | 32 | 1 | 3.1 | 4.0 | |
| Erk2f/f; nestin-cre+ | Erk2f/f; nestin-cre− | 67 | 24 | 35.8 | 3.9 |
Erk2 CKO mice display abnormal social interactions
We further investigated whether Erk2 CKO mice display abnormal social interactions. First, we investigated social memory, which, in mice, predominantly depends on olfactory cues. This ability is needed for social familiarity and can be identified as a consistent decrease in olfactory investigation during repeated encounters with the same female in the social recognition test. In the social recognition test, Erk2 CKO mice showed abnormal social memory (Fig. 7A; two-way RM ANOVA (trials 1–4), F(1,22) = 4.92, p < 0.05 (genotype), F(3,66) = 14.38, p < 0.0001 (trial), F(3,66) = 2.37, p > 0.05 (interaction between genotype and trial)). Control mice showed a significant decline in the time spent investigating a female with subsequent presentation of the same female compared with Erk2 CKO mice (Fig. 7A). This decrease was not due to a general decline in olfactory investigation because presentation of a novel female during trial five resulted in a similar amount of investigation as trial one with the original female. Therefore, it was concluded that Erk2 CKO males, with persistent interest during repeated presentations, failed to develop social memory.
Figure 7.

Erk2 CKO mice are impaired in social behaviors. A–D, Abnormal behaviors in social interaction tests. A, Olfactory investigations in Erk2 CKO mice are used for the social recognition test. Social memory by male mice is measured as the difference in investigation time (control, n = 12; Erk2 CKO, n = 12). The data depict the amount of time spent investigating the same female during each of four successive 1 min trials. A fifth trial depicts the response to a new female. B, When exposed to caged social and inanimate targets in the open field, Erk2 CKO mice show a decreased duration of interaction with the social target, and a similar duration of interaction with the inanimate object (control, n = 29; Erk2 CKO, n = 23). Percentage time is depicted in B–D. C, In the sociability test in a three-room chamber, Erk2 CKO mice spend less time than controls with the social target (control, n = 11; Erk2 CKO, n = 11). D, In the social novelty test, controls show a preference for social novelty, while Erk2 CKO mice show no preference between the novel and familiar targets. Erk2 CKO mice spend significantly less time than controls interacting with the novel target. The same set of mice is used as in C. E, Erk2 CKO mice are not significantly different from controls in the latency to find a buried treat following overnight food deprivation. The same set of mice is used as in B. F, Erk2 CKO mice show a significant decrease in interaction with a novel object in their home cages. The same set of mice is used as in B. G, Erk2 CKO mice show significant deficits in nest formation (control, 6 cages, n = 4 mice per cage; Erk2 CKO, 6 cages, n = 4 mice per cage). H, I, Representative photographs of control (H) and Erk2 CKO (I) cages, 45 min after the introduction of cotton nesting materials into each cage. Note the fluffy nest built in the control cage and the huddling of mice in this nest, in contrast to the poorly formed nests in the Erk2 CKO cage. **p < 0.01, ***p < 0.001. N.S., Not significant.
Mice are a social species and exhibit behavioral social interaction (Murcia et al., 2005; Kwon et al., 2006). Social interaction is thought to be a core paradigm to test autistic behavior in mice (Crawley, 2004; Moretti et al., 2005; Kwon et al., 2006). In the open field test for social versus inanimate preference (sociability), Erk2 CKO mice exhibited abnormal social interaction (Fig. 7B; two-way ANOVA, F(1,100) = 6.13, p < 0.05 (genotype), F(1,100) = 4.20, p < 0.05 (interaction between genotype and preference). Post hoc comparison confirmed that control mice spent significantly more time interacting with the social target than with the inanimate target (social vs inanimate, p < 0.0001), indicating that control mice showed the normal preference for a social target over an inanimate target. In contrast to the control mice, Erk2 CKO mice exhibited a significant decrease in interaction with the social target compared with controls (post hoc test, control vs Erk2 CKO, p < 0.01). Erk2 CKO mice showed no significant difference in the time they spent interacting with the social or inanimate targets (post hoc test, social vs inanimate, p > 0.05). The interaction with the inanimate target was similar in both Erk2 CKO and control groups (post hoc test, control vs Erk2 CKO, p > 0.05).
Additional social behavior tests gave similar results. In a sociability test for social versus empty preference using the three-roomed chamber, Erk2 CKO mice exhibited abnormal social interaction (Fig. 7C; two-way ANOVA, F(1,40) = 5.04, p < 0.05 (genotype), F(1,40) = 15.07, p < 0.001 (interaction between genotype and preference)). The interaction with the empty cage was similar in both the Erk2 CKO and control groups (post hoc test, control vs Erk2 CKO, p > 0.05), but Erk2 CKO mice exhibited significantly decreased interaction with the social target compared with controls (post hoc test, control vs Erk2 CKO, p < 0.0001) and spent a similar amount of time interacting with both targets (post hoc test, social vs empty, p > 0.05). The control mice spent significantly more time interacting with the social target than with the empty cage (post hoc test, social vs empty, p < 0.0001), indicating that control mice showed the normal preference for a social target over an empty cage. These results were consistent with those of the sociability test in the open field. Subsequently, we examined a preference for social novelty using the three-room chamber and found that Erk2 CKO mice exhibited abnormal behaviors in this test (Fig. 7D; two-way ANOVA, F(1,40) = 12.61, p < 0.001 (genotype), F(1,40) = 6.12, p < 0.05 (interaction between genotype and preference). When control mice were exposed to a familiar mouse versus a novel mouse, control mice showed a clear preference for the novel mouse over the familiar mouse as expected (post hoc test, novel vs familiar, p < 0.01). On the other hand, the interaction with a familiar mouse was similar in both Erk2 CKO mice and control groups (post hoc test, control vs Erk2 CKO, p > 0.05), and Erk2 CKO mice did not show a preference for social novelty (post hoc test, novel vs familiar, p > 0.05).
These results indicate that Erk2 CKO mice have pervasive deficits in social interaction. We did not attribute the abnormalities in social memory and interaction to an overt loss of olfactory function, since we did not detect any significant difference between groups in a test for olfaction (Fig. 7E; t test, t = 0.40, p > 0.05).
Erk2 CKO mice also exhibited a significant decrease compared with control mice in interaction with a novel object in a familiar cage compared with control mice (t test, t = 3.37, p < 0.01), suggesting a general disinterest in novelty (Fig. 7F). Thus the deficient social interactions might be at least partly attributable to a general lack of interest in novelty.
Additionally, we examined nest formation, a test for home cage behavior, of Erk2 CKO mice. As well as social behavior, nesting has been proposed as a core paradigm to test autistic behavior in mice (Crawley, 2004), and has been used to measure autism-like behaviors in other mutant mouse models (Moy et al., 2004; Moretti et al., 2005; Kwon et al., 2006). In contrast to the immediate activity of nest building in control mice, Erk2 CKO mice showed little nest-building activity (Fig. 7G–I). Control mice built nests from cotton nesting material that averaged >50 mm in depth, while Erk2 CKO mice built significantly shallower nests, with depths that averaged <30 mm (Fig. 7G; t test, t = 4.49, p < 0.01). The poor nest building displayed by Erk2 CKO mice also demonstrated an essential role for Erk2 in social behavior in the home cage.
Erk2 CKO mice exhibit reduced anxiety
To examine responses to an unfamiliar environment, Erk2 CKO mice were assayed in the open field test. Statistical analyses of walking velocity (Fig. 8A; t test, control vs Erk2 CKO, t = 0.13, t > 0.05) and total path-length (distance traveled) (Fig. 8B; t test, control vs Erk2 CKO, t = 0.62, t > 0.05) revealed that Erk2 CKO mice did not differ from control animals in their exploratory behavior and locomotor activity. Next to study whether ERK2 abrogation affects anxiety-related behavior, Erk2 CKO mice were subjected to the EPM test. Erk2 CKO mice showed decreased anxiety-like behavior as they spent a significantly longer time in the open arms than did the controls (Fig. 8E; t test, t = 6.96, p < 0.001). Consistently, Erk2 CKO mice showed decreased anxiety-like behavior in the open field test as indicated by prolonged localization in the central area (Fig. 8C,D). Normally, mice show a preference for the corners and peripheral areas over the central area of the open field, because they feel safer there (Fig. 8D, left). In contrast, Erk2 CKO mice spent significantly more time in the central zone (Fig. 8D, right) compared with controls (Fig. 8C; t test, control vs Erk2 CKO, t = 4.24, t < 0.001). Together with the finding that Erk1 knock-out mice did not differ significantly from controls in the amount of time spent in the open arms in the EPM (Ailing et al., 2008), our results suggested that ERK2 might play an important role in anxiety-like behavior.
Figure 8.

Erk2 CKO mice exhibit normal locomotor activity but decreased anxiety-related behaviors. A–D, Open field test (control, n = 19; Erk2 CKO, n = 20). A, The average speed without resting time is not different between control and Erk2 CKO mice. B, The 10 min total path-length traveled is not significantly different between control and Erk2 CKO mice. C, Erk2 CKO mice exhibit reduced anxiety-like behavior, because they spend significantly more time in the central zone of the open field apparatus. D, Representative tracks of control and Erk2 CKO mice in the open field chamber over 10 min. E, Erk2 CKO mice exhibit reduced anxiety-like behavior in the elevated plus-maze test (the same set of mice as in A). ***p < 0.001.
Erk2 CKO mice are impaired in long-term memory
To characterize the hippocampal-dependent and/or amygdala-dependent long-term memory in Erk2 CKO mice, these mice were subjected to contextual/cued fear conditioning. We previously reported that Erk2 knockdown mice exhibited impaired Pavlovian fear learning (Satoh et al., 2007). Other previous reports have revealed that preventing ERK activation with a MEK inhibitor impaired fear learning in mice (Atkins et al., 1998; Schafe et al., 1999). Fear memory was assessed based on the freezing behavior to the conditioned cue or the context to which the mice were previously exposed. The context freezing response of Erk2 CKO mice was significantly reduced compared with that in controls after a 24 h retention delay (Figure 9A; t test, t = 3.47, p < 0.005). The response of Erk2 CKO mice to the cued fear conditioning was also significantly reduced compared with control mice after a 48 h retention delay (Fig. 9B; t test, t = 7.72, p < 0.0001). These results suggest that ERK2 plays an important role in long-term fear memory.
Figure 9.

Erk2 CKO mice show impaired memory performance in both contextual and cued tests. A, The freezing response is measured in the context before shock (basal freezing) and in the conditioning chamber (contextual fear response) 24 h after conditioning (control, n = 12; Erk2 CKO, n = 12). B, The freezing response (the same set of mice as in A) is measured in an alternative context either without an auditory cue (basal freezing after conditioning) or with a cue 48 h after conditioning. **p < 0.01, ***p < 0.001.
Plasma Oxt levels in Erk2 CKO mice are normal
Oxt belongs to the posterior pituitary hormone family and is essential for the induction of normal labor through uterine contraction and lactation in mammals (Hatton, 1990; Argiolas and Gessa, 1991; Russell et al., 2003). Several researchers have suggested that the Oxt signaling system also plays a crucial role in social interaction and it is implicated in the etiology of developmental psychiatric disorders characterized by deficits in social behavior (Argiolas and Gessa, 1991; Takayanagi et al., 2005; Jin et al., 2007). To investigate the possibility that abrogation of the ERK2 pathway affects the plasma concentration of Oxt, we examined the plasma concentration of Oxt in Erk2 CKO mice. There was no significant difference in Oxt levels between control and Erk2 CKO mice [Fig. 10A,B; t test, t = 0.61, p > 0.05 (male); t = 0.44, p > 0.05 (female)], suggesting that the neuroendocrine system was not impaired in Erk2 CKO mice and that ERK2 may not regulate Oxt secretion.
Figure 10.

Plasma Oxt levels are not altered in Erk2 CKO mice. A, B, Levels of plasma Oxt in male (control, n = 10; Erk2 CKO, n = 10) (A), and female (control, n = 8; Erk2 CKO, n = 8) mice (B). The levels in Erk2 CKO mice are not different from those in controls.
Oxt increases ERK phosphorylation in the hippocampus
Next, we examined whether there was a differences in the activation of ERK between control and Erk2 CKO mice in response to Oxt treatment. Western blotting revealed that subcutaneous injection of Oxt (10 ng/kg body weight) significantly increased ERK1 phosphorylation levels in the hippocampus compared with in vehicle-injected mice 10 min after injection, regardless of genotype (Fig. 11A,B). Two-way ANOVA indicated a main effect of Oxt treatment (vehicle vs Oxt: F(1,12) = 39.74, p < 0.001) and of genotype (control vs Erk2 CKO: F(1,12) = 61.06, p < 0.001). Two-way ANOVA also revealed a significant interaction effect between Oxt treatment and genotype (F(1,12) = 9.27, p < 0.05). There were no differences in total ERK1 expression levels between vehicle- and Oxt-treated mice both for controls and Erk2 CKOs (Fig. 11A).
Figure 11.
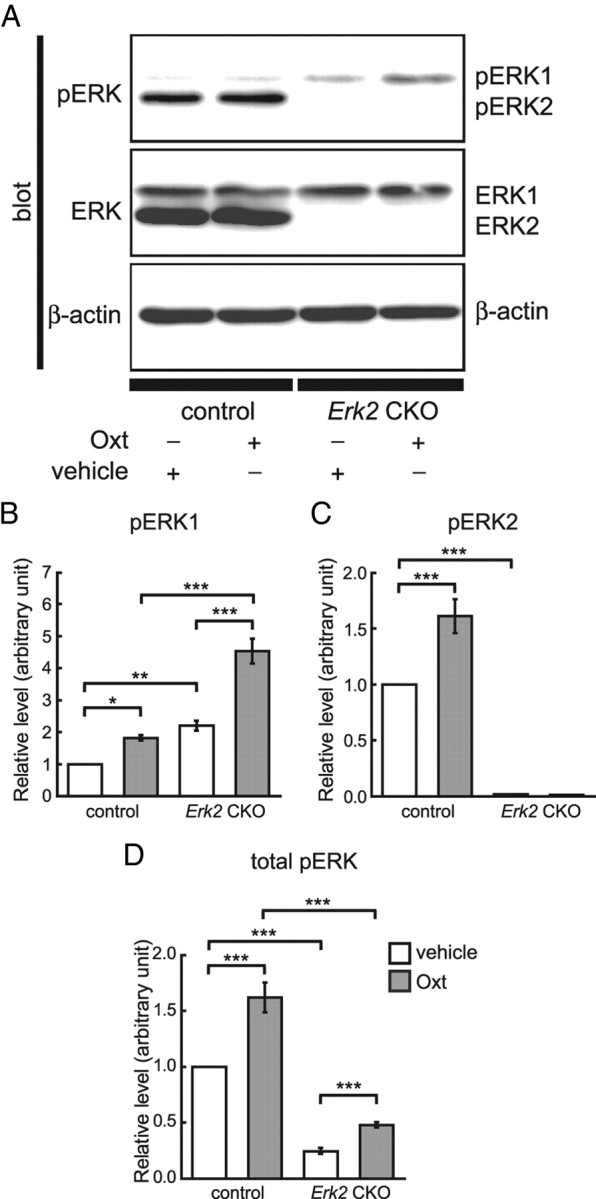
Subcutaneous injection of Oxt (10 ng/kg body weight) significantly increases phosphorylated ERK levels in the hippocampus. A, Hippocampus homogenates from vehicle-treated or Oxt-treated control (n = 4 for each) and Erk2 CKO littermates (n = 4 for each) are analyzed simultaneously for phospho-ERK1/2 (pERK1/2) and ERK1/2 by quantitative Western blotting. In control mice, ERK1 phosphorylation level is significantly elevated 10 min after Oxt injection. Similarly, ERK1 phosphorylation is significantly elevated 10 min after Oxt injection in Erk2 CKO mice. There is no difference in total ERK1 expression level among all groups. The ERK2 phosphorylation level is also elevated in control mice after Oxt injection without change of total ERK2 expression level. β-Actin serves as the control for protein loading. B–D, To evaluate phosphorylation, the intensities of the phospho-ERK1 and phospho-ERK2 bands are divided by their corresponding loading control (β-actin). Then, relative levels of ERK1 (B), ERK2 (C), and total ERK (D) phosphorylation are normalized to the mean control with vehicle injection. *p < 0.05; **p < 0.01; ***p < 0.001.
Oxt also significantly increased ERK2 phosphorylation levels in control mice compared with vehicle-treated mice (Fig. 11A,C). Two-way ANOVA indicated a main effect of Oxt treatment (vehicle vs Oxt: F(1,12) = 12.13, p < 0.01), and of genotype (control vs Erk2 CKO: F(1,12) = 218.00, p < 0.0001), and an interaction between Oxt treatment and genotype (F(1,12) = 12.32, p < 0.01). There were no significant differences in total ERK2 expression levels between vehicle- and Oxt-treated mice among controls (Fig. 11A).
These results are consistent with the notion that Oxt signals through ERK activation. Furthermore, this result suggest that lack of ERK2 activation was compensated for by ERK1 activation in Erk2 CKO mice. Although the amount of change in ERK1 phosphorylation was more robust in Erk2 CKO mice than in controls (Fig. 11B; t test, t = 4.45, p < 0.01), total phosphorylation level of ERK in response to Oxt was smaller in Erk2 CKO mice than in controls (Fig. 11D; two-way ANOVA, F(1,12) = 151.48, p < 0.001 (genotype), F(1,12) = 30.91, p < 0.001 (Oxt treatment), F(1,12) = 6.31, p < 0.05 (interaction between Oxt treatment and genotype).
Pharmacological blockade of ERK1 phosphorylation in Erk2 CKO mice does not produce additional effect on impaired long-term memory
As described above, phosphorylation level of ERK1 was increased in Erk2 CKO mice although the total ERK1 expression levels were unchanged. To identify putative ERK2 functions that could be compensated for, and to confirm that the abnormal behaviors in Erk2 CKO mice were caused by the loss of ERK2 per se rather than a secondary upregulation of ERK1 activity, we examined the effect of the MEK inhibitor SL327 in Erk2 CKO mice. SL327 (30 mg/kg) was administered intraperitoneally to subjects 1 h before sample preparation. Western blot analysis revealed that SL327 attenuated phosphorylated ERK levels (Fig. 12A,B) as described previously (Selcher et al., 1999). SL327 administration affected ERK1 phosphorylation levels 1 h later in the hippocampus (Fig. 12A). Two-way ANOVA confirmed this difference, indicating a main effect of SL327 treatment (Fig. 12B; F(1,16) = 9.28, p < 0.05). Administration of SL327 also significantly reduced ERK2 phosphorylation levels (Fig. 12A). Two-way ANOVA confirmed this difference, indicating a main effect of SL327 treatment (Fig. 12B; F(1,16) = 24.90, p < 0.01), although ERK2 phosphorylation was undetectable in Erk2 CKO mice (Fig. 12A,B). There was no concurrent decrease in ERK1 and 2 expression levels (Fig. 12A).
Figure 12.
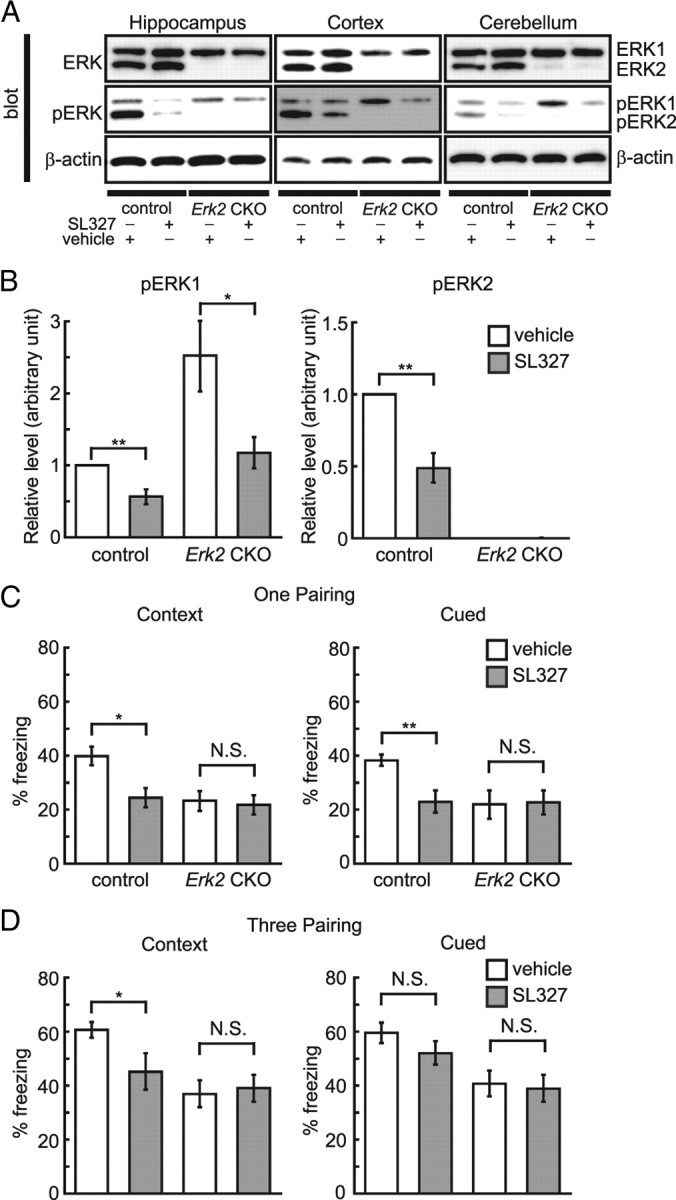
Pharmacological blockade of ERK1 phosphorylation in Erk2 CKO mice does not additionally affect long-term memory. A, ERK1 and 2 phosphorylation levels in the hippocampus, cortex, and cerebellum are significantly inhibited 60 min after injection of 30 mg/kg MEK inhibitor SL327 in the hippocampus, cortex and cerebellum. There is no concurrent decrease in the total expression level of either isoform. B, The phosphorylation levels of ERK1 and 2 in the hippocampus as analyzed by band density (control, n = 5; Erk2 CKO, n = 5). β-Actin serves as the control for protein loading. C, In control mice, SL327 attenuates freezing in the contextual and cued fear conditioning after receiving one pairing of a tone and shock, compared with vehicle (vehicle, n = 10; SL327, n = 10). However, no significant difference is observed between animals treated with SL327 or vehicle for Erk2 CKO mice (vehicle, n = 10; SL327, n = 10). D, In control mice, SL327 attenuates freezing in contextual fear conditioning after receiving three pairings of a tone and shock compared with vehicle, but the additional pairings eliminated the effect of SL327 on freezing in response to the cue (vehicle, n = 10; SL327, n = 10). There is no significant difference in freezing in response to context and cue between Erk2 CKO mice treated with SL327 or vehicle (vehicle, n = 10; SL327, n = 10). *p < 0.05, **p < 0.01. N.S., Not significant.
We investigated whether the administration of SL327 (30 mg/kg) to Erk2 CKO mice could produce additional effects in the fear conditioning test. SL327 was administered intraperitoneally to subjects 1 h before cue and contextual fear conditioning. It was reported that 30 mg/kg SL327 did not impair basal activity in control mice (Selcher et al., 1999). For these experiments, foot shock was paired either once or three times with an auditory CS. With a one pairing protocol, SL327 administration affected the contextual conditioning response after a 24 h retention delay (Fig. 12C). Two-way ANOVA indicated a main effect of SL327 administration (F(1,36) = 6.54, p < 0.05) and that the genotype effect had a significant interaction with SL327 administration (F(1,36) = 4.48, p < 0.05). In control mice, post hoc test revealed that injection of SL327 significantly reduced the contextual freezing response after a 24 h retention delay compared with vehicle (vehicle vs SL327, p < 0.05). However, in Erk2 CKO mice, there was no significant difference in freezing between animals treated with vehicle and SL327 in contextual conditioning (post hoc test, vehicle vs SL327, p > 0.05).
SL327 administration also affected the cued freezing response after a 48 h retention delay (Fig. 12C). Two-way ANOVA indicated a main effect of SL327 administration (F(1,36) = 4.44, p < 0.05), and that a genotype effect showed a significant interaction with SL327 administration (F(1,36) = 7.37, p < 0.05). In control mice, post hoc test revealed that the response of control mice to the cued fear conditioning was reduced significantly (vehicle vs SL327, p < 0.01). However, in Erk2 CKO mice, there was no significant difference in freezing between animals treated with vehicle and SL327 in the cued response (post hoc test, vehicle vs SL327, p > 0.05).
When a three pairing protocol was used, SL327 administration affected the contextual conditioning response after a 24 h retention delay (Fig. 12D). Two-way ANOVA indicated a main effect of SL327 administration (F(1,36) = 16.18, p < 0.001), and that a genotype effect had a significant interaction with SL327 administration (F(1,36) = 11.42, p < 0.01). In control mice, post hoc test revealed that SL327 reduced the contextual freezing response compared with vehicle (vehicle vs SL327, p < 0.05). In Erk2 CKO mice, there was no significant difference in freezing between animals treated with vehicle and SL327 in the contextual response (post hoc test, vehicle vs SL327, p > 0.05). On the other hand, SL327 administration did not affect the cued conditioning response (two-way ANOVA, F(1,36) = 2.48, p > 0.05 (SL327 treatment)) (Fig. 12D).
These results suggest that the deficit in ERK2 function in long-term memory was not compensated for by upregulation of ERK1 activity as a result of ERK2 deletion. Moreover, these results suggest that the deficits are probably not the result of a secondary upregulation of ERK1 activity.
Pharmacological blockade of ERK1 phosphorylation in Erk2 CKO mice does not produce additional effect on abnormal social behaviors
Next, we examined the effect of a MEK inhibitor SL327 (30 mg/kg) on social behaviors. In the sociability test in the open field, two-way ANOVA indicated that there was a significant main effect of genotypes (F(1,52) = 8.29, p < 0.01) and a significant interaction between genotype and SL327 treatment (F(1,52) = 4.23, p < 0.05), although there was no significant main effect of SL327 treatment (F(1,52) = 0.65, p > 0.05). In control mice, SL327 reduced the duration of interaction with the social target compared with vehicle (Fig. 13A). On the other hand, there was no significant difference in the duration of interaction with the social target between SL327- and vehicle-treated Erk2 CKO mice, although both SL327- and vehicle-treated Erk2 CKO mice showed reduced social interaction compared with vehicle-treated control mice (Fig. 13A). Vehicle-treated controls showed a preference for the social target, while SL327-treated control mice did not (Fig. 13A). Neither SL327- nor vehicle-treated Erk2 CKO mice showed a significant preference for the social target (Fig. 13A).
Figure 13.

Pharmacological blockade of ERK1 phosphorylation in Erk2 CKO mice does not additionally affect social behaviors. A–C, Treatment with the MEK inhibitor SL327 (30 mg/kg) reduces social behaviors in control mice, but does not additionally affect impaired social behaviors in Erk2 CKO mice. A, When SL327 in control mice are exposed to caged social and inanimate targets in the open field, the duration of interaction with the social target is significantly decreased compared with vehicle (n = 14 mice for each). On the other hand, there is no significant difference in the spent with the social target between SL327- and vehicle-treated Erk2 CKO mice (n = 14 mice for each). Percentage time is depicted in A–C. B, In the sociability test in the three-room chamber, SL327 reduces the time spent with the social target compared with vehicle in control mice. On the other hand, there is no significant difference in the time spent with the social target between SL327- and vehicle-treated Erk2 CKO mice. The same set of mice is used as in A. C, In the social novelty test, vehicle-treated controls show preference for the novel target, while SL327-treated control mice does not. Furthermore, SL327 in control mice reduces the time spent with the social target compared with vehicle. On the other hand, there is no significant difference in the time spent with the novel target between SL327- and vehicle-treated Erk2 CKO mice. The same set of mice is used as in A and B. *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-injected control mice; #p < 0.05; ###p < 0.001. N.S., Not significant.
In the sociability test in the three-room chamber (Fig. 13B), two-way ANOVA indicated that there was a significant main effect of genotype (F(1,52) = 8.24, p < 0.01) and a significant interaction between genotype and SL327 treatment (F(1,52) = 4.05, p < 0.05), although there was no significant main effect of SL327 treatment (F(1,52) = 1.21, p > 0.05). While SL327 reduced the time spent with the social target in control mice, there was no significant difference between SL327- and vehicle-treated Erk2 CKO mice (Fig. 13B). Both SL327- and vehicle-treated Erk2 CKO mice showed reduced social interaction compared with vehicle-treated control mice and did not show a significant preference for the social target (Fig. 13B).
In the social novelty test, two-way ANOVA indicated that there was a significant main effect of genotype (F(1,52) = 4.78, p < 0.05) and significant interaction between genotype and SL327 treatment (F(1,52) = 4.15, p < 0.05), although there was no significant main effect of SL327 treatment (F(1,52) = 1.11, p > 0.05). SL327 reduced the time spent with the social target compared with vehicle in control mice (Fig. 13C). On the other hand, there was no significant difference in time spent with a novel target between SL327- and vehicle-treated Erk2 CKO mice, although the time interacting with novel target was significantly less in Erk2 CKO mice than in vehicle-treated control mice (Fig. 13C). Vehicle-treated controls showed a preference for the novel target, while SL327-treated control mice did not (Fig. 13C). Neither SL327- nor vehicle-treated Erk2 CKO mice showed a preference for the novel target. These results indicate that blockade of ERK1 phosphorylation causes no additional effects on the impaired social behaviors in Erk2 CKO mice.
Discussion
The ERK signaling pathway and social behaviors
Using a conditional genetic approach, we have demonstrated important roles for the ERK2 signaling pathway in vivo: Erk2 CKO mice exhibited pervasive deficits in social behaviors as well as anomalous affective behaviors and cognitive functions. This suggests that ERK2 plays complex and multiple roles in brain function, with important implications for human psychiatric disorders characterized by deficits in social behaviors. Our findings of pervasive deficits in social behaviors in Erk2 CKO mice are consistent with those of some previous reports. For instance, it was reported that ERK was activated in the dorsal medial preoptic area neurons upon exposure to pups, leading to upregulation of several genes critical for maternal behaviors (Kuroda et al., 2007). Genetic disruption of BDNF, a downstream target of the ERK pathway, caused increased aggression (Lyons et al., 1999). ERK is activated in social behavior circuits during resident-intruder aggression tests (Trainor et al., 2010).
On the other hand, it was reported that mice with the deletion of ERK2 in the CNS (Heffron et al., 2009) did not exhibit prominent anomalies in behavior tests, although they did not perform social behavior tests. They used breeding pairs consisted of male nestin-cre+; Erk2flox/+ mated with female nestin-cre−; Erk2flox/flox mice: all mice in their study were borne and reared by control mothers. Thus, abnormal maternal behaviors of Erk2 CKO mother would not be manifested in their studies.
Implications of ERK signaling for ASD
With the obvious interpretative limitation of translating mouse phenotypes to clinical symptoms in humans, it is still interesting to note that Erk2 deficiency in the CNS caused behavioral phenotypes associated with ASD. ASD is a neuropsychiatric disorder characterized primarily by impairments in social, communicative, and behavioral functioning, although the molecular mechanisms of ASD remain largely unknown. While we found that Erk2 CKO mice exhibited pervasive deficits in social behaviors that are core features of ASD, Erk2 CKO mice also exhibited decreased general interest in novelty, deficits in nest-building and deficits in long-term memory, all of which also have great relevance to ASD. In humans, deletion of chromosome 16p11.2, on which Erk1 is located is associated with autism (Kumar et al., 2008), although Erk1 ablation in mice produced a behavioral excitement profile similar to bipolar disorder (Engel et al., 2009). Erk2 gene is located on human chromosome 22q11.2, deletion of which has also been reported to be associated with autism (Vorstman et al., 2006; Mukaddes and Herguner, 2007). With relevance to human psychiatric disorders, it might be important to dissect the brain functions of ERK1 and ERK2 in animal models to understand the functional link between the ERK cascade and neuropsychiatric disorders.
The EPM and open field test revealed that Erk2 CKO mice exhibited reduced anxiety. There is an interpretative limitation of this finding because the underlying mechanisms for anxiety in human and the anxiety-like behavior of rodents in the EPM would be different. Rodents' anxiety-like behavior in the EPM reflects the natural balance between the exploratory and escaping drives. Thus, entry into the open arm could also be conceptualized as risk-taking behavior. In our experiment, Erk2 CKO mice might not recognize the risk rather than anxiety is reduced in them. In this context, it is interesting to note that ASD patient sometimes have deficits in recognizing the risk.
Oxt signaling and ERK
Our results revealed that Erk2 CKO mice exhibited marked abnormalities in several social behaviors, including abnormal nurturing behavior and high levels of aggression, significantly similar to mice with disrupted Oxt signaling (Ferguson et al., 2000; Takayanagi et al., 2005). Mice deficient for the Oxt gene (Ferguson et al., 2000) or the Oxt receptor gene (Takayanagi et al., 2005) failed to develop social memory or maternal care, respectively. Oxt signaling system plays a crucial role in social interaction and it is implicated in the etiology of ASD (Jin et al., 2007). In humans, the Oxt signaling system regulates a wide range of social behaviors including nursing, social recognition, and pair binding in rodents and love, trust and fear in humans (Keverne and Curley, 2004; Kirsch et al., 2005; Kosfeld et al., 2005). It was also reported that individuals diagnosed with ASD showed significantly lower levels of plasma Oxt compared with age-matched healthy controls (Modahl et al., 1998). Furthermore, the Oxt signaling system is thought to have some relationship to human diseases associated with abnormal social behavior (Lim et al., 2005). Thus, one is prompted to speculate that the disruption of Oxt signal might be related to the abnormal social behaviors in Erk2 CKO mice. However, the plasma Oxt level was not altered in Erk2 CKO mice at 9–11 weeks of age. Thus, it is unlikely that ERK2 signaling regulates Oxt levels, although we cannot exclude the possibility that there are differences in Oxt levels during development or in the postpartum period that could account for some of the behavioral differences. Instead, ERK might mediate Oxt signal to induce enduring changes at the behavioral and physiological levels. Consistently, our data indicated that subcutaneous injection of Oxt increased the phosphorylation levels of ERK1 and ERK2 in control mice. Notably, in Erk2 CKO mice, the increase in ERK1 phosphorylation was larger than that in control mice, suggesting that compensatory regulation of ERK signaling occurs upon Oxt stimulation. It may mean that, in the Oxt-activated state, the compensatory mechanism tries to maintain the total amount of ERK1 and ERK2 phosphorylation. Further analysis is required to understand the role of ERK in the regulation of Oxt signaling.
Central contribution of ERK2 to brain functions
Previous reports have suggested that ERK1 is not critical for associative learning (Selcher et al., 2001), but that ERK2 might be crucially involved in learning and memory (Satoh et al., 2007; Samuels et al., 2008). In this report, we demonstrated that long-term memory was impaired in Erk2 CKO mice, consistent with these reports.
As described above, the phosphorylation level of ERK1 was greater in Erk2 CKO mice than in controls. The administration of the ERK cascade inhibitor SL327 to Erk2 CKO mice did not additionally affect long-term memory although the level of ERK1 phosphorylation was reduced. Thus, the deficiency in long-term memory in Erk2 CKO mice was caused by the loss of ERK2 per se, rather than by a secondary upregulation of ERK1 activity. It was also reported that in mice lacking ERK1, ERK2 did not only compensate for the lack of ERK1, but also exhibited stronger biological activity in some regions (Mazzucchelli et al., 2002; Tronson et al., 2008). Erk1 KO mice exhibited dramatic enhancement of striatum-dependent long-term memory likely due to enhanced activation of ERK2 (Mazzucchelli et al., 2002). These results indicated an unexpected complexity of ERK signaling. Together with these reports, our findings suggest that ERK2, but not ERK1, might provide a central and specific contribution to long-term memory. Furthermore, our study extended these findings by demonstrating pleiotropic and central involvement of ERK2 in many neuronal functions.
One possibility for the mechanism underlying the distinct roles of ERK2 and ERK1 would be resulted from the differences in the N terminus between ERK1 and ERK2, which affect the shuttling rates between the cytoplasm and the nucleus (Marchi et al., 2008). Another possibility is that ERK1 exclusively interacts with MP1, which forms a scaffold for MEK-ERK signaling proteins (Schaeffer et al., 1998). However, we could not exclude the possibility that the deficit was attributable to developmentally related structural changes, for instance in astroglial cells in the cortex.
Conclusions
Our findings demonstrated that ERK2 plays a critical role in regulating not only long-term memory but also social behaviors and anxiety and that ERK2 may be a factor in human neurodevelopmental or psychiatric disorders. Further investigation is required to dissect the roles of ERK1 and ERK2 and determine whether psychiatric disorders are linked to defects in the ERK signaling cascades.
Footnotes
This study was in part performed with support from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Naito Foundation, and Japan Foundation for Aging and Health. We thank Masako Suzuki, Yuko Ogura, and Kiyoko Takamiya for their excellent technical help; Yayoi Ichiki for her technical assistance in the electron microscopic study, Dr. Kouichi Fukuda for his assistance in animal administration, and Drs. Yuki Takayanagi, Tadashi Kimura, and Kenichi Furuya for advice on the study of maternal behaviors. The nestin-cre transgenic mice were a kind gift from Dr. R. Kageyama (University of Kyoto).
References
- Ailing F, Fan L, Li S, Manji S. Role of extracellular signal-regulated kinase signal transduction pathway in anxiety. J Psychiatr Res. 2008;43:55–63. doi: 10.1016/j.jpsychires.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiolas A, Gessa GL. Central functions of oxytocin. Neurosci Biobehav Rev. 1991;15:217–231. doi: 10.1016/s0149-7634(05)80002-8. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Calverley RK, Jones DG. Contributions of dendritic spines and perforated synapses to synaptic plasticity. Brain Res Brain Res Rev. 1990;15:215–249. doi: 10.1016/0165-0173(90)90002-6. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev. 2004;10:248–258. doi: 10.1002/mrdd.20039. [DOI] [PubMed] [Google Scholar]
- Engel SR, Creson TK, Hao Y, Shen Y, Maeng S, Nekrasova T, Landreth GE, Manji HK, Chen G. The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry. 2009;14:448–461. doi: 10.1038/sj.mp.4002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet. 2000;25:284–288. doi: 10.1038/77040. [DOI] [PubMed] [Google Scholar]
- Goldin M, Segal M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci. 2003;17:2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- Hatton GI. Emerging concepts of structure-function dynamics in adult brain: the hypothalamo-neurohypophysial system. Prog Neurobiol. 1990;34:437–504. doi: 10.1016/0301-0082(90)90017-b. [DOI] [PubMed] [Google Scholar]
- Heffron DS, Landreth GE, Samuels IS, Mandell JW. Brain-specific deletion of extracellular signal-regulated kinase 2 mitogen-activated protein kinase leads to aberrant cortical collagen deposition. Am J Pathol. 2009;175:2586–2599. doi: 10.2353/ajpath.2009.090130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature. 2007;446:41–45. doi: 10.1038/nature05526. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Curley JP. Vasopressin, oxytocin and social behaviour. Curr Opin Neurobiol. 2004;14:777–783. doi: 10.1016/j.conb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, Gruppe H, Mattay VS, Gallhofer B, Meyer-Lindenberg A. Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neurosci. 2005;25:11489–11493. doi: 10.1523/JNEUROSCI.3984-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E. Oxytocin increases trust in humans. Nature. 2005;435:673–676. doi: 10.1038/nature03701. [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Kuroda KO, Meaney MJ, Uetani N, Fortin Y, Ponton A, Kato T. ERK-FosB signaling in dorsal MPOA neurons plays a major role in the initiation of parental behavior in mice. Mol Cell Neurosci. 2007;36:121–131. doi: 10.1016/j.mcn.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28:511–527. doi: 10.1128/MCB.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, Herrup K, Stevens KE, Maccaferri G, McBain CJ, Sussman DJ, Wynshaw-Boris A. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905. doi: 10.1016/s0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- Lim MM, Bielsky IF, Young LJ. Neuropeptides and the social brain: potential rodent models of autism. Int J Dev Neurosci. 2005;23:235–243. doi: 10.1016/j.ijdevneu.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi M, D'Antoni A, Formentini I, Parra R, Brambilla R, Ratto GM, Costa M. The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One. 2008;3:e3873. doi: 10.1371/journal.pone.0003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Modahl C, Green L, Fein D, Morris M, Waterhouse L, Feinstein C, Levin H. Plasma oxytocin levels in autistic children. Biol Psychiatry. 1998;43:270–277. doi: 10.1016/s0006-3223(97)00439-3. [DOI] [PubMed] [Google Scholar]
- Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:205–220. doi: 10.1093/hmg/ddi016. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Mukaddes NM, Herguner S. Autistic disorder and 22q11.2 duplication. World J Biol Psychiatry. 2007;8:127–130. doi: 10.1080/15622970601026701. [DOI] [PubMed] [Google Scholar]
- Murcia CL, Gulden F, Herrup K. A question of balance: a proposal for new mouse models of autism. Int J Dev Neurosci. 2005;23:265–275. doi: 10.1016/j.ijdevneu.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Nakata T, Yorifuji H. Morphological evidence of the inhibitory effect of taxol on the fast axonal transport. Neurosci Res. 1999;35:113–122. doi: 10.1016/s0168-0102(99)00074-7. [DOI] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Leng G, Douglas AJ. The magnocellular oxytocin system, the fount of maternity: adaptations in pregnancy. Front Neuroendocrinol. 2003;24:27–61. doi: 10.1016/s0091-3022(02)00104-8. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Kobayashi Y, Takeuchi A, Pagès G, Pouysségur J, Kazama T. Deletion of ERK1 and ERK2 in the CNS causes cortical abnormalities and neonatal lethality: Erk1 deficiency enhances the impairment of neurogenesis in Erk2-deficient mice. J Neurosci. 2011;31:1149–1155. doi: 10.1523/JNEUROSCI.2243-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–637. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, Yanagisawa T, Kimura T, Matzuk MM, Young LJ, Nishimori K. Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci U S A. 2005;102:16096–16101. doi: 10.1073/pnas.0505312102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Trainor BC, Crean KK, Fry WH, Sweeney C. Activation of extracellular signal-regulated kinases in social behavior circuits during resident-intruder aggression tests. Neuroscience. 2010;165:325–336. doi: 10.1016/j.neuroscience.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Fischer A, Sananbenesi F, Pagès G, Pouysségur J, Radulovic J. Regulatory mechanisms of fear extinction and depression-like behavior. Neuropsychopharmacology. 2008;33:1570–1583. doi: 10.1038/sj.npp.1301550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernay B, Koch M, Vaccarino F, Briscoe J, Simeone A, Kageyama R, Ang SL. Otx2 regulates subtype specification and neurogenesis in the midbrain. J Neurosci. 2005;25:4856–4867. doi: 10.1523/JNEUROSCI.5158-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Morcus ME, Duijff SN, Klaassen PW, Heineman-de Boer JA, Beemer FA, Swaab H, Kahn RS, van Engeland H. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry. 2006;45:1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]