

Abstract
Accumulation of iron occurs in the CNS in several neurodegenerative diseases. Iron is essential for life but also has the ability to generate toxic free radicals if not properly handled. Iron homeostasis at the cellular level is therefore important to maintain proper cellular function, and its dysregulation can contribute to neurodegenerative diseases. Iron export, a key mechanism to maintain proper levels in cells, occurs via ferroportin, a ubiquitously expressed transmembrane protein that partners with a ferroxidase. A membrane-bound form of the ferroxidase ceruloplasmin is expressed by astrocytes in the CNS and regulates iron efflux. We now show that oligodendrocytes use another ferroxidase, called hephaestin, which was first identified in enterocytes in the gut. Mice with mutations in the hephaestin gene (sex-linked anemia mice) show iron accumulation in oligodendrocytes in the gray matter, but not in the white matter, and exhibit motor deficits. This was accompanied by a marked reduction in the levels of the paranodal proteins contactin-associated protein 1 (Caspr) and reticulon-4 (Nogo A). We show that the sparing of iron accumulation in white matter oligodendrocytes in sex-linked anemia mice is due to compensatory upregulation of ceruloplasmin in these cells. This was further confirmed in ceruloplasmin/hephaestin double-mutant mice, which show iron accumulation in both gray and white matter oligodendrocytes. These data indicate that gray and white matter oligodendrocytes can use different iron efflux mechanisms to maintain iron homeostasis. Dysregulation of such efflux mechanisms leads to iron accumulation in the CNS.
Introduction
Iron is an essential trace metal required for many biological functions including oxygen transport, DNA synthesis, and mitochondrial oxidation. Iron can cycle between the ferrous (Fe2+) and ferric (Fe3+) states. This ability to accept and donate electrons allows it to serve as an important cofactor for enzymes. However, ferrous iron can generate highly reactive free radicals, which can cause oxidative damage (Berg et al., 2004). Excessive iron in the CNS has been implicated in several neurodegenerative conditions (LeVine, 1997; Bakshi et al., 2001; Carrí et al., 2003; Zecca et al., 2004; Molina-Holgado et al., 2007).
In the CNS, iron levels are highest in myelin and oligodendrocytes (Connor and Menzies, 1996; Erb et al., 1996). In these cells, iron is needed for biosynthetic enzymes involved in lipid and cholesterol synthesis, as well as enzymes required for the increased metabolic demands of myelination (Hill and Switzer, 1984; LeVine and Macklin, 1990). Oligodendrocytes synthesize high levels of ferritin (Connor et al., 1994) and therefore possess a high buffering capacity for iron. Although this buffering capacity offers some protection, excessive iron load can be expected to disrupt some aspects of oligodendrocyte function. To prevent overload, iron levels in all cells, including oligodendrocytes, are tightly regulated.
One mechanism by which cells regulate intracellular iron levels is via iron efflux through the ubiquitously expressed iron exporter ferroportin (Fpn) (Donovan et al., 2000). Ferroportin partners with ferroxidases to efflux iron (Jeong and David, 2003). The multi-copper ferroxidase ceruloplasmin (Cp), a serum protein secreted by the liver, has been shown to mediate iron efflux from hepatocytes and macrophages (Osaki and Johnson, 1969; Harris et al., 1999). We have shown previously that an alternatively spliced, glycosylphosphatidylinositol (GPI)-anchored form of Cp is expressed by astrocytes (Patel and David, 1997; Patel et al., 2000) and is required for iron efflux from these cells (Jeong and David, 2003). This suggests that ferrous iron effluxed via Fpn requires oxidation by a ferroxidase such as Cp for proper iron release. Another copper-dependent ferroxidase, hephaestin (Heph), was identified in duodenal enterocytes (Vulpe et al., 1999) and was also shown to play a role in iron efflux. In the sex-linked anemia (sla) mouse, a partial deletion of the heph gene results in the expression of a truncated Heph protein that exhibits only partial ferroxidase activity (Vulpe et al., 1999; Chen et al., 2004).
Heph and Cp are expressed in retinal Muller glia and in retinal pigment epithelial cells, and are thought to mediate iron efflux from these cells (Hahn et al., 2004). To date, the mechanisms of iron efflux from nonastrocytic cells in the CNS and the possible role of Heph in this process are not known. We therefore examined the expression and function of Heph in the mammalian CNS. We provide in vitro and in vivo evidence that Heph is expressed by mature oligodendrocytes and plays a role in regulating iron efflux from these cells. In addition, our data suggest that gray and white matter oligodendrocytes can use different ferroxidases to regulate iron homeostasis.
Materials and Methods
Animals.
Sla mice were purchased from Jackson Laboratories, and C57BL/6 mice and Sprague Dawley rats were purchased from Charles River. Animals of either sex were used for all experiments. All procedures were approved by the McGill University Animal Care committee and followed the guidelines of the Canadian Council on Animal Care.
Purification of oligodendrocyte precursor cells, astrocytes, and microglia from neonatal rat cerebral cortex.
Astrocytes were purified from neonatal rat cerebral cortex as described previously (Jeong and David, 2003). Microglia and oligodendrocyte precursor cells (OPCs) were purified from neonatal rat cortex as described previously (Chen et al., 2007). Briefly, to obtain microglia, mixed glial cultures were prepared and plated in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS, Invitrogen). After 10–14 d, microglia were shaken off for 3 h and cultured in DMEM/F12 (Invitrogen) containing 10% FBS. Mixed glia cultures were shaken again overnight to detach oligodendrocytes. The detached cells were plated on uncoated Petri dishes for 3 h to remove residual microglia. The nonadherent OPCs were then plated on poly-l-lysine-coated tissue culture flasks in oligodendrocyte-specific medium (DMEM/F12 containing 5 μg/ml insulin, 50 μg/ml human transferrin, 16 μg/ml putrescine, 30 nm triiodothyronine 10 mm HEPES, 0.1% BSA, 20 nm hydrocortisone, 20 nm progesterone, 10 nm biotin, 30 nm selenium) supplemented with 2.5 ng/ml platelet-derived growth factor (PDGF)-AA and 2.5 ng/ml basic fibroblast growth factor (bFGF) (all from Sigma-Aldrich). For differentiation of OPCs, PDGF-AA and bFGF were removed.
Purification of OPCs from neonatal mouse cerebral cortex.
For work on sla and wild-type mice, neural progenitor cells were isolated from neonatal mouse cerebral cortex and differentiated into OPCs using neurosphere cultures, as previously described (Chen et al., 2007), with the following modifications: neonatal mouse cerebral cortex was triturated with a fire-polished pipette, passed through a 40 μm cell strainer, and plated onto 6-well plates (1 brain/6-well plate) in neurosphere growth medium (NPM) consisting of DMEM/F12 with B27 supplement (Invitrogen) containing 20 ng/ml epidermal growth factor and 20 ng/ml bFGF. To generate oligospheres, the neurosphere medium was gradually changed to oligosphere medium (NPM/conditioned media of B104 cells, 7:3) by replacing one-fourth of the former medium with the latter every other day. After 7 d in culture, oligospheres were mechanically dissociated and resuspended in oligosphere medium. After 2–4 d, oligospheres were again dissociated and replated. For proliferation of OPCs, the cell suspension was plated onto poly-l-lysine-coated 6-well plates in oligodendrocyte-specific medium supplemented with 5.0 ng/ml PDGF-AA and 5.0 ng/ml bFGF. For differentiation of OPCs into oligodendrocytes, PDGF-AA and bFGF were removed.
Iron efflux study.
Oligodendrocytes that were differentiated from OPCs purified from sla and wild-type mice were plated 2 d before use in a 6-well plate at a density of 1 × 106 cells/well. Cells were incubated in serum- and transferrin-free oligodendrocyte medium (SFM) for 1 h to remove any transferrin-bound iron. Radiolabeled iron (55FeCl3; PerkinElmer Life Science) was mixed with nonlabeled FeCl3 (total of 40 μm) and l-ascorbate (Sigma-Aldrich) was added (molar ratio of FeCl3 to l-ascorbate was 1:44). Cells were then incubated with SFM containing radiolabeled iron-ascorbate (1 μCi/well) for 6 h. After iron loading, cells were washed and incubated with fresh SFM without radiolabeled iron. At each time point (0, 12, and 24 h), cells were washed three times with PBS, detached, pelleted, and treated with Pronase (Calbiochem) for 1 h at 4°C to remove membrane-bound iron. Cells were lysed in 0.1N NaOH, and radioactivity was measured using a liquid scintillation counter. Sister cultures were treated in the same way without radioactive iron, and viable cell numbers were estimated by trypan blue (Sigma) exclusion. The radioactivity taken up by the cells was normalized to a value per 106 cells. The radioactivity within cells at time point 0 h was set to 100%. Measurements at each time point were done in triplicate and repeated in three separate experiments. Results are shown as mean ± SEM. Two-way ANOVA was used to determine statistical significance.
Immunofluorescence labeling.
Immunostaining was performed on cultures of neonatal rat oligodendrocytes grown on poly-l-lysine-coated 12 mm round glass coverslips. Cells were stained for 30 min at room temperature with mouse anti-A2B5 (1:100; R&D Systems), mouse anti-O4 (1:100; Millipore Bioscience Research Reagents) or mouse anti-Gal-C (1:100; Millipore Bioscience Research Reagents), and rabbit anti-Heph (1:400; Chris Vulpe, University of California, Berkeley, Berkeley, CA) or rabbit anti-Fpn (1:200; Alpha Diagnostics). Primary antibodies were visualized with rhodamine-conjugated goat anti-mouse IgG (1:400; Jackson ImmunoResearch) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:400; Invitrogen) for 30 min at room temperature. Cells were fixed in acetic acid/ethanol [5:95 (v/v)] at −20°C for 10 min and coverslipped with mounting medium containing 100 ng/ml 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories) to visualize cell nuclei and viewed with a Zeiss Axioskop 2 Plus microscope.
For immunofluorescence labeling of tissue sections, sla and wild-type mice were perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Cryostat sections (14 μm) were incubated with different combinations of the following primary antibodies: mouse anti-CC1 (1:200; Millipore Bioscience Research Reagents); rabbit anti-Fpn (1:200; Alpha Diagnostics); rabbit anti-Heph (1:400; Chris Vulpe, University of California, Berkeley, Berkeley, CA); rabbit anti-ferritin (1:200; DakoCytomation); rabbit anti-Caspr (1:800; Abcam); rabbit anti-Nogo A (1:100; Santa Cruz Biotechnology), and secondary antibodies [rhodamine-conjugated goat anti-mouse IgG (1:400, Jackson ImmunoResearch) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:400; Invitrogen)] as described previously (Jeong and David, 2006; Rathore et al., 2008). Sections were coverslipped with DAPI-containing medium and viewed with a Zeiss Axioskop 2 Plus microscope.
Quantification of immunofluorescence labeling.
Images were captured with a Retiga 1300 C digital camera (QImaging), and all sections were analyzed with the BioQuant Nova Prime image analysis system using a Zeiss Axioskop 2 microscope. Thresholds for the detection of contactin-associated protein 1 (Caspr) and reticulon-4 (NogoA) immunoreactivity were first established and then applied throughout. The pixel density of Caspr immunoreactivity was measured within a fixed area in the ventral horn and the ventral white matter in cross sections of cervical, thoracic, and lumbar spinal cord (three sections per animal, three mice; n = 3). The pixel density of Nogo A immunoreactivity was determined per CC1-positive cell in the gray matter of the lumbar spinal cord (30 cells per section, three sections per animal, three mice; n = 3). The area of each cell was measured and the pixel density per square micrometer was calculated. The data obtained for Caspr and Nogo A immunoreactivity were analyzed for statistical significance by two-way repeated-measures ANOVA, and Student's t test, respectively. When appropriate, post hoc Tukey tests were done for comparisons between groups. The data are plotted as means ± SEM.
Perl's histochemistry.
Wild-type and sla mice were perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Modified enhanced Perl's histochemistry was performed on 14-μm-thick cryostat sections of the spinal cord to detect iron accumulation, as described previously (Smith et al., 1997). Briefly, sections were incubated for 10 min with 4% potassium ferrocyanide only, followed by a 50 min incubation with 4% HCl and 4% potassium ferrocyanide (1:1). The reaction product was enhanced with diaminobenzidine (DAB). Sections were counterstained with methyl green and mounted with Entellan (all from Sigma-Aldrich).
Perfusion-Perl's method for electron microscopy.
Wild-type and sla mice were perfused with the perfusion-Perl's method, and spinal cords were prepared for electron microscopy according to a previously described protocol (Meguro et al., 2005). In brief, mice were first perfused with 0.1 m phosphate buffer, pH 7.4, containing heparin (10 U/ml), then with 4% paraformaldehyde and 1.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, followed by 4% paraformaldehyde, 1% glutaraldehyde, and 1% potassium ferrocyanide in distilled H2O, pH 1.0. Spinal cords were dissected out and postfixed overnight in 4% paraformaldehyde and 1.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. Vibratome sections (100 μm) were obtained and washed twice with PBS and once with PBS containing 0.3% H2O2 and 0.1 m NaN3. Sections were incubated for 10 min in 0.025% DAB and 0.005% H2O2 in PBS. Tissue sections were then postfixed in 2% osmium tetroxide for 1 h and processed for embedding in Epon. Thin sections (1 nm) picked up on copper grids were stained with lead citrate and viewed with a Philips CM 10 electron microscope.
Western blotting.
Total protein was extracted from spinal cord tissue, separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Bio-Rad). The membrane was incubated overnight with rabbit anti-Heph (1:600; Chris Vulpe, University of California, Berkeley, Berkeley, CA), rabbit anti-Fpn (1:1000; Alpha Diagnostics), rabbit anti-Cp (1:800; DakoCytomation) or rabbit anti-β-actin (1:400; Sigma-Aldrich). Blots were then incubated with peroxidase-conjugated anti-rabbit IgG (1:500,000; Jackson ImmunoResearch) and developed with enhanced chemiluminescence (PerkinElmer Life Sciences).
RT-PCR.
RNA was purified from whole spinal cord or cultured oligodendrocytes and Caco2 cells using the RNeasy Lipid Tissue Mini Kit (Qiagen). RT-PCR was performed using the Omniscript RT Kit (Qiagen) and the following primers: Cp_forward: 5′-TGG GAC TAT GCT TCT GGC AGT GAA-3′; Cp_reverse: 5′-TTG GCA CGC AGA ACG TAC AAA TAC-3′; divalent metal transporter 1 (DMT1)_forward: 5′-ACA AAT ATG GCT TGC GGA AGC TGG-3′; DMT1_reverse: 5′-ACA TGT TGT GCG GCA TGA TCA CAG-3′; Fpn_forward: 5′-TCA GGA CTG GCT CAG CTT TCT TGT-3′; Fpn_reverse: 5′-CAG CAA TGA CTC CTG CAA ACA GCA-3′; Heph_forward: 5′-ATG GAC CGG GAA TTT GCC TTG TTG-3′; Heph_reverse: 5′-TCA TGC CCA GCA TCT TCA CAT TGC-3′; T cell immunoglobulin and mucin domain 2 (TIM-2)_forward: 5′-ATC CCT CCA CAG AAG CCA CAG AAA-3′; TIM-2_reverse: 5′-ACT GAG GCG GTG CTG ATA CAA GAA-3′; transferrin receptor 1 (TFR1)_forward: 5′-AGC AGC ATC TGC TAA TGA GAC CCA-3′; and TFR1_reverse: 5′-TGC AAG CTT TGT CTT CCC AAC AAC-3′.
In situ hybridization.
Sla and wild-type mice were perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Cryostat sections (14 μm) were obtained, and in situ hybridization was performed with digoxygenin (DIG)-labeled locked nucleic acids (LNAs) (Exiqon) using a modified version of a protocol that has been described previously (Obernosterer et al., 2007). Briefly, tissue sections were fixed in 4% PFA for 10 min at room temperature, then washed three times for 3 min in PBS and incubated 10 min in acetylation solution (1.3% Triethanolamine, 0.25% acetic anhydride, 0.17% concentrated HCl in DEPC-treated dH2O) to reduce background binding. Sections were then washed in PBS for 5 min and incubated for 5 min with proteinase K (5 μg/ml in PBS) to increase permeability and hybridization efficiency. After three more washes for 3 min in PBS, prehybridization was performed in hybridization buffer (50% formamide, 5× SSC, 5× Denhardt's solution, 200 μg/ml yeast RNA, 0.4 g of Roche blocking reagents in DEPC-treated dH2O) for 5 h at room temperature. Sections were then incubated with 1 pm of the Cp LNA DIG-labeled probe in denaturizing hybridization buffer (hybridization buffer containing 0.25% CHAPS) overnight at 52°C. Slides were soaked in prewarmed 60°C 5× SSC, incubated in 0.2× SSC at 60°C for 1 h, and washed in B1 solution (0.1 m Tris, pH 7.5, 0.15 m NaCl) at room temperature for 10 min. After an incubation in blocking solution (1% FBS and 0.05% Tween20 in B1 solution) for 1 h at room temperature, slides were incubated with a sheep anti-DIG fluorescein-coupled antibody (1:200 in blocking solution; Roche) overnight at 4°C. Slides were washed three times for 5 min in PBST, mounted with DAPI, and viewed with a Zeiss Axioskop 2 Plus microscope. Up to the hybridization step, everything was performed with DEPC-treated dH2O under RNase-free conditions.
Rotarod assay.
Motor performance was assessed using a rotarod test (Steele et al., 1998). Mice were made to walk on the rotating rod for a maximum duration of 3 min at a constant speed of 16 rpm. Each mouse was given three trials with an intertrial interval of 5 min. Six animals per group were tested. Results are shown as a mean of the three trials ± SEM. Two-way ANOVA was used to determine statistical significance.
Results
Hephaestin and ferroportin are expressed in mature oligodendrocytes
We first assessed the expression of Heph in primary cultures of the neonatal rat CNS. Mixed cerebellar cultures exhibit Heph expression in oligodendrocytes and microglia but not in astrocytes or neurons (Fig. 1). Furthermore, quantification of Heph staining in glial cell cultures from the neonatal cerebral cortex indicates that 100% of the oligodendrocytes express Heph, whereas none of the astrocytes express this ferroxidase.
Figure 1.
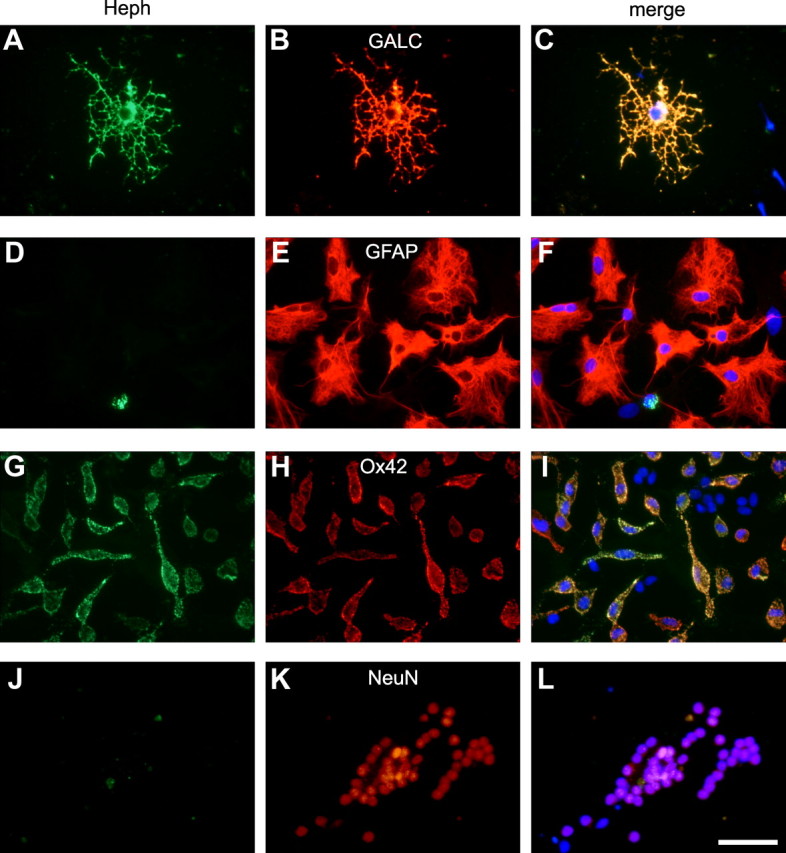
A–L, Expression of Heph in mixed cerebellar cultures. Double-immunofluorescence labeling with anti-Heph (A, D, G, J) and antibodies against the oligodendrocyte marker Gal-C (B), astrocyte maker GFAP (E), microglial marker OX42 (H), or the neuronal marker NeuN (K). Note that Heph is expressed in oligodendrocytes and microglia but not in astrocytes or neurons. C, F, I, L, Merged images showing the nuclear DAPI staining. Scale bar, 50 μm.
Oligodendrocytes in vitro go through a series of well defined maturational stages that are delineated by the expression of various cell surface antigens. One series of maturational changes involves the temporal shift in expression of A2B5 to O4 and eventually to galactocerebroside (Gal-C) antigens (Gard and Pfeiffer, 1993). We therefore assessed the stage of oligodendrocyte maturation in which Heph expression first occurs. Our immunofluorescence analysis indicates that Heph is expressed only by the more mature Gal-C-expressing oligodendrocytes (100 ± 0.0%) but not by A2B5-expressing early OPCs (1 ± 0.004%) or by O4-expressing late OPCs (2 ± 0.048%) (Fig. 2A–I). As Heph would likely partner with an iron transporter to efflux iron, we assessed the expression of Fpn at different stages of oligodendrocyte maturation. Double-immunofluorescence labeling shows that Fpn, like Heph, is expressed by the more mature Gal-C-expressing oligodendrocytes (99 ± 0.009%) but not by A2B5+ (0%) or O4+ OPCs (0%) (Fig. 2J–R).
Figure 2.
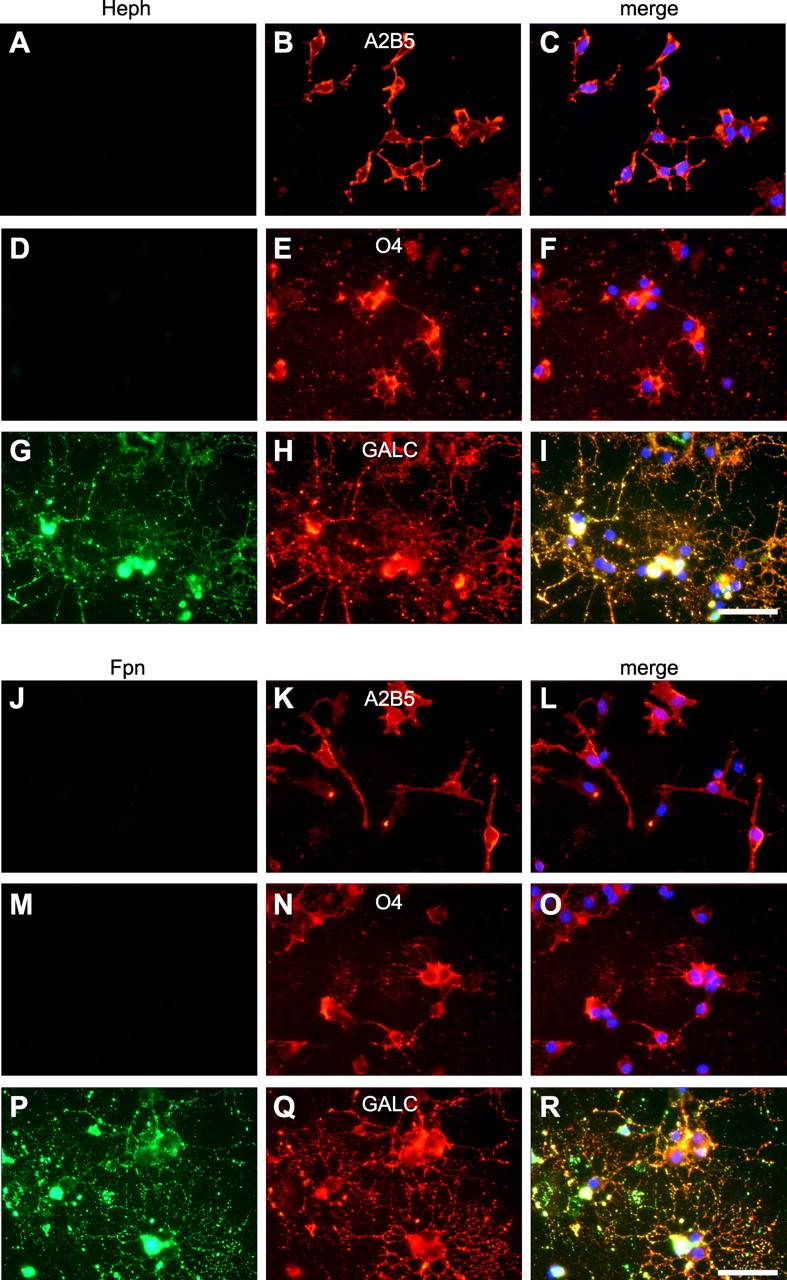
Heph and Fpn are only expressed in mature oligodendrocytes. Oligodendrocyte precursor cells were purified from neonatal rat cerebral cortex and allowed to mature in vitro, and double-immunofluorescence labeling was carried out. A–R, Staining with anti-Heph (A, D, G) or with Fpn (J, M, P) and the early OPC marker anti-A2B5 (B, K), the late OPC marker anti-O4 (E, N), or the marker for more mature oligodendrocytes anti-Gal-C (H, Q) shows that Heph and Fpn are only expressed in mature Gal-C+ oligodendrocytes. C, F, I, L, O, R, Merged figures including DAPI staining of the nuclei. Scale bars, 50 μm.
Hephaestin and ferroportin are expressed in the adult mouse spinal cord
To confirm the expression of Heph and Fpn in vivo, we performed double-immunofluorescence labeling on spinal cord tissue sections of adult mice using antibodies against Heph or Fpn combined with an antibody against the oligodendrocyte-specific marker CC1. Heph is expressed in the majority of CC1+ oligodendrocytes in the spinal cord (Fig. 3A–C). About 80% of oligodendrocytes in the gray and white matter express Heph (white matter = 83.42 ± 4.44%; gray matter = 78.98 ± 3.1). Furthermore, almost 90% of the Heph+ cells are CC1+ oligodendrocytes (white matter = 88.6 ± 1.6%; gray matter = 89.77 ± 3.25%). Quantification of the intensity of Heph immunostaining in CC1+ oligodendrocytes as an index of expression per cell showed higher Heph expression in white matter oligodendrocytes (71.47 ± 3.89 pixel density/μm2) compared with gray matter oligodendrocytes (36.95 ± 7.23 pixel density/μm2), suggesting that white matter oligodendrocytes have a greater requirement for ferroxidase activity than those in the gray matter. The efflux iron transporter Fpn, a ubiquitously expressed molecule, is expressed by both CC1+ oligodendrocytes as well as by CC1− cells (Fig. 3D–F). We have evidence that Fpn is also expressed in astrocytes and neurons (data not shown). As mentioned previously, sla mutant mice express a truncated, partially active form of Heph in non-CNS tissues (Vulpe et al., 1999; Chen et al., 2004). We therefore compared the expression of Heph and Fpn in the CNS of sla and wild-type mice by Western blot analysis. Spinal cord tissue from sla mice contains low levels of the truncated form of Heph relative to the full-length form in wild-type mice (Fig. 3G), a decrease likely due to the rapid degradation of the truncated protein in vivo. In contrast, expression of the iron exporter Fpn is similar in the spinal cords of wild-type and sla mice (Fig. 3G).
Figure 3.

In vivo expression of Heph and Fpn in the spinal cord and the role of Heph in iron efflux. A–F, Double-immunofluorescence labeling shows expression of Heph (A) and Fpn (D) in CC1-labeled oligodendrocytes (B, E) in a cross section of the adult mouse spinal cord. Arrows indicate cells that are double labeled for Heph/CC1 or Fpn/CC1. Arrowheads indicate CC1-negative cells that are Fpn+. C, F, Merged images that include the nuclear DAPI labeling. Note also that Fpn is ubiquitously expressed. Scale bars: F, 100 μm; F, inset, 20 μm. G, Western blot shows reduced expression of Heph in the spinal cord of sla mice, which carries a mutation in the heph gene compared with wild-type (WT) mice. Reprobing the blot for Fpn demonstrates equal Fpn levels in sla and WT mice. β-actin was used as a protein loading control. H, An in vitro iron efflux assay using radiolabeled iron (55Fe) was performed with oligodendrocytes obtained from neonatal sla and wild-type mice. Cells were loaded with iron-ascorbate containing 55Fe for 6 h, washed, and cultured in nonradioactive medium. The amount of radiolabeled iron in the cell pellets was measured at 0, 12, and 24 h. Marked impairment of iron efflux is seen in cultures of sla oligodendrocytes compared with wild-type cells. Measurements at each time point were done in triplicate and repeated in three separate experiments. Results are shown as mean ± SEM. *p ≤ 0.05, two-way ANOVA.
Hephaestin mediates iron efflux from oligodendrocytes
Using an in vitro iron efflux assay, we next assessed the involvement of Heph in iron efflux from oligodendrocytes. Oligodendrocyte precursor cells were first purified from the brains of neonatal sla and wild-type mice and allowed to mature in vitro. After loading with radiolabeled iron, mature oligodendrocyte cultures were incubated with fresh medium and assayed for iron retention at several time points. In oligodendrocytes from wild-type mice, ∼60% of the radiolabeled iron is effluxed after 24 h, whereas iron efflux from oligodendrocytes from sla mice is severely impaired (Fig. 3H). The iron uptake was not significantly different in wild-type and sla oligodendrocytes, similar to the finding reported for astrocytes lacking Cp (Jeong and David, 2003). These data provide direct evidence that Heph is required for iron efflux from oligodendrocytes and demonstrate that the low amount of functionally impaired Heph present in the sla mice is insufficient to mediate iron efflux.
Iron accumulates in oligodendrocytes in sla mice
Since Heph was found to be involved in iron export from oligodendrocytes in culture, we reasoned that iron would accumulate in oligodendrocytes of sla mice in vivo. Iron accumulation in the CNS of sla mice was assessed by enhanced Perl's histochemistry on tissue sections of mice aged 3, 6, and 10 months (Fig. 4A–H). Iron accumulation in the spinal cord of sla mice is seen as early as 3 months of age (Fig. 4C,F). Interestingly, cells labeled for iron are located primarily in the gray matter (Fig. 4B) and display the process-bearing morphology characteristic of oligodendrocytes (Fig. 4H). Iron accumulation in these cells increases at 6 and 10 months of age in sla mice but not in wild-type controls (Fig. 4D–H). To confirm that these iron-loaded cells are indeed oligodendrocytes, we performed double-immunofluorescence labeling for the iron-binding protein ferritin (a sensitive surrogate marker for increased cellular iron load) and the oligodendrocyte marker CC1. These studies clearly demonstrate that CC1+ oligodendrocytes in the spinal cord gray matter of sla mice, but not wild-type mice, express high levels of ferritin (Fig. 4I–N). The sla mice also show enhanced iron accumulation relative to wild-type mice in other CNS gray matter regions including the brainstem and the deep cerebellar nuclei (data not shown). We analyzed the iron deposits within these cells further, using the perfusion-Perl's enhancement technique and electron microscopy. This ultrastructural analysis revealed iron deposits in the cytosol of oligodendrocytes in the gray matter of the spinal cord in 10-month-old sla mice (Fig. 5C,D). Such deposits are not seen in oligodendrocytes of wild-type mice (Fig. 5A,B). The iron accumulation in sla mice is unlikely to be due to increased iron influx, as the mRNA expression of the iron importers TFR1 and DMT1 is not increased (Fig. 5E). Furthermore, the expression of the ferritin receptor TIM-2, which is thought to mediate influx of ferritin-bound iron into oligodendrocytes (Todorich et al., 2008), is decreased in sla mice, possibly in an effort to minimize iron overload in oligodendrocytes (Fig. 5E).
Figure 4.
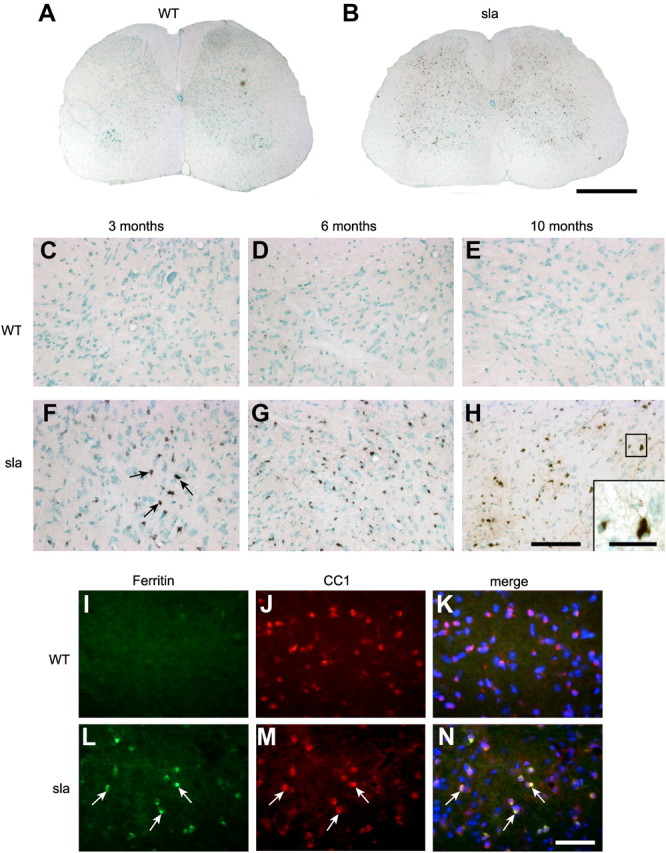
Iron accumulates in gray matter oligodendrocytes in the spinal cord of sla mice. A, Perl's histochemistry shows no labeling for iron in 6-month-old wild-type mouse spinal cord. B, In contrast, iron accumulation is abundant in the spinal cord gray matter in 6-month-old sla mouse. C–E, Higher-magnification images showing Perl's histochemistry of the ventral gray matter of the spinal cord show that there is no iron accumulation in the gray matter of wild-type mice between 3 and 10 months of age. F–H, In contrast, increasing iron accumulation is seen in sla mice with age, at 3 months (F), 6 months (G), and 10 months (H) of age. Arrows in F indicate iron-loaded cells. H, inset, An iron-containing cell that has a long processes resembling oligodendrocytes. I–N, Further confirmation that iron accumulation in the spinal cord gray matter of sla mice occurs in oligodendrocytes was obtained by double-immunofluorescence labeling with anti-ferritin (I, L) as a surrogate marker for iron, and anti-CC1 (J, M) for oligodendrocytes. Merged images including DAPI-stained nuclei are shown in K and N. L–N, Note the ferritin/CC1 double-labeled cells in the sla spinal cord, indicated by arrows. Scale bars: B, 500 μm; H, 100 μm; H, inset, 30 μm); N, 50 μm.
Figure 5.
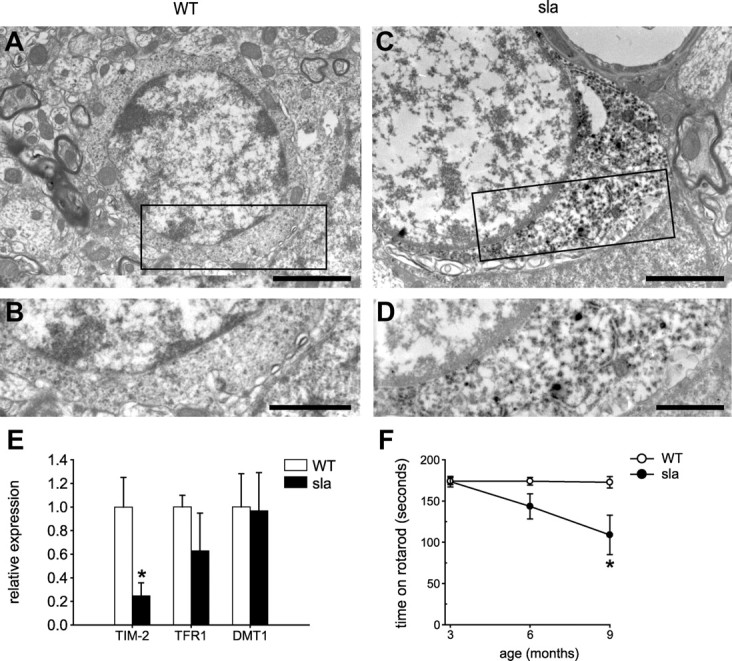
Ultrastructural evidence of iron accumulation in oligodendrocytes in sla mice, the expression of iron influx proteins and effects on motor coordination. A, Electron micrograph of oligodendrocyte from wild-type mouse shows no evidence of iron deposits in tissue prepared by perfusion Perl's histochemistry. B, Higher magnification of the area outlined in A. C, In contrast, electron micrograph of oligodendrocyte in a sla mouse shows clear evidence of iron deposits detected by perfusion Perl's histochemistry. Note the dark DAB reaction product indicating iron deposits. D, Higher magnification of the area outlined in C showing the dark deposits of iron in the cytosol. Scale bars: A, B, 2 μm; C, D, 1 μm. E, RT-PCR data showing mRNA expression of TFR1 and DMT1 are not significantly different in the sla mouse spinal cord compared with the wild-type mouse spinal cord, whereas TIM-2 mRNA levels are decreased in the sla spinal cord. Results are shown as mean ± SEM. *p ≤ 0.05, Student's t test. F, Rotarod analysis shows that motor function in sla mice is significantly impaired at 9 months of age compared with age-matched wild-type mice. The results are shown as the mean ± SEM; n = 6 for each group. *p ≤ 0.05, two-way ANOVA.
Sla mice show deficits in motor performance with age
Motor control of sla and wild-type mice was tested at 3, 6, and 9 months of age using the rotarod test. Whereas wild-type mice remain on the rotarod for ∼180 s (maximum test duration), sla mice fall off after ∼100 s at 9 months of age, suggesting that motor coordination in sla mice is impaired with age (Fig. 5F).
Expression of the paranodal proteins Caspr and Nogo A is reduced in the spinal cord of sla mice
Iron efflux from glia may be an important route for iron availability to neurons. If this were the case, impaired iron efflux in gray matter oligodendrocytes in sla mice might lead to neuronal, axonal, or synaptic abnormalities due to a lack of iron. We therefore assessed neuronal numbers in spinal cord tissue sections stained with cresyl violet. In addition, we assessed the expression of the synaptic membrane protein, synapsin, by Western blotting. No differences between wild-type and sla mice were observed in either of these neuronal indicators. Furthermore, despite the marked iron accumulation in oligodendrocytes, there is no loss of CC1+ oligodendrocytes in the gray matter of sla mice, indicating that oligodendrocytes have a high iron-buffering capacity. In addition, the overall extent of myelination as determined by Luxol fast blue staining appeared unchanged in sla mice (data not shown). To further confirm this, we also examined whether myelin thickness is affected in the sla mice by quantifying the g-ratio from electron micrographs of the spinal cord gray and white matter. The g-ratio is obtained by dividing the axon diameter with the fiber diameter (axon diameter plus myelin). This analysis revealed no differences between sla and wild-type mice (mean g-ratio in the gray matter: wild-type mice = 0.77 ± 0.01; sla mice = 0.75 ± 0.02; mean g-ratio in the white matter: wild-type mice = 0.74 ± 0.01; sla mice = 0.73 ± 0.02). We therefore assessed sla mice for subtle disruptions of myelination, such as altered expression of paranodal proteins. It has been reported that impaired expression of the axonal protein Caspr at the paranodal junction results in subtle morphological changes that lead to behavioral deficits (Berglund et al., 1999; Boyle et al., 2001). Interestingly, we found that immunofluorescence labeling for Caspr in spinal cord sections is reduced in sla mice relative to wild-type mice (Fig. 6A–D). Quantification of the immunofluorescence staining in the cervical, thoracic, and lumbar spinal cord shows a significant reduction in Caspr expression in the gray and white matter in sla mice relative to wild-type controls (Fig. 6E,F). To rule out the possibility that the reduced Caspr expression in sla mice might be due to axonal loss, we did immunofluorescence labeling for neurofilaments. Quantification of the neurofilament immunostaining shows no difference between sla (ventral gray matter: 1.30 ± 0.18 pixel density/μm2; ventral white matter: 1.73 ± 0.18 pixel density/μm2) and wild-type mice (ventral gray matter: 1.39 ± 0.31 pixel density/μm2; ventral white matter: 1.74 ± 0.30 pixel density/μm2). We next assessed the expression of Nogo A, a known partner of Caspr that is expressed by oligodendrocytes in CNS (Nie et al., 2003). Double-immunofluorescence labeling shows reduction of Nogo A expression in oligodendrocytes in the gray matter in the sla spinal cord compared with wild-type mice (Fig. 7A–F). Quantification of Nogo A immunolabeling in CC1+ oligodendrocytes in the lumbar spinal cord shows a marked decrease of Nogo A expression levels in the gray and white matter of sla mice relative to wild-type mice (Fig. 7G).
Figure 6.
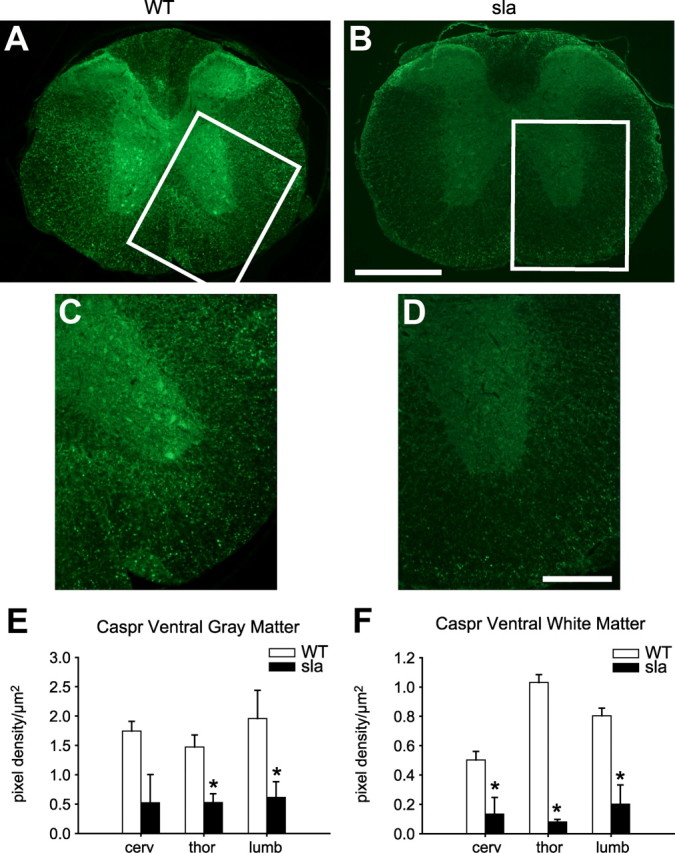
Expression of the paranodal protein Caspr is reduced in the sla spinal cord. A, Cross section of wild-type mouse spinal cord shows strong immunofluorescence labeling for Caspr in gray and white matter. B, In contrast, Caspr labeling is very weak in the sla spinal cord. The areas outlined in A and B are shown at higher magnification in C and D, respectively. E, F, Quantification of the pixel density per square micrometer in the gray matter of the ventral horn and the ventral white matter of the cervical, thoracic, and lumbar spinal cord shows a significant reduction of Caspr expression in sla mice compared with wild-type mice. Results are shown as mean ± SEM. *p ≤ 0.05, two-way repeated-measures ANOVA. Scale bars: B, 500 μm; D, 200 μm.
Figure 7.
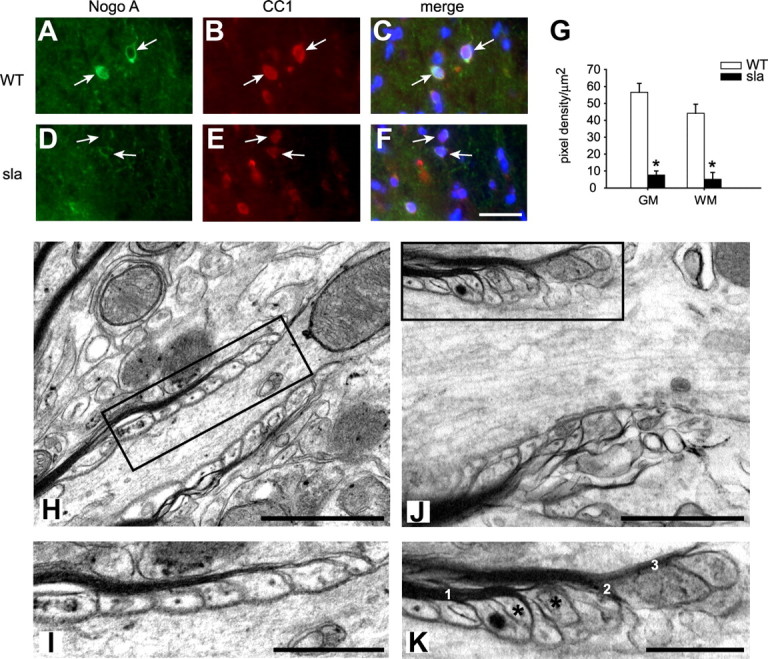
Reduced expression of Nogo A in oligodendrocytes in the sla spinal cord and ultrastructural abnormalities at the paranodal region in sla mice. A–C, Wild-type spinal cord shows strong immunofluorescence labeling for Nogo A (A, arrows) in CC1-labeled oligodendrocytes (B, arrows). D–F, In contrast, sla spinal cord shows markedly reduced labeling for Nogo A (D, arrows) in CC1-labeled oligodendrocytes (E). C and F show merged images. G, Quantification of the Nogo A pixel density per square micrometer of individual CC1-positive cells (30 per animal, n = 3 mice) in the gray and white matter of the lumbar spinal cord shows a significant reduction of Nogo A expression in sla mice compared with wild-type mice. Results are shown as mean ± SEM. *p ≤ 0.05, Student's t test. Scale bar: F, 30 μm. H, Electron micrograph of a paranodal region in the gray matter of a wild-type mouse. I, The area of the paranode outlined in the rectangle in H is shown at higher magnification. Note the regularly arranged terminal myelin loops that are closely associated with the axonal membrane of the paranodal region. J, In contrast, the paranodal region in the sla spinal cord gray matter shows abnormal structure. K, A higher magnification of the area outlined in the rectangle in J. Note the multiple loops of myelin (labeled 1, 2, and 3), and the floating paranodal loops that fail to make contact with the axonal membrane (asterisks). Scale bars: H, J, 1 μm; I, K, 0.5 μm.
Abnormal paranodal structure in the spinal cord of sla mice
Paranodal proteins at the axonal membrane and the terminal myelin loops form paranodal junctions that are important in establishing and maintaining the structure of the paranodes. We therefore assessed by electron microscopy whether the reduced expression of the paranodal proteins Caspr and Nogo A affects paranodal structure in the spinal cord gray matter of sla compared with wild-type mice. The paranodal region in sla mice showed abnormal separation of the myelin sheath into multiple distinct myelin lamellae (Fig. 7J,K), as well as floating terminal myelin loops that are not attached to the axonal membrane (Fig. 7J,K). In contrast, the paranodal loops in wild-type animals are properly aligned and in close association with the axonal membrane, and did not show splitting of the myelin at the paranodal region (Fig. 7H,I). The paranodal abnormalities seen in sla mice are similar to those reported in Caspr mutant and contactin mutant mice, which were also associated with functional defects (Berglund et al., 1999; Bhat et al., 2001; Boyle et al., 2001). These results indicate that although the overall myelination (g-ratio) is not impaired in sla mice, the structure of the paranodes is severely disrupted, which could account for the motor deficits seen in these mice.
Ceruloplasmin compensates for the loss of Heph function in white matter oligodendrocytes
It was surprising to see that iron accumulation in sla mice was limited to gray matter oligodendrocytes. We hypothesized that the absence of iron accumulation in white matter oligodendrocytes of sla mice might be due to upregulation of the other known ferroxidase, Cp. Although oligodendrocytes do not express Cp at the protein level (data not shown), RT-PCR analysis indicate that oligodendrocytes in vitro do express Cp mRNA (Fig. 8A). Interestingly, in support of our hypothesis, Western blot analysis shows that Cp is upregulated at the protein level in the spinal cord of sla mice (Fig. 8B,C). To assess whether Cp expression is upregulated in oligodendrocytes in spinal cord white matter of sla mice, we performed in situ hybridization. We see high Cp mRNA expression in the white matter of sla but not wild-type mice (Fig. 8D,E). In addition, quantification of the numbers of cells expressing Cp in the sla spinal cord showed fourfold greater induction in white matter (29.28 cells/100 μm2) compared with gray matter (7.25 cells/100 μm2) (Fig. 8F). This is not due to differences in the number of oligodendrocytes (CC1+ cells) in gray and white matter (white matter = 36.33 ± 3.04 cells/100 μm2; gray matter = 39.68 ± 0.18 cells/100 μm2). This compensatory upregulation of Cp expression further points to the greater need for ferroxidase activity in white matter oligodendrocytes. Furthermore, in situ hybridization for Cp combined with immunofluorescence labeling for oligodendrocytes using an anti-CC1 antibody shows that Cp is upregulated in CC1+ oligodendrocytes in the white matter of the spinal cord in sla mice (Fig. 8G–I) but not in wild-type mice (Fig. 8J–L).
Figure 8.

Expression of Cp is upregulated in white matter oligodendrocytes in sla mice. A, RT-PCR analysis shows that cultured oligodendrocytes express mRNA for Heph, Fpn, as well as Cp. In contrast, oligodendrocytes do not express Cp protein (data not shown). RNA isolated from the intestinal cell line Caco2 is included as a positive control for Heph and Fpn and a negative control for Cp. PPIA levels demonstrate that equal amounts of RNA were used. B, Western blot shows that Cp expression is increased in the spinal cord of sla mice compared with wild-type (WT) mice. Spinal cord protein extract of Cp KO (Cp−/−) mice was used as a negative control. The blot was reprobed for β-actin to ensure equal protein loading. C, Quantification of the Western blot data shows a significant increase of Cp expression in the spinal cord of sla mice. Results are shown as mean ± SEM (n = 3; *p ≤ 0.05, Student's t test). D, E, In situ hybridization for Cp on spinal cord cross sections shows upregulation of Cp expression in the white matter of sla mice (E) compared with wild-type mice (D). The dashed lines outline the boundary between gray (g) and white (w) matter. F, Quantification of the number of Cp-positive cells in the gray and white matter of sla spinal cord shows that Cp induction is markedly greater in the white matter compared with the gray matter. Results are shown as mean ± SEM. n = 3; *p ≤ 0.05, Student's t test. G–I, Higher magnification of the sla spinal cord white matter showing in situ hybridization for Cp (G) combined with immunofluorescence labeling with anti-CC1 for oligodendrocytes (H), indicating that Cp expression is upregulated in white matter oligodendrocytes (arrows). J–L, In contrast, there is no Cp expression (J) in CC1-positive oligodendrocytes (K) in wild-type (WT) mice. I and L show merged images. Scale bars: E, 100 μm; L, 50 μm. WM, White matter; GM, gray matter; PPIA, peptidylprolyl isomerase A.
To determine whether the compensatory upregulation of Cp in white matter oligodendrocytes in sla mice rescues them from iron accumulation, we generated Hephsla/Cp−/− double-mutant mice and performed iron histochemistry. Enhanced Perl's histochemistry of 9–12-month-old animals shows iron accumulation in both the gray and white matter of the spinal cord in the Hephsla/Cp−/− double mutants (Fig. 9A–D). The iron-loaded cells in the white matter of the double-mutant mice display the morphology of oligodendrocytes with processes surrounding axons (Fig. 9D). Immunofluorescence labeling with anti-ferritin as a surrogate marker for iron shows high ferritin levels in both the gray and white matter of the spinal cord in Hephsla/Cp−/− double mutants, but only in the gray matter in sla mice (Fig. 9E–G). To determine whether the ferritin-positive cells in the white matter of the Hephsla/Cp−/− mice were oligodendrocytes, we performed double-immunofluorescence labeling for ferritin and the oligodendrocyte marker CC1, and found high levels of ferritin predominantly in CC1+ oligodendrocytes (Fig. 9H–M). These results suggest that oligodendrocytes in the white matter are able to compensate for the loss of Heph function by upregulating Cp expression. We also examined whether the Hephsla/Cp−/− double-mutant mice show increased motor deficits on rotarod analysis. Hephsla/Cp−/− mice at 6 months of age fell off the rotating rod earlier than wild-type and sla mice [wild-type mice, mean = 174 s (n = 6); sla mice, mean = 143.5 s (n = 6); Hephsla/Cp−/− mice, mean = 80 s (n = 2)]. These data indicate that increased iron accumulation in white and gray matter in Hephsla/Cp−/− mice are associated with even earlier impairment of motor control compared with sla mice, further supporting the importance of compensatory upregulation of Cp in white matter oligodendrocytes in sla mice.
Figure 9.
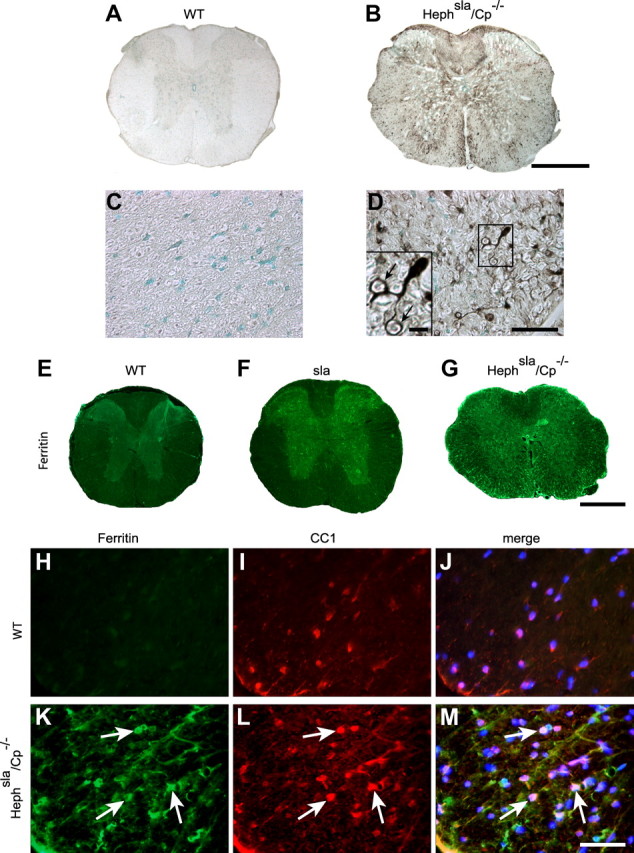
Iron accumulation occurs in spinal cord gray and white matter in Hephsla/Cp−/− double-mutant mice. A, B, Perl's histochemistry of spinal cord cross sections of 12-month-old wild-type (A) and Hephsla/Cp−/− (B) mice. Note the marked iron labeling in both the gray and white matter of the Hephsla/Cp−/− double-mutant mouse in B. C, D, Higher-magnification micrographs showing Perl's staining of the ventral white matter region of wild-type (C) and Hephsla/Cp−/− (D) spinal cord. Note there is no iron labeling in the wild-type spinal cord (C) but marked iron accumulation in the white matter in the Hephsla/Cp−/− double mutant (D). The cell indicated in the rectangle in D is illustrated at higher magnification in the inset, which shows an iron-loaded cell with the morphology of an oligodendrocyte that sends processes that appear to wrap around axons (arrows). E–G, Immunofluorescence labeling of spinal cord cross sections with anti-ferritin used as a surrogate marker for iron shows very weak ferritin immunoreactivity in the wild-type mouse (E), strong ferritin immunoreactivity in the gray matter in sla mouse (F), and strong ferritin immunoreactivity in both the gray and white matter in the Hephsla/Cp−/− mouse (G). H–M, Double-immunofluorescence labeling with anti-ferritin (H, K) and the oligodendrocyte marker anti-CC1 (I, L) shows strong ferritin immunoreactivity in white matter oligodendrocytes in Hephsla/Cp−/− mouse spinal cord (K–M, arrows) but not in the wild-type mouse (H–J). The merged images that include DAPI-stained nuclei are shown in J and M. Scale bars: B, G, 500 μm; D, M, 50 μm.
Discussion
Astrocytes efflux iron via Fpn and its interactions with the GPI-anchored form of the ferroxidase Cp (Jeong and David, 2003). Ceruloplasmin, however, is not expressed by oligodendrocytes in the normal CNS. We show here that another ferroxidase, Heph, is expressed by oligodendrocytes and plays an important role in iron efflux from these cells.
Heph mediates iron efflux from duodenal enterocytes into the circulation via interactions with Fpn (Wessling-Resnick, 2006; Yeh et al., 2009). We also found expression of Fpn in oligodendrocytes and show that expression of Fpn and Heph is restricted to mature oligodendrocytes. The lack of Fpn and Heph expression in earlier stages of the oligodendrocyte lineage suggests that iron efflux is a feature of mature oligodendrocytes. OPCs might have a greater need to acquire and retain iron, as iron is required for the proliferation and differentiation of OPCs, as well as for myelination (Morath and Mayer-Pröschel, 2001; Schonberg and McTigue, 2009; Todorich et al., 2009). After myelination is complete, oligodendrocytes likely require less iron and maintain stable iron levels through iron efflux. In support of this idea, oligodendrocytes also downregulate expression of the iron importer TFR1 after differentiation is completed (Hill et al., 1985; Han et al., 2003). Nevertheless, OPCs and mature oligodendrocytes express ferritin receptors that permit uptake of extracellular ferritin-bound iron (Todorich et al., 2008), suggesting that mature oligodendrocytes also require basal levels of iron influx.
Cells handle increased intracellular iron load in a variety of ways. One mechanism is the upregulation of the iron-binding protein ferritin in the cytosol. The age-associated increase in ferritin immunostaining seen in oligodendrocytes in sla mice suggests an increased iron load. Another way in which cells control high intracellular iron levels is by decreasing iron influx. However, despite the high iron levels in oligodendrocytes in sla mice, TFR1 mRNA level in the spinal cord remains unchanged from normal levels. As expression of TFR1 in mature oligodendrocytes is generally low (Hill et al., 1985; Han et al., 2003), a further decrease in TFR1 expression via the iron regulatory protein-iron response element system (Pantopoulos, 2004) may be undetectable in sla mice. However, expression of the ferritin receptor TIM-2, which is likely to function as an iron importer in mature oligodendrocytes, is reduced in sla mice. These data suggest that oligodendrocytes in sla mice may be attempting to minimize iron uptake in response to high intracellular iron levels; however, this compensatory mechanism appears to be insufficient as iron overload continues to occur with age in sla oligodendrocytes.
Cells also handle increased intracellular iron through efflux of excess iron. Our in vitro studies show that the reduction of Heph in sla mice leads to impaired iron efflux from oligodendrocytes. Heph likely regulates iron efflux from oligodendrocytes by converting ferrous iron transported via Fpn into its ferric form. The immediate oxidation of ferrous iron appears to be necessary for its efflux, as has been shown for Cp and Fpn in astrocytes (Jeong and David, 2003). In vitro studies with astrocytes and C6 glioma cells indicate that, in the absence of GPI-Cp, cell surface Fpn gets internalized and rapidly degraded (De Domenico et al., 2007). In contrast to this, we have evidence that Fpn remains on the cell surface of oligodendrocytes from sla mice (data not shown). Whether this is due to the presence of low levels of mutant Heph in the membrane is currently not known. Consistent with the role of Heph in iron efflux from oligodendrocytes in cell culture, we found iron accumulation in oligodendrocytes in vivo in the CNS of sla mice with age. Our data indicate that the residual activity of Heph in sla mice is insufficient to mediate effective iron efflux from oligodendrocytes in vitro and in vivo.
Interestingly, iron accumulation in sla mice occurs only in gray matter oligodendrocytes, suggesting differential requirements for the management of iron homeostasis in gray and white matter oligodendrocytes. Gray and white matter oligodendrocytes differ in their metabolic function and in their capacity to handle metabolic stress (D'Amelio et al., 1990; Bauer et al., 2002). Because white matter oligodendrocytes are involved in myelination of long fiber tracts in the CNS, they may require higher iron levels than their counterparts in the gray matter. Thus, white matter oligodendrocytes may need to be equipped with additional iron efflux mechanisms to prevent intracellular iron overload and potential damage to white matter tracts. Interestingly, we observed upregulation of Cp expression in white matter oligodendrocytes in sla mice, indicating that Cp compensates for the loss of Heph function. Additionally, the extensive iron accumulation in white matter oligodendrocytes in the Hephsla/Cp−/− double-mutant mice further supports the proposed view that expression of Cp in white matter oligodendrocytes in sla mice protects these cells from iron accumulation. The severe motor defects in Hephsla/Cp−/− double-mutant mice further emphasize the importance of Cp upregulation in white matter oligodendrocytes in sla mice. The ability of Cp to function as a fail-safe mechanism has also been demonstrated in sla mice in the gut, where it is thought to rescue these mice from early-onset microcytic anemia (Bannerman and Cooper, 1966; Cherukuri et al., 2005).
We show here that motor coordination of sla mice is impaired, suggesting that some aspect of neuronal function is affected. Unlike myelin-producing white matter oligodendrocytes, the function of gray matter oligodendrocytes remains incompletely understood. However, perineuronal oligodendrocytes have been shown to provide trophic factor support for neurons (Du and Dreyfus, 2002). We did not observe any loss in the number of neurons in the sla spinal cord, nor was there an increase in the number of apoptotic cells, as assessed by annexin V staining (data not shown). Similarly, no differences were seen in the expression of the synaptic protein, synapsin, in the spinal cord of sla and wild-type mice (data not shown). Despite increased iron accumulation in gray matter oligodendrocytes in sla mice, their numbers were not diminished. This is in contrast to the marked loss of astrocytes in Cp-null mice which exhibit iron accumulation in astrocytes (Jeong and David, 2006). Collectively, these findings suggest that oligodendrocytes, which have a higher requirement for iron, appear to have a better capacity to buffer increased iron load and to remain viable relative to astrocytes. Also myelination overall is not affected as assessed by Luxol fast blue staining and g-ratio analysis. However, there is decreased expression of the paranodal proteins Nogo A and Caspr in the spinal cord of sla mice. Nogo A is expressed by oligodendrocytes and is localized to the paranodal loops of myelin where it partners with the axonal protein Caspr (Nie et al., 2003). Caspr is a main component of the paranodal region, and plays an important role in establishing and maintaining the paranodal junctions (Einheber et al., 1997). These axoglial junctions form a critical barrier between the voltage-gated Na+ channels at the node and the delayed rectifier K+ channels in the juxtaparanodal region, and are therefore crucial for the propagation of action potentials via saltatory conduction (Waxman and Ritchie, 1993; Denisenko-Nehrbass et al., 2002). The reduction of Caspr and Nogo A in sla mice was observed in the gray as well as white matter. Since Caspr is a neuronal/axonal protein, its reduction in the white matter might be due to the involvement of the neuronal cell bodies of projecting neurons that are located in the gray matter in other regions of the CNS. We observed iron accumulation in gray matter regions throughout the CNS of sla mice (data not shown). The reduced expression of Caspr in sla mice may also be due to the loss of its binding partner Nogo A in oligodendrocytes, which may result from indirect effects of increased iron load. The reduction of the two paranodal proteins Caspr and NogoA is likely to result in the structural abnormalities of the paranodes that were observed in sla mice. Alterations of the paranodal loops seen by electron microscopy may contribute to the motor deficits in these animals. This is supported by previous work demonstrating functional deficits caused by reduction of Caspr and ultrastructural abnormalities of the paranodal loops (Bhat et al., 2001; Boyle et al., 2001).
In summary, our data show that Heph is expressed by mature oligodendrocytes and plays a role in iron efflux from these cells. The findings presented here also indicate that white and gray matter oligodendrocytes can regulate iron efflux differently. We show that in the absence of Heph, white matter oligodendrocytes upregulate the expression of Cp, likely as a fail-safe mechanism. The lack of such compensatory upregulation of Cp in gray matter oligodendrocytes results in severe iron accumulation. We show that although oligodendrocytes have a high capacity to buffer excess iron, the increased iron load in gray matter oligodendrocytes leads to paranodal abnormalities that likely contribute to the motor deficits seen in the sla mutants. The present study reveals the first clear demonstration of a ferroxidase that is required for iron efflux from oligodendrocytes. This knowledge will help in future studies on iron homeostasis in various neurodegenerative diseases in which iron accumulation occurs.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to S.D. K.S. has received supported from the CIHR Neuroinflammation Training Program and is currently supported by a studentship from the Multiple Sclerosis Society of Canada. We thank Ourania Tsatas for technical assistance and Margaret Attiwell for help with preparing the illustrations. We also acknowledge Dr. Suh Young Jeong for helpful discussions at the early stages of this work, and Dr. Jennifer Berard for help with editing this manuscript.
References
- Bakshi R, Dmochowski J, Shaikh ZA, Jacobs L. Gray matter T2 hypointensity is related to plaques and atrophy in the brains of multiple sclerosis patients. J Neurol Sci. 2001;185:19–26. doi: 10.1016/s0022-510x(01)00477-4. [DOI] [PubMed] [Google Scholar]
- Bannerman RM, Cooper RG. Sex-linked anemia: a hypochromic anemia of mice. Science. 1966;151:581–582. doi: 10.1126/science.151.3710.581. [DOI] [PubMed] [Google Scholar]
- Bauer J, Bradl M, Klein M, Leisser M, Deckwerth TL, Wekerle H, Lassmann H. Endoplasmic reticulum stress in PLP-overexpressing transgenic rats: gray matter oligodendrocytes are more vulnerable than white matter oligodendrocytes. J Neuropathol Exp Neurol. 2002;61:12–22. doi: 10.1093/jnen/61.1.12. [DOI] [PubMed] [Google Scholar]
- Berg D, Youdim MB, Riederer P. Redox imbalance. Cell Tissue Res. 2004;318:201–213. doi: 10.1007/s00441-004-0976-5. [DOI] [PubMed] [Google Scholar]
- Berglund EO, Murai KK, Fredette B, Sekerková G, Marturano B, Weber L, Mugnaini E, Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Carrí MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull. 2003;61:365–374. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- Chen H, Attieh ZK, Su T, Syed BA, Gao H, Alaeddine RM, Fox TC, Usta J, Naylor CE, Evans RW, McKie AT, Anderson GJ, Vulpe CD. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood. 2004;103:3933–3939. doi: 10.1182/blood-2003-09-3139. [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005;2:309–319. doi: 10.1016/j.cmet.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Connor JR, Boeshore KL, Benkovic SA, Menzies SL. Isoforms of ferritin have a specific cellular distribution in the brain. J Neurosci Res. 1994;37:461–465. doi: 10.1002/jnr.490370405. [DOI] [PubMed] [Google Scholar]
- D'Amelio F, Eng LF, Gibbs MA. Glutamine synthetase immunoreactivity is present in oligodendroglia of various regions of the central nervous system. Glia. 1990;3:335–341. doi: 10.1002/glia.440030504. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007;26:2823–2831. doi: 10.1038/sj.emboj.7601735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisenko-Nehrbass N, Faivre-Sarrailh C, Goutebroze L, Girault JA. A molecular view on paranodal junctions of myelinated fibers. J Physiol Paris. 2002;96:99–103. doi: 10.1016/s0928-4257(01)00085-7. [DOI] [PubMed] [Google Scholar]
- Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- Du Y, Dreyfus CF. Oligodendrocytes as providers of growth factors. J Neurosci Res. 2002;68:647–654. doi: 10.1002/jnr.10245. [DOI] [PubMed] [Google Scholar]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, Peles E, Salzer JL. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol. 1997;139:1495–1506. doi: 10.1083/jcb.139.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb GL, Osterbur DL, LeVine SM. The distribution of iron in the brain: a phylogenetic analysis using iron histochemistry. Brain Res Dev Brain Res. 1996;93:120–128. doi: 10.1016/0165-3806(96)00020-x. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC-oligodendrocyte progenitors. Dev Biol. 1993;159:618–630. doi: 10.1006/dbio.1993.1269. [DOI] [PubMed] [Google Scholar]
- Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci U S A. 2004;101:13850–13855. doi: 10.1073/pnas.0405146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Day JR, Connor JR, Beard JL. Gene expression of transferrin and transferrin receptor in brains of control vs. iron-deficient rats. Nutr Neurosci. 2003;6:1–10. [PubMed] [Google Scholar]
- Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96:10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Switzer RC., 3rd The regional distribution and cellular localization of iron in the rat brain. Neuroscience. 1984;11:595–603. doi: 10.1016/0306-4522(84)90046-0. [DOI] [PubMed] [Google Scholar]
- Hill JM, Ruff MR, Weber RJ, Pert CB. Transferrin receptors in rat brain: neuropeptide-like pattern and relationship to iron distribution. Proc Natl Acad Sci U S A. 1985;82:4553–4557. doi: 10.1073/pnas.82.13.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, David S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. 2003;278:27144–27148. doi: 10.1074/jbc.M301988200. [DOI] [PubMed] [Google Scholar]
- Jeong SY, David S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J Neurosci. 2006;26:9810–9819. doi: 10.1523/JNEUROSCI.2922-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine SM. Iron deposits in multiple sclerosis and Alzheimer's disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/s0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- LeVine SM, Macklin WB. Iron-enriched oligodendrocytes: a reexamination of their spatial distribution. J Neurosci Res. 1990;26:508–512. doi: 10.1002/jnr.490260415. [DOI] [PubMed] [Google Scholar]
- Meguro R, Asano Y, Odagiri S, Li C, Iwatsuki H, Shoumura K. The presence of ferric and ferrous iron in the nonheme iron store of resident macrophages in different tissues and organs: histochemical demonstrations by the perfusion-Perls and -Turnbull methods in the rat. Arch Histol Cytol. 2005;68:171–183. doi: 10.1679/aohc.68.171. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Hider RC, Gaeta A, Williams R, Francis P. Metals ions and neurodegeneration. Biometals. 2007;20:639–654. doi: 10.1007/s10534-006-9033-z. [DOI] [PubMed] [Google Scholar]
- Morath DJ, Mayer-Pröschel M. Iron modulates the differentiation of a distinct population of glial precursor cells into oligodendrocytes. Dev Biol. 2001;237:232–243. doi: 10.1006/dbio.2001.0352. [DOI] [PubMed] [Google Scholar]
- Nie DY, Zhou ZH, Ang BT, Teng FY, Xu G, Xiang T, Wang CY, Zeng L, Takeda Y, Xu TL, Ng YK, Faivre-Sarrailh C, Popko B, Ling EA, Schachner M, Watanabe K, Pallen CJ, Tang BL, Xiao ZC. Nogo-A at CNS paranodes is a ligand of Caspr: possible regulation of K(+) channel localization. EMBO J. 2003;22:5666–5678. doi: 10.1093/emboj/cdg570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Osaki S, Johnson DA. Mobilization of liver iron by ferroxidase (ceruloplasmin) J Biol Chem. 1969;244:5757–5758. [PubMed] [Google Scholar]
- Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. 1997;272:20185–20190. doi: 10.1074/jbc.272.32.20185. [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. 2000;275:4305–4310. doi: 10.1074/jbc.275.6.4305. [DOI] [PubMed] [Google Scholar]
- Rathore KI, Kerr BJ, Redensek A, López-Vales R, Jeong SY, Ponka P, David S. Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J Neurosci. 2008;28:12736–12747. doi: 10.1523/JNEUROSCI.3649-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberg DL, McTigue DM. Iron is essential for oligodendrocyte genesis following intraspinal macrophage activation. Exp Neurol. 2009;218:64–74. doi: 10.1016/j.expneurol.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele PM, Medina JF, Nores WL, Mauk MD. Using genetic mutations to study the neural basis of behavior. Cell. 1998;95:879–882. doi: 10.1016/s0092-8674(00)81712-2. [DOI] [PubMed] [Google Scholar]
- Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor JR. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem. 2008;107:1495–1505. doi: 10.1111/j.1471-4159.2008.05678.x. [DOI] [PubMed] [Google Scholar]
- Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: the role of iron. Glia. 2009;57:467–478. doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Ritchie JM. Molecular dissection of the myelinated axon. Ann Neurol. 1993;33:121–136. doi: 10.1002/ana.410330202. [DOI] [PubMed] [Google Scholar]
- Wessling-Resnick M. Iron imports. III. Transfer of iron from the mucosa into circulation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1–6. doi: 10.1152/ajpgi.00415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh KY, Yeh M, Mims L, Glass J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am J Physiol Gastrointest Liver Physiol. 2009;296:G55–G65. doi: 10.1152/ajpgi.90298.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]