

Abstract
In mammals, the increased secretion of arginine–vasopressin (AVP) (antidiuretic hormone) and oxytocin (natriuretic hormone) is a key physiological response to hyperosmotic stress. In this study, we examined whether chronic hyperosmotic stress weakens GABAA receptor-mediated synaptic inhibition in rat hypothalamic magnocellular neurosecretory cells (MNCs) secreting these hormones. Gramicidin-perforated recordings of MNCs in acute hypothalamic slices prepared from control rats and ones subjected to the chronic hyperosmotic stress revealed that this challenge not only attenuated the GABAergic inhibition but actually converted it into excitation. The hyperosmotic stress caused a profound depolarizing shift in the reversal potential of GABAergic response (EGABA) in MNCs. This EGABA shift was associated with increased expression of Na+–K+–2Cl− cotransporter 1 (NKCC1) in MNCs and was blocked by the NKCC inhibitor bumetanide as well as by decreasing NKCC activity through a reduction of extracellular sodium. Blocking central oxytocin receptors during the hyperosmotic stress prevented the switch to GABAergic excitation. Finally, intravenous injection of the GABAA receptor antagonist bicuculline lowered the plasma levels of AVP and oxytocin in rats under the chronic hyperosmotic stress. We conclude that the GABAergic responses of MNCs switch between inhibition and excitation in response to physiological needs through the regulation of transmembrane Cl− gradients.
Introduction
In the mammalian CNS, GABA typically acts as an inhibitory neurotransmitter (Macdonald and Olsen, 1994). However, there are certain conditions in which GABA can function to excite the postsynaptic neuron. For example, in the developing nervous system, it is well established that GABA can depolarize neurons to generate action potentials (Ben-Ari, 2002). Moreover, under certain pathological conditions, such as epilepsy, there is increasing evidence for GABA-evoked excitation that occurs as part of the pathology of disease (De Koninck, 2007). Finally, there is evidence for GABA-mediated excitation in the normal adult CNS (Wagner et al., 1997; Choi et al., 2008; Herbison and Moenter, 2011), but the functional significance has been elusive.
We have been exploring this issue in magnocellular neurosecretory cells (MNCs) in the hypothalamic supraoptic (SON) and paraventricular nuclei (PVN) that secrete the neurohormones oxytocin and arginine–vasopressin (AVP). The secretion of these hormones is an essential part of the physiological response to hyperosmotic stress (Levine, 2000; Antunes-Rodrigues et al., 2004), and these cells are densely innervated by GABAergic afferents (Decavel and Van den Pol, 1990). In the present study, we sought to determine whether this inhibitory input is weakened when a rat is subjected to chronic hyperosmotic stress, a condition in which a sustained increase in AVP and oxytocin secretion is needed. Here we report that, under this physiologically demanding condition, GABA switches from functioning as an inhibitory transmitter to evoking an excitatory response in MNCs. This switch contributes significantly to the increased secretion of AVP and oxytocin. Furthermore, we present evidence that the upregulation of the Cl− importer Na+–K+–2Cl− cotransporter 1 (NKCC1) in the postsynaptic neuron, driven by the activation of central oxytocin receptors, is the mechanism underlying the inhibitory-to-excitatory switch in GABAergic transmission.
Materials and Methods
Animal care.
Male Sprague Dawley rats (age, 6–20 weeks) from Orient Bio were used in the current study. They were housed in a temperature-controlled vivarium (22–24°C) with a 12 h light/dark cycle for at least 1 week before experiments. The experimental procedures described below were approved by the Korea University College of Medicine Animal Research Policies Committee and conformed to the guidelines of the Council of the Korean Physiological Society and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All possible efforts were made to minimize the number of animals as well as suffering.
Drinking water.
The rats were supplied with one of the following as solutions for drinking water before hypothalamic slices were prepared: (1) tap water for their lifetimes (T group), (2) 2% NaCl solution for 1 d (S1 group), (3) 2% NaCl solution for 7 d (S7 group), and (4) tap water for 1, 3, or 7 d after 2% NaCl solution for 1 week (ST1, ST3, and ST7 groups).
Hypothalamic slice preparation.
Brain slices were prepared in a manner similar to the one described previously (Kim et al., 2005). In brief, the animal was anesthetized with sodium pentobarbitone (100 mg/kg body weight, i.p.), and the brain was quickly excised from the skull and submerged in ice-cold artificial CSF (ACSF) (in mm: 124 NaCl, 1.3 MgSO4, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, and 10 glucose). After being chilled for 1–2 min, the brain was trimmed to a block containing the hypothalamus. With the use of a vibroslicer (Campden Instruments), coronal slices (300–450 μm), containing the SON and/or the PVN, were cut from the tissue block in ice-cold ACSF. The slices were transferred to a gas interface recording chamber that was perfused with warm (34–35°C) aerated (95% O2/5% CO2) ACSF, at a rate of 0.5–1 ml/min, by a peristaltic pump-driven or gravity-fed bath-perfusion system (Choi et al., 2008). Warm (34–35°C), humidified 95% O2/5% CO2 gas mixture was continuously blown over the slices to further ensure adequate oxygenation of cells in the tissue.
Intracellular recording.
Current- or voltage-clamp recordings were obtained from neurons in the SON or PVN of hypothalamic slices equilibrated for 1–12 h in the recording chamber. Micropipettes (tip diameter, 1.5–2.0 μm; 3–6 MΩ) pulled from borosilicate tubings (P-97; Sutter Instruments) and filled with gramicidin (50 μg/ml)-containing solution (in mm: 143 K-gluconate, 2 KCl, 10 HEPES, and 0.5 EGTA, pH 7.2–7.3) were used for recording in a perforated configuration (Rhee et al., 1994). Our use of the low chloride (2 mm) internal solution along with gramicidin provided a clear readout of when the patch was ruptured, resulting in a whole-cell recording configuration (i.e., GABAergic EPSPs/EPSCs were converted into IPSPs/IPSCs). Stable perforated recording condition was usually achieved within 10–25 min after a seal was formed. Those recordings having steady series resistances (range, 30–90 MΩ) and action potential amplitudes of ≥40 mV (measured from spike threshold) were the only ones included in the data pool; the EGABA and the reversal potential of muscimol-evoked response (Emuscimol) were estimated only with the recordings that had a series resistance of <60 MΩ (typically 30–50 MΩ). The voltage errors resulting from the series resistance were compensated offline for voltage-clamp recordings and online for current-clamp recordings by using the bridge circuit. Neurons sampled in the SON and the magnocellular division of the PVN, which responded to a depolarizing current pulse delivered at a holding potential between −70 and −80 mV with repetitive spikes having a delayed onset, were taken as MNCs (Tasker and Dudek, 1991). The signals from neurons amplified by Axoclamp-2B amplifier (bandwidth filter set at 10 kHz for current-clamp and 1 kHz for voltage-clamp recordings) were digitized and sampled at 50 μs intervals (Digidata 1320, pClamp 8.0; Molecular Devices).
Extracellular recording.
Extracellular single-unit recordings were obtained from MNCs in the SON and PVN slices. Micropipettes (tip diameter, 1.5–2.0 μm; 4–7 MΩ) pulled from borosilicate tubings (P-97; Sutter Instruments) and filled with the slice perfusion medium (i.e., ACSF) were used for recording. The voltage signals from recording electrodes were fed serially into an Axoclamp-2B amplifier (Molecular Devices) and a differential amplifier (AM 502; Tektronix) for amplification, alternating current coupling, and bandpass filtering (200 Hz to 1 kHz). The processed signals were digitized and sampled at 50 μs intervals (Digidata 1320A, pClamp 8.0; Molecular Devices). The criteria used to identify single units were consistent spike waveform and amplitude.
Electrical stimulation to evoke GABAergic postsynaptic responses.
To elicit GABAA receptor-mediated synaptic responses from MNCs in the PVN and SON, a site midway between the PVN and the suprachiasmatic nucleus and a site dorsomedial to the SON were stimulated, respectively. For this, constant current (0.1–0.7 mA) or voltage (5–50 V) pulses (biphasic square wave, 0.5 ms duration) were applied to these sites through custom-made bipolar tungsten electrodes.
Correction of EGABA and Emuscimol values.
The reported EGABA and Emuscimol values are the ones corrected for the difference in Cl− concentration (127.8 ± 0.4 mm, n = 5) between slice perfusion medium and the CSF of rats used for experiments (T, 122.6 ± 1.2 mm, n = 5; S1, 126.8 ± 1.6 mm, n = 4; S7, 151.9 ± 3.9 mm, n = 9; ST1, 116.3 ± 2.7 mm, n = 3; ST3, 117.5 ± 2.8 mm, n = 6; ST7, 121.8 ± 0.3 mm, n = 4). For this correction, we used the following equation: EGABA (or Emuscimol) = (RT/F)ln[([Cl−]e + 0.2[HCO3−]e)/([Cl−]i + 0.2[HCO3−]i)], where R is the gas constant, T is the absolute temperature on the Kelvin scale, and F is the Faraday constant. We assumed that the HCO3−-to-Cl− permeability ratio for GABA channel is 0.2 and that [HCO3−]i and [HCO3−]e are 16 and 26 mm, respectively, at intracellular and extracellular pH of 7.2 and 7.4 and equal intracellular and extracellular [CO2].
Drugs.
We purchased all the drugs and chemicals used in the current study from Sigma-Aldrich, except muscimol (GABAA receptor agonist; Ascent Scientific), gabazine (GABAA receptor antagonist; Tocris Bioscience) and oxytocin and AVP antagonists (gift from Dr. M. Manning at the University of Toledo College of Medicine, Toledo, OH). We prepared the solutions of muscimol, bicuculline methiodide (GABAA receptor antagonist), gabazine, and dl-2-amino-5-phosphonopentanoic acid (AP-5; NMDA receptor antagonist) by dissolving these drugs in ACSF, the standard slice perfusion medium, and 6,7-dinitroquinoxaline-2,3-dione (DNQX; non-NMDA receptor antagonist) solution by diluting its dimethylsulfoxide-based stock solution with ACSF (final concentrations of dimethylsulfoxide, 0.05%). The solution of bumetanide (NKCC blocker) was prepared by dissolving this agent in AP-5 and DNQX-containing ACSF. These drug solutions were applied to hypothalamic slices by bath perfusion. In some experiments, muscimol was also applied focally by “Y-tube” method (Murase et al., 1989). Bicuculline methiodide solution for intravenous injection was prepared by dissolving the agent in 0.9% NaCl solution. Oxytocin and AVP receptor antagonist solutions for intracerebroventricular infusion were prepared by dissolving these peptides in ACSF.
Western blot.
The SON was excised from hypothalamic slices prepared as described above. The SON tissues from three rats were pooled to form a single sample. Each sample was lysed in triple-detergent lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.02% sodium azide, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1 mm phenylmethylsulfonyl fluoride). After the determination of protein concentration of total tissue lysate with Bradford assay, the tissue lysate was denatured with SDS-reducing buffer and heat (70–80°C, 4 min). The denatured lysate and prestained molecular size markers were resolved by 8% SDS-PAGE and transferred to nitrocellulose membrane. After being incubated in 5% skimmed milk/Tris-buffered saline for 1 h at room temperature, the membrane was reacted sequentially with primary antibodies [rabbit polyclonal anti-NKCC1 (1:1000; Millipore), rabbit polyclonal anti-KCC2 (1:500; Millipore), or mouse monoclonal anti-β-actin (1:2000; Santa Cruz Biotechnology)] and HRP-conjugated secondary antibodies (anti-rabbit or anti-mouse IgG; 1:2000; Assay Designs). Immunoreactive protein signals were detected with the enhanced chemiluminescence detection system (Thermo Fisher Scientific).
Immunohistochemistry.
The animals were anesthetized by intraperitoneal injection of urethane (1.25 g/kg) and perfused transcardially with phosphate buffer (0.12 m, pH 7.4) containing 4% paraformaldehyde. Their brains were postfixed in the same fixative overnight and cryoprotected in phosphate buffer containing 30% sucrose for at least 3 d at 4°C. Brains were cut coronally in 30 μm thickness on a sliding microtome, and the sections were stored in anti-freezing solution (30% glycerol and 30% ethylene glycol in 0.02 m phosphate buffer) at −20°C until additional processing. Every fourth section of the hypothalamus containing the SON and/or the PVN was used for the study. After being preincubated for 1 h in 0.12 m PBS containing 0.25% Triton X-100, 0.1% gelatin, and 0.1% sodium azide (PBSGTA), the sections were incubated overnight with mouse anti-NKCC monoclonal antibody (clone T4, 1:500; Developmental Studies Hybridoma Bank) (Lytle et al., 1995). The sections were further incubated with goat fluorescent-conjugated anti-mouse IgG (1:200; Alexa Fluor 488 H+L; Invitrogen) for 2 h after washing with PBSGTA. Then, they were mounted on the slide glass in anti-fade reagent (ProLong Gold; Invitrogen). NKCC-positive signals were scrutinized with a confocal fluorescent microscope (Carl Zeiss LSM 510). The SON and PVN contained NKCC-immunoreactive cells and processes, whereas the lateral hypothalamus (LH) had no obvious NKCC-positive signals. The brain sections that were processed by this method, omitting the primary antibody, did not show detectable fluorescent signal. Images taken under the same settings of the microscope were converted to grayscale images and then inverted. Using Scion Image program and Wacom tablet, we traced the SON boundary in each of the images and measured the mean gray value within the boundary. The same tracing frame was dragged to the LH above the SON and measured the mean gray value, which represented the background immunoreactivity level. NKCC immunoreactivity in the SON was presented as the ratio of mean gray values in the SON and LH. The mean gray values in the LH were not significantly different among different experimental groups of rats. NKCC immunoreactivity was measured in the boundaries of individual SON neurons as well. The ratios of these gray values measured in the individual neurons to the mean gray value in the LH were taken as the NKCC immunoreactivities in the cell membrane.
Intracerebroventricular infusion of AVP and oxytocin receptor antagonists.
Before use for electrophysiology, some of rats in T and S7 groups received a 10 d intracerebroventricular infusion of one of four treatments: (1) desGly-NH2,d(CH2)5[d-Tyr2, Thr4]OVT (selective oxytocin receptor antagonist; Manning et al., 1995); (2) d(CH2)5[Tyr(Me)2, Ala-NH29]AVP (antagonist selective for V1a with respect to V2 AVP receptor, but whose possible anti-oxytocin effect has not been tested; Manning et al., 1992); (3) a combination of both antagonists; or (4) vehicle (ACSF). The surgery to implant an osmotic minipump (infusion rate, 0.5 μl/h; Alzet model 2002) and an infusion cannula (Brain Infusion kit 2; Alzet) targeting the right lateral ventricle (anteroposterior, −1.0 mm from the bregma; mediolateral, +1.6 mm; dorsoventral, +4 mm below the surface of the skull; Paxinos and Watson, 1998) was performed under the anesthesia induced by an intraperitoneal injection of a combination of ketamine (43 mg/kg body weight) and xylazine (8.7 mg/kg body weight). The pump was positioned subcutaneously in the scapular region and attached to the cannula via polyethylene tubing. Before the implantation, the pump had been filled with a solution containing either drugs or vehicle control and placed in warm (36°C) saline overnight for priming.
ELISA.
For blood sampling and intravenous injection of bicuculline, the jugular vein of the rat was cannulated under a brief gas anesthesia (4% enflurane). After 3 h recovery period, 150 μl blood samples were taken at 0, 10, 20, and 30 min and replaced with heparinized saline. Bicuculline methiodide (2 mg/kg body weight in 100 μl of saline) was administered immediately after the first blood sample was taken at 0 min. AVP and oxytocin concentrations in the plasma were determined by ELISA using Arg8–vasopressin and oxytocin enzyme immunoassay kits (Assay Designs). The intra-assay coefficients of variation for the assay for AVP and oxytocin were 11.7 and 27.9%, respectively, and the sensitivities were 3.4 and 6.4 pg/ml, respectively.
Statistical analysis.
Numerical data are expressed as the mean ± SEM. Student's t test and Mann–Whitney U test were used for the comparison of two independent datasets with and without normal distribution, respectively. One-way ANOVA and pairwise comparison with Holm–Sidak method were performed to compare multiple independent datasets with normal distributions, whereas Kruskal–Wallis one-way ANOVA on ranks and pairwise comparison with Tukey's test or Dunn's method were performed to compare datasets without normal distributions. Comparison of multiple datasets collected with repeated measurement was performed with the generalized estimating equations (GEE) method. Fisher's exact test was performed to see whether there was a contingency between the two kinds of classification. p < 0.05 was considered significant.
Results
GABA normally functions as an inhibitory transmitter in MNCs
We first examined GABAergic PSPs/PSCs of MNCs recorded in the acute SON and PVN slices prepared from rats supplied with normal drinking water. The GABAergic PSPs/PSCs were isolated from glutamatergic ones by including AP-5 (100 μm) and DNQX (20 μm) in the recording medium and monitored at various holding potentials with the use of gramicidin-perforated recording technique, which preserves the intracellular Cl− concentration ([Cl−]i) of the recorded cell (Rhee et al., 1994). We could detect no physiological differences between the MNCs recorded in the SON and PVN regions and therefore pooled the resulting data for this study. The GABAergic PSPs/PSCs detected in the MNCs (n = 117) of rats maintained under these control conditions (n = 42) were either inhibitory (i.e., hyperpolarizing relative to action potential threshold; Fig. 1a, top trace) or excitatory (i.e., depolarizing relative to action potential threshold), with the former being the predominant of the two (73.5 vs 26.5%). The reversal potentials of these synaptic events (EGABA values) averaged −51.6 ± 2.3 mV (n = 21; range, −19 to −70 mV) and were not significantly different from the Emuscimol values (−50.3 ± 1.8 mV, n = 8), the reversal potentials of the responses elicited by focal application of muscimol (5 μm, 50 ms; GABAA receptor agonist) (Fig. 2). In many of the control neurons having GABAergic IPSPs, the EGABA was similar to or significantly more positive than the resting membrane potential (RMP; −58.3 ± 2.1 mV, n = 21). Therefore, GABAergic IPSPs in these cells were either buried in the noise (5 of 18 cells) or detected as depolarizing responses at RMP (7 of 18 cells). To ensure that the GABAergic PSPs occurring in the control neurons are mostly inhibitory, we next examined the effects of the GABAA receptor antagonist gabazine on the firing rates of these cells. Because most of the MNCs were electrically silent, we injected intracellularly constant levels of (+) direct current to induce firing while testing the effects of gabazine. Bath application of gabazine (3 μm) increased the firing rate in a reversible manner in most cases (Fig. 1d); the firing rates before, during, and after the application of gabazine were 3.5 ± 0.6 (n = 14), 4.9 ± 0.6 (n = 14), and 3.5 ± 0.8 impulses/s (n = 13), respectively (overall difference, p = 0.0031, before vs during, p = 0.0017; during vs after, p = 0.0046; before vs after, p = 0.8635; GEE methods). Thus, our physiological data suggest that, when rats were held in normal drinking water, GABA functions typically as an inhibitory transmitter in the MNCs.
Figure 1.

GABAA receptor-mediated inhibition and excitation in MNCs. a, Spontaneous GABAergic IPSPs (○, top trace) and EPSPs (●, bottom trace) recorded in the MNCs of control and hyperosmolar rats, respectively. RMP, −52 mV (cell with IPSPs), −54 mV (cell with EPSPs). b, Reversible blockade by bath-applied bicuculline methiodide (BIC, 30 μm) of EPSPs recorded in a cell from hyperosmolar rat (RMP, −51 mV). Action potentials in a and b are truncated. c, Muscimol (5 μm, 50 ms focal application)-induced depolarization and spike generation in a cell from hyperosmolar rat (RMP, −54 mV), which shows spontaneous GABAergic EPSPs. d, Effects of GABAA receptor blockade by gabazine on the firing rates of MNCs. Top and middle rows, Voltage traces showing the firing of MNCs of control and hyperosmolar rats before, during, and after bath application of gabazine (3 μm). The duration of gabazine application (5 min) is indicated with a horizontal bar. The numbers associated with arrows denote the levels of membrane potential (in millivolts). RMP, −58 mV (control), −55 mV (hyperosmolar). Bottom, Graphic summary of the results from MNCs tested with gabazine as in the top and middle rows. The symbols connected by lines denote data from the same cells.
Figure 2.

Hyperosmolar conditions cause a depolarizing shift in the reversal potentials for GABAA receptor-evoked responses in MNCs. a1, b1, GABAergic PSCs evoked in the presence of AP-5 (100 μm) and DNQX (20 μm) in the MNCs of control and hyperosmolar rats. They were evoked at various holding potentials (VH) by electrical stimulation (▴) at a site near recorded cells. RMP, −57 mV (cell in a1), −50 mV (cell in b1). Double arrowheads, Regenerative inward current (truncated). c1, Amplitudes of the GABAergic PSCs illustrated in a1 and b1 are plotted against VH. The PSC that elicited the regenerative inward current (b1) was not included in the plot. Linear regression was used to fit the data points. The intersections (↓, ↑) of the regression lines with the abscissa were taken as the reversal potentials of GABAergic PSCs (i.e., EGABA values). The validity of the method of determining the EGABA with the extrapolated regression line as in (c1, hyperosmolar) was confirmed through a supplementary experiment, in which muscimol (GABAA receptor agonist)-elicited currents were recorded in the MNCs of control and hyperosmolar rats at various VH between −90 and −20 mV after blocking action currents with tetrodotoxin (a2, b2); the Emuscimol values estimated on the basis of these currents (c2) (control, −50.3 ± 1.8 mV, n = 8; hyperosmolar, −39.3 ± 1.6 mV, n = 9) was not significantly different from the EGABA values estimated with synaptic currents (c1) (control, −51.6 ± 2.3 mV, n = 21; hyperosmolar, −34.0 ± 1.6 mV, n = 25).
Chronic hyperosmotic stress converts GABAergic inhibition into excitation in MNCs
In contrast, when rats (n = 28) were given a 2% NaCl solution in their drinking water for 7 d, the GABAergic PSPs/PSCs recorded in the MNCs were almost always excitatory (75 of 76 neurons; Fig. 1a, bottom trace). Under these conditions, the EGABA was −34.0 ± 1.6 mV (n = 25; Figs. 2, 3), with a range between −9 to −45 mV. This value was not statistically different from the Emuscimol (−39.3 ± 1.6 mV, n = 9, p > 0.072) and considerably less negative than the RMP (−57.5 ± 1.4 mV, n = 25). Gabazine (3 μm, n = 15) and another GABAA receptor antagonist, bicuculline (30 μm, n = 15; Fig. 1b), reversibly blocked the EPSPs/EPSCs occurring in the MNCs of the hyperosmolar rats. Moreover, gabazine (3 μm) decreased the firing rates of these neurons (Fig. 1d); the firing rates before, during, and after the application of gabazine were 1.8 ± 0.3 (n = 15), 0.4 ± 0.2 (n = 15), and 1.5 ± 0.3 impulses/s (n = 13), respectively (overall difference, p < 0.0001; before vs during, p < 0.0001; during vs after, p = 0.0007; before vs after, p = 0.0778; GEE methods). Conversely, muscimol (5 μΜ, 50 ms focal application) depolarized the membrane (15.8 ± 6.2 mV, n = 20) and elicited action potentials (20 of 20 neurons) (Fig. 1c). These results indicate that the chronic hyperosmotic stress causes the EGABA to depolarize beyond the action potential threshold in MNCs and thus converts GABAA receptor-mediated inhibition into excitation in these neurons.
Figure 3.

Time course of depolarizing shift of EGABA. Groups of rats (n = 3–8 per group) were held under durations of high Na+ drinking water (2% NaCl). Control rats were supplied with tap water as drinking water (T group). The S1 group was exposed to the salty water for 1 d, whereas the S7 group drank salty water for 7 d. ST1, ST3, and ST7, Rats supplied with tap water as drinking water for 1, 3, and 7 d, respectively, after the high Na+ water for 1 week. a, Histogram shows the EGABA for each of the groups. The number of cells used in calculating the mean is given in parentheses. The mean values denoted with the same symbols were not significantly different from each other, but they were different from the means indicated with different symbols (pairwise comparisons with Holm–Sidak method after one-way ANOVA). b, Histogram plots of the plasma osmolality values obtained for each of the groups. *p < 0.005, **p < 0.001 when compared with the mean of T group (pairwise comparisons with Holm–Sidak method after one-way ANOVA); †indicates difference from values denoted by asterisks.
GABAergic excitation takes days to develop and is reversible
In the next set of experiments, we sought to determine the time course of the EGABA change (Fig. 3). We measured plasma osmolality and the EGABA for groups of rats held for 1 and 7 d with high Na+ drinking water (2% NaCl) and then 1, 3, and 7 d after the return to tap water. One day after the rats were switched to the high Na+ drinking water, the plasma osmolality had already significantly increased (p < 0.05 compared with controls), but the EGABA was still the same as control. By 7 d on the high Na+ drinking water, both plasma osmolality and the EGABA were significantly changed (p < 0.05). To determine whether this switch in EGABA was reversible, rats that had been exposed to high Na+ drinking water for 7 d were then returned to regular water for 1, 3, and 7 d. The EGABA shift was reversed after 7 d but not 1 or 3 d. These results indicate that the depolarizing EGABA shift takes days to develop and is reversible.
Additional control experiments
Recent studies have found that GABAergic excitation in immature CNS neurons can be reversed by adding energy sources to the recording medium and thus raises the possibility that GABAergic excitation is an artifact of metabolic condition (Rheims et al., 2009; Holmgren et al., 2010). To determine whether GABAergic excitation in MCNs could be reversed with energy supplements, we investigated the effect of replacing the slice-perfusing ACSF by an energy-enriched medium containing ketone bodies and pyruvate (Holmgren et al., 2010). Under these energy-enriched conditions, GABAergic PSPs/PSCs of the MNCs (n = 22) of hyperosmolar rats (n = 3) were still excitatory. Moreover, the EGABA of the MNCs recorded in the energy-enriched medium (−34.5 ± 2.7 mV, n = 22) was essentially the same as that of the cells recorded in the ACSF (−34.0 ± 1.6 mV, n = 25). These results argue against the idea that the GABAergic excitation in the MNCs is an artifact of the metabolic conditions and fit with recent studies on GABAergic excitation in developing neurons (Kirmse et al., 2010; Ruusuvuori et al., 2010; Tyzio et al., 2011).
The osmolality and Cl− concentration of the standard perfusion medium (i.e., ACSF) used in this study were 300.5 ± 0.5 mOsm/kg H2O (n = 4) and 127.8 ± 0.4 mm (n = 5), respectively, whereas those of the CSF of hyperosmolar rats were 347.3 ± 2.1 mOsm/kg H2O (n = 3) and 151.9 ± 3.9 mm (n = 9). To exclude the possibility that the exposure of slices from hyperosmolar rats to an acute osmolar change at the time of recording led to the emergence of GABAergic excitation in MNCs, we raised the osmolality and Cl− concentration of the slice perfusion medium to the in vivo levels (i.e., 348 mOsm/kg H2O and 152 mm) by adding NaCl. Under these conditions, the GABAergic PSPs/PSCs were still mostly excitatory (20 of 21 neurons, n = 3 rats), just as those observed in the standard perfusion medium. Furthermore, their EGABA values (−32.5 ± 2.9 mV, n = 21) were not significantly different from those of the GABAergic PSCs recorded in the standard perfusion medium [−34.0 ± 1.6 and −30.5 ± 1.6 mV (n = 25); corrected and uncorrected values, respectively, for the difference in Cl− concentration between the ACSF and the CSF of rats used]. Thus, these results rule out the possibility that the GABAergic excitation is an artifact resulting from the exposure of slices to an acute osmolar change.
Last, to ensure that the GABAergic EPSPs/EPSCs were not an artifact of gramicidin-perforated recording technique, we obtained extracellular single-unit recordings from the MNCs and examined the effect of muscimol (10 μm, 10 ms focal application) on their firing rates in the presence of AP-5 (100 μm) and DNQX (20 μm). Muscimol increased the firing rate of all neurons (25 of 25 cells; Fig. 4a) recorded in the slices from hyperosmolar rats (n = 2). In contrast, muscimol decreased the firing rate in a majority of neurons (26 of 29 cells; Fig. 4b) recorded in slices from control rats (n = 4); in the remaining cells, it increased the firing rate. The proportion of cells excited or inhibited by muscimol was significantly different between control and hyperosmolar rats (p < 0.001, Fisher's exact test). Bath application of bicuculline (30 μm) prevented both the excitatory (10 of 10 neurons) and inhibitory (6 of 6 neurons) effects of muscimol (data not shown). Thus, chronic hyperosmotic stress produces excitatory effects of GABA even when experiments are performed with a technique that keeps the membrane of the recorded cell intact.
Figure 4.

Excitatory effect of muscimol on the extracellularly recorded single-unit electrical activities of MNCs of hyperosmolar rats. a, Responses to muscimol (10 μm, 10 ms focal application once every 30 s, indicated by ▴) of a silent MNC recorded in slice from a hyperosmolar rat. b, Responses to muscimol (10 μm, 10 ms focal application once a minute, indicated by ▴) of a spontaneously active MNC recorded in slice from a control rat.
GABAergic PSPs in parvocellular neurons of the PVN are not altered by chronic hyperosmotic stress
To determine whether chronic hyperosmotic stress alters GABAergic signaling in neurons other than MNCs, we examined the GABAergic PSPs of parvocellular neurons recorded in brain slices. These cells were sampled from the medial portion of the PVN that is highly packed with corticotrophin-releasing hormone/AVP colocalizing neurons that are stress-activated (Tramu et al., 1983; Mouri et al., 1993; Kovacs and Sawchenko, 1996; Jiang et al., 2004). The GABAergic PSPs in these neurons were mostly inhibitory regardless of the osmolar condition of the rat (control, 22 of 25 cells; hyperosmolar, 13 of 18 cells; p = 0.247, Fisher's exact test, n = 3 rats per group). These results suggest that the change in GABAergic PSP/PSC profile is a specific physiological adaption within the osmoregulatory circuit to chronic hyperosmotic stress.
NKCC1 contributes to the depolarizing shift of EGABA and the resultant emergence of GABAergic excitation
Previous work suggests that the Cl−-importing transporter NKCC1 plays a critical role in GABAergic excitation in neurons (Ben-Ari, 2002; Choi et al., 2008; Irwin and Allen, 2009; Belenky et al., 2010). To examine the role of this transporter in the depolarizing shift of EGABA in MNCs (Fig. 4), we first substituted NaCl in the slice perfusion medium with Tris-Cl. This experimental manipulation reduces NKCC1 activity because the transporter requires external Na+ (Russell, 2000). We then determined that reversal potential (Emuscimol) of the response elicited by the focal application of the GABAA receptor agonist muscimol (5 μm). In the MNCs of hyperosmolar rats (n = 7), the Emuscimol was estimated to be −24.3 ± 2.4 mV (n = 13) under control condition and −43.9 ± 2.1 mV (n = 13) when sodium was replaced. Returning these cells to the original medium caused the Emuscimol to return to −25.5 ± 5.3 mV (n = 7) (overall difference, p < 0.0001; before vs during the perfusion with low-Na+ medium, p < 0.0001; during vs after the perfusion with low-Na+ medium, p = 0.0079; before vs after the perfusion with low-Na+ medium, p = 0.0933; GEE method) (Fig. 5a1,b1,c1). Meantime, in the MNCs of control rats (n = 3), the Emuscimol values estimated before, during, and after the application of the low-Na+ medium were −56.1 ± 3.0 mV (n = 9), −64.2 ± 2.1 mV (n = 9), and −53.6 ± 1.8 mV (n = 4), respectively (overall difference, p < 0.0001; before vs during the perfusion with low-Na+ medium, p = 0.0103; during vs after the perfusion with low-Na+ medium, p = 0.0351; before vs after the perfusion with low-Na+ medium, p = 0.1189; GEE method) (Fig. 5a2,b2,c2). Thus, inhibiting NKCC1 by sodium replacement caused reversible hyperpolarizing shifts in Emuscimol in the MNCs of both control and hyperosmolar rats, with this effect being greater in the latter (8.1 ± 1.7 mV, n = 9 vs 19.6 ± 3.0 mV, n = 13; Mann–Whitney U test, p = 0.002). These results suggest that NKCC1 plays a role in setting EGABA in both resting and chronically active MNCs. More importantly, they suggest that chronic hyperosmotic stress increases NKCC1 activity in MNCs, making EGABA depolarize and driving GABAergic EPSPs.
Figure 5.
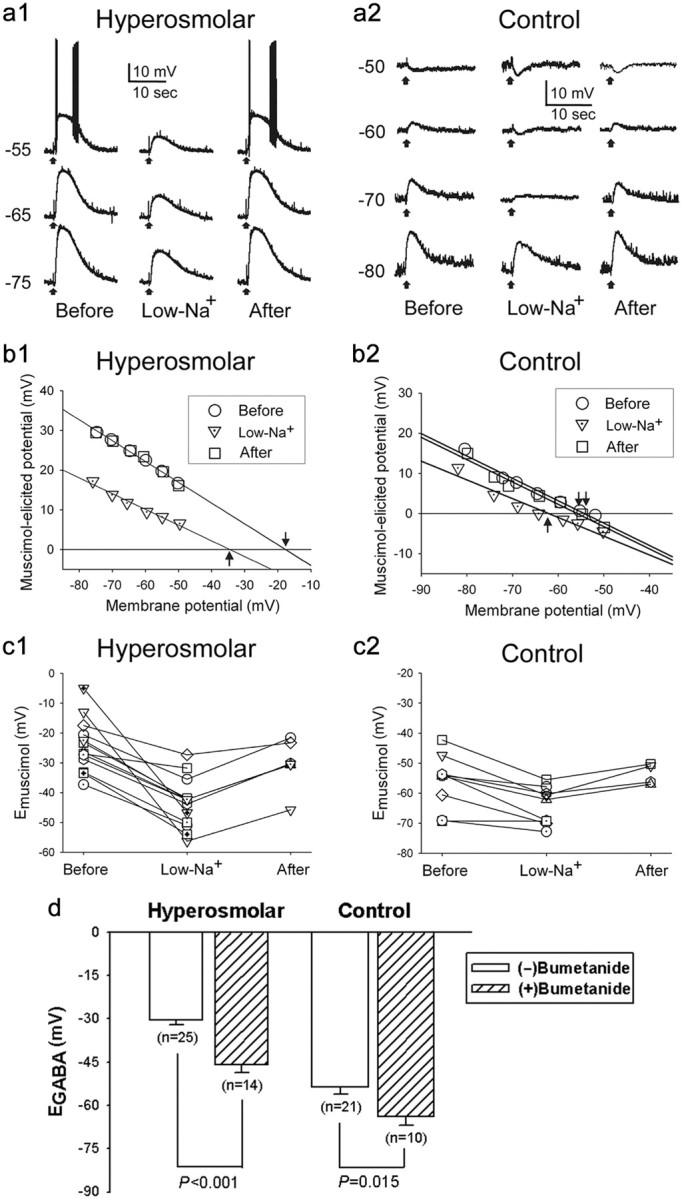
Both low-Na+ recording solution and the NKCC1 blocker bumetanide prevent the depolarizing shifts in Emuscimol and EGABA. a, Responses to muscimol (5 μm, 50 ms focal application, ↑) of MNCs (a1, hyperosmolar; a2, control) recorded in current-clamp mode before, during, and after the application of low-Na+ medium. The numbers to the left of the voltage traces denote holding potentials (in millivolts). b1, b2, Plots against holding potentials of the amplitudes of muscimol-elicited responses in a1 and a2. Responses not illustrated in a1 and a2 are also included in these plots. The intersections with the abscissa of the regression lines used to fit the data points (arrows) were taken as Emuscimol values. c, Graphic summary of the low-Na+ medium-induced changes in Emuscimol detected in different MNCs of hyperosmolar (c1) and control (c2) rats. The symbols connected by lines denote data from the same cells. d, Graphic summary of the bumetanide effects on the EGABA of MNCs of hyperosmolar and control rats.
To obtain additional evidence for the role of NKCC1, we next examined the effect of bumetanide (NKCC inhibitor) on EGABA by comparing EGABA values measured in the presence and the absence of the drug. Bumetanide was bath applied at 10 μm, a concentration that can be used to block NKCC without significantly affecting the K+–Cl− cotransporter (KCC) (Payne et al., 2003). We found that the proportion of MNCs of hyperosmolar rats that exhibited GABAergic EPSPs/EPSCs was significantly smaller in the presence of bumetanide (7 of 14 vs 25 of 25, p < 0.001, Fisher's exact test). In addition, the application of bumetanide caused a significant negative shift in the EGABA measured in both hyperosmolar and control conditions (Fig. 5d). Conversely, bumetanide treatment did not alter RMP and input resistance of the MNC measured in control and hyperosmolar rats (control, −56.6 ± 2.1 mV, n = 10 vs −58.3 ± 2.1 mV, n = 21 and 370 ± 64 MΩ, n = 10 vs 398 ± 34 MΩ, n = 21; hyperosmolar, −61.3 ± 2.4 mV, n = 14 vs −57.5 ± 1.4 mV, n = 25 and 262 ± 29 MΩ, n = 14 vs 266 ± 26 MΩ, n = 25). These results again suggest that NKCC1 plays a role in setting EGABA in the MNCs in both control and hyperosmolar rats. Furthermore, the results suggest that intracellular Cl− accumulation by NKCC1 is critical for the depolarizing shift of EGABA and the consequent emergence of GABAergic excitation in the MNCs of hyperosmolar rat.
Chronic hyperosmotic stress upregulates NKCC1 expression
Previous work suggests that NKCC1 is expressed weakly in the MNCs (Kanaka et al., 2001). We sought to determine whether the chronic hyperosmotic stress would change this expression. First, we used the Western blot technique to measure NKCC1 and KCC2 from tissue samples of the SON (Fig. 6). We were able to detect both Cl− transporters from the samples. Interestingly, the chronic hyperosmotic stress produced by 7 d of high Na+ drinking water produced a profound increase in the level of NKCC1 protein (137.0 ± 23.1%, n = 8) in the SON. This increase in protein expression was fully reversible after 7 d in normal water. The hyperosmotic stress also produced a modest increase in KCC2 levels (34.1 ± 6.3%, n = 12), although this increase did not reverse after return to normal drinking water. These suggest that an upregulation in the levels of NKCC1 expression underlies the dynamic change in EGABA in the MNCs of hyperosmolar rats.
Figure 6.
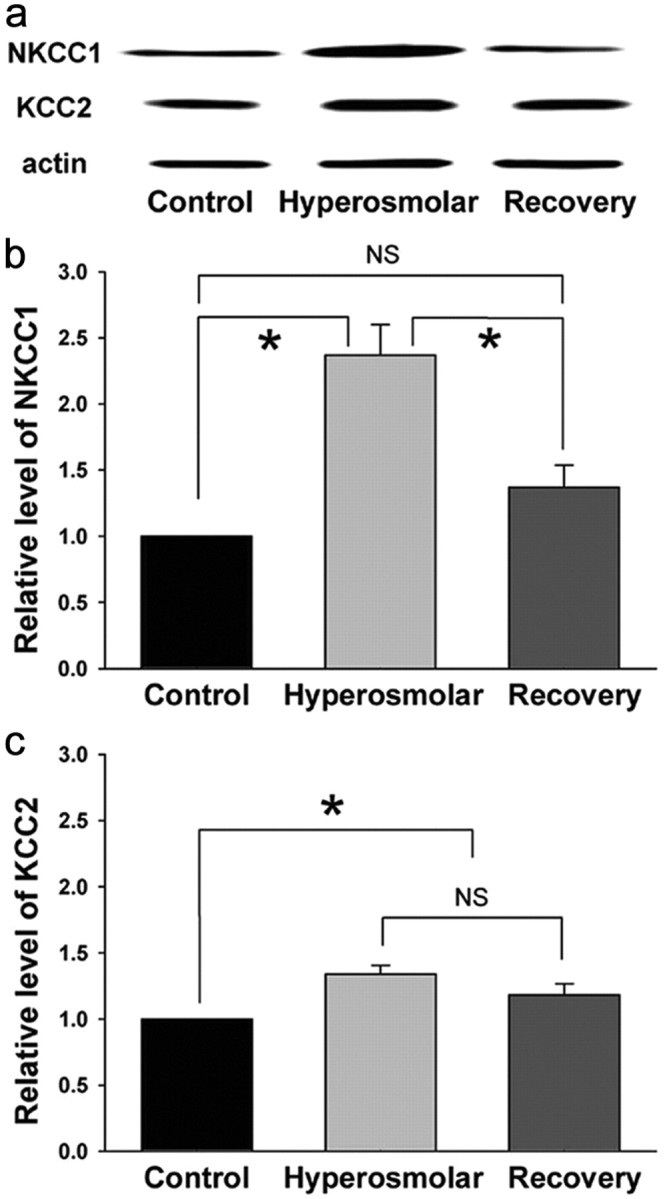
Chronic hyperosmotic stress increases the NKCC1 protein level in the SON. a, NKCC1, KCC2, and actin bands recognized by Western blot for the SON tissue samples obtained from rats under different experimental conditions. b, c, Summary graphs showing the relative levels of NKCC1 (n = 8) and KCC2 (n = 12) in the SONs of rats under different experimental conditions. For each experiment, the values were normalized to the samples collected from the control rats. NKCC1 and KCC2 levels were normalized to actin to control for loading. *p < 0.05; NS, statistically not different (pairwise comparison with Tukey's test or Dunn's method after Kruskal–Wallis one-way ANOVA on ranks). Recovery, Rats supplied with tap water as drinking water for 7 d after 2% NaCl drink for 1 week.
Next we used immunohistochemical techniques to better determine the anatomical location of NKCC1 expression (Fig. 7). In both the SON and PVN, the NKCC1 immunoreactivities were localized in MNCs with soma diameters of 20–30 μm (Fig. 7a,b). In response to hyperosmotic stress, NKCC1 immunoreactivity robustly increased in both brain regions. Densitometric analyses of the intensities of NKCC1-positive signals revealed that NKCC1 expression in the SON as well as the cell perimeters (presumably the membranes) of SON neurons were significantly greater in hyperosmolar animals (Fig. 7c,d). We were unable to detect NKCC1 immunoreactivity in the parvocellular division of the PVN. These results again support the hypothesis that the upregulation of NKCC1 contributes to the depolarization of EGABA and the consequent emergence of GABAergic excitation.
Figure 7.

Chronic hyperosmotic stress increases immunohistochemically detectable NKCC1 signals in the SON and PVN. a, b, NKCC1-positive signals in the SON and PVN. Bottom rows, Magnified views of the areas in the top rows indicated with dotted boxes. Scale bar, 300 μm. 3V, Third ventricle; OC, optic chiasm. c, d, NKCC1 levels in the SON and the cell perimeters (presumably the membranes) of SON neurons. These levels were normalized to that in the LH located above the SON. The numbers in parentheses in c and d denote the numbers of rats and cells included in the analyses, respectively. *p < 0.05 (pairwise comparison with Holm–Sidak method after one-way ANOVA in c and pairwise comparison with Dunn's method after Kruskal–Wallis one-way ANOVA on ranks in d). Recovery, Rats supplied with tap water as drinking water for 7 d after 2% NaCl drink for 1 week.
Centrally released oxytocin may link chronic hyperosmotic stress to the EGABA shift
Hyperosmotic stress is a good stimulus for the release of AVP and oxytocin from the dendrites of MNCs in the PVN and SON (Ludwig, 1998), and it is possible that these neuropeptides could drive the changes in EGABA. In the next set of experiments (Fig. 8), we examined the effects of intracerebroventricular infusion of desGly-NH2,d(CH2)5 [d-Tyr2, Thr4]OVT (highly selective oxytocin receptor antagonist) on the depolarizing shift of EGABA and the emergence of GABAergic EPSCs in MNCs. The oxytocin receptor antagonist was infused (2.4 μg/d) (Teruyama et al., 2008) for 10 d while the rats were exposed to the high Na+ drinking water. The vehicle (i.e., ACSF) infusion served as the control. Brain slices were then prepared, and EGABA in the MNCs was determined as described previously. The pretreatment with the selective oxytocin receptor antagonist significantly reduced the depolarizing EGABA shift in the MNCs of hyperosmolar rats (ACSF, −30.2 ± 1.7 mV, n = 36 vs oxytocin receptor antagonist, −39.4 ± 2.5 mV, n = 29, p < 0.05; Fig. 8a, left). The antagonist also significantly reduced the proportion of MNCs showing GABAergic EPSCs (ACSF, 36 of 36 vs oxytocin receptor antagonist, 19 of 29, p < 0.003; Fig. 8b, left). For comparison, we also examined the impact of the intracerebroventricular infusion (10 d, 5.184 μg/d) (Dobruch et al, 2005) of the AVP receptor antagonist d(CH2)5[Tyr(Me)2, Ala-NH29]AVP. This antagonist had no significant effect on the depolarizing EGABA shift occurring in the MNCs of hyperosmolar rats (ACSF, −30.2 ± 1.7 mV, n = 36 vs AVP receptor antagonist, −36.4 ± 3.0 mV, n = 35, p > 0.05; Fig. 8a, left), although it reduced significantly the proportion of MNCs exhibiting GABAergic EPSCs (ACSF, 36 of 36 vs AVP receptor antagonist, 28 of 35, p = 0.015; Fig. 8b, left). In control rats, neither of the AVP and oxytocin receptor antagonists had effect on the EGABA and GABAergic PSC profile (Fig. 8a,b, right). These results suggest that central oxytocin receptor activation is part of the signal that links the chronic hyperosmotic stress to shift to GABAergic excitation in the PVN and SON.
Figure 8.

Intracerebroventricularly administered oxytocin receptor antagonist prevents the depolarizing shift in EGABA in MNCs. The selective oxytocin receptor antagonist desGly-NH2,d(CH2)5[d-Tyr2, Thr4]OVT was administered intracerebroventricularly for 10 d while the rats were exposed to the high Na+ drinking water. The impact of the intracerebroventricular infusion of the AVP receptor antagonist d(CH2)5[Tyr(Me)2, Ala-NH29]AVP is shown for comparison. The impact of the combination of the two antagonists was also determined and is labeled “Cocktail.” The ACSF infusion served as the control. Brain slices were prepared and EGABA was estimated with PSCs evoked in the presence of AP-5 (100 μm) and DNQX (20 μm). a, Histograms show the EGABA under control and hyperosmolar conditions. b, Histograms show the percentage of neurons expressing GABA-mediated excitatory currents under each condition. The number of MNCs sampled in each experiment (each group contains 3–5 rats) is given in parentheses. *p < 0.05, significant difference from control as determined by Kruskal–Wallis one-way ANOVA on ranks followed by pairwise comparisons with Dunn's method. NS, Statistically not different.
The lack of effect of the AVP receptor antagonist on EGABA can be interpreted as suggesting that AVP has no significant role in the depolarizing EGABA shift and the resultant emergence of GABAergic excitation. Alternatively, under the assumption that this antagonist has an anti-oxytocin as well as anti-AVP efficacy, it can be interpreted as suggesting that AVP and oxytocin have opposite effects. To test this possibility more directly, we next examined the effect of the mixture of the AVP and oxytocin receptor antagonists. Our rationale was that, if indeed AVP has an effect opposite to that of oxytocin, the mixture of AVP and oxytocin receptor antagonists would exert smaller effects on EGABA and GABAergic PSCs profile than oxytocin receptor antagonist alone. The results (Fig. 8a,b, left) show that the mixture had essentially the same effect as the oxytocin receptor antagonist alone, indicating that AVP has little or no role in linking hyperosmotic stress to depolarizing EGABA shift.
GABAergic excitation contributes to the secretion of AVP and oxytocin in rats under chronic hyperosmotic stress
Finally, to assess the functional significance of GABAergic neurotransmission in MNCs, we blocked GABAA receptors and examined the plasma levels of AVP and oxytocin (Fig. 9). In hyperosmolar rats, blocking GABAA receptors with an intravenous injection of bicuculline (2 mg/kg body weight: subconvulsive dose) caused a significant decrease in AVP level after 30 min of the injection; the decrease in oxytocin level was also seen at 10 and 20 min after the injection. Conversely, the bicuculline injection did not produce any significant changes in the levels of AVP and oxytocin in control rats.
Figure 9.

GABAA receptor blockade lowers plasma levels of AVP and oxytocin under hyperosmolar conditions. Plasma levels of AVP (left) and oxytocin (right) before and after intravenous injection (↑) of bicuculline methiodide (BIC) in the control (○) and hyperosmolar (■) rats, which were measured with ELISA. AVP and oxytocin concentrations are normalized to the level at 0 min. The number of rats used in each experiment is given to the right of the corresponding graph. *p < 0.005, **p < 0.0001 compared with the value at 0 min (GEE method; overall difference, p < 0.0001).
Discussion
The present results demonstrate that, under resting conditions, the GABAA receptor-mediated transmission in MNCs is inhibitory in ∼75% of the neurons. Importantly, GABAergic transmission becomes excitatory in virtually all MNCs when there is a sustained physiological demand for increased secretion of AVP and oxytocin. The hyperosmotic stress caused a profound depolarizing shift in the reversal potential of GABAergic response (EGABA) in MNCs. This EGABA shift was associated with increased expression of NKCC1 in MNCs and was prevented by treatments that blocked NKCC activity. Blocking central oxytocin receptors prevented the shift to GABAergic excitation under chronic hyperosmotic stress. These results suggest that GABAergic responses of MNCs are not consistently inhibitory but switch between inhibition and excitation in response to physiological needs.
The present study is not the first that demonstrates the emergence of excitatory actions of GABA in mature CNS neurons under particular conditions or states. Previous studies have shown that pathological conditions such as epilepsy, axonal injury, and neuropathy can cause a depolarizing shift in EGABA in neurons related to the pathologies, reducing the magnitude of GABAergic inhibition in these cells and, in some cases, even producing GABAergic excitation (De Koninck, 2007). Similarly, a recent study has shown that acute restraint stress weakens GABAA receptor-mediated inhibition in putative corticotrophin-releasing hormone neurons in the PVN by causing EGABA to depolarize (Hewitt et al., 2009). Furthermore, this study has demonstrated that, after the stress, repetitive activation at high frequencies of GABAergic inputs to these cells produces GABAA receptor-mediated excitation. However, the EGABA shifts in these studies were not large enough such that a single stimulation of GABAergic afferent seldom elicited an action potential. In contrast, in the current study, a single stimulation of GABAergic inputs evoked action potentials in essentially all of the MNCs, by virtue of the large depolarization of EGABA. To the best of our knowledge, the results from the current study provide the first demonstration that a physiological state or environmental challenge can induce a profound EGABA change in a specific population of adult CNS neurons so that GABA functions as an excitatory neurotransmitter in these cells.
The EGABA shifts in MNCs did not follow the time course of osmolality shifts during the chronic hyperosmotic treatment; they took days to establish and recover. The relatively slow depolarization of EGABA might be related to that, at the early stages of hyperosmotic stress, changes in glutamatergic or other excitatory input to MNCs are sufficient for the secretion of AVP and oxytocin needed for coping with the stress. Meanwhile, the slow recovery of EGABA may represent a delayed disarmament for possible re-attack of hyperosmotic stress.
The results of the present study suggest that the upregulation of the Cl− importer NKCC1 is a key mechanism contributing to the depolarizing EGABA shift and the resultant inhibitory-to-excitatory switch in GABAergic transmission in the MNCs of rats subjected to chronic hyperosmotic stress. Previous work found that NKCC1 is expressed at a low level in the PVN (Kanaka et al., 2001). In the PVN and SON, we found low basal levels of NKCC1 protein that could be dynamically upregulated in response to osmotic stress. The techniques used do not allow us to determine the relative roles of transcription or trafficking of the protein in the increased expression. The immunohistochemistry results suggest that more NKCC1 is localized in the cell perimeter presumably in the membrane. NKCC1 activity has also been found to be critical for the GABAergic excitation observed in immature cortical neurons (Ben-Ari, 2002) and gonadotropin-releasing hormone neurons (DeFazio et al., 2002) as well as in the suprachiasmatic nucleus (Belenky et al., 2008, 2010; Choi et al., 2008).
Downregulation of the activity of the Cl−-extruding transporter KCC2 is another mechanism that has been postulated to contribute to GABAergic excitation (Coull et al., 2003; Hewitt et al., 2009). However, we found that the level of KCC2 expression was increased, not decreased, by the chronic hyperosmotic stress. Expression of a protein as measured by Western blot is not always linked to activity of the transporter, and future work on the role of KCC2 is required. Interestingly, previous work suggests that KCC2 may play other physiological roles besides Cl− transport (Li et al., 2007). It is possible that the upregulated KCC2 contributes to the structural remodeling of MNCs and their synapses that occurs under chronically hyperosmolar condition (Theodosis et al., 1995; Hatton, 1997). Our finding that the KCC2 level in the SON remained elevated after return to normal drinking water, unlike the changes in NKCC1 and EGABA, is consistent with this hypothesis.
The results of the current study suggest that oxytocin is a key signal that links chronic hyperosmotic stress to the depolarizing EGABA shifts in MNCs. Conversely, they do not support a role for AVP; if this neuropeptide were important for the EGABA shifts or had an effect opposite to that of oxytocin, the mixture of the AVP and oxytocin receptor antagonists would have exerted an effect different from that of oxytocin receptor antagonist alone. In this study, we have not determined the source of oxytocin and the location of its receptors responsible for the EGABA shifts. Whether the activation of oxytocin receptors in MNCs (Freund-Mercier et al., 1994) or nearby glial cells (Di Scala-Guenot and Strosser, 1992; Di Scala-Guenot et al., 1994) by oxytocin from the dendrites of MNCs (Ludwig, 1998) leads to the shifts is open for additional investigation.
Our results (Fig. 5c,d) indicate that EGABA was different between control and hyperosmolar rats after NKCC1 blockade with bumetanide and in low-Na+ conditions. It may be that these manipulations did not completely block the NKCC1 transporter. Alternatively, it may be that other transporters/factors also influence EGABA. However, KCC2 is unlikely to be an explanation for the more positive EGABA values in the MNCs of hyperosmolar rats, because its expression was increased by chronic hyperosmotic stress.
The secretion of AVP and oxytocin from MNCs depends on the electrical activities of these cells (Poulain and Wakerley, 1982; Renaud and Bourque, 1991). In this study, we found that GABAA receptor activation by muscimol increased the firing rate of the MNCs of hyperosmolar rats, whereas GABAA receptor blockade by gabazine did the opposite. Moreover, we discovered that intravenously injected bicuculline (GABAA receptor antagonist) lowered the plasma levels of AVP and oxytocin in these animals. There was a difference in the measured time course of the effects that is likely explained by the differences between the half-lives of these hormones in the blood (AVP, 2–17 min vs oxytocin, 1–2 min; Janáky et al., 1982; Gimpl and Fahrenholz, 2001). The absence of bicuculline-induced release of AVP and oxytocin in control rats might be attributable to that the amount of bicuculline injected (2 mg/kg body weight: subconvulsive dose) was too small to remove significantly the strong GABAergic inhibition. Together, these results indicate that the GABAA receptor-mediated transmission in the MNCs promotes the secretion of AVP and oxytocin from these cells by increasing their firing rates during chronic hyperosmotic stress.
In summary, this study demonstrates that, in response to physiological needs, GABA can switch from functioning as an inhibitory transmitter to evoking an excitatory response in MNCs. Most neurons are embedded in circuits in which their intrinsic firing is regulated by the balance of excitatory and inhibitory transmission; we have found evidence that the postsynaptic neuron can shift this balance through the regulation of the Cl− uptake mechanism NKCC1. The activation of central oxytocin receptors may provide the trigger that causes this switch. It remains to be seen whether this novel form of synaptic plasticity is specific to these hypothalamic neurons or is widespread throughout the CNS.
Footnotes
This work was supported by National Research Foundation of Korea Grants R0804721, R0804722, and R0804723 (Y.I.K.) Korea Science and Engineering Foundation Grant R13-2002-020-03002-0 (Y.-W.C.). Y.I.K., H.C.H., W.B.K., Y.-B.K., S.W.L., and H.-J.C. were supported by The Brain Korea 21 Project in 2008–2011. Authors are grateful for the editorial help of April M. Kim.
References
- Antunes-Rodrigues J, de Castro M, Elias LL, Valença MM, McCann SM. Neuroendocrine control of body fluid metabolism. Physiol Rev. 2004;84:169–208. doi: 10.1152/physrev.00017.2003. [DOI] [PubMed] [Google Scholar]
- Belenky MA, Yarom Y, Pickard GE. Heterogeneous expression of gamma-aminobutyric acid and gamma-aminobutyric acid-associated receptors and transporters in the rat suprachiasmatic nucleus. J Comp Neurol. 2008;506:708–732. doi: 10.1002/cne.21553. [DOI] [PubMed] [Google Scholar]
- Belenky MA, Sollars PJ, Mount DB, Alper SL, Yarom Y, Pickard GE. Cell-type specific distribution of chloride transporters in the rat suprachiasmatic nucleus. Neuroscience. 2010;165:1519–1537. doi: 10.1016/j.neuroscience.2009.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Lee CJ, Schroeder A, Kim YS, Jung SH, Kim JS, Kim do Y, Son EJ, Han HC, Hong SK, Colwell CS, Kim YI. Excitatory actions of GABA in the suprachiasmatic nucleus. J Neurosci. 2008;28:5450–5459. doi: 10.1523/JNEUROSCI.5750-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2872–2891. doi: 10.1210/me.2002-0163. [DOI] [PubMed] [Google Scholar]
- De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol. 2007;7:93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Di Scala-Guenot D, Strosser MT. Oxytocin receptors on cultured astroglial cells. Kinetic and pharmacological characterization of oxytocin-binding sites on intact hypothalamic and hippocampic cells from foetal rat brain. Biochem J. 1992;284:491–497. doi: 10.1042/bj2840491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Scala-Guenot D, Mouginot D, Strosser MT. Increase of intracellular calcium induced by oxytocin in hypothalamic cultured astrocytes. Glia. 1994;11:269–276. doi: 10.1002/glia.440110308. [DOI] [PubMed] [Google Scholar]
- Dobruch J, Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E. Enhanced involvement of brain vasopressin V1 receptors in cardiovascular responses to stress in rats with myocardial infarction. Stress. 2005;8:273–284. doi: 10.1080/10253890500456287. [DOI] [PubMed] [Google Scholar]
- Freund-Mercier MJ, Stoeckel ME, Klein MJ. Oxytocin receptors on oxytocin neurones: histoautoradiographic detection in the lactating rat. J Physiol. 1994;480:155–161. doi: 10.1113/jphysiol.1994.sp020349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- Hatton GI. Function-related plasticity in hypothalamus. Annu Rev Neurosci. 1997;20:375–397. doi: 10.1146/annurev.neuro.20.1.375. [DOI] [PubMed] [Google Scholar]
- Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABAA receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J Neuroendocrinol. 2011;23:557–569. doi: 10.1111/j.1365-2826.2011.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Holmgren CD, Mukhtarov M, Malkov AE, Popova IY, Bregestovski P, Zilberter Y. Energy substrate availability as a determinant of neuronal resting potential, GABA signaling and spontaneous network activity in the neonatal cortex in vitro. J Neurochem. 2010;112:900–912. doi: 10.1111/j.1471-4159.2009.06506.x. [DOI] [PubMed] [Google Scholar]
- Irwin RP, Allen CN. GABAergic signaling induces divergent neuronal Ca2+ responses in the suprachiasmatic nucleus network. Eur J Neurosci. 2009;30:1462–1475. doi: 10.1111/j.1460-9568.2009.06944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janáky T, László FA, Sirokmán F, Morgat JL. Biological half-life and organ distribution of [3H]8-arginine-vasopressin in the rat. J Endocrinol. 1982;93:295–303. doi: 10.1677/joe.0.0930295. [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Kawashima H, Iwasaki Y, Uchida K, Sugimoto K, Itoi K. Differential effects of forced swim-stress on the corticotropin-releasing hormone and vasopressin gene transcription in the parvocellular division of the paraventricular nucleus of rat hypothalamus. Neurosci Lett. 2004;358:201–204. doi: 10.1016/j.neulet.2004.01.041. [DOI] [PubMed] [Google Scholar]
- Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience. 2001;104:933–946. doi: 10.1016/s0306-4522(01)00149-x. [DOI] [PubMed] [Google Scholar]
- Kim DY, Choi HJ, Kim JS, Kim YS, Jeong DU, Shin HC, Kim MJ, Han HC, Hong SK, Kim YI. Voltage-gated calcium channels play crucial roles in the glutamate-induced phase shifts of the rat suprachiasmatic circadian clock. Eur J Neurosci. 2005;21:1215–1222. doi: 10.1111/j.1460-9568.2005.03950.x. [DOI] [PubMed] [Google Scholar]
- Kirmse K, Witte OW, Holthoff K. GABA depolarizes immature neocortical neurons in the presence of the ketone body β-hydroxybutyrate. J Neurosci. 2010;30:16002–16007. doi: 10.1523/JNEUROSCI.2534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács KJ, Sawchenko PE. Sequence of stress-induced alterations in indices of synaptic and transcriptional activation in parvocellular neurosecretory neurons. J Neurosci. 1996;16:262–273. doi: 10.1523/JNEUROSCI.16-01-00262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JE. The hypothalamus as a major integrating center. In: Conn PM, Freeman ME, editors. Neuroendocrinology in physiology and medicine. Totowa, NJ: Humana; 2000. pp. 75–93. [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Ludwig M. Dendritic release of vasopressin and oxytocin. J Neuroendocrinol. 1998;10:881–895. doi: 10.1046/j.1365-2826.1998.00279.x. [DOI] [PubMed] [Google Scholar]
- Lytle C, Xu JC, Biemesderfer D, Forbush B., 3rd Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol. 1995;2691:C1496–C1505. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- Manning M, Stoev S, Bankowski K, Misicka A, Lammek B, Wo NC, Sawyer WH. Synthesis and some pharmacological properties of potent and selective antagonists of the vasopressor (V1-receptor) response to arginine-vasopressin. J Med Chem. 1992;35:382–388. doi: 10.1021/jm00080a027. [DOI] [PubMed] [Google Scholar]
- Manning M, Miteva K, Pancheva S, Stoev S, Wo NC, Chan WY. Design and synthesis of highly selective in vitro and in vivo uterine receptor antagonists of oxytocin: comparisons with Atosiban. Int J Pept Protein Res. 1995;46:244–252. doi: 10.1111/j.1399-3011.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- Mouri T, Itoi K, Takahashi K, Suda T, Murakami O, Yoshinaga K, Andoh N, Ohtani H, Masuda T, Sasano N. Colocalization of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of the human hypothalamus. Neuroendocrinology. 1993;57:34–39. doi: 10.1159/000126339. [DOI] [PubMed] [Google Scholar]
- Murase K, Ryu PD, Randic M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- Paxino G, Watson C. The rat brain in stereotactic coordinates. San Diego: Academic; 1998. [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Poulain DA, Wakerley JB. Electrophysiology of hypothalamic magnocellular neurones secreting oxytocin and vasopressin. Neuroscience. 1982;7:773–808. doi: 10.1016/0306-4522(82)90044-6. [DOI] [PubMed] [Google Scholar]
- Renaud LP, Bourque CW. Neurophysiology and neuropharmacology of hypothalamic magnocellular neurons secreting vasopressin and oxytocin. Prog Neurobiol. 1991;36:131–169. doi: 10.1016/0301-0082(91)90020-2. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Ebihara S, Akaike N. Gramicidin perforated patch-clamp technique reveals glycine-gated outward chloride current in dissociated nucleus solitarii neurons of the rat. J Neurophysiol. 1994;72:1103–1108. doi: 10.1152/jn.1994.72.3.1103. [DOI] [PubMed] [Google Scholar]
- Rheims S, Holmgren CD, Chazal G, Mulder J, Harkany T, Zilberter T, Zilberter Y. GABA action in immature neocortical neurons directly depends on the availability of ketone bodies. J Neurochem. 2009;110:1330–1338. doi: 10.1111/j.1471-4159.2009.06230.x. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Ruusuvuori E, Kirilkin I, Pandya N, Kaila K. Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J Neurosci. 2010;30:15638–15642. doi: 10.1523/JNEUROSCI.3355-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE. Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol. 1991;434:271–293. doi: 10.1113/jphysiol.1991.sp018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruyama R, Lipschitz DL, Wang L, Ramoz GR, Crowley WR, Bealer SL, Armstrong WE. Central blockade of oxytocin receptors during mid-late gestation reduces amplitude of slow afterhyperpolarization in supraoptic oxytocin neurons. Am J Physiol Endocrinol Metab. 2008;295:E1167–E1171. doi: 10.1152/ajpendo.90620.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT, el Majdoubi M, Gies U, Poulain DA. Physiologically-linked structural plasticity of inhibitory and excitatory synaptic inputs to oxytocin neurons. Adv Exp Med Biol. 1995;395:155–171. [PubMed] [Google Scholar]
- Tramu G, Croix C, Pillez A. Ability of the CRF immunoreactive neurons of the paraventricular nucleus to produce a vasopressin-like material. Immunohistochemical demonstration in adrenalectomized guinea pigs and rats. Neuroendocrinology. 1983;37:467–469. doi: 10.1159/000123595. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Allene C, Nardou R, Picardo MA, Yamamoto S, Sivakumaran S, Caiati MD, Rheims S, Minlebaev M, Milh M, Ferré P, Khazipov R, Romette JL, Lorquin J, Cossart R, Khalilov I, Nehlig A, Cherubini E, Ben-Ari Y. Depolarizing actions of GABA in immature neurons depend neither on ketone bodies nor on pyruvate. J Neurosci. 2011;31:34–45. doi: 10.1523/JNEUROSCI.3314-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H, Yarom Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature. 1997;387:598–603. doi: 10.1038/42468. [DOI] [PubMed] [Google Scholar]