

Abstract
Mental retardation (MR) and autism are highly heterogeneous neurodevelopmental disorders. IL-1-receptor accessory protein-like 1 (IL1RAPL1) is responsible for nonsyndromic MR and is associated with autism. Thus, the elucidation of the functional role of IL1RAPL1 will contribute to our understanding of the pathogenesis of these mental disorders. Here, we showed that knockdown of endogenous IL1RAPL1 in cultured cortical neurons suppressed the accumulation of punctate staining signals for active zone protein Bassoon and decreased the number of dendritic protrusions. Consistently, the expression of IL1RAPL1 in cultured neurons stimulated the accumulation of Bassoon and spinogenesis. The extracellular domain (ECD) of IL1RAPL1 was required and sufficient for the presynaptic differentiation-inducing activity, while both the ECD and cytoplasmic domain were essential for the spinogenic activity. Notably, the synaptogenic activity of IL1RAPL1 was specific for excitatory synapses. Furthermore, we identified presynaptic protein tyrosine phosphatase (PTP) δ as a major IL1RAPL1–ECD interacting protein by affinity chromatography. IL1RAPL1 interacted selectively with certain forms of PTPδ splice variants carrying mini-exon peptides in Ig-like domains. The synaptogenic activity of IL1RAPL1 was abolished in primary neurons from PTPδ knock-out mice. IL1RAPL1 showed robust synaptogenic activity in vivo when transfected into the cortical neurons of wild-type mice but not in PTPδ knock-out mice. These results suggest that IL1RAPL1 mediates synapse formation through trans-synaptic interaction with PTPδ. Our findings raise an intriguing possibility that the impairment of synapse formation may underlie certain forms of MR and autism as a common pathogenic pathway shared by these mental disorders.
Introduction
Mental retardation (MR) and autism are highly heterogeneous neurodevelopmental disorders. MR, defined as a failure to develop cognitive abilities, is the most frequent cause of serious handicap in children and young adults (Chelly and Mandel, 2001). A number of genes associated with X-linked MR have been identified by positional-cloning strategies (Chelly et al., 2006; Ropers, 2006; Chiurazzi et al., 2008). Autism is comprised of a clinically heterogeneous group of disorders, collectively termed “autism spectrum disorders (ASDs),” that are characterized by severe deficits in socialization, impaired communication, and a limited range of interests and behavior (Abrahams and Geschwind, 2008; Levy et al., 2009). Although ASD is highly heritable, the identification of susceptibility genes has been hindered by the heterogeneity of the syndrome. Recently, rare variants in candidate genes have been reported (Pinto et al., 2010; Gilman et al., 2011; Levy et al., 2011; Sanders et al., 2011; Voineagu et al., 2011).
Nonsyndromic MR is characterized by reduced cognitive function without any other clinical features, thus providing the most direct approach to specifically study the neurobiology of cognition and pathogenesis of MR. IL1-receptor accessory protein-like 1 (IL1RAPL1) was initially identified as the product of an X-linked gene responsible for a nonsyndromic form of MR (Carrié et al., 1999). Subsequently, the IL1RAPL1 gene was also shown to be associated with autism (Piton et al., 2008). The observation that mutations in IL1RAPL1 may lead to MR, autism, or both is in line with recent studies noting an overlap of genetic loci in susceptibility to these disorders (Jamain et al., 2003; Durand et al., 2007; Kim et al., 2008; Berkel et al., 2010; Laumonnier et al., 2010). In fact, cognitive impairment is common in autism, and ∼70% of autistic individuals suffer from MR (Fombonne, 1999). Although the underlying causes of these mental disorders are extremely heterogeneous, molecular alterations in monogenic disorders may identify common pathogenic pathways shared by MR or autism or both (Bill and Geschwind, 2009). Thus, the elucidation of functional roles of IL1RAPL1 will contribute to our understanding of the pathogenesis of MR and autism. IL1RAPL1 regulates N-type voltage-gated calcium channel and neurite elongation in PC12 cells through the neuronal calcium sensor-1 (Bahi et al., 2003; Gambino et al., 2007) and synaptic localization of PSD-95 by controlling c-Jun terminal kinase (JNK) activity (Pavlowsky et al., 2010). The JNK/PSD-95 pathway was implicated in impaired hippocampal long-term potentiation and the decrease of hippocampal dendritic spines observed in IL1RAPL1 knock-out mice (Pavlowsky et al., 2010). On the other hand, we found that in zebrafish presynaptic IL1RAPL1 regulates synapse formation in vivo of olfactory sensory neurons by stimulating both synaptic vesicle accumulation and morphological remodeling of the axon terminals (Yoshida and Mishina, 2008). Here, we examined the possible role of IL1RAPL1 in synapse formation of mammalian cortical neurons. Our results suggest that IL1RAPL1 mediates synapse formation through trans-synaptic interaction with presynaptic protein tyrosine phosphatase (PTP) δ. These results imply that the impairment of synapse formation may underlie the pathogenesis of certain forms of MR and autism.
Materials and Methods
Construction of expression vectors.
The entire coding sequences of mouse IL1RAPL1, IL-1R type 1 (IL-1R1), N-cadherin, neuroligin (NLGN) 1, PTPδ, PTPδA6, leukocyte antigen-related (LAR) A6B−, LARA−, LARA−B−, PTPσA−B−, PTPσA−, and netrin-G ligand (NGL)-3 were cloned into pcDNA3 vector (Invitrogen) or pcDNA3-TOPO vector (Invitrogen) to yield pIL1RAPL1, pIL-1R1, pN-cadherin, pNLGN1, pPTPδ, pPTPδA6, pLARA6B−, pLARA−, pLARA−B−, pPTPσA−B− pPTPσA−, and pNGL-3, respectively. Coding sequences of PTPδB−, PTPδA6B−, PTPδA3, PTPδA3B−, PTPδA−, PTPδA−B−, and LARA6 were generated by PCR-based mutagenesis using pPTPδ, pPTPδA6, or pLARA6B− as templates. The coding sequence of pre-protrypsin signal peptide followed by FLAG tag was ligated with the coding sequence of IL1RAPL1, IL-1R1, and NLGN1 lacking signal peptide to obtain FLAG-IL1RAPL1, FLAG-IL-1R1, and FLAG-NLGN1 coding sequences, respectively. The coding sequence of cytoplasmic domain of FLAG-IL1RAPL1 was swapped with that of FLAG-IL-1R1 to produce the FLAG-cytoplasmic domain (CPD)-swap coding sequence. The coding sequence of extracellular and transmembrane domains of FLAG-IL1RAPL1 was swapped with that of FLAG-IL-1R1 to produce the FLAG-extracellular domain (ECD)-swap coding sequence. The coding sequences of Toll/IL-1 receptor (TIR) and C-terminal domains of FLAG-IL1RAPL1 were swapped with those of FLAG-IL-1R1 to produce FLAG-TIR-swap and FLAG-CT-swap coding sequences, respectively. The coding sequences of IL1RAPL1, FLAG-IL1RAPL1, FLAG-IL-1R1, FLAG-CPD-swap, FLAG-ECD-swap, FLAG-TIR-swap, FLAG-CT-swap, FLAG-NLGN1, and N-cadherin were cloned into the multicloning site of pEGFP-CAGMCS (Uemura et al., 2010) to yield pEGFP-CAG-IL1RAPL1, pEGFP-CAG-FLAG-IL1RAPL1, pEGFP-CAG-FLAG-IL-1R1, pEGFP-CAG-FLAG-CPD-swap, pEGFP-CAG-FLAG-ECD-swap, pEGFP-CAG-FLAG-TIR-swap, pEGFP-CAG-FLAG-CT-swap, pEGFP-CAG-FLAG-NLGN1 and pEGFP-CAG-N-cadherin, respectively. The entire coding sequences of PTPδ and PTPδA−B− were cloned into the multicloning site of pRFP-CAGMCS (Uemura et al., 2010) to yield pRFP-CAG-PTPδ and pRFP-CAG-PTPδA−B−, respectively. The coding sequences of mouse IL1RAPL1 and NGL-3, except for signal peptides, were cloned into pDisplay vector (Invitrogen) to yield the expression vectors for hemagglutinin (HA)-tagged mouse IL1RAPL1 and NGL-3 (pDisplay-IL1RAPL1 and pDisplay-NGL-3), respectively. DNA fragments encoding the extracellular domain of mouse IL1RAPL1 and that of NGL-3 were amplified by PCR with primers, 5′-CTCGAGATCTGAAGGTTGTGACTAAAAGAGG-3′ and 5′-AGCGGCCGCTCGTTTATGAAGGAGAACACTGG-3′, and 5′- CTCGAGATGCTGGTGGGGGCGGTG-3′ and 5′-GCGGCCGCGGTGGTCTTCATGACATCG-3′ using pIL1RAPL1 and pNGL-3, respectively, as templates. The amplified fragments were cloned into the XhoI–NotI sites of pEB6-Igκ-Fc (Uemura and Mishina, 2008) to yield pEB6-IL1RAPL1-ECD-Fc and pEB6-NGL-3-ECD-Fc. Oligonucleotides 5′-GGCCCATCACCATCACCATCACTGA-3′ and 5′-GGCCTCAGTGATGGTGATGGTGATG-3′ were annealed and ligated into the NotI site of pEB6-IL1RAPL1-ECD-Fc to yield pEB6-IL1RAPL1-ECD-His. DNA fragments encoding the extracellular domains of PTPδ and PTPδA−B− were amplified with primers, 5′-GCTAGCCACCATGCCGTTGACAAACTGCAG-3′ and 5′-AAGCTTCCTCTTCGTCTGTGATTGG-3′, using pPTPδ and pPTPδ-A−B− as templates, and were cloned into NheI–Eco47III sites of pAP-tag5 vector (GenHunter Corporation) to yield pPTPδ-ECD-MH and pPTPδA−B−-ECD-MH, respectively.
Animals.
Mice were fed ad libitum with standard laboratory chow and water in standard animal cages under a 12 h light/dark cycle. Mice of either sex were used for the experiments unless otherwise specified. All animal procedures were approved by the Animal Care and the Use Committee of Graduate School of Medicine, the University of Tokyo (Approval no. 1721T062).
Cell cultures, transfection, and coculture assay.
Primary cortical and hippocampal cultures were prepared from mice at postnatal day 0 (P0) or embryonic day 16 essentially as described previously (Uemura et al., 2004). The cortical and hippocampal cells were placed on coverslips coated with 30 μg/ml poly-l-lysine and 10 μg/ml mouse laminin at a density of 2–5 × 105 and 2 × 105 cells/well, respectively. The cells were cultured in Neurobasal-A supplemented with 2% B-27 supplement (Invitrogen), 5% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.5 mm l-glutamine for 24 h, and then were cultured in the same medium without FCS. Cultures of HEK293T cells were maintained as described previously (Uemura et al., 2004). Expression vectors were transfected to HEK293T cells using FuGene6 transfection reagent (Roche). After 2 d of culture, the transfected cells were washed with PBS containing 2 mm EDTA and incubated with the same buffer at 37°C for 10 min. Dispersed cells were plated onto cortical neurons at day in vitro (DIV) 14. After 24 h of coculture, cells were fixed for immunostaining. IL1RAPL1-ECD-Fc, IL-1R1-ECD-Fc, and Fc proteins were bound to Protein A-conjugated magnetic particles (smooth surface, 4.0–4.5 μm diameter; Spherotech). Beads coupled with IL1RAPL1-ECD-Fc, IL-1R1-ECD-Fc, or Fc proteins were added to cortical and hippocampal neurons (DIV8). After 24 h, cultures were fixed for immunostaining. Cultured neurons at DIV12 were transfected with expression vectors using NeuroMag transfection reagent (OZ Biosciences) according to the manufacturer's instructions and were fixed at DIV14 for immunostaining. For neuron–neuron coculture assay, cortical neurons from ICR mice at P0 were first transfected with pEGFP-CAGMCS or pEGFP-CAG-FLAG-IL1RAPL1 by electroporation using Nucleofector and mouse Nucleofector kit (program G-013; Amaxa Biosystems) and placed on coverslips at the density of 1 × 103 cells/well. Then cortical neurons from Ptprd+/+ or Ptprd−/− mice at P0 were placed on the transfected cells at a density of 3 × 105 cells/well. The cultures were fixed at DIV9 for immunostaining.
Immunostaining.
Immunostaining of primary neuronal cultures and neuron–fibroblast cocultures was performed as described previously (Uemura et al., 2004; Uemura and Mishina, 2008) using chicken anti-FLAG (Kamiya Biomedical), goat anti-IL1RAPL1 (R&D Systems), rabbit anti-IL1RAPL1 (custom-made antibody, Sigma), rabbit anti-pan-cadherin (Sigma), rabbit anti-MAP2 (Millipore Bioscience Research Reagents), mouse anti-gephyrin (Synaptic Systems), rabbit anti-Synapsin 1 (Millipore), mouse anti-Tau1 (Millipore Bioscience Research Reagents), rabbit anti-vesicular glutamate transporter (VGluT) 1 (Miura et al., 2006), mouse anti-Bassoon (Stressgen), goat anti-vesicular GABA transporter (VGAT) (Miyazaki et al., 2003), rabbit anti-Shank1 (provided by Dr. Y. Hata, Tokyo Medical and Dental University, Tokyo, Japan) and rabbit anti-Shank2 (provided by Dr. M. Watanabe, Hokkaido University, Sapporo, Japan) antibodies as primary antibodies. For secondary antibodies, Alexa Fluor 488-, 555-, and 647-conjugated anti-IgG antibodies (Invitrogen) and Cy3- and Cy5-conjugated anti-IgG antibodies (Jackson ImmunoResearch) were used. For detection of magnetic beads coated with Fc fusion proteins, FITC-conjugated anti-human Fcγ antibody (Jackson ImmunoResearch) was used.
Small interfering RNA-mediated knockdown.
siRNAs against Il1rapl1 were custom synthesized (Stealth RNAi, Invitrogen). The sense sequences of siRNAs were as follows: siIl1rapl1–1, 5′-CAGUGGAGAUGUCAGUCCUUUAAUU-3′; siIl1rapl1–2, 5′-UAGAUCAAAGUAAGCGGCUGAUUAU-3′. For control siRNA, Stealth RNAi Negative Control kit (Invitrogen) was used. Expression vector for res-FLAG-IL1RAPL1 (pEGFP-CAG-res-FLAG-IL1RAPL1) was generated by substituting the Il1rapl1 coding sequences 5′-CAGTGGAGATGTCAGTCCTTTAATT-3′ and 5′-TAGATCAAAGTAAGCGGCTGATTAT-3′ of pEGFP-CAG-FLAG-IL1RAPL1 with 5′-TTCAGGCGACGTATCACCGCTGATC-3′ and 5′-TGGACCAGTCCAAAAGATTAATCAT-3′, respectively. Cultured neurons at DIV10 were transfected with a mixture of siRNAs (60 pmol total/well) and expression vectors (1 μg/well) using NeuroMag transfection reagent, and were fixed at DIV14 for immunostaining.
Affinity chromatography.
Brains of mice at P8–P20 were homogenized in buffer A (0.1 m NaCl, 4 mm KCl, 5 mm CaCl2, 2.5 mm EDTA, 20 mm NaHCO3, 20 mm Tris-HCl, pH 7.5, 0.1 g/l PMSF, 1 mg/L leupeptin, 1 mg/L pepstatin A, and 1 mg/L aprotinin). CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate) was added to a final concentration of 2% (w/v) and dissolved under stirring at 4°C for 1 h. The extract containing ∼1 g of protein was diluted with an equal volume of buffer A and chromatographed on the affinity columns (IL1RAPL1-ECD-Fc and Fc attached to Protein A-Sepharose). Columns were washed with buffer A and eluted with a gradient of NaCl from 0.1 to 2.0 m using AKTApurifier (GE Healthcare). Fractions were analyzed by SDS-PAGE, negative staining, and immunoblotting. For mass spectrometry analysis, proteins eluted at ∼0.8 m NaCl were separated by SDS-PAGE and negatively stained. Bands were cut and subjected to in-gel reduction, alkylation, and digestion with trypsin followed by analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS), as described previously (Uemura et al., 2010).
Preparation of Sindbis viral particles.
The entire coding sequence of enhanced yellow fluorescent protein (EYFP) was inserted immediately before the stop codon of mouse IL1RAPL1 to produce the coding sequence of IL1RAPL1-EYFP. The coding sequence of pre-protrypsin signal peptide followed by FLAG tag was ligated with the coding sequence of PTPδ lacking signal peptide to obtain FLAG-PTPδ coding sequence. The coding sequences of EGFP, EYFP, IL1RAPL1-EYFP, and FLAG-PTPδ were cloned into pSinRep5 (Invitrogen) to yield pSinRep-EGFP, pSinRep-EYFP, pSinRep-IL1RAPL1-EYFP, and pSinRep-FLAG-PTPδ, respectively. These vectors were then used as the template for in vitro transcription using SP6 RNA polymerase (Invitrogen). The RNA transcript and the helper RNA from a DH (26S) cDNA template (Invitrogen) were cotransfected into baby hamster kidney cells by electroporation. Twenty-four hours after transfection, the culture medium was harvested to obtain the infectious particles of Sindbis virus.
Coimmunoprecipitation.
Cultured cortical neurons at DIV9 (5 × 106 cells) were infected with Sindbis virus for FLAG-PTPδ or that for EGFP. After 24 h of infection, cells were lysed with radioimmunoprecipitation (RIPA) buffer (50 mm Tris-HCl, pH8.0, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40) containing protease inhibitors (Complete EDTA-free; Roche). Soluble fractions were incubated with anti-FLAG M2 agarose (Sigma), and the agarose was washed five times with RIPA buffer. Proteins were eluted from the agarose by boiling in SDS sample buffer, separated by SDS-PAGE, and analyzed by Western blotting with goat anti-IL1RAPL1 and rabbit anti-FLAG (Sigma) antibodies.
Cell surface binding assay.
Expression vectors pPTPδ, pPTPδB−, pPTPδA6, pPTPδA6B−, pPTPδA3, pPTPδA3B−, pPTPδA−, pPTPδA−B−, pLARA6, pLARA6B−, pLARA−, pLARA−B−, pPTPσA−B−, and pPTPσA− were cotransfetced with pEGFP-C1 (Clontech) into HEK293T cells. Transfected cells were incubated with IL1RAPL1-ECD-Fc (35 μg/ml), NGL-3-ECD-Fc (60 μg/ml), or Fc (15 μg/ml) in HBSS containing 2 mm CaCl2 and 1 mm MgCl2 for 1 h at room temperature. After washing, cells were fixed with 4% PFA, immunostained with goat anti-PTPδ (Santa Cruz Biotechnology) and rabbit anti-human IgG (Rockland), and incubated with Alexa Fluor 555-conjugated donkey anti-rabbit IgG and Alexa Fluor 647-conjugated donkey anti-goat IgG (Invitrogen). For binding assay of IL1RAPL1 and NGL-3 to PTPδ, HEK293T cells transfected with pPTPδ were incubated with 0.1 μm IL1RAPL1-ECD-His in the presence of 0.03, 0.1, 0.3, 1.0, or 3.0 μm NGL-3-ECD-Fc or IL1RAPL1-ECD-Fc for 1 h. After washing, cells were fixed with 4% PFA, incubated with goat anti-PTPδ and mouse anti-His (Millipore) antibodies and immunostained with Alexa Fluor 555-conjugated donkey anti-mouse IgG and Alexa Fluor 647-conjugated donkey anti-goat IgG antibodies.
Cell aggregation assay.
HEK293T cells were transfected with coexpression vectors pEGFP-CAG-IL1RAPL1 pRFP-CAG-PTPδ, and pRFP-CAG-PTPδA−B−. After 2 d, the cells were washed with PBS followed by incubating with PBS containing 2 mm EDTA at 37°C for 10 min. Carefully triturated cells were mixed at a density of 4.0 × 106 cells/ml. Cells were incubated in 1.5 ml microcentrifuge tubes at room temperature with rotation in a total volume of 400 μl of HBSS containing 2 mm CaCl2 and 1 mm MgCl2. After 20 min, fluorescence images were taken.
Preparation of recombinant proteins and surface plasmon resonance binding analysis.
Soluble recombinant IL1RAPL1-ECD-Fc, NGL-3-ECD-Fc, Fc, PTPδ-ECD-MH, and PTPδA−B−-ECD-MH were prepared as described previously (Uemura and Mishina, 2008) by transfection of respective expression vectors into the Freestyle 293 cells (Invitrogen). PTPδ-ECD-MH and PTPδA−B−-ECD-MH were purified from culture medium by Talon metal affinity resin (Clontech) and dialyzed against HBSS or artificial CSF [(in mm) 10 sucrose, 150 NaCl, 3 KCl, 1.4 CaCl2, 0.8 MgCl2, 0.8 Na2HPO4, 0.2 NaH2PO4]. Surface plasmon resonance (SPR) binding assay was conducted on a Biacore 3000 biosensor equipped with a sensor chip CM5 (GE Healthcare). Anti-human Fc antibodies were covalently immobilized over independent flow cells using Human Antibody Capture Kit (GE Healthcare). IL1RAPL-ECD-Fc was captured in flow cell 2 to generate the IL1RAPL-ECD-Fc sensor chips. Untreated flow cell 1 served as a reference. Binding analysis was performed at 25°C in a running buffer of 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% surfactant P20, 2 mm CaCl2, and 1 mm MgCl2. PTPδ-ECD-MH or PTPδA−B−-ECD-MH in the running buffer was injected in the order of increasing concentration. Each sample was flowed over the chip surface for 3 min at a flow rate of 25 μl/min, followed by a 2 min dissociation phase. Data analysis was performed using BIAevaluation software, version 4.1. The responses were analyzed by steady-state kinetics to calculate the dissociation constant (KD).
RT-PCR and sequencing analyses of splice variants of PTPδ, LAR, and PTPσ.
RT-PCR was performed using mRNAs prepared from P11 mouse brains with primers 5′-GGTACCAAGCTGCTGGTTCATGCCG-3′ and 5′-GGGCTATTGCTTCAATGACC-3′ for PTPδ, 5′-AGACCCTGAGATCTCTTGG-3′, and 5′-CTCTTTGGTCAGTTCCTCG-3′ for LAR, and 5′-CCATTGACTTTGACGAGAGC-3′ and 5′-GGAGTTGACAGCTGACACC-3′ for PTPσ. Amplified DNA fragments were cloned into pCRII-TOPO vector (Invitrogen), and 50 clones randomly picked up for each gene were sequenced to estimate the populations of splice variants.
Structure modeling.
The structural model was generated with SWISS-MODEL (Arnold et al., 2006) (http://swissmodel.expasy.org/). The amino acid sequence of Ig-like domains (amino acid residues 21–321) of PTPδ was submitted to SwissModel Automatic Modeling Mode (http://swissmodel.expasy.org/workspace/index.php?func=modeling_simple1). The structure of the I-BAND fragment I67–I69 from TITIN (PDB ID 2rikA) (von Castelmur et al., 2008) was selected as a template for modeling. The obtained alignment was imported into Swiss-Pdb Viewer 4.0.1 (Guex and Peitsch, 1997) (http://www.expasy.org/spdbv/) and manually optimized.
DiI labeling.
DiI labeling was performed essentially according to Kim et al. (2006). Briefly, brains were fixed with 1.5% PFA and coronally sectioned into 200 μm slices. Solid DiI crystals were applied onto layer 2/3 of the somatosensory cortex. The slices were incubated in PBS at room temperature for 12 h, fixed again with 4% PFA for 30 min, and washed in PBS three times. Basal dendrites of the pyramidal neurons in cortical layer 2/3 were randomly sampled and imaged with a confocal laser-scanning microscope (TCS SP5; Leica, zoom setting; 5, z step; 0.17 μm) using a Leica 63 × water lens (numerical aperture, 1.20). Dendritic spines were identified and counted in the 3D projection images. When dendritic spines were too crowded to separate them from each other, we turned to serial stack images to delineate individual spines. By scrolling through the stack of different optical sections, individual spine heads could be identified. All dendritic protrusions with a clearly recognizable neck were counted as spines.
Injection of recombinant proteins and viruses in vivo.
The guide cannula (26 gauge; Plastics One) was implanted above the somatosensory cortex (anteroposterior −1.0 mm, mediolateral +2.2 mm from bregma) of mice at P15 under anesthesia with 2.5% isoflurane. Animals were monitored postoperatively until mobile and were allowed to recover for 2 d. We injected recombinant proteins (16 μg of IL1RAPL1-ECD-Fc and 7 μg of Fc in 2–8 μl of artificial CSF) into the somatosensory cortex of mice at P17 through the internal cannula (33 gauge; Plastics One) via polyethylene tubing to microsyringes at a flow rate of 0.1 μl/min using a microinjection pump (CMA/100; Carnegie Medicin). Injected mice were fixed for DiI labeling at 24 h after injection. DiI crystals were applied onto layer 2/3 of the somatosensory cortex within 500 μm from the injection site. Two microliters of a virus solution with or without recombinant proteins (16 μg of PTPδ-ECD-MH, PTPδA−B−-ECD-MH, or AP-MH) was injected into the somatosensory cortex of mice at P17 at a constant flow rate of 0.3 μl/min, and the glass micropipette was left in place for an additional 3 min before being slowly withdrawn. For animals injected with both virus solution and recombinant proteins at P17, recombinant proteins (16 μg of PTPδ-ECD-MH, PTPδA−B−-ECD-MH, or AP-MH) were injected again into the same sites with the glass micropipette using the same procedures at P18. Injected mice were fixed for immunohistochemistry with anti-VGluT1 antibody at 48 h after injection of virus particles.
Image acquisition and quantification.
Images were collected from at least three separate experiments. Images of cell surface binding assay and fibroblast–neuron or beads–neuron coculture experiments were obtained with a confocal laser-scanning microscope (TCS SP5; Leica) under constant conditions as to laser power, iris, gain, z-steps, and zoom setting throughout the experiments, and all quantitative measurements were performed with ImageJ 1.36b software. For the quantification of accumulation of immunostaining signals of presynaptic marker proteins in cocultures, optical mean densities within a circle of 30 μm diameter enclosing transfected HEK293T cells or those within a circle of 7 μm diameter enclosing coated beads were measured. Then, the optical mean densities of the surrounding regions within circles of 40 and 14 μm diameters were measured and subtracted as background signals for HEK293T cells and beads, respectively. We confirmed that the background signals for presynaptic proteins were comparable among experiments to monitor the culture conditions. For quantification of the numbers of dendritic protrusions and Bassoon and Shank2 puncta, z-series optical sections of cortical or hippocampal neurons were projected by the brightest point method, and the thickest dendrite was chosen for measurements. The thickness of dendrites was comparable in each condition. Numbers of dendritic protrusions ranging from 0.5 to 8 μm and Bassoon and Shank2 puncta on EGFP-labeled dendritic shafts and protrusions were counted manually in a blind manner with respect to expression vectors transfected. For the quantification of immunostaining signals of VGluT1 on the basal dendrites of cortical layer 2/3 pyramidal neurons infected with Sindbis virus, the ratio of the optical mean density of staining signals within rectangles (6 × 20 μm) enclosing the basal dendrite and that of surrounding regions in cortical layer 2/3 was measured. In all experiments in vivo, image acquisition and quantification were performed in a blind manner with respect to genotypes of mice and viruses injected. Statistical significance was evaluated by Student's t test or one-way ANOVA followed by post hoc Tukey's test.
Results
IL1RAPL1 mediates synapse formation in cultured cortical neurons
Immunocytochemical staining with anti-IL1RAPL1 antibody showed that endogenous IL1RAPL1 clusters were distributed in the dendrites of cultured cortical neurons and were partially overlapped with postsynaptic Shank1 clusters (Fig. 1A). On the other hand, only weak staining signals were partly observed in the axons of cortical neurons (data not shown). We examined whether the knockdown of endogenous IL1RAPL1 by siRNAs affects the synapse formation. We generated siRNAs directed against the mouse Il1rapl1 and tested their efficacy by immunostaining of endogenous IL1RAPL1 of cortical neurons (Fig. 1B). When the siRNAs against Il1rapl1 were transfected into cultured cortical neurons at DIV10, we observed strong reduction of both punctate staining signals for Bassoon along the dendrites and the number of dendritic protrusions (Fig. 1C). There were significant differences in the staining signals of Bassoon and the number of protrusions between siRNA- and control siRNA-transfected neurons (p < 0.01) (Fig. 1D,E). Transfection of an expression vector for an siRNA-resistant form of mouse IL1RAPL1 tagged with FLAG epitope at the N terminus (res-FLAG-IL1RAPL1) together with the siRNAs restored the synapse formation of cortical neurons as estimated by the accumulation of punctate staining signals for presynaptic Bassoon and the density of dendritic protrusions (p < 0.01) (Fig. 1C–E). Consistently, expression of FLAG-IL1RAPL1 in cultured cortical neurons increased the staining signals of Bassoon and Synapsin 1 and the number of protrusions from the dendrites of cortical neurons (p < 0.01) (Fig. 1G–I) (data not shown). Similar results were obtained in cultured hippocampal neurons (data not shown). These results suggest that postsynaptic IL1RAPL1 plays a role in presynaptic differentiation and dendritic spine formation in cortical and hippocampal neurons.
Figure 1.
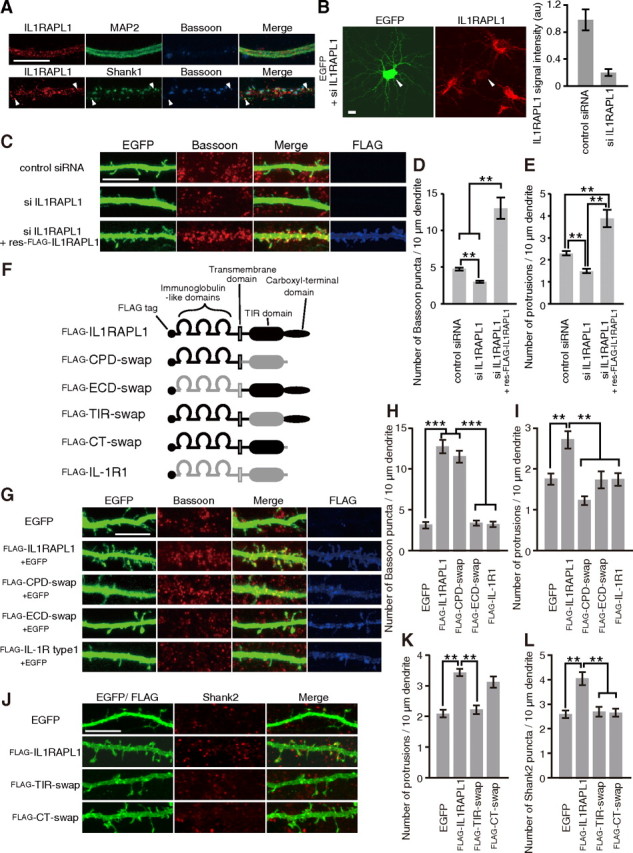
Effects of IL1RAPL1 on synapse formation. A, Immunostaining of cultured cortical neurons with antibodies against IL1RAPL1, MAP2, and Bassoon (top) and with those against IL1RAPL1, Shank1, and Bassoon (bottom). B, Efficacy of siRNAs against Il1rapl1 in cultured cortical neurons. Cortical neurons were transfected with siRNAs against Il1rapl1 together with an expression vector for EGFP and were immunostained for IL1RAPL1. siRNAs against Il1rapl1 reduced IL1RAPL1 staining signal intensity by 80% compared with control siRNA. C, Reduction of numbers of Bassoon puncta and dendritic protrusions of cultured cortical neurons by siRNAs against Il1rapl1 and rescue by res-FLAG-IL1RAPL1. D, E, Effects of siRNA treatments on the numbers of Bassoon puncta (D) and dendritic protrusions (E) along dendrites of cortical neurons. n = 52, 52, and 20 neurons for control siRNA, siRNA against Il1rapl1 mRNA, and siRNA against Il1rapl1 mRNA with res-FLAG-IL1RAPL1, respectively. F, Schematic structures of FLAG-IL1RAPL1, FLAG-IL-1R1, and their swap mutants. G, Effects of IL1RAPL1/IL-1R1 swap mutants on dendritic protrusions and Bassoon puncta in cultured cortical neurons. H, I, Numbers of Bassoon puncta (H) and dendritic protrusions (I) along dendrites of cortical neurons transfected with EGFP, FLAG-IL1RAPL1, FLAG-CPD-swap, FLAG-ECD-swap, and FLAG-IL-1R1 (n = 28–40 neurons). J, Effects of TIR and C-terminal domain-swap mutants on dendritic protrusions and Shank2 puncta in cultured cortical neurons. K, L, Numbers of dendritic protrusions (K) and Shank2 puncta (L) along dendrites of cortical neurons transfected with EGFP, FLAG-IL1RAPL1, FLAG-TIR-swap, and FLAG-CT-swap (n = 19–30 neurons). All values represent mean ± SEM. **p < 0.01, ***p < 0.001, Tukey's test. Scale bars, 10 μm.
IL1RAPL1 belongs to a novel class of the TIR family (Carrié et al., 1999). IL1RAPL1 protein consists of three Ig-like domains, a single transmembrane segment, the TIR domain, and the C-terminal domain. Mouse IL-1R1 sharing similar domain structures and 27% amino acid sequence identity with IL1RAPL1 (Sims et al., 1988) exerted little effect on the accumulation of Bassoon and the number of dendritic protrusions. We constructed domain-swap mutants between IL1RAPL1 and IL-1R1 to examine whether the distinct domains of IL1RAPL1 mediate the increase in the number of protrusions and enhanced accumulation of Bassoon (Fig. 1F). Swapping the CPD of IL1RAPL1 with that of IL-1R1 abolished the stimulatory effect on the number of protrusions but retained the enhancement effect on Bassoon accumulation (Fig. 1G–I). On the other hand, swapping both the ECD, comprising three Ig-like domains, and the transmembrane segment of IL1RAPL1 with those of IL-1R1 abolished the stimulatory effect on both the accumulation of Bassoon and the number of protrusions. Thus, the stimulatory effect of IL1RAPL1 on the presynaptic differentiation requires its ECD, while both the ECD and CPD are indispensable for the increment of dendritic protrusions.
The CPD of IL1RAPL1 comprises the TIR and C-terminal domains. Swapping the TIR domain of IL1RAPL1 with that of IL-1R1 abolished the stimulatory effect on the number of dendritic protrusions (Fig. 1J,K). On the other hand, swapping the C-terminal domain of IL1RAPL1 with that of IL-1R1 hardly affected the enhancement effect on the number of dendritic protrusions (Fig. 1J,K). Expression of FLAG-IL1RAPL1 in cultured cortical neurons also increased the staining signals of excitatory postsynaptic marker Shank2 (Fig. 1J,L). Swapping either the TIR or C-terminal domain abolished the enhancement effect on Shank2 accumulation (Fig. 1J,L). Thus, the TIR domain in the CPD of IL1RAPL1 is responsible for the regulation of dendritic protrusions, while both the TIR and C-terminal domains are required for the accumulation of postsynaptic Shank2.
The ECD of IL1RAPL1 is sufficient to trigger excitatory presynaptic differentiation
We further examined the synaptogenic activity of IL1RAPL1 by a neuron-fibroblast coculture assay (Scheiffele et al., 2000). HEK293T cells transfected with IL1RAPL1 were seeded on the top of cultured cortical neurons. After 24 h of coculture, we detected numerous punctate signals for Bassoon and VGluT1 on the surface of HEK293T cells expressing IL1RAPL1 (Fig. 2A,B). Immunostaining signals for IL1RAPL1 and Bassoon were well merged on the surface of transfected HEK293T cells. There were significant differences in the intensities of staining signals for Bassoon and VGluT1 between control and IL1RAPL1-expressing cells (p < 0.01) (Fig. 2C,D). However, staining signals for VGAT on the surface of HEK293T cells expressing IL1RAPL1 were as marginal as those of control cells (Fig. 2E). In contrast, HEK293T cells expressing FLAG-NLGN1 showed punctate staining signals for both VGluT1 and VGAT on their surface. These punctate signals of axon terminals were hardly detectable on the surface of HEK293T cells expressing N-cadherin and control HEK293T cells expressing EGFP alone. These results suggest that IL1RAPL1 triggers excitatory presynaptic differentiation in vitro. The ECD of IL1RAPL1 fused to the Ig constant region (IL1RAPL1-ECD-Fc) was coated onto beads of 4–4.5 μm diameter. Coated beads were applied to cultured cortical neurons. After incubation for 24 h, we detected numerous punctate staining signals for Bassoon and VGluT1 on the surface of IL1RAPL1-ECD-Fc-coated beads, but staining signals for VGAT on the surface of IL1RAPL1-ECD-Fc-coated beads were as marginal as those of control beads (Fig. 2F–K). Similar results were also obtained in cultured hippocampal neurons (data not shown). These results suggest that the ECD of IL1RAPL1 is sufficient to induce presynaptic differentiation.
Figure 2.

IL1RAPL1 triggers excitatory presynaptic differentiation. A, B, Cocultures of cortical neurons and HEK293T cells expressing EGFP, IL1RAPL1, FLAG-NLGN1, or N-cadherin were immunostained for Bassoon (red) and each effector molecule (blue) (A) and for VGluT1 (red) and VGAT (blue) (B). C–E, Intensity of staining signals for Bassoon (C), VGluT1 (D), and VGAT (E) on the surface of HEK293T cells expressing EGFP, IL1RAPL1, FLAG-NLGN1, or N-cadherin (n = 15–20 HEK293T cells). F–H, Cocultures of cortical neurons and Fc- or IL1RAPL1-ECD-Fc-coated beads were immunostained for Bassoon (F), VGluT1 (G), or VGAT (H). I–K, Intensities of staining signals for Bassoon (I), VGluT1 (J), and VGAT (K) on the surface of Fc- and IL1RAPL1-ECD-Fc-coated beads (n = 10–42 beads). All values represent mean ± SEM. **p < 0.01, Tukey's test; ***p < 0.001, Student's t test. Scale bars: A, B, 10 μm; F–H, 5 μm.
Isolation of IL1RAPL1-ECD interacting protein
Then, the question arises how IL1RAPL1 exerts its synaptogenic activity. We speculate that IL1RAPL1 may exert its synaptogenic activity by interacting with presynaptic proteins since its ECD is required and sufficient to induce presynaptic differentiation. To examine the issue, we isolated IL1RAPL1-ECD binding proteins by affinity chromatography using IL1RAPL1-ECD-Fc or Fc protein immobilized on Protein A-Sepharose beads. Total mouse brain proteins solubilized in CHAPS were loaded onto the affinity columns. Columns were extensively washed and eluted with gradient of NaCl from 0.1 to 2.0 m. SDS-PAGE and negative staining of the fractions revealed IL1RAPL1-ECD-Fc binding proteins of ∼80 and 90 kDa (Fig. 3A, asterisks). These proteins were digested with trypsin, subjected to LC-MS/MS, and identified as the phosphatase subunit (P-subunit) and extracellular subunit (E-subunit) of PTPδ based on the matching of 17 and 19 peptides, respectively (Fig. 3B). Immunoblot analysis using an antibody against the C terminal of PTPδ confirmed that the protein band of ∼80 kDa in the eluate from IL1RAPL1-ECD-Fc column corresponds to the P-subunit of PTPδ.
Figure 3.
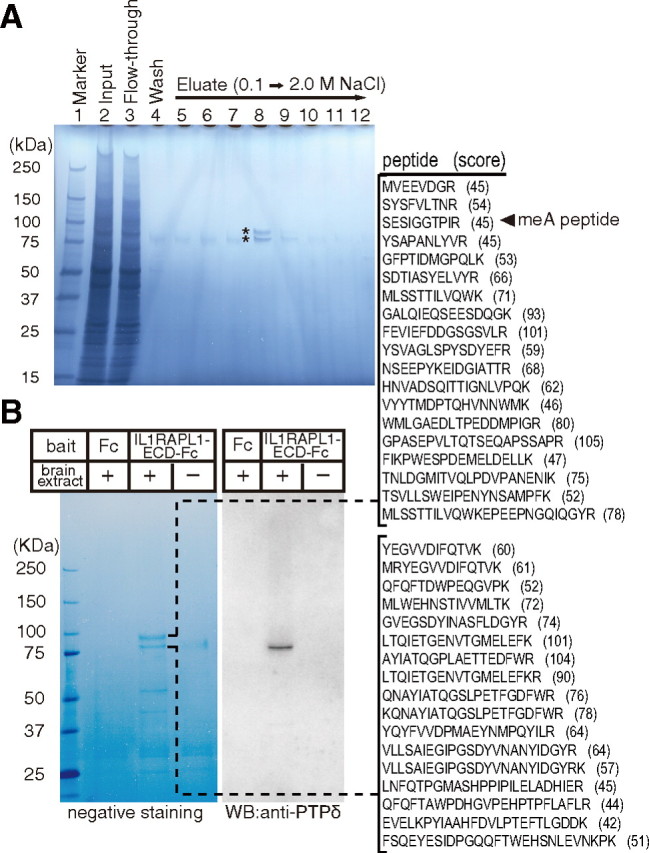
Affinity purification of IL1RAPL1-ECD-interacting protein. A, SDS-PAGE and negative staining of total mouse brain proteins solubilized in CHAPS (lane 2), flow-through fraction (lane 3), last wash fraction (lane 4), and elution fractions with 0.1 to 2.0 m NaCl (lanes 5–12) of IL1RAPL1-ECD-Fc column. B, Negative staining and immunoblot analysis of purified proteins (elution fraction corresponding to lane 8 in A) with anti-PTPδ antibody. Numbers on the left indicate the size of molecular weight markers. Slices of negatively stained SDS-PAGE gel were digested with trypsin and analyzed by LC-MS/MS. Identified peptide sequences of E- and P-subunits of PTPδ and their scores of Mascot searches are shown.
We examined the interaction between IL1RAPL1 and PTPδ by cell surface binding assay. HEK293T cells transfected with expression vectors for PTPδ and EGFP were incubated with soluble IL1RAPL1-ECD-Fc to test their ability to interact with IL1RAPL1. We found robust immunofluorescent signals for IL1RAPL1-ECD-Fc on the surface of HEK293T cells transfected with PTPδ but not on those expressing EGFP alone (Fig. 4A). Furthermore, when cultured cortical neurons were transfected with PTPδ tagged with FLAG epitope (FLAG-PTPδ), endogenous IL1RAPL1 protein was coimmunoprecipitated together with FLAG-PTPδ by an antibody against FLAG (Fig. 4B).
Figure 4.
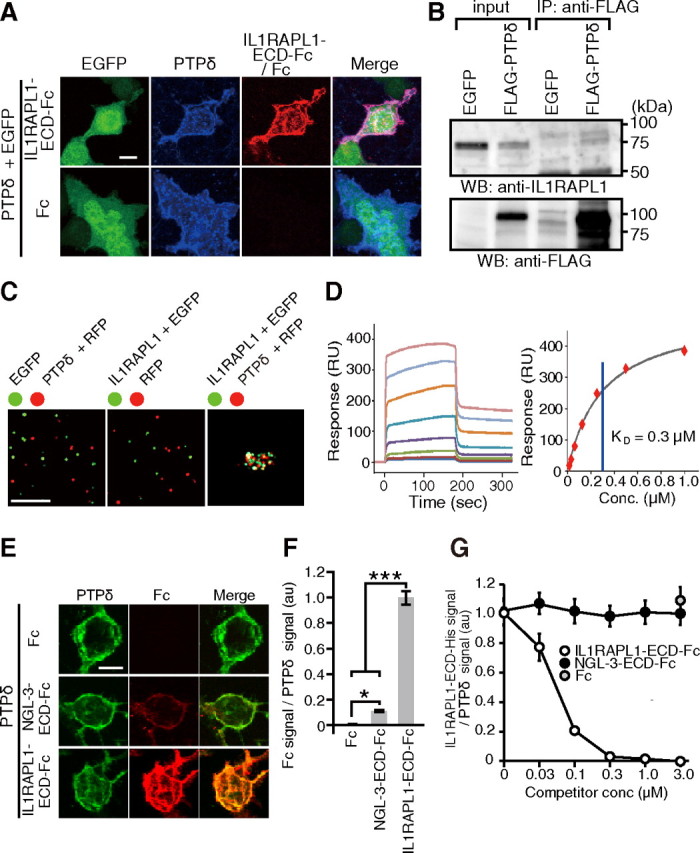
Characterization of interaction between IL1RAPL1 and PTPδ. A, Cell surface binding assay. HEK293T cells transfected with PTPδ were incubated with soluble IL1RAPL1-ECD-Fc (top) or Fc (bottom) and immunostained for anti-PTPδ and anti-Fcγ antibodies. B, Interaction of IL1RAPL1 with FLAG-PTPδ in cultured cortical neurons. Lysates of cultured cortical neurons infected with Sindbis viruses for FLAG-PTPδ or EGFP were immunoprecipitated with anti-FLAG antibody. Input lanes contain 2% of lysates for immunoprecipitation experiments. C, Cell aggregation assay. Representative images of mixtures of HEK293T cells transfected with EGFP and those with RFP and PTPδ (left), HEK293T cells transfected with EGFP and IL1RAPL1 and those with RFP (middle), and HEK293T cells transfected with EGFP and IL1RAPL1 and those with RFP and PTPδ (right). More than three independent experiments were performed for B and C. D, SPR analysis. Interaction kinetics was measured by passing various concentrations (7.8, 15.6, 31.3, 62.5, 125, 250, 500, 1000 nm) of purified PTPδ-ECD-MH over IL1RAPL1-ECD-Fc captured on the surface of a sensor chip (left). Steady-state kinetics of interaction between PTPδ-ECD-MH and IL1RAPL1-ECD-Fc (right). E, Binding of NGL-3-ECD-Fc and IL1RAPL1-ECD-Fc to HEK293T cells transfected with PTPδ. F, Ratios of staining signals for Fc fusion proteins and PTPδ on the surface of HEK293T cells in E (n = 24 each). G, Competition assay. HEK293T cells transfected with PTPδ were incubated with 0.1 μm IL1RAPL1-ECD-His in the presence of 0–3.0 μm IL1RAPL1-ECD-Fc or NGL-3-ECD-Fc and immunostained with anti-His and anti-PTPδ antibodies (n = 20 each). All values represent mean ± SEM. *p < 0.05, ***p < 0.001, Tukey's test. Scale bars: A, E, 10 μm; C, 200 μm.
To determine whether heterophilic binding between the ECD of IL1RAPL1 and the ECD of PTPδ mediates cell adhesion, we incubated HEK293T cells transfected with IL1RAPL1 and EGFP and those transfected with PTPδ and RFP (Fig. 4C). The transfected cells aggregated into large clumps, which suggests that IL1RAPL1 and PTPδ function as heterophilic cell adhesion molecules. Neither HEK293T cells expressing IL1RAPL1 nor those expressing PTPδ showed homophilic aggregation.
To quantify the interaction between IL1RAPL1 and PTPδ, we used SPR binding assay. Increasing concentrations of PTPδ-ECD tagged with Myc and His epitopes at the C terminus (PTPδ-ECD-MH) were injected over the IL1RAPL1-ECD-Fc-tethered chip surface (Fig. 4D). Analysis of the steady-state kinetics of the sensorgrams showed a KD of 0.3 μm, indicating a high-affinity interaction between IL1RAPL1 and PTPδ.
We compared the binding abilities of IL1RAPL1 and NGL-3 to PTPδ since it was reported that NGL-3 interacts with PTPδ in addition to LAR and PTPσ (Woo et al., 2009; Kwon et al., 2010). HEK293T cells transfected with an expression vector for PTPδ were incubated with soluble IL1RAPL1-ECD-Fc or NGL-3-ECD-Fc. We detected immunofluorescent signals for both IL1RAPL1-ECD-Fc and NGL-3-ECD-Fc on the surface of HEK293T cells expressing PTPδ (Fig. 4E). However, the intensity of staining signals for IL1RAPL1-ECD-Fc was much stronger than that for NGL-3-ECD-Fc (p < 0.001) (Fig. 4F). Moreover, NGL-3-ECD-Fc hardly affected the interaction between PTPδ and IL1RAPL1-ECD tagged with His epitope (IL1RAPL1-ECD-His) (Fig. 4G).
Mini-exon peptides in PTPδ determine interaction with IL1RAPL1
Cloning and sequencing analyses of the Ptprd cDNA prepared from the brains of mice at P11 revealed the existence of at least six splice variants in the Ig-like domains of PTPδ (Fig. 5A). All variants have four fibronectin type III-like domains, consistent with a previous report (Mizuno et al., 1994). The most abundant form (PTPδ) contains both mini-exon (me) A and meB peptides as reported for human PTPδ (Pulido et al., 1995), while some variants contain 6 or 3 aa instead of meA peptide (PTPδA6 or PTPδA3), and others lack meA, meB, or both (PTPδA−, PTPδA3B−, or PTPδA−B−). Comparison of cDNA and genomic DNA of the mouse Ptprd gene revealed that meA peptide (ESIGGTPIR) is derived from two exons encoding 3 aa (ESI) and 6 aa (GGTPIR). We expressed eight possible splice variants in HEK293T cells and examined their binding ability to IL1RAPL1. Strong staining signals for IL1RAPL1-ECD-Fc were detected on the surface of HEK293T cells expressing PTPδ, PTPδB−, or PTPδA6, but not on the surface of those expressing other splice variants (Fig. 5B). The staining intensity was significantly stronger on the surface of PTPδ-expressing cells than PTPδB−- or PTPδA6-expressing cells (Fig. 5C). These results suggest that the meA and meB peptides of PTPδ are critical for the interaction with IL1RAPL1. Consistently, HEK293T cells transfected with IL1RAPL1 failed to form aggregates with those transfected with PTPδA−B− (data not shown). In SPR binding assay, no significant responses were detectable between IL1RAPL1-ECD-Fc and PTPδA−B−-ECD-MH (data not shown). In fact, LC-MS/MS identified the meA peptide in PTPδ protein purified by the affinity chromatography with IL1RAPL1-ECD (Fig. 3B). In a structure model of Ig-like domains of PTPδ, the meA and meB peptides appear to be prominent from the domains (Fig. 5D).
Figure 5.
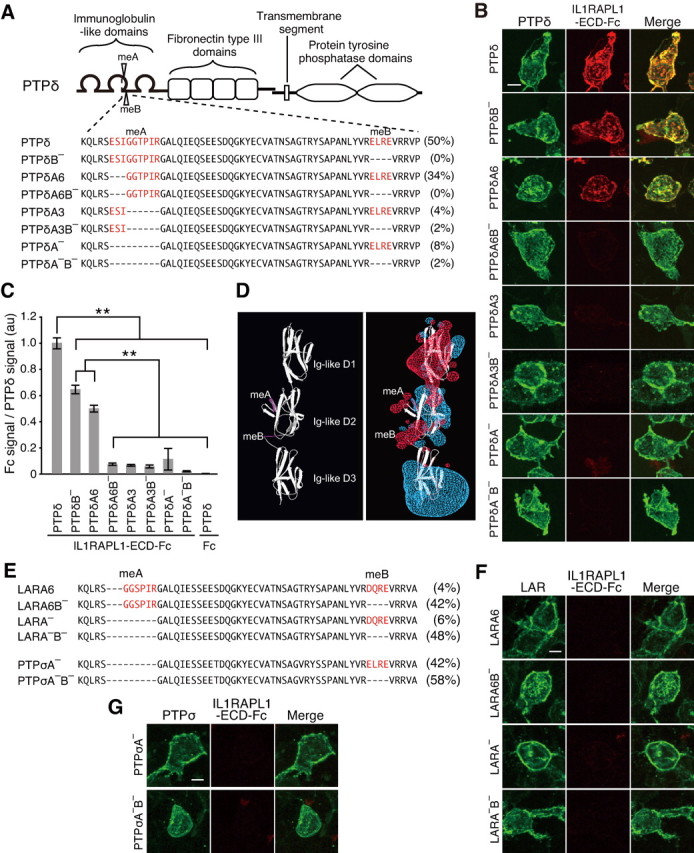
Selective interaction of IL1RAPL1 with PTPδ variants. A, Schematic structure of PTPδ and amino acid sequences of splice variants in Ig-like domains. Populations of these splice variants in brains of mice at P11 are indicated on the right. B, Binding of IL1RAPL1-ECD-Fc to HEK293T cells transfected with PTPδ splice variants. C, Ratios of staining signals for IL1RAPL1-ECD-Fc and PTPδ splice variants in B (n = 10 HEK293T cells each). D, Structure models of Ig-like domains of PTPδ. meA and meB peptides are indicated in magenta in ribbon diagram (left). In the model with electrostatic potential (right) (red, positive potential; blue, negative potential), vicinities of the meA and meB peptides are electrostatically positive. E, Alignment of deduced amino acid sequences of second Ig-like domain of splice variants of LAR and PTPσ cloned from mRNAs prepared from brains of mice at P11. Populations of these splice variants are indicated on the right. F, G, HEK293T cells transfected with splice variants of LAR (F) and PTPσ (G) were incubated with IL1RAPL1-ECD-Fc. All values represent mean ± SEM. **p < 0.01, Tukey's test. Scale bars, 10 μm.
PTPδ, PTPσ, and LAR share similar domain structures and constitute a 2A subfamily of receptor-like PTPs (2A type RPTPs) (Tonks, 2006). In the developing mouse brain, predominant variants of PTPσ are lacking meA peptide, and those of LAR are without meB peptide (Fig. 5E). These PTPσ and LAR variants expressed in the brain failed to show any significant binding signals for IL1RAPL1-ECD-Fc (Fig. 5F,G). Thus, IL1RAPL1 appears to interact selectively with PTPδ in the brain.
IL1RAPL1 requires PTPδ for induction of presynaptic differentiation
To examine directly the role of IL1RAPL1–PTPδ interaction in synapse formation, we seeded HEK293T cells transfected with IL1RAPL1 and EGFP on the top of cultured cortical neurons prepared from PTPδ knock-out mice (Uetani et al., 2000). After 24 h of coculture, cells were immunostained with antibody against Bassoon. We detected numerous punctate staining signals for Bassoon on the surface of HEK293T cells expressing IL1RAPL1 when cocultured with cortical neurons from wild-type mice (Fig. 6A). However, these punctate signals were hardly detectable for cortical neurons prepared from Ptprd−/− mice. Furthermore, the intensity of signals was reduced in cortical neurons prepared from Ptprd+/− mice (p < 0.01) (Fig. 6D). Consistently, IL1RAPL1-ECD-Fc-coated beads failed to induce the accumulation of punctate Bassoon signals around the beads when cocultured with cortical neurons from Ptprd−/− mice (data not shown). On the other hand, staining signals on the surface of HEK293T cells expressing FLAG-NLGN1 were comparable among Ptprd+/+, Ptprd+/−, and Ptprd−/− cultures (Fig. 6B,D). HEK293T cells expressing N-cadherin showed marginal staining signals on the surface when cocultured with cortical neurons prepared from any of these mice (Fig. 6C,D). These results suggest that IL1RAPL1 requires PTPδ for the induction of presynaptic differentiation.
Figure 6.

PTPδ is required for induction of presynaptic differentiation by IL1RAPL1. A–C, Immunostaining for Bassoon of cocultures of cortical neurons derived from Ptprd+/+, Ptprd+/−, or Ptprd−/− mice and HEK293T cells expressing IL1RAPL1 (A), FLAG-NLGN1 (B), or N-cadherin (C). D, Intensity of staining signals for Bassoon on the surface of HEK293T cells expressing IL1RAPL1, FLAG-NLGN1, and N-cadherin (n = 21–24). E, Immunostaining for Synapsin1 of cocultures of cortical neurons derived from Ptprd+/+ or Ptprd−/− mice and HEK293T cells expressing HA-IL1RAPL1 or HA-NGL-3. F, Intensities of staining signals for Synapsin1 on the surface of HEK293T cells expressing HA-IL1RAPL1 and HA-NGL-3 (n = 30–32). G, Expression levels of HA-IL1RAPL1 and HA-NGL-3 on the surface of HEK293T cells. HEK293T cells transfected with HA-IL1RAPL1 or HA-NGL-3 were immunostained with anti-HA under nonpermeabilized conditions. Intensities of fluorescent staining signals were quantified (n = 32 and 35 for HA-IL1RAPL1 and HA-NGL-3, respectively). All values represent mean ± SEM. **p < 0.01, ***p < 0.001, Tukey's test in D and F, and Student's t test in G. Scale bars, 10 μm.
We next compared the synaptogenic activities of IL1RAPL1 and NGL-3. HA-tagged IL1RAPL1 (HA-IL1RAPL1) and HA-tagged NGL-3 (HA-NGL-3) were expressed in HEK293T cells, and these cells were cocultured with cortical neurons form Ptprd+/+ and Ptprd−/− mice. Staining signals for Synapsin1 accumulated around HEK293T cells expressing HA-NGL-3, but the intensities of staining signals were comparable between Ptprd+/+ and Ptprd−/− cortical neurons. Furthermore, the intensity of Synapsin1 signals around HEK293T cells expressing HA-NGL-3 was weaker than that around HEK293T cells expressing HA-IL1RAPL1 (p < 0.001) (Fig. 6E,F), despite that the amount of HA-NGL-3 expressed on the surface of HEK293T cells was much higher than that of HA-IL1RAPL1 (p < 0.001) (Fig. 6G). These results suggest that the synaptogenic activity of NGL-3 is much weaker than IL1RAPL1 and is not dependent on PTPδ in contrast to IL1RAPL1.
IL1RAPL1 requires PTPδ for induction of dendritic protrusions and postsynaptic differentiation
To examine whether presynaptic PTPδ is involved in postsynaptic IL1RAPL1-mediated induction of dendritic protrusions and postsynaptic differentiation, we labeled cortical neurons from wild-type mice by electroporation with an expression vector for EGFP and FLAG-IL1RAPL1 or that for EGFP at DIV0 and sparsely plated them on dishes. The labeled neurons were then cocultured for 9 d with cortical neurons from Ptprd+/+ or Ptprd−/− mice. Expression of FLAG-IL1RAPL1 significantly increased the number of dendritic protrusions and Shank2 staining signals of the transfected neurons when cocultured with cortical neurons from Ptprd+/+ mice (p < 0.01 and < 0.01) (Fig. 7A–C). However, FLAG-IL1RAPL1 failed to increase dendritic protrusions and Shank2 staining signals in the coculture with cortical neurons from Ptprd−/− mice. These results suggest that postsynaptic IL1RAPL1 requires presynaptic PTPδ for the induction of dendritic protrusions and postsynaptic differentiation.
Figure 7.
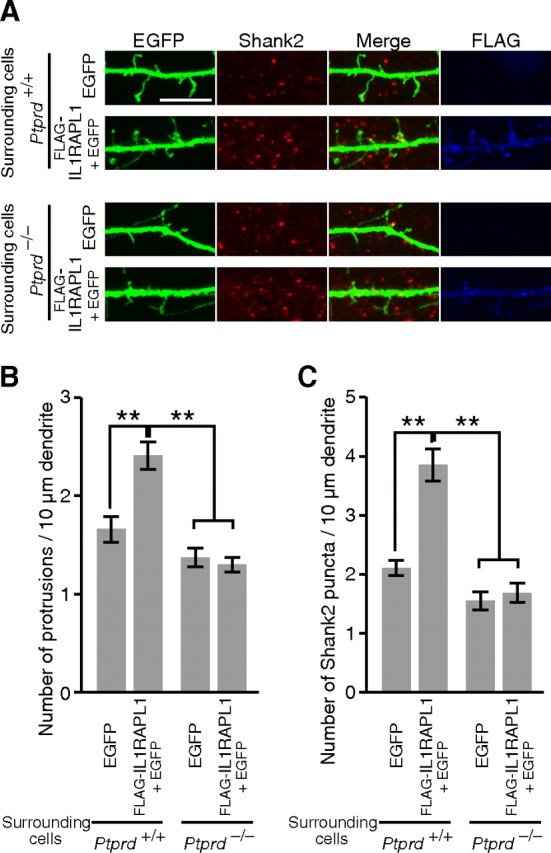
PTPδ is required for increments of dendritic protrusions and Shank2 puncta by IL1RAPL1. A, FLAG-IL1RAPL1 expressed in cortical neurons failed to increase dendritic protrusions and Shank2 puncta in coculture with cortical neurons from Ptprd−/− mice. B, C, Numbers of dendritic protrusions (B) and Shank2 puncta (C) of EGFP-transfected and EGFP- and FLAG-IL1RAPL1-transfected cortical neurons surrounded by cortical neurons from Ptprd+/+ or Ptprd−/− mice (n = 25–29 neurons). All values represent mean ± SEM. **p < 0.01, Tukey's test. Scale bar, 10 μm.
We further examined the ability of PTPδ to induce postsynaptic differentiation by fibroblast–neuron mixed culture assay. HEK293T cells expressing PTPδ were cocultured with cortical neurons, and were immunostained for Shank2 and inhibitory postsynaptic marker gephyrin. PTPδ induced the accumulation of staining signals for Shank2 but not for gephyrin (Fig. 8A–D). The addition of IL1RAPL1-ECD-Fc significantly suppressed the PTPδ-induced accumulation of Shank2 staining signals (p < 0.001) (Fig. 8E,F). In contrast, NGL-3-ECD-Fc hardly affected the PTPδ-mediated postsynaptic differentiation. These results suggest that PTPδ has an activity to induce excitatory postsynaptic differentiation through IL1RAPL1.
Figure 8.
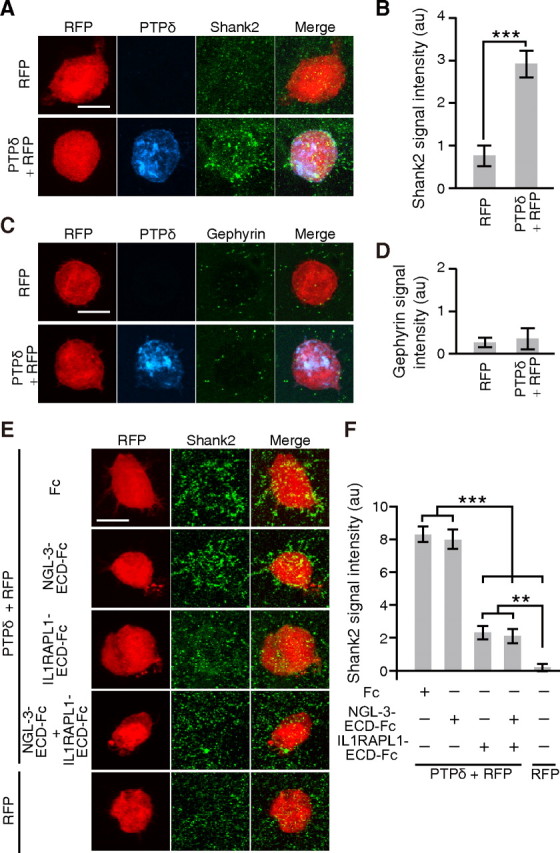
PTPδ induces excitatory postsynaptic differentiation through IL1RAPL1. A, C, Cocultures of cortical neurons and HEK293T cells expressing PTPδ and RFP were immunostained for Shank2 (A) or gephyrin (C). HEK293T cells expressing RFP alone serve as controls. B, D, Intensity of staining signals for Shank2 (B) and gephyrin (D) on the surface of HEK293T cells (n = 15 HEK293T cells each). E, Cocultures of cortical neurons and HEK293T cells expressing PTPδ and RFP in the presence of IL1RAPL1-ECD-Fc, NGL-3-ECD-Fc or Fc, or both NGL-3-ECD-Fc and IL1RAPL1-ECD-Fc were immunostained for Shank2. HEK293T cells expressing RFP alone served as controls. F, Quantification of Shank2 signals in E (n = 36–47). All values represent mean ± SEM. **p < 0.01, ***p < 0.001, Student's t test in B and Tukey's test in F. Scale bars, 10 μm.
IL1RAPL1-PTPδ interaction is required for synapse formation in vivo
To examine the significance of the interaction between IL1RAPL1 and PTPδ in synapse formation in vivo, we injected recombinant IL1RAPL1-ECD-Fc proteins into the developing cerebral cortex of wild-type mice at P17. The injection of soluble IL1RAPL1-ECD-Fc protein into the cerebral cortex of wild-type mice significantly reduced the spine density of basal dendrites of cortical layer 2/3 pyramidal neurons proximal to the injection site as indicated by DiI labeling (p < 0.001) (Fig. 9A,B). Notably, the extent of the decrease in the spine density by the injection of IL1RAPL1-ECD-Fc was comparable to that observed in PTPδ knock-out mice (Fig. 9C,D). Furthermore, the suppressive effect of IL1RAPL1-ECD-Fc on synapse formation in vivo was abolished in PTPδ knock-out mice (Fig. 9A,B).
Figure 9.

Interaction of IL1RAPL1 and PTPδ mediates synapse formation in vivo. A, PTPδ-dependent decrease in the spine density of basal dendrites of cortical layer 2/3 pyramidal neurons by injection of IL1RAPL1-ECD-Fc. B, Spine densities of basal dendrites of cortical layer 2/3 pyramidal neurons of wild-type and Ptprd−/− mice injected with Fc and IL1RAPL1-ECD-Fc (n = 25–37 neurons from two or three mice). C, Decrease in spine density of basal dendrites of cortical layer 2/3 pyramidal neurons in Ptprd−/− mice. D, Spine densities of basal dendrites of cortical layer 2/3 pyramidal neurons of Ptprd+/+, Ptprd+/−, and Ptprd−/− mice (n = 37–48 neurons from three animals each). E, Immunostaining signals for VGluT1 along the basal dendrites of cortical layer 2/3 pyramidal neurons expressing IL1RAPL1-EYFP in Ptprd+/+ and Ptprd−/− mice. F, Relative densities of immunostaining signals for VGluT1 along the basal dendrites of cortical layer 2/3 pyramidal neurons expressing IL1RAPL1-EYFP in Ptprd+/+ and Ptprd−/− mice (n = 15 neurons from one animal each). Immunostaining signals along the dendrites of the pyramidal neurons expressing EYFP in Ptprd+/+ mouse serve as control (n = 15 neurons from one animal). G, Immunostaining signals for VGluT1 along the basal dendrites of cortical layer 2/3 pyramidal neurons expressing IL1RAPL1-EYFP in the presence of AP-MH, PTPδA−B−-ECD-MH, or PTPδ-ECD-MH. Immunostaining of the pyramidal neurons expressing EYFP serve as control. H, Relative densities of immunostaining signals for VGluT1 along the basal dendrites of cortical layer 2/3 pyramidal neurons expressing IL1RAPL1-EYFP in the presence of AP-MH, PTPδA−B−-ECD-MH or PTPδ-ECD-MH (n = 24–26 neurons from two mice each). Immunostaining signals along the dendrites of the pyramidal neurons expressing EYFP serve as control (n = 13 neurons from one animal). All values represent mean ± SEM. **p < 0.01, and ***p < 0.001, Student's t test in B and Tukey's test in D, F, and H. Scale bars, 5 μm.
We further examined whether IL1RAPL1 stimulates synapse formation in vivo using a Sindbis virus-mediated expression system. To monitor the infected neurons, we used IL1RAPL1-EYFP, which was capable of stimulating the accumulation of presynaptic marker proteins in cultured neurons (data not shown). As shown in Figure 9, E and F, Sindbis virus-mediated expression of IL1RAPL1-EYFP protein in vivo in cortical layer 2/3 pyramidal neurons of wild-type mouse strongly enhanced VGluT1 staining signals around the basal dendrites of infected neurons. However, the in vivo synaptogenic activity of IL1RAPL1-EYFP was abolished in PTPδ knock-out mice (p < 0.001). Consistently, the injection of soluble PTPδ-ECD-MH protein into the cortical layer 2/3 significantly suppressed the enhancement effect of IL1RAPL1-EYFP on VGluT1 staining signals around the dendrites of cortical layer 2/3 pyramidal neurons (Fig. 9G,H). PTPδA−B−-ECD-MH lacking the activity to interact with IL1RAPL1 failed to suppress the stimulatory effect of IL1RAPL1-EYFP on synapse formation. These results suggest that the interaction of IL1RAPL1 and PTPδ is required for synapse formation in vivo.
Discussion
Both MR and ASD are highly heritable neurodevelopmental disorders characterized by marked genetic heterogeneity. IL1RAPL1 is responsible for MR (Carrié et al., 1999) and is also associated with autism (Piton et al., 2008). Interestingly, some genetic loci are also overlapping in susceptibility to these disorders (Jamain et al., 2003; Laumonnier et al., 2004; Durand et al., 2007; Kim et al., 2008; Berkel et al., 2010; Laumonnier et al., 2010). Thus, the elucidation of functional roles of IL1RAPL1 will provide an important clue for identifying common pathogenic pathways shared by MR and autism (Bill and Geschwind, 2009). Previously, we showed that presynaptic IL1RAPL1 plays a role in presynaptic differentiation of zebrafish olfactory sensory neurons in vivo by controlling both synaptic vesicle accumulation and morphological remodeling of axon terminals, which are mediated by IP3-dependent Ca2+/calmodulin-protein kinase A signaling and neuronal activity-dependent Ca2+/calmodulin signaling, respectively (Yoshida and Mishina, 2005, 2008; Yoshida et al., 2009). Here, we provide evidence that postsynaptic IL1RAPL1 mediates synapse formation in the forebrain through trans-synaptic interaction with presynaptic PTPδ. Evidence is fivefold. First, knockdown of endogenous IL1RAPL1 reduced the intensity of punctate staining signals for active zone protein Bassoon and the number of dendritic protrusions. Consistently, the expression of IL1RAPL1 in cultured cortical and hippocampal neurons stimulated the accumulation of Bassoon and increased the number of dendritic protrusions. Second, IL1RAPL1 showed the activity to induce presynaptic differentiation of cortical neurons in a neuron-fibroblast coculture assay. Notably, the synaptogenic activity of IL1RAPL1 was specific for excitatory synapses. The ECD of IL1RAPL1 was required and sufficient for the induction of presynaptic differentiation. Third, we identified PTPδ as a major IL1RAPL1-ECD interacting protein by affinity chromatography followed by LC-MS/MS. In fact, PTPδ was essential for IL1RAPL1 to induce presynaptic differentiation since the synaptogenic activity of IL1RAPL1 was abolished in primary neurons from PTPδ knock-out mice. Fourth, IL1RAPL1 transfected into the cortical neurons of developing mouse brain showed robust synaptogenic activity, and this in vivo synaptogenic activity of IL1RAPL1 was not detectable in PTPδ knock-out mice. Finally, both the ECD and CPD of IL1RAPL1 were indispensable for the induction of dendritic protrusions and postsynaptic differentiation. Furthermore, IL1RAPL1 requires presynaptic PTPδ for the induction of spinogenesis and postsynaptic differentiation. These results all together suggest that trans-synaptic interaction between IL1RAPL1 and PTPδ bidirectionally controls synapse formation of cortical and hippocampal pyramidal neurons (Fig. 10). Synapse formation is the key step in the development of neuronal networks. Precise synaptic connections between nerve cells in the brain provide the basis of perception, learning, memory, and cognition. Our results suggest a link between synapse formation and mental disorders. Consistently, failure in neuronal development is implicated in the pathogenesis of mental disorders including MR and autism (McAllister, 2007; Ramocki and Zoghbi, 2008; Guilmatre et al., 2009). Thus, it is worthwhile to note that the PTPRD encoding the trans-synaptic binding partner of IL1RAPL1 could be a member of candidate genes susceptible to MR and autism.
Figure 10.

A proposed model for trans-synaptic interaction between postsynaptic IL1RAPL1 and presynaptic PTPδ. Ig, Ig-like domain; FN, fibronectin type III domain; D1 and D2, protein tyrosine phosphatase domains; CT, C-terminal domain.
Interestingly, Pavlowsky et al. (2010) reported a significant decrease in dendritic spine density and excitatory synapses in the hippocampus of IL1RAPL1 knock-out mice. They showed that IL1RAPL1 interacts with PSD-95 through the C terminus and regulates the synaptic localization of PSD-95 by controlling JNK activity and PSD-95 phosphorylation. Thus, the alterations of the JNK pathway in IL1RAPL1 knock-out mice was suggested to lead to a mislocalization of PSD-95 and a reduction in dendritic spine density and excitatory synapses. On the contrary, they also reported that the interaction of IL1RAPL1 with PSD-95 is not required for exogenous IL1RAPL1-dependent increases of the density of spines and synapses. We here showed that the TIR domain but not the C-terminal domain of IL1RAPL1 was responsible for the spinogenesis. Although it is known that the juxtaposition of TIR domains of IL-1 receptor family proteins induced by ligand binding recruits canonical IL-1 signaling molecules IRAK and MyD88 to activate the transcription factor NF-κB and the mitogen-activated protein kinase (MAPK) pathways (Dunne and O'Neill, 2003), the dimerization of the TIR domain of IL1RAPL1 does not activate NF-κB and MAPK pathways (Born et al., 2000; Khan et al., 2004). Thus, the TIR domain of IL1RAPL1 may recruit signaling molecules other than the canonical IL-1 signaling molecules. Furthermore, presynaptic PTPδ is required for the postsynaptic IL1RAPL1 to exert spinogenic activity. Notably, the injection of IL1RAPL1-ECD into the cortices decreased the spine density to a similar extent as observed in PTPδ knock-out mice. The synapse-organizing role of IL1RAPL1 through the interaction with PTPδ well explains the reduction of dendritic spine density and excitatory synapses in IL1RAPL1 knock-out mice.
Recent studies showed that mutations in some genes, including NLGN3, NLGN4X, NRXN1, SHANK2, and SHANK3 in addition to IL1RAPL1, are associated with autism and MR, noting the overlap of genetic loci in susceptibility to these mental disorders (Jamain et al., 2003; Laumonnier et al., 2004; Durand et al., 2007; Kim et al., 2008; Berkel et al., 2010). NLGNs are involved in proper synapse maturation and function, although they are dispensable for the initial formation of synaptic contacts (Südhof, 2008). Neurexins (NRXNs) interact with NLGNs as trans-synaptic cell adhesion molecules (Südhof, 2008), and SH3 and multiple ankyrin repeat domains (SHANKs) are scaffolding proteins in excitatory synapses (Sheng and Kim, 2000). The role of NRXNs in synapse formation in vivo is indicated by our recent finding that the trans-synaptic interaction between NRXN and glutamate receptor δ2 through Cbln1 mediates synapse formation in the cerebellum (Uemura et al., 2010). Shank proteins promote the maturation of dendritic spines (Sala et al., 2001). Thus, we suggest that synapse formation and synapse maturation processes represent one of the common pathogenic pathways shared by MR and autism.
We showed that the developing mouse brain expressed at least six variants of PTPδ derived from alternative splicing at three small exons encoding meA and meB peptides. Analysis of binding activities of all possible variants revealed that meA and meB peptides in the Ig-like domains of PTPδ were critical for the interaction with IL1RAPL1. Interestingly, the meA and meB peptides appear to form positively charged prominences from the domains in a structure model of Ig-like domains of PTPδ according to Titin (Fig. 5D). PTPδ shares similar domain structures with PTPσ and LAR, constituting 2A type RPTPs (Tonks, 2006). PTPσ and LAR expressed in the brain were lacking meA or meB peptide and failed to show any significant binding activity for IL1RAPL1. These results suggest that IL1RAPL1 selectively interacts with specific variants of PTPδ among 2A type PTPs and the mini-exon peptides of PTPδ represent protein codes for the specificity of synapse formation through the trans-synaptic interaction between IL1RAPL1 and PTPδ.
In contrast to the highly selective interaction of IL1RAPL1 with specific variants of PTPδ among 2A type RPTPs, NGL-3 interacts with all members of 2A type RPTPs and induces presynaptic differentiation of cultured rat hippocampal neurons (Woo et al., 2009; Kwon et al., 2010). Under the conditions we used, however, the synaptogenic activity of NGL-3 was considerably smaller than that of IL1RAPL1 and appeared to be independent of PTPδ since the activity was unaltered in cortical neurons from PTPδ knock-out mice. Although NGL-3 might influence the interaction of IL1RAPL1 and PTPδ, NGL-3-ECD hardly affected the binding of IL1RAPL1-ECD to PTPδ expressed in HEK293T cells. Recently, Takahashi et al. (2011) reported that neurotrophin receptor tyrosine kinase (Trk) C selectively interacts with PTPσ among 2A type RPTPs and induces excitatory synapse formation bidirectionally. Furthermore, PTPσ regulates the synapse number of zebrafish olfactory sensory neurons (Chen et al., 2011). Interestingly, the interaction of TrkC and PTPσ was partly affected by the insertion of meB peptide into the Ig-like domains of PTPσ (Takahashi et al., 2011). On the other hand, the first two fibrinectin-3-like domains of LAR and PTPσ were responsible for the interaction with NGL-3 (Kwon et al., 2010). These studies, together with our findings, suggest that diverse forms of 2A type RPTPs may provide presynaptic molecular codes for synapse formation by interacting with diverse postsynaptic cell adhesion molecules in a subtype- and variant-specific manner. Recent studies also revealed that presynaptic NRXNs interact with LRRTM2 and GluRδ2 in addition to NLGNs to mediate synapse formation in a variant-specific manner (Südhof, 2008; de Wit et al., 2009; Ko et al., 2009; Uemura et al., 2010). The diversity of synaptogenic molecules would be required for the selective formation of the huge numbers of synapses in the brain. An intriguing question is whether these molecules involved in the pathogenesis of mental disorders affect primarily specific networks in the brain.
Footnotes
This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Pharmacological Research Foundation, Tokyo. We thank M. Watanabe for antibodies against VGluT1, Shank2, and VGAT; and Y. Hata for anti-Shank1 antibody. We are grateful to K. Kanemaru for advice on preparation of Sindbis virus particles; T. Shimizu and S. Tokuoka for use of the Biacore 3000 biosensor; T. Shiroshima, A. Kakihara, and A. Maeda for technical assistance; and Y. Nakajima for help in preparation of the manuscript.
The authors declare no competing financial interests.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Bahi N, Friocourt G, Carrié A, Graham ME, Weiss JL, Chafey P, Fauchereau F, Burgoyne RD, Chelly J. IL1 receptor accessory protein like, a protein involved in X-linked mental retardation, interacts with neuronal calcium sensor-1 and regulates exocytosis. Hum Mol Genet. 2003;12:1415–1425. doi: 10.1093/hmg/ddg147. [DOI] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, Bonin M, Riess A, Engels H, Sprengel R, Scherer SW, Rappold GA. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Bill BR, Geschwind DH. Genetic advances in autism: heterogeneity and convergence on shared pathways. Curr Opin Genet Dev. 2009;19:271–278. doi: 10.1016/j.gde.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born TL, Smith DE, Garka KE, Renshaw BR, Bertles JS, Sims JE. Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family. Delineation of a new class of IL-1R-related proteins based on signaling. J Biol Chem. 2000;275:29946–29954. doi: 10.1074/jbc.M004077200. [DOI] [PubMed] [Google Scholar]
- Carrié A, Jun L, Bienvenu T, Vinet MC, McDonell N, Couvert P, Zemni R, Cardona A, Van Buggenhout G, Frints S, Hamel B, Moraine C, Ropers HH, Strom T, Howell GR, Whittaker A, Ross MT, Kahn A, Fryns JP, Beldjord C, et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet. 1999;23:25–31. doi: 10.1038/12623. [DOI] [PubMed] [Google Scholar]
- Chelly J, Mandel JL. Monogenic causes of X-linked mental retardation. Nat Rev Genet. 2001;2:669–680. doi: 10.1038/35088558. [DOI] [PubMed] [Google Scholar]
- Chelly J, Khelfaoui M, Francis F, Chérif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet. 2006;14:701–713. doi: 10.1038/sj.ejhg.5201595. [DOI] [PubMed] [Google Scholar]
- Chen X, Yoshida T, Sagara H, Mikami Y, Mishina M. Protein tyrosine phosphatase σ regulates the synapse number of zebrafish olfactory sensory neurons. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07411.x. [DOI] [PubMed] [Google Scholar]
- Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. Eur J Hum Genet. 2008;16:422–434. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;171:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E. The epidemiology of autism: a review. Psychol Med. 1999;29:769–786. doi: 10.1017/s0033291799008508. [DOI] [PubMed] [Google Scholar]
- Gambino F, Pavlowsky A, Béglé A, Dupont JL, Bahi N, Courjaret R, Gardette R, Hadjkacem H, Skala H, Poulain B, Chelly J, Vitale N, Humeau Y. IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca2+-channel and neurite elongation. Proc Natl Acad Sci U S A. 2007;104:9063–9068. doi: 10.1073/pnas.0701133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, Layet V, Rosier A, Briault S, Bonnet-Brilhault F, Laumonnier F, Odent S, Le Vacon G, Joly-Helas G, David V, Bendavid C, Pinoit JM, Henry C, Impallomeni C, Germano E, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JA, Brint EK, O'Neill LA, Tong L. Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem. 2004;279:31664–31670. doi: 10.1074/jbc.M403434200. [DOI] [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol. 2006;198:401–415. doi: 10.1016/j.expneurol.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, Troxell R, Kaplan L, Volkmar F, Klin A, Tsatsanis K, Harris DJ, Noens I, et al. Disruption of neurexin1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SK, Woo J, Kim SY, Kim H, Kim E. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase δ (PTPδ), and PTPσ via specific domains regulate excitatory synapse formation. J Biol Chem. 2010;285:13966–13978. doi: 10.1074/jbc.M109.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthélémy C, Moraine C, Briault S. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, Ropers HH, Ronce N, Blesson S, Moraine C, Gécz J, Raynaud M. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry. 2010;15:767–776. doi: 10.1038/mp.2009.14. [DOI] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, Wigler M. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Levy SE, Mandell DS, Schultz RT. Autism. Lancet. 2009;374:1627–1638. doi: 10.1016/S0140-6736(09)61376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Fukaya M, Sato T, Sugihara K, Asano M, Yoshioka K, Watanabe M. Expression and distribution of JNK/SAPK-associated scaffold protein JSAP1 in developing and adult mouse brain. J Neurochem. 2006;97:1431–1446. doi: 10.1111/j.1471-4159.2006.03835.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre–Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Hasegawa K, Ogimoto M, Katagiri T, Yakura H. Developmental regulation of gene expression for the MPTPδ isoforms in the central nervous system and the immune system. FEBS Lett. 1994;355:223–228. doi: 10.1016/0014-5793(94)01188-5. [DOI] [PubMed] [Google Scholar]
- Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M, Valnegri P, Rezai X, Bassani S, Brambilla D, Kumpost J, Blahos J, Roux MJ, Humeau Y, Chelly J, Passafaro M, Giustetto M, Billuart P, Sala C. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol. 2010;20:103–115. doi: 10.1016/j.cub.2009.12.030. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, Champagne N, Lafrenière RG, Hamdan FF, Joober R, Fombonne E, Marineau C, Cossette P, Dubé MP, Haghighi P, Drapeau P, Barker PA, Carbonetto S, Rouleau GA. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet. 2008;17:3965–3974. doi: 10.1093/hmg/ddn300. [DOI] [PubMed] [Google Scholar]
- Pulido R, Krueger NX, Serra-Pagès C, Saito H, Streuli M. Molecular characterization of the human transmembrane protein-tyrosine phosphatase δ. J Biol Chem. 1995;270:6722–6728. doi: 10.1074/jbc.270.12.6722. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH. X-linked mental retardation: many genes for a complex disorder. Curr Opin Genet Dev. 2006;16:260–269. doi: 10.1016/j.gde.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Davis Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113:1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- Sims JE, March CJ, Cosman D, Widmer MB, MacDonald HR, McMahan CJ, Grubin CE, Wignall JM, Jackson JL, Call SM. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science. 1988;241:585–589. doi: 10.1126/science.2969618. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Arstikaitis P, Prasad T, Bartlett TE, Wang YT, Murphy TH, Craig AM. Postsynaptic TrkC and presynaptic PTPσ function as a bidirectional excitatory synaptic organizing complex. Neuron. 2011;69:287–303. doi: 10.1016/j.neuron.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Uemura T, Mishina M. The amino-terminal domain of glutamate receptor δ2 triggers presynaptic differentiation. Biochem Biophys Res Commun. 2008;377:1315–1319. doi: 10.1016/j.bbrc.2008.10.170. [DOI] [PubMed] [Google Scholar]
- Uemura T, Mori H, Mishina M. Direct interaction of GluRδ2 with Shank scaffold proteins in cerebellar Purkinje cells. Mol Cell Neurosci. 2004;26:330–341. doi: 10.1016/j.mcn.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Uetani N, Kato K, Ogura H, Mizuno K, Kawano K, Mikoshiba K, Yakura H, Asano M, Iwakura Y. Impaired learning with enhanced hippocampal long-term potentiation in PTPδ-deficient mice. EMBO J. 2000;19:2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Castelmur E, Marino M, Svergun DI, Kreplak L, Ucurum-Fotiadis Z, Konarev PV, Urzhumtsev A, Labeit D, Labeit S, Mayans O. A regular pattern of Ig super-motifs defines segmental flexibility as the elastic mechanism of the titin chain. Proc Natl Acad Sci U S A. 2008;105:1186–1191. doi: 10.1073/pnas.0707163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J, Kwon SK, Choi S, Kim S, Lee JR, Dunah AW, Sheng M, Kim E. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat Neurosci. 2009;12:428–437. doi: 10.1038/nn.2279. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Mishina M. Distinct roles of calcineurin-nuclear factor of activated T-cells and protein kinase A-cAMP response element-binding protein signaling in presynaptic differentiation. J Neurosci. 2005;25:3067–3079. doi: 10.1523/JNEUROSCI.3738-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Mishina M. Zebrafish orthologue of mental retardation protein IL1RAPL1 regulates presynaptic differentiation. Mol Cell Neurosci. 2008;39:218–228. doi: 10.1016/j.mcn.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Uchida S, Mishina M. Regulation of synaptic vesicle accumulation and axon terminal remodeling during synapse formation by distinct Ca2+ signaling. J Neurochem. 2009;111:160–170. doi: 10.1111/j.1471-4159.2009.06309.x. [DOI] [PubMed] [Google Scholar]