

Abstract
The mechanisms governing the recruitment of functional glutamate receptors at nascent excitatory postsynapses following initial axon-dendrite contact remain unclear. We examined here the ability of neurexin/neuroligin adhesions to mobilize AMPA-type glutamate receptors (AMPARs) at postsynapses through a diffusion/trap process involving the scaffold molecule PSD-95. Using single nanoparticle tracking in primary rat and mouse hippocampal neurons overexpressing or lacking neuroligin-1 (Nlg1), a striking inverse correlation was found between AMPAR diffusion and Nlg1 expression level. The use of Nlg1 mutants and inhibitory RNAs against PSD-95 demonstrated that this effect depended on intact Nlg1/PSD-95 interactions. Furthermore, functional AMPARs were recruited within 1 h at nascent Nlg1/PSD-95 clusters assembled by neurexin-1β multimers, a process requiring AMPAR membrane diffusion. Triggering novel neurexin/neuroligin adhesions also caused a depletion of PSD-95 from native synapses and a drop in AMPAR miniature EPSCs, indicating a competitive mechanism. Finally, both AMPAR level at synapses and AMPAR-dependent synaptic transmission were diminished in hippocampal slices from newborn Nlg1 knock-out mice, confirming an important role of Nlg1 in driving AMPARs to nascent synapses. Together, these data reveal a mechanism by which membrane-diffusing AMPARs can be rapidly trapped at PSD-95 scaffolds assembled at nascent neurexin/neuroligin adhesions, in competition with existing synapses.
Introduction
In the developing brain, synaptogenesis is a multistep process at sites of axodendritic or axosomatic contacts, initiated by adhesion proteins and followed by the recruitment of scaffold proteins and receptor channels in a precise temporal order (Friedman et al., 2000; Bresler et al., 2001, 2004; Gerrow et al., 2006).
The transmembrane adhesion proteins neurexins (Nrxs) and neuroligins (Nlgs) are key players in synapse initiation and validation (Südhof, 2008). These molecules form a bridge between presynaptic and postsynaptic membranes through high affinity recognition between ectodomains, in an isoform- and splice variant-specific manner (Craig and Kang, 2007). At the presynapse, Nrxs bind the multimodal scaffolding protein CASK (Mukherjee et al., 2008), and have an essential function in coupling calcium channels to the release machinery (Missler et al., 2003). At excitatory postsynapses, neuroligin-1 (Nlg1) binds the major scaffold protein PSD-95 (Irie et al., 1997), which interacts directly with NMDA glutamate receptors (NMDAR), and indirectly with AMPA glutamate receptors (AMPAR) through binding to the auxiliary subunit stargazin and related transmembrane AMPAR-associated proteins (TARPs) (Bats et al., 2007; Shi et al., 2009). The importance of Nlgs in nervous system function is highlighted by the facts that Nlg knock-out (KO) mice show altered NMDA-mediated synaptic responses (Chubykin et al., 2007), deficits in long-term potentiation (Jung et al., 2010), and reduced network activity in respiratory centers (Varoqueaux et al., 2006).
Studies in neuronal cultures showed that overexpressing Nlgs increases the number and size of synapses (Levinson et al., 2005; Ko et al., 2009), whereas downregulating Nlgs does the opposite (Chih et al., 2005). In addition, primary neurons form functional presynaptic terminals onto cocultured heterologous cells expressing Nlgs (Scheiffele et al., 2000), and develop postsynaptic PSD-95 scaffolds onto cocultured fibroblasts expressing Nrx1β (Graf et al., 2004). These effects can be reproduced using microspheres coated with either purified Nlg (Dean et al., 2003) or Nrx (Graf et al., 2004; Heine et al., 2008b), indicating that clustering adhesion molecules is sufficient to trigger presynaptic or postsynaptic differentiation, respectively.
One important issue for the establishment of functional synapses is how glutamate receptors are recruited at nascent excitatory postsynapses following initial axon/dendrite contact. Although both NMDA and AMPA receptors accumulate at novel Nrx/Nlg adhesions (Graf et al., 2004; Nam and Chen, 2005; Heine et al., 2008b; Barrow et al., 2009), the underlying mechanisms are still unclear. Several processes contribute to the synaptic delivery of glutamate receptors, including exocytosis (Kennedy et al., 2010; Thyagarajan and Ting, 2010), transport of preassembled packets (Washbourne et al., 2002), or surface diffusion (Groc et al., 2006; Bats et al., 2007). We tested here the hypothesis that surface AMPARs may accumulate at Nrx/Nlg contacts through a diffusion/trap mechanism. We addressed this issue in primary neurons using live imaging, immunocytochemistry, and electrophysiology experiments, upon selective formation or perturbation of Nrx/Nlg adhesions. We show that Nrx/Nlg contacts, in competition with preexisting synapses, assemble a PSD-95 scaffold which captures surface-diffusing AMPARs.
Materials and Methods
Molecular constructs
The pcDNA PSD-95:GFP and Homer1c:GFP were gifts from S. Okabe (Tokyo University, Japan). For the PSD-95:mCherry construct, mCherry was amplified by PCR with primers containing KpnI/BsrGI sites. It was then inserted at the C terminus of PSD-95:GFP in place of GFP using these restriction sites. pcDNA dimer DsRed Homer1c was generated by ligation of PCR-amplified dimer DsRed in frame to replace the existing EGFP using HindIII and BsrGI sites. N-terminal HA-tagged Nlg1 constructs WT, ΔC (truncated for the last 72 AA of the C terminus tail) and Swap (acetylcholine-like extracellular region swapped with regular acetylcholine esterase) were gifts from P. Scheiffele (Biozentrum, Basel, Switzerland). To make SEP:GluA1 and SEP:GluA2, the SEP (superecliptic pHluorin) sequence was amplified by PCR with primers containing AgeI/NheI sites. It was then inserted after the signal peptide of GluA1 or GluA2 cloned in eukaryotic expression vectors (respectively, prk5 and pcDNA). shRNA against PSD-95 (shPSD-95) and a control plasmid containing the same shRNA against endogenous PSD-95 but also expressing a recombinant PSD-95:GFP insensitive to the shRNA (replPSD-95) were gifts from O. Schlüter (Stanford University, Palo Alto, CA). For pSuper Neo GFP sh rat SAP-97, an annealing of the primers sh rat SAP-97 2281 XhoI 5′-gatccccgatatccaggagcataaatttcaag agaatttatgctcctggatatctttttc-3′ and sh rat SAP-97 2281 XhoI As 5′-tcgagaaaaagatatccaggagcataaattctcttgaaatttatgctcctggatatcggg-3′ was first performed. The obtained double-stranded DNA was then inserted at BglII/XhoI sites in pSuper Neo GFP (generously provided by P. Worley, Johns Hopkins University, Baltimore, MD). The efficiency of the construct in inhibiting SAP-97 expression was assessed in COS cells transfected with rat SAP-97:GFP (a gift from U. Thomas and A. H. Chishti, Magdeburg), or cotransfected with SAP-97:GFP and shSAP-97, followed by cell lysis, SDS-PAGE and immunoblot using an antibody against SAP-97 (Stressgen) (data not shown).
Purified recombinant proteins
The pcDNA neomycin β-Neurexin δ4-human Fc (abbreviated Nrx1β-Fc) plasmid received from P. Scheiffele was subcloned between sites HindIII/XhoI in pcDNAhygro(+) vector to select a stable hygromycin-resistant HEK cell line producing Nrx1β-Fc. Recombinant Nrx1β-Fc protein was expressed in HEK cells and purified from conditioned medium as described previously (Heine et al., 2008b), to a concentration of 0.8 mg/ml. The pFab7-SP encoding GluA2/4-specific antibody Fab fragment (called Fab anti-GluA2) plasmid was a gift from K. Keinänen (University of Helsinki, Finland). It was expressed in E. coli Origami strain (Novagen) and proteins were extracted by sonication and centrifugation. Fab was purified on protein G affinity column to a concentration of 0.6 mg/ml. Goat anti-human Fc (Jackson ImmunoResearch) was conjugated to either Cy3 or Cy5 monoreactive dyes using coupling kits (GE Healthcare), followed by purification on size exclusion Sepharose columns (Bio-Rad).
Cell culture and transfection
Dissociated hippocampal neurons from embryonic day 18 rat embryos of either sex were plated on 18 mm polylysine-coated glass coverslips at a density of 10′000 cells/cm2 in MEM containing 10% horse serum (Invitrogen) for 3 h, then cultured in Neurobasal medium supplemented with B27 on a layer of glial cells (Goslin et al., 1991). Neurons were transfected at 4–7 DIV using Effectene (Qiagen) and processed 2–5 d later. The same protocol was used to culture dissociated hippocampal neurons from postnatal day 0 (P0) Nlg1 KO, GluA2 KO or WT littermate mice pups of either sex. To obtain neurons with a Nlg1-null background, we bred heterozygotes Nlg1 mice (Varoqueaux et al., 2006) generously given by N. Brose and F. Varoqueaux (Max Planck Institute, Goettingen, Germany). Newborn littermate pups were genotyped at P0, allowing selection of WT and KO animals for dissociated hippocampal cultures, performed as above. The same protocol was used to obtain GluA2 KO cultures from GluA2 heterozygote mice (gift from R. Sprengel, Max Planck Institute, Heidelberg, Germany).
Single nanoparticle tracking
1 μl of 655 nm Quantum dots (Qdots) conjugated to goat (Fab′)2 anti-mouse or anti-rabbit IgG (Invitrogen) were incubated with 1 μl of Fab anti-GluA2, anti-NCAM, or rabbit anti-GFP, in 7 μl of PBS for 20 min at room temperature. Qdot conjugates are ∼20 nm in size and antibodies add another 10 nm layer (Wu and Bruchez, 2004). Nonspecific binding was blocked by adding 1 μl of 10% casein solution for 15 min (Vector Laboratories), and this solution was kept at 4°C throughout the experiment. Neurons were incubated for 10 min at 37°C in 1 ml of culture medium containing 1 μl of the anti-GluA2 or anti-NCAM-coated Qdot solution, or 0.05 μl of anti-GFP-coated Qdots. Cells were rinsed and mounted in an aluminum chamber containing Tyrode's solution, and then observed on a Nikon microscope (Eclipse TE 2000-U), thermostated to 37°C using an objective heater (Bioptechs) and an air blower (Precision instruments). Qdots and GFP or DsRed tagged proteins were illuminated by a mercury lamp and detected using a 100×/1.4 oil objective with appropriate excitation/emission filters (Nikon FITC filter set: Ex 482/35; DM 506; Em 536/40, Nikon TRITC filter set: Ex 543/22; DM 562; Em 593/40, and HCRed1 filter set from Chroma Technology: Ex HQ 575/50; DM Q610LP, Em HQ640/50), allowing separation of the three channels. A series of 1000 images were recorded with an integration time of 50 ms, using an EMCCD camera (Quantem, Roper Scientific). Qdots were followed on randomly selected dendritic regions for up to 20 min. Qdot images were processed with the MetaMorph software (Universal Imaging Corp). The global instantaneous diffusion coefficient, D, was calculated for each trajectory, from linear fits of the first 8 points of the mean-square-displacement (MSD) versus time function using MSD (t) = 4Dt (Groc et al., 2007). The synaptic and extrasynaptic diffusion coefficients were also calculated for stretches of trajectories identified as inside or outside synapses or PSD-95 clusters, by reference to the Homer1c:GFP or PSD-95:GFP labelings, respectively.
The monodispersity of the Qdot stock solution is guaranteed by the manufacturer. However, it is possible that Qdot aggregation occurs after antibody conjugation. We checked that single Qdots were observed by analyzing their fluorescence emission over time. The blinking behavior is a photo-physical characteristics of Qdots and a signature of the fact that they are isolated entities (Groc et al., 2007). However, even if Qdots form small aggregates, their diffusion coefficient should be reduced by a factor proportional to the logarithm of the aggregate size (Saint-Michel et al., 2009), thus making a small contribution to the measurement of AMPAR diffusion properties.
Nrx1β-Fc cluster formation and surface SEP:GluA2 X-link
2 μg of soluble Nrx1β-Fc were mixed with 1 μg of anti-Human-Fc into 100 μl of culture medium or observation medium containing 0.3% globulin-free BSA (Sigma) to avoid nonspecific binding. Depending on the application, we used either Cy3-conjugated, Cy5-conjugated, or unlabeled anti-Fc.
To block GluA2 surface diffusion, live neurons transfected with recombinant SEP:GluA2 were treated with a 1:100 dilution of polyclonal anti-GFP (Invitrogen) at 37°C in culture medium during 10 min.
Immunocytochemistry
To measure endogenous AMPAR and NCAM recruitment, a 10 min live labeling of surface proteins was performed at 37°C with 1:100 antibody dilution in warm medium containing 0.3% of BSA. We respectively used a polyclonal anti-GluA1 (a gift from M. D. Ehlers, Duke University, Durham, NC) and a polyclonal anti-NCAM (a gift from R. M. Mège, INSERM, Paris). Cells were fixed for 10 min in warm 4% paraformaldehyde-4% sucrose in PBS, and remaining active sites were saturated with 50 mm NH4Cl in PBS for 15 min. Secondary Alexa Fluor 488-conjugated goat anti-rabbit antibody (2 mg/ml, Invitrogen) was added 1:1000 for 30 min at room temperature. Coverslips were then mounted in Mowiol (Calbiochem).
To measure recombinant SEP:GluA1 or SEP:GluA2 recruitment, cells were fixed and stained with 1:400 mouse anti-GFP (Roche) followed by 1:1000 Alexa Fluor 488-conjugated goat anti-mouse antibody (2 mg/ml, Invitrogen) for 30 min at room temperature.
To measure synapse number, cells expressing Nlg1 mutants were fixed and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Nonspecific binding was blocked with PBS containing 1% BSA. Presynapses were labeled with 1:400 mouse anti-synapsin (Synaptic Systems) followed by 1:1000 Alexa Fluor 568-conjugated goat anti-mouse antibody (2 mg/ml, Invitrogen).
To localize active presynapses in live experiments, cells were labeled using a 1:100 dilution of anti-synaptotagmin1 lumenal domain conjugated to Oyster 550 fluorophore (Synaptic Systems) in observation medium containing 50 mm KCl.
To quantify the level of Nlg1 surface expression, living neurons were incubated with soluble Nrx1β-Fc (1:50 dilution in culture medium), fixed, and then stained with 1:300 goat anti-Human Fc, conjugated to cy3 using a coupling kit from GE Healthcare.
To measure the level of overexpression or knock-down of PSD-95, neurons transfected with shPSD-95 or replPSD-95 were fixed, permeabilized and labeled with 1:500 primary monoclonal anti-PSD-95 (UC Davis/NIH NeuroMab Facility) followed by 1:1000 Alexa Fluor 568-conjugated goat anti-mouse antibody (Invitrogen).
To visualize the colocalization between Nlg1 and PSD-95, neurons cotransfected with Nlg1WT and PSD-95:GFP were live stained with 1:100 rat anti-HA (Roche), then fixed and incubated with 1:1000 anti-rat Alexa Fluor 568.
Immunostainings were visualized on a Leica DM R epifluorescence microscope equipped with a 63×/1.32 NA objective and appropriate filter sets. Images were acquired with a CCD camera (HQ CoolSnap, Roper Scientific), using the MetaMorph software (Universal Imaging Corp.).
Image analysis of cluster density and apposition
To measure enrichment factors of PSD-95 or AMPARs to Nrx1β-Fc clusters the Cy5 (Nrx1β-Fc) images were treated by a segmentation program written within MetaMorph (Groc et al., 2007), allowing precise detection of clusters. The corresponding contours were transferred to the PSD-95 or AMPAR image, and the signal intensity was measured in these regions, and normalized by that on a control area. To count the number of synapses per surface area of neurite, synapsin images were treated by a segmentation program allowing clusters to be detected as single objects, which were then automatically counted. An intensity threshold was applied to define the neurite contour and calculate its area. To compute the degree of apposition between presynaptic elements (synapsin or synaptotagmin stainings) and postsynaptic clusters (Homer1c:GFP or PSD-95:GFP labelings), a plugin was developed within ImageJ. Two binary images, one for each type of staining, were first generated by a wavelet segmentation program, allowing identification of synaptic structures as individual objects. Two spots were taken as being apposed if the distance between them was smaller or equal to 1 pixel. These apposed regions were used to determine the percentage of postsynaptic clusters which are apposed to a presynapse. The reverse analysis is difficult to do because the presynaptic staining labels all cells, making the fraction of synapsin puncta apposed to transfected PSD-95:GFP clusters very small and hard to use for a quantification.
Live imaging of PSD-95:GFP
Neurons (8–10 DIV) expressing Nlg1WT and PSD-95:GFP were mounted on an open chamber containing Tyrode's solution (containing, in mm: 30 d-glucose, 120 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 25 HEPES, pH 7.4) supplemented with 0.3% BSA. Cells were then observed on an Olympus microscope (IX70) using a 100×/1.4 oil objective, illumination with a mercury lamp, and appropriate GFP and Cy5 filter sets. At time 0, Cy5-Nrx1β antibody aggregates were added, and the PSD-95:GFP images were recorded every 20 s for 30 min. At the end of this period, unbound Nrx1β-Fc was rinsed out, and a single Cy5 image was taken to localize Nrx1β clusters. To measure the depletion in PSD-95:GFP fluorescence, PSD-95:GFP clusters present before incubation with Nrx1β aggregates were isolated using a segmentation program written within MetaMorph (Groc et al., 2007). The fluorescence intensity of isolated clusters was measured throughout the image acquisition stack, and was corrected for the global photobleaching. Because we analyzed only clusters that can be followed throughout the whole image stack, independently of newly forming clusters, this value is actually an underestimation.
Electrophysiology
Iontophoresis experiments.
The extracellular solution contained in millimolar concentrations: 145 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES and 10 d-glucose, pH 7.4 supplemented with 10 μm bicuculline (Tocris Bioscience), 10 μm APV and 1 μm TTX to block GABA-A receptors, NMDA receptors and sodium channels, respectively. Borosilicate pipettes (Clark Electromedical) were pulled with a micropipette puller (Sutter Instruments) to produce patch electrodes with resistances of 3–5 MΩ. The intracellular solution contained in millimolar concentrations: 130 CH3CsSO3, 2 MgCl2, 1 CaCl2, 2 NaATP, 10 EGTA, 10 HEPES, and 0.4 GTP, pH = 7.25. Micropipette positioning was achieved through 3-axes micromanipulators (Luigs-Neumann, Germany), under an inverted epifluorescence microscope equipped with a 60×/1.35 objective (Zeiss). Recordings were performed with an EPC 10 double patch-clamp amplifier (HEKA Electronics). Data were acquired and stored using Patch-Master software version 2.0 (HEKA Electronics) and analyzed with IGOR (WaveMetrics) and GraphPad Prism software. Local activation of receptors was performed by glutamate iontophoresis, using pipettes with a resistance of 40–60 MΩ filled with 150 mm sodium glutamate pH 7.4 and connected to an amplifier from NPI Electronic. A small retaining current was needed to keep glutamate inside the pipette (usually between 10 and 50 nA). Current pulses of 80–150 nA and 1 ms duration were applied to evoke glutamate receptor-mediated currents. At least 10 recorded current traces were averaged.
To identify synapses and new PSD-95 clusters, a fluorescent image of a PSD-95:GFP-expressing cell was taken before incubation with Nrx1β cross-linked with anti-human Fc antibodies conjugated to Cy3, and 30–60 min after. We checked in the Cy3 fluorescence channel that the new PSD-95:GFP clusters were associated to Nrx1β spots. After the establishment of the whole-cell patch configuration, the iontophoretic pipette was navigated toward preselected spots of PSD-95:GFP that appeared during the incubation period, or were there before as control. The distance between the iontophoretic pipette and the recorded cell was controlled by the rise time of glutamate-evoked currents. If the rise time was longer than 3 ms, recordings were excluded from analysis.
Cluster experiments in cell culture.
Hippocampal neurons (5 DIV) were transfected with Nlg1WT and PSD-95:GFP plasmids. After 3–4 d of expression, cells were perfused with extracellular solution composed of the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 4 CaCl2, 4 MgCl2, 25 glucose and saturated with 95% O2/5% CO2. The patch pipette was loaded with the intracellular solution composed of the following (in mm): 140 Cs-gluconate, 10 HEPES, 10 EGTA, 4 Mg-ATP, 6 NaCl, and 0.4 GTP. The extracellular and intracellular solutions were almost equimolar (300–305 mOsm). Cells were recorded in whole-cell configuration at a −70 mV holding potential, close to the chloride inversion potential making GABA currents undetectable. To isolate AMPA mEPSCs, 1 μm TTX and 50 μm APV were added to the bath. For each coverslip, the first transfected cell was recorded for 15 min as the control condition. Then cross-linked Nrx1β was added for 10 min (without perfusion), and allowed for 20 min more (with perfusion) the formation of clusters. After this incubation, miniature events from transfected cells (1–2 per coverslip) were recorded for 15 min.
Slice experiments.
Electrophysiological recordings of CA1 pyramidal cells were performed using parasagittal hippocampal slices from 8- and 9-d-old WT and Nlg1 KO mice of either sex. Briefly, AMPA miniature EPSCs (mEPSCs) recordings (4–5 MΩ electrodes) were made at 33°C from pyramidal cells of the hippocampal CA1 field visualized by infrared video-microscopy. Slices were perfused with extracellular solution composed of the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 4 CaCl2, 4 MgCl2, 10 glucose and saturated with 95% O2/5% CO2. TTX (0.5 μm) was added to the bath to block action potential generation. The intracellular solution was composed of following (in mm): 140 Cs-gluconate, 10 HEPES, 10 EGTA, 4 Mg-ATP, 6 NaCl, and 0.4 GTP. Cells were kept at holding potential of −60 mV for 10 min, then mEPSCs were recorded for 15 min. We included in the comparison only cells which had similar membrane resistance, which reveals proper cell opening and seal quality.
Detection and analysis of AMPA mEPSCs.
mEPSCs recordings were analyzed with IGOR (WaveMetrics), using an automated macro modified from Hwang and Copenhagen (1999). This program automatically detected events which corresponded to AMPA mEPSCs, given defined characteristics, such as rise time, decay time, duration, and a 7 pA detection threshold.
Immunostaining of hippocampal sections
Nlg1 WT and Nlg1 KO P9 littermate pups of either sex were killed by cervical dislocation. Brains were quickly removed, snap frozen in liquid nitrogen, and stored at −80°C before sectioning. Eight to 12 hippocampal coronal sections (of 14 μm width) per animal were generated using a cryostat (Microm). Sections were double-stained with mouse anti-SAP102 (1:500, UC Davis/NIH NeuroMab Facility) and rabbit anti-phosphorylated GluA1 (1:500, Abcam) on gelatin-coated slides for 96 h. Primary antibody staining was followed by overnight incubation with Alexa Fluor 488-labeled anti-rabbit and Alexa Fluor 568-labeled anti-mouse (both 1:1000, Invitrogen) secondary antibodies. The stratum radiatum area of the CA1 region was observed using an HCX PL Apo CS 100×/1.4 NA oil objective on a spinning disk microscope (Leica DMI6000) equipped with a confocal Scanner Unit CSU-X1 (Yokogawa). GluA1 and SAP-102 stainings were illuminated using 491 and 561 nm diode lasers, respectively, and captured by an Evolv EMCCD camera (Roper Scientific). z-stacks with a 0.2 μm interval were taken in a single field of view using a galvanometric stage (Leica Microsystems), giving ∼10–20 images per section. Images were acquired from 7 Nlg1 WT and 9 Nlg1 KO mice. Camera aperture, magnification, light power, and exposure time were fixed for all images. Images were later processed as described below. Immunostainings of PSD-95 yielded weak and heterogeneous signals (data not shown), consistent with low PSD-95 expression levels in young animals (Sans et al., 2000).
To count the number of SAP-102 or GluA1 clusters per surface area in the stratum radiatum region, and to measure their integrated intensity, images were treated by a segmentation program allowing clusters to be detected as single objects, which were then automatically counted. An intensity threshold was applied to define the stratum radiatum region contour (excluding cell bodies) and calculate its area. For each cluster detected as an object, a region mask was created and transferred onto the original image to measure the integrated intensity after background correction.
Statistical analyses
For Qdot experiments, non-Gaussian distributions of diffusion coefficients were represented as median ± percentile (25–75%), and compared using nonparametric statistical tests (Mann–Whitney for two conditions, and Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test to compare more than two conditions).
The recruitment of postsynaptic proteins at Nrx clusters was quantified by enrichment factors. The data at different time points were represented by their mean ± SEM, and compared using parametric two-way ANOVA, and Bonferroni's parametric post hoc test. When only two conditions were compared at a single time point, an unpaired t test was used.
For electrophysiology experiments, the amplitude and frequency of AMPA currents were represented as mean ± SEM, and compared using unpaired t tests.
For immunocytochemistry and live imaging experiments, involving the quantification of object density, fluorescence intensity levels, or percentage of colocalization or apposition, data were compared using parametric tests (unpaired t test to compare 2 independent conditions, and one-way ANOVA followed by Bonferroni's post hoc test to compare multiple conditions).
Results
To characterize the mechanisms of AMPAR recruitment at Nrx/Nlg adhesions, we performed multidimensional manipulations. We mostly used primary rat neurons but, in some cases, we generated cultures from WT versus Nlg1 KO mice. Nlg1 and PSD-95 levels were controlled through overexpression, downregulation, or rescue experiments, and the connection between Nlg1 and PSD-95 was altered using mutant Nlg1 constructs. The assembly of novel Nrx/Nlg adhesions was triggered using antibody aggregates of purified Nrx1β. Several types of experiments were then carried out: single nanoparticle tracking of both endogenous and recombinant AMPAR subunits, live imaging of PSD-95, immunostainings, and electrophysiological recordings.
Nlg1 overexpression reduces the surface diffusion of GluA2-containing AMPARs
To explore the role of Nrx/Nlg adhesion on AMPAR surface diffusion, we first overexpressed Nlg1 in 8–10 DIV primary rat hippocampal neurons, together with GFP or Homer1c:GFP as a postsynaptic marker (Kuriu et al., 2006), and monitored AMPAR surface diffusion by single nanoparticle tracking. We used Quantum dots (Qdots) conjugated with Fabs directed against the GluA2 subunit, to label both GluA1/GluA2 and GluA2/GluA3 heteromers which form the majority of endogenous AMPAR complexes in hippocampal neurons (Wenthold et al., 1996; Lu et al., 2009). We checked that Qdots attached specifically to GluA2-containing receptors, by either overexpressing recombinant SEP:GluA2 in rat neurons, or using cultures from GluA2 KO mice, and counting the number of bound Qdots (Fig. 1A,B). Dilution was adjusted to yield sufficiently isolated Qdots, to allow for clear trajectory reconstruction, and the lateral motion of single Qdots was captured at 20 Hz (Fig. 1C,D). Custom software provided calculation of the instantaneous diffusion coefficient for each trajectory (Saint-Michel et al., 2009), and the distributions of diffusion coefficients were plotted for hundreds of Qdots, allowing comparison between conditions. Global AMPAR diffusion was decreased by a factor of 2 upon overexpression of Nlg1WT, compared with control cells (Fig. 1C,D). In contrast, tracking Qdots directed against the control nonsynaptic adhesion molecule NCAM revealed that NCAM diffusion was increased, suggesting that the response to Nlg1 expression was specific to synaptic receptors (Fig. 1C,D).
Figure 1.
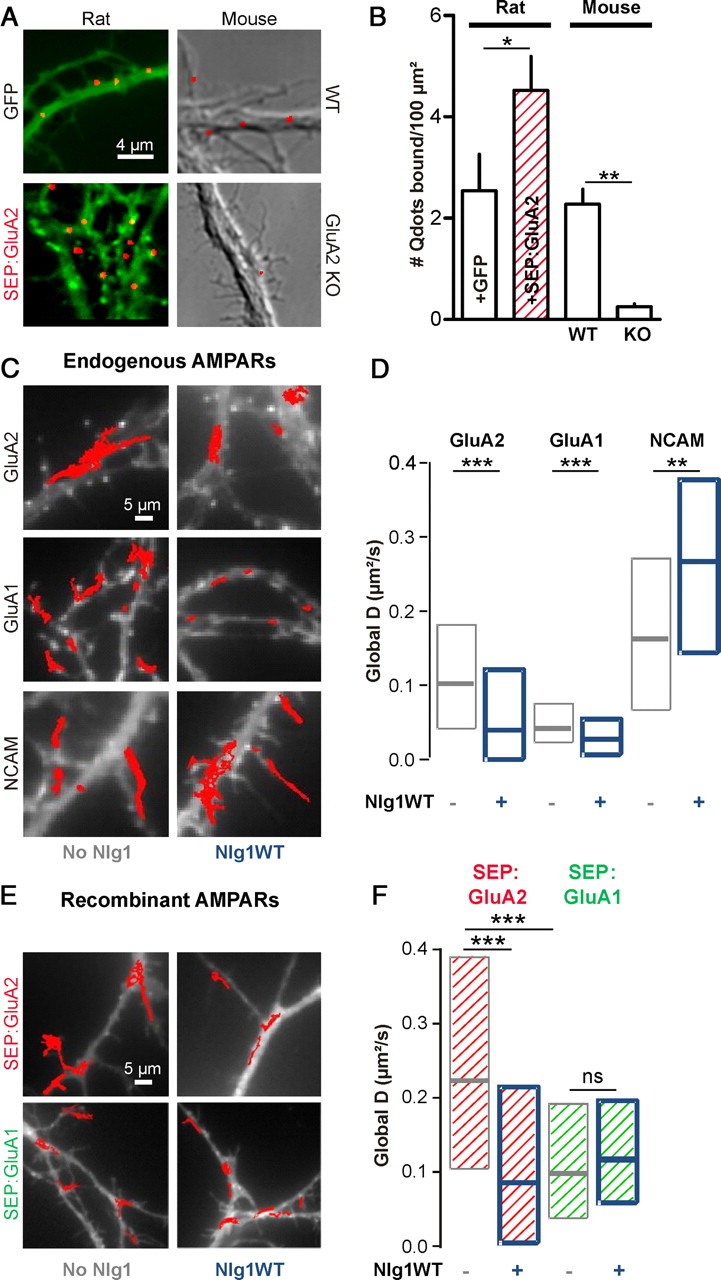
Nlg1 overexpression decreases AMPAR membrane diffusion. A, Neurons (10 DIV) incubated for 10 min with Qdots conjugated with Fab anti-GluA2. Rat neurons (left) were transfected with GFP (7 cells) or SEP:GluA2 (5 cells), and compared with untransfected neurons from either WT (9 cells) or GluA2 KO (12 cells) mice cultures (right). B, Number of Qdots bound per dendritic surface area (mean ± SEM), in all conditions. C, Surface mobility of anti-GluA2, anti-GluA1, or anti-NCAM-conjugated Qdots in 8–10 DIV rat neurons transfected with Homer1c:GFP (white staining) alone (left), or with Homer1c:GFP and Nlg1 (right). Red traces represent individual Qdots trajectories over a 50 s period. D, The global diffusion coefficient was calculated for 200–350 trajectories per condition for 5 independent experiments (anti-GluA2), 500–1200 trajectories from 3 experiments (anti-GluA1), and 200–700 trajectories from 2 experiments (anti-NCAM). E, SEP-tagged GluA1 or GluA2 subunits transfected in neurons with or without Nlg1 were tracked using anti-GFP-conjugated Qdots. F, Distributions of global diffusion coefficients for 70–400 trajectories per condition from 2 experiments. Statistical P-values: *p < 0.05, **p < 0.01, ***p < 0.001.
Immobilization of AMPARs by Nlg1 requires the GluA2 subunit
To assess whether the impact of Nlg1 overexpression on AMPAR diffusion depended on subunit composition, we used Qdots conjugated to anti-GluA1 antibodies, which essentially labels GluA1/GluA2 heteromers since the proportion of GluA1 homomers is only 8% in these neurons (Wenthold et al., 1996; Lu et al., 2009). The diffusion coefficient of endogenous GluA1-containing AMPARs was also strongly decreased by Nlg1 overexpression (Fig. 1C,D). Intriguingly, the basal mobility of anti-GluA1-conjugated Qdots was twice lower than that of anti-GluA2-coated Qdots, reflecting either a valency difference between entire anti-GluA1 antibodies compared with anti-GluA2 Fabs (Saint-Michel et al., 2009), and/or an intrinsically higher diffusion of GluA2/GluA3 versus GluA1/GluA2 heteromers, as reported (Kropf et al., 2008).
To further identify a potential specificity in the diffusion properties of GluA1 or GluA2 receptors upon Nlg1 expression, we transfected recombinant SEP-tagged GluA1 and GluA2 subunits, and tracked their movement using anti-GFP-conjugated Qdots (Fig. 1E,F). Recombinant GluA1 and GluA2 are most likely forming GluA1/GluA1 and GluA2/GluA2 homomers, respectively (Shi et al., 2001). Indeed, SEP:GluA1 expression in hippocampal neurons leads to inward-rectifying AMPA currents (Heine et al., 2008a), lying half way between those measured in untransfected neurons (no rectification), and those obtained in GluA1-expressing HEK cells (strong rectification) (Derkach et al., 1999), showing that there is precisely one GluA1 homomer for one AMPAR in SEP:GluA1-expressing neurons. Since immunostaining with antibodies against GluA1 revealed that SEP:GluA1 was overexpressed twofold above endogenous GluA1 levels (90 ± 12 vs 48 ± 9 fluorescence levels in transfected vs untransfected cells, 10 cells, *p < 0.05 by unpaired t test), we deduce that SEP:GluA1 essentially form GluA1 homomers in our cultures. SEP:GluA2 are also likely to form homomers, since they are overexpressed threefold above endogenous GluA2 levels (340 ± 70 vs 113 ± 25 fluorescence levels in transfected vs untransfected cells, 6 cells, *p < 0.05 by unpaired t test) and show little heteromerization with endogenous GluA2 subunits (Kropf et al., 2008).
Both SEP:GluA1 and SEP:GluA2 subunits were approximately twice more mobile than endogenous GluA1- and GluA2-containing AMPARs, respectively (Fig. 1E,F), likely because of a change in the ratio between overexpressed receptors and available scaffold. However, the basal mobility of SEP:GluA1 subunits was twice lower than that of SEP:GluA2 receptors, revealing an intrinsic differential mobility of GluA1 vs GluA2 homomers, possibly linked to a differential association with scaffold proteins. Strikingly, the mobility of SEP:GluA2 receptors was reduced by a factor of two upon Nlg1 overexpression, whereas that of SEP:GluA1 receptors was unaffected (Fig. 1E,F). Together, these data indicate that AMPAR immobilization by Nlg1 is governed by the GluA2 subunit.
Nlg1 level bidirectionally affects AMPAR surface diffusion
To assess whether AMPAR mobility was also sensitive to a downregulation of Nlg1 expression level, we used both KO and shRNA strategies. Surface labeling with soluble Nrx1β-Fc was approximately tenfold higher in rat neurons expressing Nlg1 than in untransfected counterparts, associated with twofold and threefold increases in the size and density of Nlg1 clusters, respectively (Fig. 2A,B). In contrast, mouse hippocampal neurons treated with shRNA against Nlg1 or cultured from Nlg1 KO animals, showed a similar 85% reduction in Nrx1β labeling compared with untransfected WT cells (Fig. 2A,B). These data indicate that Nlg1 is the major endogenous receptor for Nrx1β in those cultures, despite the potential expression of other Nrx receptors such as Nlgs2/3 or LRRTM2 (Graf et al., 2004; de Wit et al., 2009; Poulopoulos et al., 2009).
Figure 2.
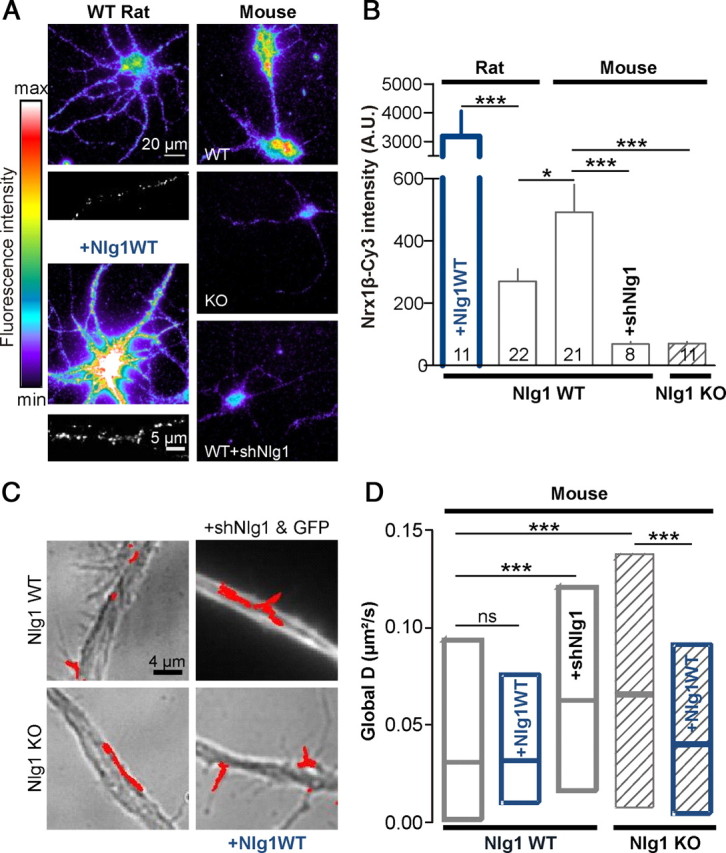
Downregulating Nlg1 expression increases AMPAR membrane diffusion. A, Live labeling of endogenous surface Nlg1 using soluble Nrx1β-Fc in 1- week-old rat or mouse neurons, upon Nlg1 overexpression or downregulation. The clustered appearance of overexpressed Nlg1 was comparable to that of endogenous Nlg1 (insets). Nlg1 cluster density was 0.24 ± 0.03/μm2 for transfected cells versus 0.08 ± 0.01/μm2 for nontransfected cells, and cluster size was 0.51 ± 0.02 μm2 versus 0.24 ± 0.02 μm2, respectively (n = 7 neurons). B, Quantification of the Cy3-conjugated anti-Fc labeling in arbitrary fluorescence units. The number of cells examined in each condition is indicated in the columns (2 independent experiments). C, AMPAR diffusion was measured using anti-GluA2-conjugated Qdots in primary 9–10 DIV neurons obtained from either WT or Nlg1 KO mouse pups. In some experiments, KO cultures were transfected with Nlg1WT, and WT cultures with Nlg1WT or shRNA against Nlg1. D, Diffusion coefficients were computed for 150–2400 trajectories from 6 independent experiments. Statistical P-values: *p < 0.05; ***p < 0.001.
We then measured global AMPAR diffusion using anti-GluA2-coated Qdots. Hippocampal cultures from Nlg1 KO mice showed an approximately twofold increase in AMPAR mobility compared with cells from WT animals (Fig. 2C,D). Accordingly, expression of shRNA against Nlg1 in neurons from WT mice increased AMPAR mobility to similar levels as those obtained in KO cultures, whereas expression of Nlg1 in KO cultures decreased AMPAR mobility to a low level similar to that of cultures from WT littermates (Fig. 2A,B). We noted that AMPAR mobility was lower in mice cultures than in rat cultures at the same age in vitro (9 DIV) (compare Figs. 1D and 2D), closely corresponding to the twofold higher Nlg1 membrane level in mice cultures compared with rat cultures (Fig. 2A,B). Thus, the Nlg1 expression level inversely correlates with AMPAR diffusion. Interestingly, in mouse neurons having higher levels of surface Nlg1, overexpression of Nlg1WT did not induce a significant decrease of AMPAR diffusion as observed in rat cultures (Fig. 2B,D), suggesting that a twofold difference in Nlg1 level is enough to reach a maximal effect on AMPAR diffusion.
The reduction of AMPAR diffusion by Nlg1 requires the binding between Nlg1 and PSD-95
Overexpression of the synaptogenic protein Nlg1 in neurons is known to approximately double the number of synapses, an effect mediated by the Nlg1 ectodomain (Chih et al., 2005; Levinson et al., 2005; Ko et al., 2009). To examine whether the reduction of AMPAR mobility caused by Nlg1 overexpression was linked to Nlg1/PSD-95 or Nrx1β/Nlg1 interactions, we used two Nlg1 mutants, respectively (Fig. 3A): (1) Nlg1ΔC, with a C-terminal truncation preventing it to interact with PSD-95, but still able to establish synaptic contacts through its ectodomain; and (2) Nlg1Swap, with a swapped extracellular domain unable to interact with Nrx, but still able to recruit PSD-95 (Scheiffele et al., 2000). As assessed by immunocytochemistry, expression of Nlg1ΔC increased by twofold the number of synapsin puncta apposed to transfected neurons, as obtained for Nlg1WT, but these new synapses lacked PSD-95 (Fig. 3B,C). In parallel, AMPAR diffusion tracked with either anti-GluA1 or anti-GluA2-coated Qdots, was unaffected by Nlg1ΔC expression (Fig. 3E–G). This indicates that the drop in AMPAR diffusion upon Nlg1 overexpression is not linked to an increase in synapse number, but rather to the specific interaction between Nlg1 and PSD-95. Accordingly, Nlg1Swap did not increase synapse density but instead formed new extrasynaptic PSD-95 clusters (Fig. 3A–D), and diminished AMPAR diffusion similarly to Nlg1WT (Fig. 3E–G). Thus, the interaction between the Nlg1 intracellular tail and PSD-95 is necessary to immobilize AMPARs.
Figure 3.
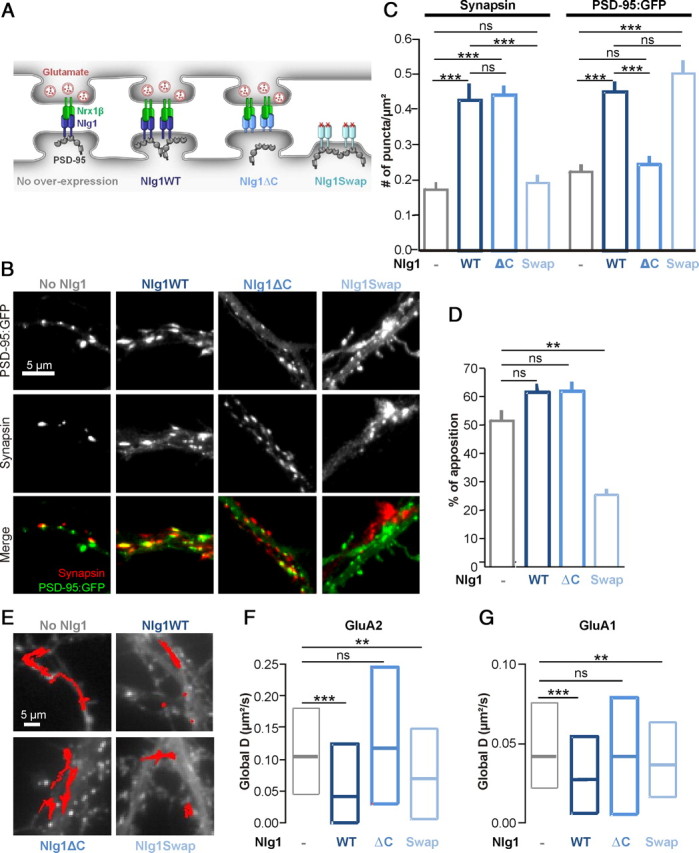
Effects of Nlg1 mutants on synaptic differentiation and AMPAR mobility. A, Schematic diagram of Nlg1 mutants. B–D, Rat hippocampal neurons expressing PSD-95:GFP and Nlg1 mutants were fixed and labeled for the presynaptic marker synapsin. C, Quantification of the number of synapsin and PSD-95:GFP puncta per μm2 (2 independent experiments, n = 8–19 cells). D, Analysis of the fraction of PSD-95:GFP clusters apposed to synapsin spots. E–G, AMPAR diffusion was monitored using anti-GluA2 or anti-GluA1 Qdots in 8–10 DIV rat neurons coexpressing Homer1c:GFP and Nlg1 mutants (no Nlg1, Nlg1WT, Nlg1ΔC, or Nlg1Swap). E, Examples of 50 s trajectories of anti-GluA2 Qdots for each condition. F, Distributions of the global diffusion coefficient of anti-GluA2-conjugated Qdots for each Nlg1 mutant (100–350 trajectories from 6 independent experiments) G, Distributions of the global diffusion coefficient for anti-GluA1-coated Qdots, for each Nlg1 mutant (450–1200 trajectories from 3 experiments). The values of the control condition and Nlg1WT expression for anti-GluA2 and anti-GluA1-coated QDots presented in Figure 1D belong to the same set of experiments. Statistical P-values: **p < 0.01, ***p < 0.001.
AMPAR immobilization by Nlg1 requires the presence of PSD-95
To confirm the requirement of PSD-95 in Nlg1-dependent AMPAR immobilization, we transfected rat hippocampal neurons with an shRNA construct to acutely knock-down PSD-95 expression (shPSD-95), or a control plasmid replacing endogenous PSD-95 by recombinant PSD-95:GFP (replPSD-95) (Schlüter et al., 2006). We estimated by immunostaining that shPSD-95 diminished by 70% the expression level of endogenous PSD-95, while repl-PSD-95 increased PSD-95 level by 20-fold (Fig. 4A,B). In cells expressing only shPSD-95, the lateral diffusion of AMPARs was not changed (Fig. 4C), indicating either a compensation by other scaffold proteins, or a sufficient residual level of PSD-95 (30%) to stabilize AMPARs. In contrast, expression of replPSD-95 caused a major reduction in global AMPAR mobility (Fig. 4C). This phenomenon is likely linked to an increase in the local concentration and size of PSD-95 clusters, acting as trapping elements for AMPARs through their auxiliary subunits such as stargazin (Bats et al., 2007; Opazo et al., 2010).
Figure 4.

AMPAR immobilization by Nlg1 requires the presence of PSD-95. A, Representative images of 10 DIV rat hippocampal neurons expressing an shRNA against rat PSD-95 (shPSD-95) and GFP, or a construct replacing endogenous PSD-95 by PSD-95:GFP (replPSD-95), immunolabeled for endogenous PSD-95. The green channel represents GFP and the red channel the PSD-95 staining. B, Quantification of the PSD-95 immunofluorescence signal in dendrites from untransfected cells or cells expressing the shPSD-95 or the replPSD-95 constructs (two independent experiments, the number of cells in each condition is indicated in the columns). C, Global diffusion of endogenous AMPARs monitored using anti-GluA2 Qdots in rat neurons expressing GFP, shPSD-95 and GFP, replPSD-95, or shSAP-97 (120–1130 trajectories per condition, two independent experiments). D, E, Mobility of GluA2-containing AMPARs in neurons transfected with no Nlg1, Nlg1WT or Ng1Swap, in combination with either GFP, shPSD-95, shSAP-97, or replPSD-95. D, Representative GluA2 trajectories in conditions of downregulation or overexpresssion of PSD-95. E, Distributions of the global diffusion coefficient for each condition (100–1350 trajectories from 3 independent experiments). Statistical P-values: *p < 0.05, ***p < 0.001.
When shPSD-95 was coexpressed with either Nlg1WT or Nlg1Swap (Fig. 4D,E), normal mobility of AMPARs was rescued, showing that the presence of PSD-95 was important for AMPAR immobilization mediated by Nlg1 overexpression. As a control, the immobilization of AMPARs caused by Nlg1WT or Nlg1Swap was not further affected by the coexpression of replPSD-95 construct (Fig. 4D,E). To assess the role of other scaffold molecules, we used an shRNA directed against SAP-97 (shSAP-97), a scaffold molecule which directly binds the GluA1 C terminus (Leonard et al., 1998). When expressed alone, shSAP-97 had no effect on global AMPAR diffusion (Fig. 4C), as observed for shPSD-95. However, when coexpressed with Nlg1, shSAP-97 was unable to rescue the immobilization of AMPARs (Fig. 4E), confirming that GluA1 was not a major player in this process. Together, these experiments indicate a specific role for PSD-95 in the regulation of AMPAR surface diffusion by Nlg1.
AMPARs specifically stop at Nlg1/PSD-95 clusters
We then analyzed in detail the local AMPAR diffusion within or outside PSD-95 versus Homer1c clusters. We observed a specific confinement of AMPARs at clusters containing both Nlg1 and PSD-95 (Fig. 5A,B). Quantitatively, the local AMPAR diffusion coefficient was significantly reduced at PSD-95 clusters, indicating that AMPARs were specifically trapped at Nlg1/PSD-95 clusters (Fig. 5C). In cells coexpressing Homer1c and Nlg1WT, synaptic AMPAR diffusion was strongly reduced at Homer1c locations (Fig. 5D), consistent with the fact that Nlg1 recruits PSD-95 at synapses. In contrast, overexpression of Nlg1Swap, which caused a global reduction of AMPAR mobility (Fig. 3E–G), had no effect on the synaptic diffusion of AMPARs at Homer1c puncta (Fig. 5D), while AMPAR mobility was instead reduced at PSD-95:GFP puncta, albeit not significantly (p = 0.2) (Fig. 5C). This suggests a functional dissociation between Homer1c and PSD-95, in agreement with the differential dynamics of these two proteins (Kuriu et al., 2006), and confirms that PSD-95 is the primary interactor with surface diffusing AMPARs. Indeed, the fraction of PSD-95 clusters colocalizing with Homer1c was significantly less in cells expressing Nlg1Swap compared with Nlg1WT (Fig. 5E,F), indicating that in the Nlg1Swap condition, new PSD-95 clusters are formed at extrasynaptic locations, dissociated from synaptic Homer1c puncta. To unify these data, we plotted the effects of Nlg1 mutants on AMPAR mobility in relation to the density of either Homer1c:GFP or PSD-95:GFP clusters (Fig. 5G). We found a striking inverse correlation between AMPAR diffusion and PSD-95 cluster density, and a very weak correlation with Homer1c spot density, confirming that AMPARs interact primarily with Nlg1/PSD-95 clusters, whether synaptic or extrasynaptic.
Figure 5.

Nlg1 immobilizes AMPARs at PSD-95 clusters. A, Representative trace showing specific confinement of anti-GluA2-conjugated Qdot on PSD-95 clusters (red pixels) for a neuron transfected with replPSD-95, Homer1c:DsRed, and Nlg1WT. B, Representative image of cells cotransfected with PSD-95:GFP (green) and Nlg1WT, immunostained with anti-HA (red). C, Distributions of local AMPAR diffusion coefficients measured either inside (on PSD-95) or outside PSD-95 clusters (Extra-PSD-95), or inside Homer1c clusters (on Homer), in cells coexpressing replPSD-95, Homer1c:DsRed, and either Nlg1WT or Nlg1Swap (70–1320 trajectories and 2 independent experiments). D, In cells expressing Nlg1 mutants and Homer1c:GFP, the diffusion coefficient was computed for stretches of trajectories specifically colocalized with Homer1c:GFP staining (40–120 trajectories, from 5 experiments). E, Representative images of cells cotransfected with Homer1c:DsRed (red), PSD-95:GFP (green), and either Nlg1WT or Nlg1Swap. F, Quantification of the colocalization between PSD-95:GFP and Homer1c:DsRed, upon either Nlg1WT or Nlg1Swap overexpression (9 cells per condition from 3 independent experiments). G, Plot of the median GluA2 diffusion coefficient versus the density of either Homer1c:GFP or PSD-95:GFP clusters, upon expression of the various Nlg1 mutants. Diffusion coefficients were taken from Figure 3F and PSD-95 cluster densities were taken from Figure 3C. Homer1c cluster density was measured using the Homer1c:GFP images taken for QDot experiments with Nlg1 mutants. Correlation coefficients were evaluated by linear regression. Statistical P-values: *p < 0.05, ***p < 0.001.
Cross-linking Nlg1 by Nrx1β multimers induces functional AMPAR-containing postsynapses
To monitor AMPAR accumulation at newly formed Nrx/Nlg contacts, we incubated younger neurons (6–7 DIV) transfected with Nlg1WT with purified Nrx1β-Fc cross-linked by Cy5 labeled secondary antibodies (Fig. 6A) (Barrow et al., 2009; Poulopoulos et al., 2009). This resulted in a rapid and massive redistribution of both Nlg1 and PSD-95 from diffuse membrane pools into micron-scale clusters, colocalizing with Nrx1β-Fc aggregates (Fig. 6B). In 30 min, the density of PSD-95 clusters increased by 60%. In control experiments, Nrx1β-Fc aggregates bound weakly to neurons transfected with Nlg1Swap (data not shown), whereas they bound strongly to neurons transfected with Nlg1ΔC but did not recruit PSD-95 (Fig. 6E), demonstrating specificity of the protein interactions.
Figure 6.

Nrx1β aggregates recruit PSD-95 and functional AMPARs. A, Schematic diagram of the Nrx/Nlg cross-linking experiment. B, Rat hippocampal neurons (6–7 DIV) expressing Nlg1WT and PSD-95:GFP were incubated for 30 min with purified Nrx1β-Fc cross-linked by Cy5-labeled secondary antibodies. Green arrowheads point to preexisting PSD-95:GFP clusters, and red arrows to new PSD-95 clusters colocalized with Nrx1β. C–E, Neurons expressing Nlg1WT and PSD-95:mCherry were incubated with Cy5-labeled Nrx1β aggregates. C, Endogenous AMPARs were live stained with antibodies against the GluA1 extracellular domain. D, The control protein NCAM was live stained with a polyclonal antibody. E, Quantification of the enrichment of GluA1, NCAM, and PSD-95:mCherry at Nrx1β aggregates (400–5000 clusters from 2 independent experiments, 7–30 cells per condition and per time point). PSD-95:mCherry and GluA1 enrichment levels over time were compared with NCAM (***p < 0.001). To analyze the recruitment of PSD-95 and AMPAR at Nrx1β clusters with either Nlg1WT or Nlg1ΔC (empty squares), data were compared for the same 30 min time point (***p < 0.0001). In parallel experiments, the enrichment level of PSD-95:mCherry, GluA1, and NCAM was measured at native synapses (i.e., PSD-95:mCherry puncta), in the absence of Nrx1β clusters. These data are represented by closed circles on the left of the graph (synapses). F, G, Representative examples of AMPA currents elicited upon glutamate iontophoresis at either preexisting synaptic PSD-95 clusters (green arrowhead) or newly formed PSD-95 clusters induced by cross-linked Nrx1β-Fc (red arrow). H, Average current amplitudes obtained from 8 neurons (number of clusters is indicated in the columns, **p < 0.01).
Before addition of cross-linked Nrx1β-Fc, ∼60% of preexisting PSD-95 clusters were apposed to presynaptic puncta labeled with synaptotagmin, as reported (Gerrow et al., 2006). After Nrx1β-Fc addition, this proportion significantly fell to 50%, suggesting that new PSD-95 clusters were mostly nonsynaptic hemi-synapses (data not shown). The enrichment level of PSD-95 in the new clusters increased sharply over time and reached by 1 h a plateau value corresponding to approximately half the enrichment of PSD-95 at endogenous synapses (Fig. 6E). We checked by immunocytochemistry that endogenous PSD-95 was recruited to similar levels as recombinant PSD-95:mCherry (enrichment at 1 h = 1.60 ± 0.03, n = 432 clusters from 5 cells.
Nrx1β clusters also contained endogenous AMPARs, live stained with anti-GluA1 antibodies, which accumulated to lower levels than PSD-95 (Fig. 6C,E). The adhesion molecule NCAM, labeled similarly, did not show any enrichment at new Nrx1β clusters (Fig. 6D,E), demonstrating specificity of the assay. AMPARs did not accumulate at Nrx1β clusters in neurons transfected with Nlg1ΔC (Fig. 6E), indicating that PSD-95 had to be present at Nrx/Nlg adhesions to recruit AMPARs. Newly accumulated AMPARs were functional, as assessed by patch-clamp recordings upon glutamate iontophoresis (Fig. 6F,G). The amplitude of the elicited AMPA currents at Nrx1β-Fc clusters was approximately half that measured at native synapses (Fig. 6G,H), closely corresponding to the difference in AMPAR enrichment level at Nrx1β-Fc clusters versus endogenous PSD-95:mCherry spots (Fig. 6E). Thus, the newly formed Nrx1β/Nlg1 adhesions recruited PSD-95 and functional AMPARs.
Recruitment of AMPARs at Nrx/Nlg contacts relies on membrane diffusion
To assess the role of membrane diffusion in AMPAR recruitment at novel Nrx/Nlg adhesions, we first measured the mobility of Qdots conjugated with Fab anti-GluA2 with or without cross-linked Nrx1β-Fc, in cells overexpressing Nlg1WT. Addition of Nrx1β-Fc resulted in a significant reduction of the global AMPAR diffusion coefficient (Fig. 7B). As a control, the mobility of NCAM was not affected by treatment with cross-linked Nrx1β-Fc (Fig. 7B). Specifically, AMPARs frequently stopped at Nrx/Nlg clusters, as they did at Homer1c puncta in control conditions (Fig. 7A). In both cases, AMPAR trapping at these clusters was characterized by a dramatic drop in diffusion coefficient (Fig. 7B).
Figure 7.

Lateral diffusion is necessary for AMPAR recruitment at novel Nrx/Nlg adhesions. A, B, Rat hippocampal neurons (8 DIV) coexpressing Nlg1WT and Homer1c:GFP were incubated or not with purified Nrx1β-Fc cross-linked by Cy3 labeled secondary antibodies. The surface mobility of either AMPARs (A, B) or NCAM (B) was monitored using anti-GluA2 or anti-NCAM-conjugated Qdots, respectively. A, Representative Qdot trajectories showing the immobilization of AMPARs at Homer1c:GFP labeled synapses in untreated cells, or at Nrx1β-Fc aggregates (red pixels). B, Quantification of the global diffusion coefficients of AMPARs (450–700 trajectories) and NCAM (100–400 trajectories), and of the local AMPAR diffusion coefficient at either Homer1c:GFP spots or Nrx1β-Fc aggregates (41 and 366 trajectories, respectively). C, Neurons (8 DIV) coexpressing Nlg1 and SEP:GluA2 in control condition (left), or after surface cross-linking of SEP:GluA2 using anti-GFP antibody (right). The red traces represent GluA2-coupled Qdot trajectories. D, Quantification of the global SEP:GluA2 diffusion in control condition, or after surface cross-linking (255 and 107 trajectories, respectively, from 2 independent experiments). Statistical P-values: *p < 0.05, ***p < 0.0001. E–H, Recruitment of recombinant AMPAR subunits at Nrx1β aggregates in 6–7 DIV neurons. Neurons were cotransfected with Nlg1WT and either SEP:GluA1 (G) or SEP:GluA2 (E, F). After a 30 min or 1 h incubation period with Nrx1β aggregates, cells were fixed and surface receptors were labeled with anti-GFP antibodies (E–G). In some experiments, neurons expressing Nlg1WT and SEP:GluA2 were pretreated with a polyclonal anti-GFP antibody to cross-link surface SEP:GluA2 to block their membrane diffusion, then exposed to Nrx1β aggregates. Cells were fixed and surface SEP:GluA2 receptors were stained with a monoclonal anti-GFP antibody followed by a secondary anti-mouse Alexa Fluor 488. H, Comparison of the specific enrichment of each subunit at Nrx/Nlg1 clusters (300–3500 clusters, 8–30 cells per condition and per time point). Data were analyzed comparing SEP:GluA2 enrichment to SEP:GluA1, or X-link SEP:GluA2 to SEP:GluA2. Statistical P-values: ***p < 0.001.
We then blocked the lateral mobility of surface AMPARs by antibody cross-linking, in neurons cotransfected with Nlg1WT and SEP-tagged GluA2 subunits and treated with a polyclonal antibody to GFP (Heine et al., 2008a). This treatment induced the formation of many SEP:GluA2 clusters, and a dramatic reduction of AMPAR surface diffusion (Fig. 7C,D). In the absence of antibody cross-link, SEP:GluA2 accumulated at Nrx/Nlg clusters (Fig. 7E,H) with a similar time course as endogenous AMPARs (Fig. 6E). Antibody cross-linking resulted in a significant decrease in the enrichment level of SEP:GluA2 at Nrx1β-Fc clusters (Fig. 7F,H). In fact, antibody-induced SEP:GluA2 clusters were not colocalized with Nrx1β-Fc puncta (Fig. 7F). Interestingly, recombinant SEP:GluA1 receptors, whose mobility was reduced compared with SEP:GluA2 (Fig. 1E,F), did not accumulate at Nrx1β-Fc clusters (Fig. 7G,H). Together, these data strongly support the concept that surface diffusion was required for AMPAR recruitment at newly formed Nrx/Nlg contacts.
A competition between new Nrx/Nlg contacts and preexisting synapses
To assess whether the recruitment of PSD-95 and AMPARs at nascent Nrx/Nlg adhesions involved a competition with native synapses, we first monitored the PSD-95 content in preexisting clusters upon addition of cross-linked Nrx1β-Fc, using time lapse imaging of PSD-95:GFP in neurons coexpressing Nlg1WT. PSD-95 clusters progressively disassembled, translating into a 35% fluorescence decay in 30 min (Fig. 8A,B). This suggests that the formation of Nrx1β/Nlg1 adhesions displaces the dynamic equilibrium between extrasynaptic and synaptic PSD-95, PSD-95 being dislodged from native synapses. This effect was specific of the Nrx1β/Nlg1/PSD-95 interactions since the fluorescence loss was significantly lower when Nrx1β-Fc was replaced by Fc, or when Nlg1WT was replaced by Nlg1ΔC (Fig. 8B,C). Since the population of preexisting PSD-95 puncta represent a combination of endogenous synapses and extrasynaptic clusters (Gerrow et al., 2006), we examined the influence of synaptic activity on PSD-95 depletion. Treating neurons with tetrodotoxin (TTX) to prevent action potentials and APV to block NMDA receptors during the recordings resulted in a major increase in the depletion rate (Fig. 8B,C). We also labeled active presynapses by uptake of a fluorescent antibody to the synaptotagmin luminal domain (Fig. 8C). Depletion rates were lower for PSD-95 clusters apposed to active presynapses than for the whole population of PSD-95 clusters (Fig. 8C). Together, these data suggest that during the induction of new Nrx/Nlg contacts, inactive synapses are more prone to lose their PSD-95 content than active ones.
Figure 8.

Recruitment of PSD-95 and AMPARs at novel Nrx/Nlg adhesions competes with preexisting synapses. A–C, Rat hippocampal neurons (9 DIV) expressing either Nlg1WT or Nlg1ΔC, together with PSD-95:GFP were incubated with purified Nrx1β-Fc (or human Fc as a control) cross-linked by Cy5 labeled anti-Fc antibodies. In some experiments, neurons were treated throughout the recordings with TTX/APV, to inhibit synaptic activity. The distribution of PSD-95:GFP was monitored in living cells during a 30 min. A, Representative time course showing the progressive disappearance of PSD-95:GFP from preexisting clusters (red arrowheads), concomitant to the formation of new clusters (white arrows) upon Nrx1β-Fc addition. B, Quantification of the fluorescence decay of preexisting PSD-95:GFP clusters, for 3 conditions (control Fc, Nrx1β-Fc untreated, and Nrx1β-Fc treated with TTX/APV). C, Depletion rates were quantified by fitting a linear relationship through the fluorescence decay for each cluster (mean ± SEM of 27–92 clusters from 6–16 cells). In some experiments, active synapses were labeled using a synaptotagmin antibody uptake, and PSD-95:GFP clusters apposed to synaptotagmin puncta (inset) were pooled as a separate group. D, E, Neurons expressing Nlg1WT and PSD-95:GFP either without treatment or after a 1 h incubation with cross-linked Nrx1β-Fc, were subjected to patch-clamp recording, in the presence of TTX and APV to isolate AMPA mEPSCs. D, Images of a patched neuron in DIC and in fluorescence (PSD-95:GFP in green, Nrx1β-Fc Cy5 clusters in red). E, Representative time sequence and average AMPA mEPSCs traces. F, Double staining of endogenous AMPAR with anti-GluA1 antibodies (green), and presynapses with anti-synapsin (red), in 10 DIV neurons expressing either Nlg1WT or Nlg1Swap. Transfected cells were detected by coexpression of GFP (data not shown). G, Analysis of the fraction of GluA1 clusters apposed to synapsin spots, either upon Nlg1WT or Nlg1Swap expression. (n = 20 and 15 cells, respectively, from 2 independent experiments). H, Neurons (10 DIV) expressing either Nlg1WT or Nlg1Swap and PSD-95:GFP, were live-labeled for endogenous AMPAR with anti-GluA1 antibody, and with anti-synapsin to stain presynapses (data not shown). The graph represents the intensity of GluA1 in puncta apposed to presynapses or colocalizing with PSD-95:GFP clusters nonapposed to synapsin (n = 12 cells for each condition from 2 independent experiments). Statistical P-values: *p < 0.05, **p < 0.01, ***p < 0.001.
To assess whether AMPARs also left endogenous synapses in this process, we recorded AMPA miniature EPSCs (mEPSCs) by patch-clamp experiments, in neurons cotransfected with Nlg1 and PSD-95 and treated with cross-linked Nrx1β-Fc for 1 h, or untreated neurons (Fig. 8D,E). We observed a significant 15% drop in the amplitude of AMPA mEPSCs in the group of neurons treated with Nrx1β clusters (16.1 ± 0.9 pA, n = 10), compared with untreated neurons (18.8 ± 0.8 pA, n = 13, *p < 0.05 by unpaired t test). This indicates that AMPARs were also removed from endogenous synapses upon triggering new Nrx/Nlg contacts, albeit to a lower level than PSD-95.
We then asked whether the formation of extrasynaptic PSD-95 clusters induced by expression of the Nlg1Swap mutant (Fig. 3A–D) was also accompanied by a depletion of synaptic AMPARs. We thus measured the degree of apposition between AMPAR puncta revealed by live labeling of GluA1, and presynapses labeled with synapsin, in conditions of Nlg1WT or Nlg1Swap expression (Fig. 8F). As expected, the percentage of GluA1 puncta apposed to presynapses was significantly higher for Nlg1WT than for Nlg1Swap (Fig. 8G). Moreover, GluA1 staining was higher at nonsynaptic PSD-95:GFP clusters when compared with synaptic puncta upon Nlg1Swap overexpression, whereas the reverse was observed for Nlg1WT (Fig. 8H). Together, these data show that both PSD-95 and AMPAR are displaced from existing synapses upon triggering extrasynaptic Nlg1 adhesions.
Altered AMPAR distribution and function in developing Nlg1 KO mice
Because of such striking effects of Nlg1 depletion on AMPAR diffusion and AMPAR targeting at Nrx1/Nlg1 adhesions in primary neurons, we examined AMPAR distribution and synaptic transmission in Nlg1 KO mice. We focused on a developmental window of active synaptogenesis (P8–P9) (Groc et al., 2002), during which the Nlg1 expression level gradually increases (Varoqueaux et al., 2006). In young animals, PSD-95 is expressed at low levels while SAP-102, a structurally similar scaffold protein, dominates and functionally replaces PSD-95 in AMPAR recruitment at synapses (Sans et al., 2000; Elias et al., 2006). We therefore performed immunofluorescence labeling of SAP-102 and GluA1 in hippocampal slices, comparing Nlg1 KO and littermate WT mice (Fig. 9A). We observed significant decreases of the density of both SAP-102 and GluA1 puncta (21% and 24%, respectively), and of the fluorescent intensity of GluA1 clusters (17%), in slices from Nlg1 KO compared with Nlg1 WT mice (Fig. 9B,C). Interestingly, the degree of colocalization between SAP-102 and GluA1 clusters (∼45%) was similar for Nlg1 WT and Nlg1 KO slices, suggesting that the absence of Nlg1 does not induce the formation of postsynapses lacking AMPARs.
Figure 9.

Reduced AMPAR content at developing postsynapses. A, Representative images of the stratum radiatum area of the CA1 region from hippocampal slices of P9 Nlg1 WT or KO pups. Slices were double-labeled for SAP-102 and GluA1. Positive staining appears in white and pyramidal cells' nuclei in black. Bottom panels shows SAP-102 and AMPAR clusters at higher magnification and for the same intensity scaling. B, Density of SAP-102 or GluA1 clusters. C, Integrated fluorescence intensity of either SAP-102 or GluA1 clusters. For each animal (number indicated in the graphs columns), several hippocampal slices were cut, labeled, and quantified. The values of cluster density and intensity obtained for each slice were averaged to give the mean per animal. D–F, AMPA mEPSCs were recorded in CA1 pyramidal neurons of acute hippocampal slices from P8–P9 WT (5 animals) or Nlg1 KO (6 animals) pups. D, Representative electrophysiological sequences of 10 s for both conditions. Events of amplitude >7 pA were automatically detected. E, The mEPSC frequency was calculated for each cell as the total number of events per 15 min recording, and averaged. The number of recorded cells is indicated in the bars. F, The median of the non-Gaussian distribution of mEPSC amplitudes per cell was averaged over 12–14 cells per condition. Statistical P-values: *p < 0.05, **p < 0.01.
Moreover, to characterize the role of Nlg1 in AMPAR-mediated synaptic transmission, we performed whole-cell patch-clamp recordings of miniature AMPA currents in CA1 pyramidal cells from acute hippocampal slices (Fig. 9D). We found a 54% decrease in frequency and a 13% decrease in amplitude of AMPA mEPSCs in Nlg1 KO versus control animals (Fig. 9E,F). Together, these data indicate that Nlg1 deficiency inhibits the recruitment of AMPARs at nascent postsynapses in vivo, by decreasing both the number of postsynapses and the unitary AMPAR content at each synapse.
Discussion
We demonstrated that functional AMPARs accumulate at PSD-95 scaffolds assembled by newly formed Nrx/Nlg adhesions, through a diffusion/trap mechanism in competition with preexisting synapses, supporting an important function for diffusive extrasynaptic AMPARs in postsynapse assembly (Fig. 10).
Figure 10.

Model of the recruitment of postsynaptic components to novel Nrx1β/Nlg1 contacts. Triggering the formation of new Nrx/Nlg adhesions (1) induces the initial recruitment of PSD-95 from both extrasynaptic pools and synapses (2). These novel PSD-95 slots serve as anchors for trapping surface diffusing AMPARs coming from both extrasynaptic compartments and synapses (3).
Nrx/Nlg adhesions assemble postsynapses containing PSD-95 and AMPARs
We observed the recruitment of PSD-95 and functional AMPARs at novel Nrx/Nlg adhesions in 0.5–1 h, a time course compatible with previous studies (Friedman et al., 2000; Bresler et al., 2001, 2004; Barrow et al., 2009), and prevented AMPARs accumulation at these clusters by blocking surface diffusion. We did not focus on the directed motion of preexisting extrasynaptic Nlg1/PSD-95 clusters since only 10% of these are mobile (Gerrow et al., 2006; Barrow et al., 2009; Gutiérrez et al., 2009), but instead observed a rapid coalescence of diffusible PSD-95 at Nrx/Nlg clusters. PSD-95 and AMPARs in Nrx/Nlg contacts reached half the levels at endogenous synapses, supporting a major role of Nrx1/Nlg1 interaction in driving excitatory synapse assembly, but also revealing that other molecules (e.g., LRRTMs) and/or mechanisms are involved for synaptic maturation (de Wit et al., 2009; Linhoff et al., 2009). Although we do not exclude other processes (Garner et al., 2002) such as motor-driven transport of AMPAR packets (Washbourne et al., 2002), or exocytosis of AMPAR vesicles (Thyagarajan and Ting, 2010), we suggest that membrane diffusion is important for AMPAR accumulation at Nrx/Nlg contacts.
The fact that PSD-95 was more enriched than AMPARs at Nrx/Nlg adhesions suggests that PSD-95 experiences a stronger driving force than AMPARs, cytosolic PSD-95 molecules being rapidly recruited by direct interaction with aggregated Nlg1, then membrane AMPARs being trapped through a slower 2D diffusion process. Alternatively, if AMPARs and PSD-95 are recruited as preformed complexes, then the higher PSD-95 enrichment may reflect a stoichiometry of 2 PSD-95 molecules for 1 AMPAR. This ratio is compatible with structural data showing that AMPARs can form complexes with 1–4 TARPs mediating PSD-95 binding (Kim et al., 2010), and with the estimates of 100–400 AMPARs and 200–450 PSD-95 molecules per synapse (Cottrell et al., 2000; Sugiyama et al., 2005; Tanaka et al., 2005). At established synapses, AMPAR enrichment reached a similar level as PSD-95 (∼2.2), revealing that either all PSD-95 slots become progressively occupied by AMPARs, or that approximately half AMPARs associate with other scaffold molecules, such as SAP-102 (Elias et al., 2006), SAP-97 (Leonard et al., 1998), S-SCAM (Iida et al., 2004), or the adhesion protein N-cadherin (Saglietti et al., 2007).
Novel Nrx/Nlg adhesions compete with existing synapses
Triggering Nrx/Nlg adhesions both increased extrasynaptic PSD-95 content and depleted synaptic PSD-95, suggesting a competition between novel postsynaptic scaffolds and native synapses. This concept of synaptic sharing is consistent with the facts that PSD-95 remodels within synapses and exchanges between synapses (Gray et al., 2006; Kuriu et al., 2006; Tsuriel et al., 2006; Blanpied et al., 2008; Steiner et al., 2008). The turnover rate of PSD-95 at synapses measured by FRAP is ∼50–60% in 30 min (Kuriu et al., 2006; Sharma et al., 2006; Tsuriel et al., 2006; Thoumine et al., 2008), indicating that novel Nrx/Nlg adhesions take up half this fraction of dynamic PSD-95. Interestingly, we found that depletion rates were higher for chronically inactive synapses. Since synaptic potentiation also increases PSD-95 turnover (Tsuriel et al., 2006; Steiner et al., 2008), PSD-95 remodeling seems implicated in synaptic adaptation to bidirectional changes in activity. Possible regulatory mechanisms could involve kinase and phosphatase signaling linked to Nrx/Nlg interactions (Lim et al., 2009) and/or PSD-95 (Steiner et al., 2008).
Addition of Nrx multimers reduced both AMPAR diffusion and AMPA mEPSCs, suggesting that PSD-95 loss at synapses causes AMPARs to diffuse out and get trapped extrasynaptically. The turnover rate of 40–50% in 5–10 min measured by photobleaching or photo-activation experiments (Ashby et al., 2006; Steiner et al., 2008; Frischknecht et al., 2009) can easily accommodate this moderate AMPAR flux. The differential synaptic escape of AMPARs and PSD-95 may be explained by interactions with other scaffold proteins, or again by a stoichiometry of 2 PSD-95 molecules carrying away 1 AMPAR.
Equilibrium AMPAR dynamics altered by Nlg1 and PSD-95 expression
At equilibrium, AMPARs switch within seconds from extrasynaptic to synaptic compartments (Borgdorff and Choquet, 2002; Tardin et al., 2003), by an interaction with PSD-95 mediated by the auxiliary subunit stargazin (Bats et al., 2007; Opazo et al., 2010). Due to steric hindrance, Qdots have little access to the core of the synaptic cleft (Howarth et al., 2008), but nevertheless probe in sufficient detail the synaptic area to reveal anchoring to scaffold elements (Groc et al., 2007). Nlg1 overexpression created synapses containing PSD-95 and reduced AMPAR diffusion, consistent with AMPARs being trapped by PSD-95. The rescue of normal AMPAR diffusion with shPSD-95 showed that PSD-95 was the major molecule involved in AMPAR trapping at synapses induced by Nlg1 expression. It was surprising to see little impact of PSD-95 knock-down alone on AMPAR diffusion. However, scaffold proteins are likely to be in excess of Nlg1, expressed at rather low levels in embryonic cultures (Varoqueaux et al., 2006). Thus, upon PSD-95 knock-down, other PDZ domain-containing proteins including SAP-102 and S-SCAM may compensate for PSD-95 to bind Nlg1 (Irie et al., 1997; Iida et al., 2004). A lack of effect of PSD-95 knock-out on AMPAR synaptic transmission was reported in slices, where the missing scaffold was replaced by SAP-102 upregulation (Elias et al., 2006). In contrast, when Nlg1 was overexpressed, it was likely to capture most available scaffold molecules, serving as templates for AMPAR trapping. Then, PSD-95 knock-down directly translated into a decrease in AMPAR binding slots, hence a relative increase in AMPAR diffusion. Conversely, overexpressing PSD-95 strongly immobilized AMPARs. This is consistent with the increase in AMPA EPSCs amplitude in CA1 cells upon PSD-95 overexpression, while shRNA against PSD-95 has no effect (Elias et al., 2008).
In addition, Nlg1ΔC allowed neither AMPAR immobilization nor recruitment at synapses or Nrx clusters, supporting that PSD-95 was a requisite for AMPAR trapping by diffusion. These findings agree with the fact that Nlg1ΔC reduces both frequency and amplitude of AMPA mEPSCs (Nam and Chen, 2005). Intriguingly, the effects of Nlg1 on AMPAR synaptic transmission in organotypic slices might not be mediated by the Nlg1 PDZ binding motif, but by a noncanonical sequence located within the Nlg1 intracellular domain (Shipman et al., 2011). The difference may be due to heteromerization between mutated Nlg1 molecules and endogenous Nlg1 molecules (Shipman et al., 2011). This is unlikely in our system because overexpression of Nlg1ΔC is dominant over endogenous Nlg1. Finally, Nlg1Swap created new extrasynaptic PSD-95 clusters, reduced AMPAR level at synapses, and slowed down AMPAR mobility only extrasynaptically. Together, these findings support that AMPAR trapping is intrinsically associated to the PSD-95 scaffold, be it synaptic, extrasynaptic, or newly assembled by Nrx/Nlg adhesion.
The effects of Nlg1 and PSD-95 expression on AMPAR mobility were not linear. The tenfold Nlg1 overexpression translated into modest twofold increases in Nlg1 and PSD-95 cluster densities (Chih et al., 2005; Levinson et al., 2005; Ko et al., 2009), and the 20-fold overexpression of PSD-95 induced only a doubling of synapse and PSD sizes (Nikonenko et al., 2008). Based on these data, we built a theoretical model showing that the number and size of PSD scaffolds, independently of absolute protein levels, bidirectionally control AMPAR diffusion by acting as trapping elements (K. Czöndör, M. Mondin, and O. Thoumine, unpublished observation).
Subunit specificity in the diffusion and recruitment of AMPAR at Nrx/Nlg adhesions
Trapping of endogenous AMPARs at Nrx/Nlg adhesions was dictated by the GluA2 subunit. Recombinant GluA1 homomers diffused slowly and did not accumulate at Nrx/Nlg adhesions, whereas GluA2 homomers showing higher diffusion were recruited constitutively. Since the PDZ binding domains of neither GluA1 nor GluA2 subunits are involved in synaptic AMPAR trapping (Bats et al., 2007; Opazo et al., 2010), AMPARs must bind indirectly to PSD-95 via TARPs. Different TARPs could associate with various stoichiometries to either GluA1 or GluA2 subunits (Shi et al., 2009), explaining the differential anchoring of AMPARs to PSD-95. Interestingly, the recruitment of GluA1 recombinant receptors at Nrx/Nlg adhesions depends on synaptic activity (Nam and Chen, 2005; Heine et al., 2008b). In parallel, AMPAR trapping at synapses also requires activity (Ehlers et al., 2007), specifically postsynaptic calcium entry which triggers CaMKII activation and stargazin phosphorylation (Heine et al., 2008a; Opazo et al., 2010). Although stargazin binds equally well GluA1 and GluA2 subunits (Chen et al., 2000), a differential association between TARPs and GluA1 versus GluA2 may thus be dynamically regulated by synaptic activity (Tomita et al., 2004; Morimoto-Tomita et al., 2009).
Nlg1 affects AMPAR distribution and synaptic transmission in vivo
Confirming the strong effects of Nlg1 in primary cultures, we found a reduction of both the number of synapses containing AMPARs and the AMPAR content at synapses, in newborn Nlg1 KO mice. In previous studies of Nlg KO phenotypes, no major effect on AMPAR function was reported (Varoqueaux et al., 2006; Chubykin et al., 2007). However, nonsignificant decreases in both the frequency and amplitude of the AMPA mPSCs were shown in cultured neocortical neurons of Nlg1–3 triple KO compared with triple WT animals (Varoqueaux et al., 2006). Furthermore, AMPAR-mediated transmission was not really assessed, since the stimulation intensity was adjusted to elicit a reference AMPAR response, then NMDA currents were analyzed (Chubykin et al., 2007). Interestingly, decreases in the amplitude and frequency of AMPA mEPSCs upon knock-down of Nlgs1–3 in primary hippocampal cultures were also reported (Chih et al., 2005), while a significant drop in AMPAR-mediated synaptic transmission upon knockdown of Nlgs1–3, and a twofold increase upon overexpression of Nlg1 or Nlg3 were found in CA1 cells (Shipman et al., 2011). A reduction in synaptic density and AMPA receptor mEPSC frequency was also reported upon disrupting Nrx-Nlg1 function by Nlg1 morpholinos in embryonic Xenopus brain (Chen et al., 2010). These reports agree with our findings to suggest an important role of Nrx/Nlg adhesions in AMPAR accumulation at nascent synapses. However, the fact that Nlg1 KO only partially inhibits AMPAR recruitment suggests that other synaptogenic molecules may be involved. Knock-down of the Nrx1β receptor LRRTM2 also reduces of AMPA transmission in CA1 cells (de Wit et al., 2009), suggesting a redundant role of LRRTM2 and Nlg1 in PSD-95 and AMPAR recruitment (Siddiqui et al., 2010). Proper synaptogenesis could thus be achieved by controlling the developmental expression profiles of specific adhesion molecules (Brose, 2009).
Footnotes
The research leading to these results has received funding from the European Union's Seventh Framework Program under Grant agreement number 232942 Nano-Dyn-Syn, Centre National de la Recherche Scientifique, Agence Nationale pour la Recherche (Grant Neuroligation), Conseil Régional Aquitaine, and Fondation pour la Recherche Médicale. We thank P. Scheiffele for the generous gifts of Nlg1 plasmids and scientific insight, O. Schlüter for the gift of PSD-95 shRNA constructs, N. Brose and F. Varoqueaux for the gift of Nlg1 KO mice and Nlg1 antibodies, R. Sprengel and P. Seeburg for GluA2 KO mice, M. D. Ehlers for the gift of anti-GluA1 and critical reading of this manuscript, R. M. Mège for the anti-NCAM antibody, K. Keinänen for the Fab anti-GluA2, A. Vimeney, A. Lacquement, and E. Normand for help on animal care and genotyping, P.S. Pinheiro and F. Lanore for electrophysiology experiments, J.B. Sibarita for help with image analysis, D. Bouchet, C. Breillat, A. Frouin, N. Retailleau, and Axel Athané for cell culture, P. Gonzales and R. Sterling for technical assistance, and G. Giannone, L. Groc, P. Opazo, M. Sainlos, and C. Mulle for scientific discussions.
References
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow SL, Constable JR, Clark E, El-Sabeawy F, McAllister AK, Washbourne P. Neuroligin1: a cell adhesion molecule that recruits PSD-95 and NMDA receptors by distinct mechanisms during synaptogenesis. Neural Dev. 2009;4:17. doi: 10.1186/1749-8104-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Kerr JM, Ehlers MD. Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci U S A. 2008;105:12587–12592. doi: 10.1073/pnas.0711669105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- Bresler T, Ramati Y, Zamorano PL, Zhai R, Garner CC, Ziv NE. The dynamics of SAP90/PSD-95 recruitment to new synaptic junctions. Mol Cell Neurosci. 2001;18:149–167. doi: 10.1006/mcne.2001.1012. [DOI] [PubMed] [Google Scholar]
- Bresler T, Shapira M, Boeckers T, Dresbach T, Futter M, Garner CC, Rosenblum K, Gundelfinger ED, Ziv NE. Postsynaptic density assembly is fundamentally different from presynaptic active zone assembly. J Neurosci. 2004;24:1507–1520. doi: 10.1523/JNEUROSCI.3819-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N. Synaptogenic proteins and synaptic organizers: “many hands make light work.”. Neuron. 2009;61:650–652. doi: 10.1016/j.neuron.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chen SX, Tari PK, She K, Haas K. Neurexin-neuroligin cell adhesion complexes contribute to synaptotropic dendritogenesis via growth stabilization mechanisms in vivo. Neuron. 2010;67:967–983. doi: 10.1016/j.neuron.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell JR, Dubé GR, Egles C, Liu G. Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. J Neurophysiol. 2000;84:1573–1587. doi: 10.1152/jn.2000.84.3.1573. [DOI] [PubMed] [Google Scholar]
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A. 2008;105:20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Frischknecht R, Heine M, Perrais D, Seidenbecher CI, Choquet D, Gundelfinger ED. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci. 2009;12:897–904. doi: 10.1038/nn.2338. [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 2002;25:243–251. doi: 10.1016/s0166-2236(02)02152-5. [DOI] [PubMed] [Google Scholar]
- Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Ed 2. Cambridge, MA: MIT; 1991. pp. 339–370. [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E. Spontaneous unitary synaptic activity in CA1 pyramidal neurons during early postnatal development: constant contribution of AMPA and NMDA receptors. J Neurosci. 2002;22:5552–5562. doi: 10.1523/JNEUROSCI.22-13-05552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A–2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Lafourcade M, Heine M, Renner M, Racine V, Sibarita JB, Lounis B, Choquet D, Cognet L. Surface trafficking of neurotransmitter receptor: comparison between single-molecule/quantum dot strategies. J Neurosci. 2007;27:12433–12437. doi: 10.1523/JNEUROSCI.3349-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez RC, Flynn R, Hung J, Kertesz AC, Sullivan A, Zamponi GW, El-Husseini A, Colicos MA. Activity-driven mobilization of post-synaptic proteins. Eur J Neurosci. 2009;30:2042–2052. doi: 10.1111/j.1460-9568.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008a;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A. 2008b;105:20947–20952. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M, Liu W, Puthenveetil S, Zheng Y, Marshall LF, Schmidt MM, Wittrup KD, Bawendi MG, Ting AY. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nat Methods. 2008;5:397–399. doi: 10.1038/nmeth.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TN, Copenhagen DR. Automatic detection, characterization, and discrimination of kinetically distinct spontaneous synaptic events. J Neurosci Methods. 1999;92:65–73. doi: 10.1016/s0165-0270(99)00095-3. [DOI] [PubMed] [Google Scholar]
- Iida J, Hirabayashi S, Sato Y, Hata Y. Synaptic scaffolding molecule is involved in the synaptic clustering of neuroligin. Mol Cell Neurosci. 2004;27:497–508. doi: 10.1016/j.mcn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Südhof TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- Jung SY, Kim J, Kwon OB, Jung JH, An K, Jeong AY, Lee CJ, Choi YB, Bailey CH, Kandel ER, Kim JH. Input-specific synaptic plasticity in the amygdala is regulated by neuroligin-1 via postsynaptic NMDA receptors. Proc Natl Acad Sci U S A. 2010;107:4710–4715. doi: 10.1073/pnas.1001084107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Yan D, Tomita S. Assembly and stoichiometry of the AMPA receptor and transmembrane AMPA receptor regulatory protein complex. J Neurosci. 2010;30:1064–1072. doi: 10.1523/JNEUROSCI.3909-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, Südhof TC. Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 2009;28:3244–3255. doi: 10.1038/emboj.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropf M, Rey G, Glauser L, Kulangara K, Johnsson K, Hirling H. Subunit-specific surface mobility of differentially labeled AMPA receptor subunits. Eur J Cell Biol. 2008;87:763–778. doi: 10.1016/j.ejcb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Kuriu T, Inoue A, Bito H, Sobue K, Okabe S. Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms. J Neurosci. 2006;26:7693–7706. doi: 10.1523/JNEUROSCI.0522-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Levinson JN, Chéry N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- Lim SH, Kwon SK, Lee MK, Moon J, Jeong DG, Park E, Kim SJ, Park BC, Lee SC, Ryu SE, Yu DY, Chung BH, Kim E, Myung PK, Lee JR. Synapse formation regulated by protein tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J. 2009;28:3564–3578. doi: 10.1038/emboj.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhoff MW, Laurén J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Morimoto-Tomita M, Zhang W, Straub C, Cho CH, Kim KS, Howe JR, Tomita S. Autoinactivation of neuronal AMPA receptors via glutamate-regulated TARP interaction. Neuron. 2009;61:101–112. doi: 10.1016/j.neuron.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Sharma M, Urlaub H, Bourenkov GP, Jahn R, Südhof TC, Wahl MC. CASK Functions as a Mg2+-independent neurexin kinase. Cell. 2008;133:328–339. doi: 10.1016/j.cell.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonenko I, Boda B, Steen S, Knott G, Welker E, Muller D. PSD-95 promotes synaptogenesis and multiinnervated spine formation through nitric oxide signaling. J Cell Biol. 2008;183:1115–1127. doi: 10.1083/jcb.200805132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Saint-Michel E, Giannone G, Choquet D, Thoumine O. Neurexin/neuroligin interaction kinetics characterized by counting single cell-surface attached quantum dots. Biophys J. 2009;97:480–489. doi: 10.1016/j.bpj.2009.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Sharma K, Fong DK, Craig AM. Postsynaptic protein mobility in dendritic spines: long-term regulation by synaptic NMDA receptor activation. Mol Cell Neurosci. 2006;31:702–712. doi: 10.1016/j.mcn.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lu W, Milstein AD, Nicoll RA. The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron. 2009;62:633–640. doi: 10.1016/j.neuron.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, Nicoll RA. Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat Neurosci. 2011;14:718–726. doi: 10.1038/nn.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Matsuzaki M, Tarusawa E, Momiyama A, Molnar E, Kasai H, Shigemoto R. Number and density of AMPA receptors in single synapses in immature cerebellum. J Neurosci. 2005;25:799–807. doi: 10.1523/JNEUROSCI.4256-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, Bats C, Lounis B, Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22:4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoumine O, Ewers H, Heine M, Groc L, Frischknecht R, Giannone G, Poujol C, Legros P, Lounis B, Cognet L, Choquet D. Probing the dynamics of protein-protein interactions at neuronal contacts by optical imaging. Chem Rev. 2008;108:1565–1587. doi: 10.1021/cr078204m. [DOI] [PubMed] [Google Scholar]
- Thyagarajan A, Ting AY. Imaging activity-dependent regulation of neurexin-neuroligin interactions using trans-synaptic enzymatic biotinylation. Cell. 2010;143:456–469. doi: 10.1016/j.cell.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tomita S, Fukata M, Nicoll RA, Bredt DS. Dynamic interaction of stargazin-like TARPs with cycling AMPA receptors at synapses. Science. 2004;303:1508–1511. doi: 10.1126/science.1090262. [DOI] [PubMed] [Google Scholar]
- Tsuriel S, Geva R, Zamorano P, Dresbach T, Boeckers T, Gundelfinger ED, Garner CC, Ziv NE. Local sharing as a predominant determinant of synaptic matrix molecular dynamics. PLoS Biol. 2006;4:e271. doi: 10.1371/journal.pbio.0040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bruchez MP. Labeling cellular targets with semiconductor quantum dot conjugates. Methods Cell Biol. 2004;75:171–183. doi: 10.1016/s0091-679x(04)75007-4. [DOI] [PubMed] [Google Scholar]