

Abstract
The cation-chloride cotransporter NKCC1 plays a fundamental role in the central and peripheral nervous systems by setting the value of intracellular chloride concentration. Following peripheral nerve injury, NKCC1 phosphorylation-induced chloride accumulation contributes to neurite regrowth of sensory neurons. However, the molecules and signaling pathways that regulate NKCC1 activity remain to be identified. Functional analysis of cotransporter activity revealed that inhibition of endogenously produced cytokine interleukin-6 (IL-6), with anti-mouse IL-6 antibody or in IL-6−/− mice, prevented chloride accumulation in a subset of axotomized neurons. Nerve injury upregulated the transcript and protein levels of IL-6 receptor in myelinated, TrkB-positive sensory neurons of murine lumbar dorsal root ganglia. Expression of phospho-NKCC1 was observed mainly in sensory neurons expressing IL-6 receptor and was absent from IL-6−/− dorsal root ganglia. The use of IL-6 receptor blocking-function antibody or soluble IL-6 receptor, together with pharmacological inhibition of Janus kinase, confirmed the role of neuronal IL-6 signaling in chloride accumulation and neurite growth of a subset of axotomized sensory neurons. Cell-specific expression of interleukin-6 receptor under pathophysiological conditions is therefore a cellular response by which IL-6 contributes to nerve regeneration through neuronal NKCC1 phosphorylation and chloride accumulation.
Introduction
Plasma membrane cation-Cl− cotransporters are important for setting the intracellular chloride concentration in neurons, and the regulation of their expression or activity has major consequences under physiological and pathophysiological conditions (Blaesse et al., 2009). In the adult peripheral nervous system, preferential expression of the inwardly directed Na+-K+-Cl− cotransporter, NKCC1, is responsible for the high intracellular chloride concentration, [Cl−]i, compared with central neurons (Price et al., 2009). Under physiological conditions, the high [Cl−]i confers depolarizing effects to the ligand-activated chloride channel GABAA, which is involved in the inhibitory control of pain (Price et al., 2009). Following inflammation, occurrence of neurogenic pain is attributed to a further increase [Cl−]i, leading to both retrograde and anterograde propagation of electrical activity (Cervero et al., 2003). In a peripheral nerve injury model, an increase in [Cl−]i among regenerating sensory neurons accelerates growth rate of neurites, suggesting that chloride homeostasis plays a major role in nerve repair (Pieraut et al., 2007).
In peripheral neurons, transcriptional modifications, phosphorylation and protein trafficking have been identified as cellular mechanisms responsible for upregulation or downregulation of [Cl−]i (Morales-Aza et al., 2004; Galan and Cervero, 2005; Pieraut et al., 2007; Geng et al., 2009). Consistent with the role of cation-Cl− cotransporters in pain processing, inflammatory mediators such nerve growth factor, bradykinin or prostaglandin all induce accumulation of chloride (Nakajima et al., 2007; Funk et al., 2008). Following nerve injury, the signaling molecules that regulate chloride homeostasis of axotomized sensory neurons are unknown. The proinflammatory cytokine, interleukin-6 (IL-6), contributes to the regeneration of afferent fiber, and is thus a potential candidate for regulating chloride homeostasis. Indeed, studies using IL-6 knock-out mice which show defects and delayed regeneration of sensory axons after crush injury, have demonstrated a beneficial function of IL-6 (Zhong et al., 1999). Combined overexpression of IL-6 and its α receptor IL-6R, IL-6R, resulted in faster regeneration of the hypoglossal nerve (Hirota et al., 1996). Furthermore, peripheral injury-induced IL-6 is able to promote regeneration within the CNS (Cafferty et al., 2004; Cao et al., 2006). Since the IL-6 signal-transducing β receptor, gp130, is ubiquitously expressed in sensory neurons, the specificity of IL-6 signaling is mainly mediated by tissue-specific constitutive IL-6 α receptor, IL-6R, expression and/or local actions of soluble IL-6R (Gardiner et al., 2002; Andratsch et al., 2009). Although the effects of IL-6 are well established in nerve regeneration, the cellular expression of IL-6R in the dorsal root ganglia is unknown. Here, we studied the cellular expression of IL-6R in dorsal root ganglia following nerve injury, and focused on the effects of IL-6 signaling on chloride accumulation, cation-Cl− cotransporter expression and phosphorylation.
Materials and Methods
Surgery and cell culture.
Adult Swiss and C57BL/6 female mice (6–8 weeks old, CERJ) and IL-6−/− mice (a gift from S. Hopkins and R. Grencis, University of Manchester, Manchester, UK) (Kopf et al., 1994), were housed in cages with a 12 h light/dark cycle and fed and water ad libitum. The care and use of the mice conformed with institutional policies and guidelines. Mice were deeply anesthetized by isoflurane inhalation. The left sciatic nerve was exposed at the mid-thigh, sectioned, and a 2–3 mm fragment removed. Five days after surgery, mice were killed by CO2 inhalation followed by cervical dislocation, and their left lumbar L4-L5 DRGs were removed. Neuronal cultures were established from L4-L5 dorsal root ganglia, as previously described (André et al., 2003). Dissociated neurons were plated on dl-polyornithine (0.5 mg/ml)-laminin (5 μg/ml)-coated glass coverslips and were incubated at 37°C in an incubator with a humidified 95% air/ 5% CO2 atmosphere.
Genotyping.
Genotyping was performed using PCR on extracted DNA. For IL-6−/− mice, the same forward primer (5′-TTCCATCCAGTTGCCTTCTTGG-3′) was used in combination with reverse primer for the control (5′-TTCTCATTTCCACGATTTCCCAG-3′) or the mutant gene (5′-CCGGAGAACCTGCGTGCAATCC-3′).
Measurement of intracellular chloride concentration.
Electrophysiological recordings of the ionotropic chloride GABAA current in DRG neurons at 1 d in vitro (DIV) were used to determine reversal potential of chloride current at room temperature as previously reported (Pieraut et al., 2007). Briefly, the bathing solution contained the following (in mm): 140 tetraethylammonium chloride, 3.5 MgCl2, 10 HEPES, and 10 glucose, at pH 7.4 (adjusted with CsOH). Recording pipettes were filled with the following solution (in mm): 145 CsCl, 10 HEPES, 2 Mg-ATP, 0.5 Na2-GTP, at pH 7.35 (adjusted with CsOH). Gramicidin-perforated technique was used to determine [Cl−]i. Gramicidin A (50 μg/ml) (Fluka) was added to the pipette solution. The progress of gramicidin perforation was evaluated by on-line monitoring, with pClamp version 8.2 software (Dipsi Industrie), the capacitive current transient produced by a 10 ms depolarizing voltage step (10 mV) from a −80 mV holding potential. When the access resistance had dropped to 50 MΩ, puff application of 50 μm muscimol activated a chloride current attributable to GABAA receptor activation. The [Cl−]i was calculated using the Nernst equation: EGABAA = RT/ZF × (log ([Cl−]i/[Cl−]e), where RT/ZF = 58 mV at room temperature and [Cl−]e = 147 mm. All recordings were made using an Axopatch 200B amplifier (Dipsi Industrie).
Interleukin-6 quantification by ELISA.
At 1 DIV, supernatant of cultured DRG cells was removed and put at −80°C. The presence of murine IL-6 in the supernatant was determined by specific ELISAs (BD Biosciences) as previously described (Djouad et al., 2007). Quantification was performed at least twice. After removal of the supernatant, the glass coverslips were submerged in lysis buffer (50% Ca- and Mg-free PBS and 50% Laemmli lysis buffer) to collect neuronal and non-neuronal cells for protein quantification using photodetection at 280 nm (Biophotometer, Eppendorf).
Immunostaining.
Primary antibodies used included the following: mouse anti-glutamine synthetase (Millipore Bioscience Research Reagents; 1:500); goat anti-mouse IL-6 receptor (R&D Systems; extracellular domain, 10 μg/ml); rabbit anti-mouse IL-6 receptor (Santa Cruz Biotechnology; C terminus, 1:50); rabbit anti-Neurofilament 200 (Sigma; 1:100 for DRG slice and 1:1000 for culture); rabbit anti-parvalbumin (SWANT;1:10,000). Rabbit anti-phospho-NKCC1 (1:500), directed against the regulatory phosphothreonines Thr212 and Thr217 of human NKCC1, was a gift from Dr. B. Forbush (Yale University, New Haven, CT), and specific staining in DRG neurons was demonstrated previously (Pieraut et al., 2007); rabbit anti-TrkB (Millipore; 1:1000); and rabbit anti-TrkA (Millipore; 1:500); mouse anti-β-III tubulin (Sigma; 1:500). Anti-parvalbumin, TrkB and TrkA antibodies were kindly provided by Dr. Patrick Carroll (Institute for Neurosciences, Montpellier, France) (Bourane et al., 2009). Secondary antibody incubations were performed with Alexa Fluor-594 or Alexa Fluor-488-conjugated secondary antibodies (Invitrogen; 1:1000).
For immunohistochemistry, L4-L5 DRGs were dissected in PBS, fixed for 2 h in 4% paraformaldehyde, immerged overnight in 25% sucrose, and embedded in OCT compound. Frozen DRG sections (14 μm) were cut, and each was washed with PBS, blocked with 10% donkey serum, and incubated overnight at 4°C with primary antibodies. For phospho-NKCC1 immunostaining, an epitope-demasking procedure was performed by immersion of sections for 15 min at 68°C in sodium citrate buffer (10 mm sodium citrate, 0.05% Tween 20 at pH 6). After a wash in PBS, DRG sections were incubated for 1 h at room temperature with secondary antibody, and then mounted in Mowiol. They were then overnight incubated at 4°C with the primary antibodies.
Images were collected using either a PL-Apochromat 20× or 40× objective mounted on an upright Zeiss microscope equipped with a CCD camera AxioCam MRm, or an LSM5 confocal microscope (Zeiss). Image acquisition and analysis was done using AxioVision (Zeiss).
Neurite growth analysis.
Neurons were seeded in 4-well chambers at a density of 500 neurons per well and maintained in culture medium at 37°C. To make accurate measurements of primary neurite length, analyses were performed 8 h after plating. We previously showed that significant neurite growth at 8 h is exhibited by 55% of neurons after a conditioning lesion compared with 8% of controls (Pieraut et al., 2007). Cultures were fixed for 15 min in 4% paraformaldehyde in PBS, and incubated for 20 min in 15% donkey serum in PBS. They were then incubated 2 h at room temperature with the primary antibodies (pan-neuronal β-III-tubulin for neurite length measurement and NF-200 as mechano-proprioceptive marker). After a wash in PBS, cultures were incubated for 1 h at room temperature with secondary antibodies, and were mounted in Mowiol. Images were collected using a PL-Apochromat 10× objective and ImageJ was used for somatic diameter and neurite analysis. Soma that extended neurites (primary neurite) greater than two cell body diameters were analyzed. Measurements were calculated from at least three wells, with three mice per experimental group.
Quantification of mRNA expression levels by reverse transcriptase-quantitative PCR.
Total mRNA was extracted from lumbar L4-L5 DRGs of adult mice using the RNeasy Mini Kit (Qiagen) in accordance with the manufacturer's instructions. Briefly, DRGs were harvested in RNAlater stabilization buffer (Qiagen) and homogenized by passing the lysate 10 times through a 20 gauge needle. Lysates were then mixed with an equal volume of 70% ethanol, and total mRNA was separated from other cellular components using RNeasy minispin columns. The eluted mRNA was quantified using spectrophotometry (Nanodrop; Labtech). Reverse transcription was performed with 1 μg of mRNA using the Quantitect RT kit (Qiagen) and the following thermal cycling protocol: 60 min at 37°C; 5 min at 95°C. cDNA was then collected and stored at −20°C until further use. Primers were designed with Primers 3.0 software. Sequences for all primers are shown in Table 1. Quantitative PCR was performed at least three times on duplicate samples of cDNA diluted to 1:10 using SYBR Green (Qiagen) for detection on the LightCycler system (Roche Diagnostics). After an initial activation step of 15 min at 95°C, 45 amplification cycles were performed consisting of 94°C for 15 s, 59°C for 20 s and 72°C for 35 s. After PCR amplification, a melting curve analysis was generated to check the specificity of the PCR. The identity of reverse transcriptase (RT)-PCR-amplified products was confirmed by sequencing (MilleGen). Polymerase (RNA) II polypeptide J, Polr2J, levels were used to normalize amounts of cDNA (Pieraut et al., 2007). The ΔCt was calculated as the difference between the Ct values, determined using the equation 2−ΔCt.
Table 1.
Sequence of primers used in real-time PCR, amplicon sizes, and their melting temperatures
| Target genes | Sequences | Amplicon length | Tm (°C) |
|---|---|---|---|
| Slc12a2 (NKCC1) | F: AAGGGCAGCGAGGAAGCCAA | 135 | 61 |
| R: GCCGTTCTGGAAGCTCACGTT | |||
| Slc12a4 (KCC1) | F: AACGAGGTCATTGTCACGCGCT | 147 | 62 |
| R: ACGCACCAACAACACCCGTT | |||
| Slc12a5 (KCC2) | F: TGCCATCAAACGGCTCAGGTGT | 68 | 62 |
| R: TTGAGCCAAGCAACAGGGCAGT | |||
| Slc12a6 (KCC3) | F: AGTGAAGATGCTCGCGCTTGGA | 170 | 62 |
| R: AGCATGCCCCCATCATGCACAA | |||
| Slc12a7 (KCC4) | F: AACTGGCGTCCACGCTTCAAGT | 122 | 62 |
| R: CCGGCAATGAGCATGGCGAAAA | |||
| Il6 | F: ACAACCACGGCCTTCCCTACTT | 100 | 62 |
| R: AGCCTCCGACTTGTGAAGTGGT | |||
| Il6ra | F: TGTTGAAGACTCTGCCAACCACGA | 145 | 62 |
| R: ACAGAGAAGCAACCCAAACGCCAA | |||
| R: TGGTCAAATCTTTGCGCAGCCC | |||
| Polr2j | F: ACCACACTCTGGGGAACATC | 160 | 59 |
| R: CTCGCTGATGAGGTCTGTGA |
Slc12a, solute carrier 12 family; Il6, interleukin 6; Il6ra, interleukin 6 receptor α chain; Polr2j, polymerase (RNA) II polypeptide J; corresponding protein is in parentheses. F, Forward; R, reverse.
Drugs.
AG490 and cucurbitacin were from Sigma, and INCB 018424 (ruxolitinib) was from Axon Medchem. Blocking-function mouse IL-6 antibody and recombinant mouse IL-6R [soluble IL-6R obtained from a DNA sequence encoding the extracellular domain of mouse IL-6R (Sugita et al., 1990)] were from R&D Systems. Blocking-function human IL-6 antibody was from Diaclone.
Statistical analysis.
All values are reported as mean ± SEM. Statistical significance was evaluated using one-way or two-way ANOVA (followed by Bonferroni's test), Student's t test or χ2 test as indicated. Mann–Whitney U test was used for RT-quantitative (q)PCR experiments. p < 0.05 was considered as significant (GraphPad Prism 5).
Results
Endogenously produced IL-6 regulates intracellular chloride concentration of axotomized sensory neurons in vitro
Following nerve injury, we previously reported an increase in [Cl−]i, of axotomized sensory neurons having an elongated neurite growth (Pieraut et al., 2007). To determine whether IL-6 was involved in chloride accumulation, we first analyzed the effects of nerve injury on Il6 gene expression in DRG. At 3 d postinjury, the normalized expression of IL-6 mRNA in injured L4-L5 DRGs (1.45 ± 0.06, n = 4) was approximately three orders of magnitude higher than in control L4-L5 DRGs (0.50 ± 0.08, n = 4, Fig. 1A). We next quantified its production in the culture supernatant of DRG neurons using ELISA. At 1 DIV, the amount of IL-6 in the culture medium of axotomized DRG cultures (n = 6), was 78 ± 12 pg/ml (Fig. 1B). We failed to detect IL-6 secretion in DRG primary cultures from IL-6−/− mice, thus demonstrating the specificity of the ELISA test (n = 2, data not shown). DRG primary cultures contain both neurons and non-neuronal cells. To assess the neuronal contribution to IL-6 secretion relative to non-neuronal cells, we performed IL-6 measurements in axotomized culture at 4 DIV in the absence and the presence of 1 μm (β-d-arabinofuranosyl)cytosine, AraC, to reduce non-neuronal cell proliferation. In agreement with a continuous IL-6 production and secretion in vitro with time (Cao et al., 2006), the amount of IL-6 was increased to 397 ± 33 pg/ml (n = 3) at 4 DIV. In the presence of AraC, it was reduced to 277 ± 10 pg/ml (n = 3) (Fig. 1B). Consistent with cell proliferation and growth, protein content was significantly increased at 4 DIV compared with 1 DIV, and AraC decreased it to level similar to 1 DIV (Fig. 1C). From these data, it appeared that probably both neurons and non-neuronal cells contribute to IL-6 production in vitro.
Figure 1.

Endogenously produced IL-6 regulates intracellular chloride concentration in sensory neurons. A, RT-qPCR shows a significant increase in mRNA expression of Il6 gene in L4–L5 DRG 3 d after sciatic nerve section. (n, number of experiments with duplicate measure of each gene; *p < 0.05, Mann–Whitney t test). B, Quantitative ELISA measurements of IL-6 production in primary cultures of axotomized neurons from L4–L5 DRGs were performed at days 1 and 4 in vitro. The AraC-sensitive IL-6 production accounted for approximately 30% of total secretion at 4 DIV (**p < 0.01, ***p < 0.001; one-way ANOVA). C, Corresponding protein content measurements show a significant protein content increase at 4 DIV which was prevented in the presence of 1 μm AraC. (*p < 0.05; one-way ANOVA). D, Gramicidin-perforated patch recordings were used to determine [Cl−]i in sensory neurons. Ramp protocols from −80 mV to +40 mV were applied every 5 s in a Ca-free, Na-free, K-free, 147 mm tetraethylammonium-Cl extracellular solution. Activation of chloride current GABAA with puff application of 50 μm muscimol allowed for determination of reversal potential, EGABAA, of the chloride current and to calculate intracellular chloride concentration (see Materials and Methods). EGABAA was −25 mV in an axotomized sensory neuron, and calculated [Cl−]i was 54 mm. E, Following axotomy, sensory neurons increase their [Cl−]i from 28.3 ± 1.1 mm to 52.3 ± 2.8 mm (***p < 0.001, t test). Incubation of axotomized neurons with 10 μg/ml anti-mouse IL-6 antibody, mIL-6 Ab, significantly decreased [Cl−]i to 30.0 ± 2.8 mm. Incubation with 10 μg/ml hIL-6 Ab, which does not cross-react with the murine IL-6, had no effect on the [Cl−]i of axotomized neurons that amounted to 55.8 ± 5.2 mm (***p < 0.001; one-way ANOVA between axotomized conditions). F, EGABAA recordings from IL-6+/+ and IL-6−/− sensory neurons show no significant difference in [Cl−]i under control conditions, which averaged 29 ± 2 mm and 23 ± 2 mm, respectively. The main effect of IL-6 deletion was associated with axotomy. [Cl−]i was significantly higher in axotomized IL-6+/+ sensory neurons (54.5 ± 3.2 mm) than in IL-6−/− axotomized sensory neurons (35 ± 3 mm) (**p < 0.01, ***p < 0.001; two-way ANOVA). Number of experiments is in brackets.
We next assessed whether the [Cl−]i, was controlled by endogenously produced IL-6 by measuring the reversal potential of the ligand-gated Cl− current GABAA, EGABAA in sensory neurons(Fig. 1D). We first focused our study on neurons having a somatic diameter >30 μm to discriminate mechano-proprioceptors from nociceptors. As previously shown, EGABAA of axotomized sensory neurons was shifted toward depolarized potentials following nerve injury (from −41.8 ± 1.0 mV, n = 16, in control neurons to −26.5 ± 1.3 mV, n = 14, in axotomized neurons). This corresponded to a twofold increase in [Cl−]i (Fig. 1E).
To verify whether IL-6 was involved in the [Cl−]i increase observed in axotomized neurons, we performed experiments using a blocking-function IL-6 antibody to neutralize secreted IL-6 in conditioned cultures. Following dorsal root ganglia dissociation, antibodies raised against mouse IL-6 (mIL-6) or human IL-6 (hIL-6) were added to the culture (10 μg/ml each) and incubated overnight before electrophysiological recordings. The reversal potential of GABAA current of axotomized neurons was significantly shifted to hyperpolarized values in cultures treated with mIL-6 antibody (−41.2 ± 2.5 mV, n = 11, Fig. 1E). However it did not change in axotomized neurons treated with hIL-6 antibody (−25.1 ± 2.5 mV, n = 7). Analysis of chloride homeostasis in IL-6−/− mice shows no significant differences in the reversal potential of the GABAA current in control neurons from IL-6+/+ (−41.3 ± 1.9 mV, n = 6) and IL-6−/− mice (−47.3 ± 2.6 mV, n = 6, Fig. 1F). Experiments performed on axotomized neurons showed that EGABAA was significantly less depolarized in IL-6−/− neurons (−36.6 ± 1.9 mV, n = 13) than in IL-6+/+ neurons (−25.4 ± 1.5 mV, n = 10, p < 0.001). Following nerve injury, the increase in [Cl−]i was significantly less in IL-6−/− neurons than in wild-type neurons (Fig. 1F). These findings indicate that endogenously produced IL-6 regulates [Cl−]i in regenerating axotomized sensory neurons.
Nerve injury upregulates IL-6 α receptor expression in dorsal root ganglia neurons
To examine whether specific changes in transcript levels of IL-6 α receptor, IL-6R, were associated with the increase in IL-6 following sciatic nerve injury in vivo, we conducted RT-qPCR on control and axotomized L4-L5 DRG. Our data show that Il6ra mRNA levels varied with injury. Remarkably, injury induced a twofold increase in mRNA expression of Il6ra in DRG (from 0.75 ± 0.05 to 1.49 ± 0.04, n = 7, Fig. 2A).
Figure 2.

Nerve injury upregulates IL-6R expression in adult mice DRG neurons. A, RT-qPCR shows a significant increase in mRNA expression of Il6ra gene in L4–L5 DRGs 3 d after sciatic nerve section (n, number of experiments with duplicate measure of each gene; **p < 0.001, Mann–Whitney t test). B, Images of transverse sections of L4–L5 DRGs immunolabeled with an antibody generated against a recombinant extracellular domain of mouse IL-6R (10 μg/ml; red, Alexa 594). In control DRG, minor staining of IL-6R is evident at/close to the neuronal plasma membrane (arrowhead). In axotomized DRG, a plasma membrane staining is observed more frequently in a subset of sensory neurons (arrowhead). C, Preincubation of 10 μg/ml anti-IL-6R antibody with 100 μg/ml soluble IL-6R for 2 h at 4°C prevented the plasma membrane staining of IL-6R in axotomized DRG (left). Right, labeling of axotomized DRG with an anti-IL6R antibody raised on the intracellular C-terminal is also observed at/close to the neuronal plasma membrane (arrowhead). D, Confocal images of transverse section of axotomized L4–L5 DRGs costained with anti-IL-6R (extracellular) and anti-glutamine synthetase (GS, a satellite cell marker) antibodies. DAPI shows the nucleus of cells surrounding neurons (note that neuronal nucleus is not observed at this stack level). The merge image shows satellite cells staining surrounding neuronal IL-6R staining (inset is 3× magnification of membrane portion, shown with an asterisk). Scale bars, 30 μm.
We next addressed with immunofluorescence microscopy whether expression of IL-6R protein was also increased in axotomized DRG. Immunohistochemistry using an antibody raised against the extracellular domain of mouse IL-6R revealed staining close to the plasma membrane in 5% of neurons from control DRG (60/1142, Fig. 2B). In contrast, the number of neurons expressing a clear IL-6R stained ring was substantially increased in axotomized DRG (305/1303, 23%, p < 0.01, χ2 test, Fig. 2B). The specificity of staining in axotomized DRG was assessed by incubating the antibody with the soluble IL-6R before immunostaining. Under this condition, no signal could be detected at the level of plasma membrane (Fig. 2C). Experiments using a second IL-6R antibody raised on the C-terminal domain confirmed signal specificity at the level of plasma membrane (Fig. 2C). Although the expression of IL-6R is well established among glial cells, neuronal expression of IL-6R has not been reported. To determine the cellular identity of IL-6R-positive cells, we performed double labeling experiments with anti-IL-6R antibody and anti-glutamine synthetase antibody, a marker of satellite glial cells. These experiments revealed that glutamine synthetase staining was surrounding IL-6R staining which supports a neuronal expression pattern for IL6-R at/close to the neuronal membrane (Fig. 2D). These results demonstrate that nerve injury upregulates expression of IL-6R transcript and induces IL-6R protein expression at/close to the neuronal plasma membrane of a subset of DRG neurons.
IL6-R is expressed in a subset of mechano- and proprioceptive sensory neurons in vivo
To identify the class of sensory neurons expressing IL-6R in axotomized DRG, we first performed a series of double labeling experiments by monitoring the coexpression of IL-6R with NF-200, a biochemical marker of myelinated sensory neurons. Most IL-6R-positive neurons expressed NF-200 (92%, 56/61), thus identifying them as part of the myelinated population (Fig. 3A). To further confirm the functional identity of this neuronal subpopulation, we performed double labeling experiments with TrkB, the high affinity receptor for brain-derived neurotrophic factor and neurotrophin-4. TrkB is expressed in subsets of myelinated mechanoreceptors. Most IL-6R-positive neurons coexpressed TrkB (67/84, 80%) (Fig. 3B). As we failed to get a clear staining with rabbit anti-TrkC antibody, we used anti-Parvalbumin antibody and found that approximately 30–40% of IL-6R coexpressed this proprioceptive marker (37/107) (Fig. 3C). In agreement with IL-6R expression among mechano-proprioreceptors, no overlap was observed between IL-6R and TrkA, a high affinity receptor for nerve growth factor that specifically marks a subset of nociceptive neurons (1/61, 2%) (Fig. 3D). From these results, we conclude that nerve injury induces expression of IL6-R in vivo among mechano- and proprioceptive sensory neurons.
Figure 3.
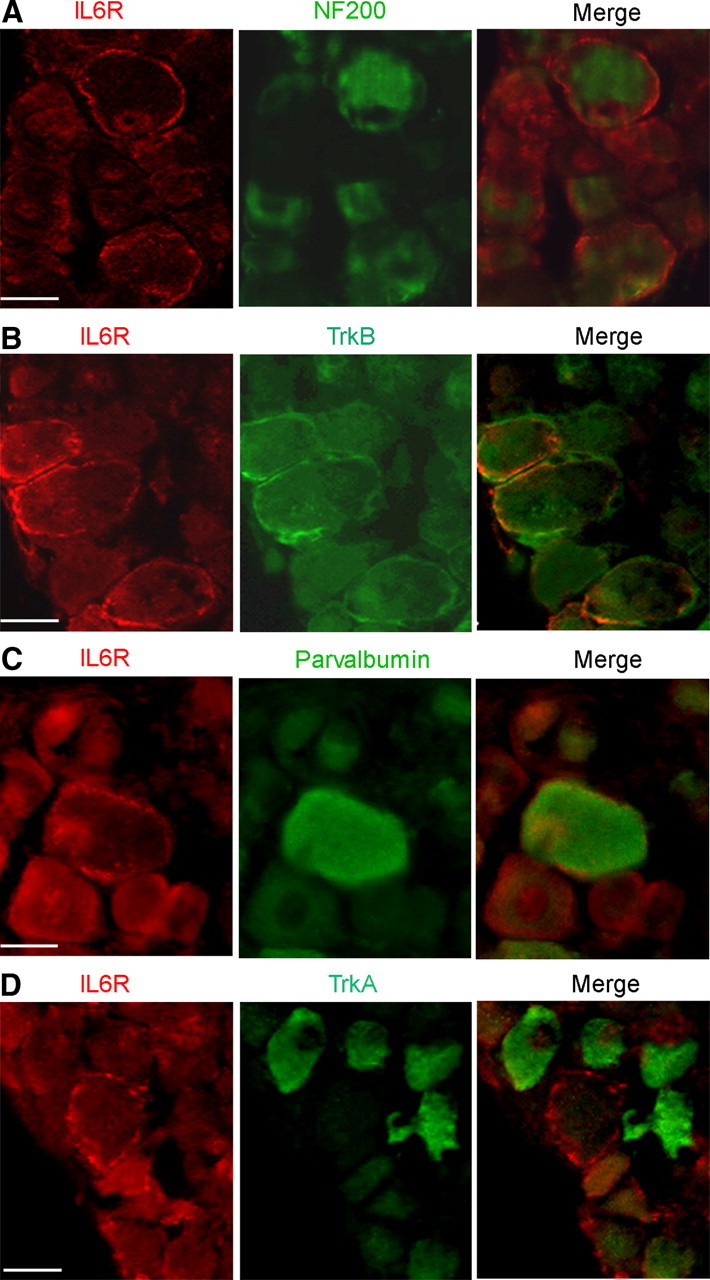
IL-6R is expressed in a subset of myelinated sensory neurons. A, Apotome images of transversal slices of axotomized DRG double stained with anti-IL-6R (red) and anti-NF-200 (green) antibodies. The merge image shows colocalization of IL-6R with NF-200-positive sensory neurons. B, Double immunochemistry using anti-TrkB antibody (green) shows IL-6R colocalization with TrkB-positive neurons (merge). C, Double immunochemistry using anti-Parvalbumin antibody (green) shows IL-6R colocalization with parvalbumin-positive neurons (merge). D, Double immunochemistry using anti-TrkA antibody (green) demonstrates no IL6-R colocalization with TrkA-positive neurons (merge). Scale bar, 30 μm.
IL-6R expression in large somatic-sized axotomized neurons is responsible for intracellular chloride regulation through the JAK/STAT pathway
To test whether the effects of IL-6 were mediated through neuronal expression of its cognate receptor IL-6R, we assessed IL-6R expression in neuronal primary cultures from axotomized DRG. Immunocytochemistry with the anti-IL-6R antibody confirmed the expression of IL-6R among medium- to large-sized axotomized sensory neurons (Fig. 4A). In agreement with the in situ immunofluorescence results, the population of stained neurons having in vitro a somatic diameter >30 μm is representative of mechano-proprioceptive sensory neurons and accounted for 34% of the total population (228/883). Expression of IL-6R was not observed among most of the nociceptors that are characterized in vitro by a somatic diameter smaller than 30 μm. Moreover, no expression was observed among non-neuronal cells.
Figure 4.
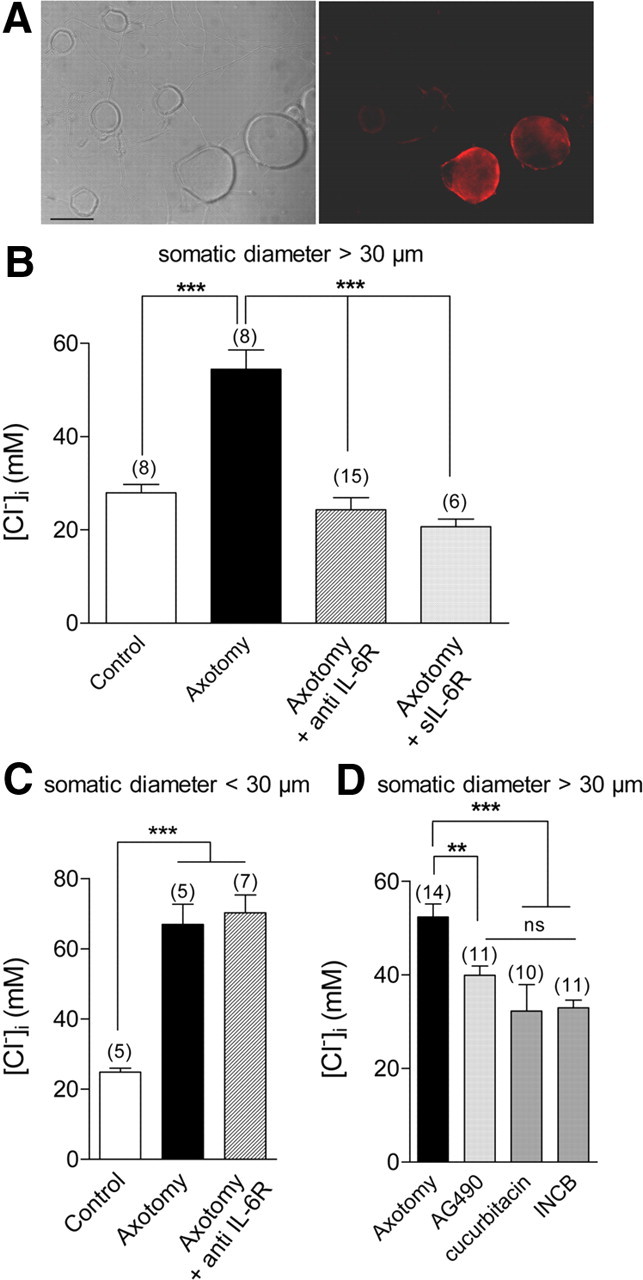
Neuronal expression of IL-6R in vitro regulates the intracellular chloride concentration of medium-sized sensory neurons. A, Differential interface contrast image of axotomized neurons at 1 d in vitro, and corresponding fluorescence image of IL-6R protein using anti-IL-6R antibody. Neurons with a somatic diameter >30 μm are IL-6R-positive (scale bar, 30 μm). B, Following overnight incubation of axotomized primary culture with 10 μg/ml anti-IL-6-R antibody, the [Cl−]i of axotomized neurons with a somatic diameter >30 μm was significantly lower (24.3 ± 2.6 mm) than nontreated, axotomized neurons (54.5 ± 4.1 mm), and no different from control values (28.0 ± 1.7 mm). Axotomized neurons incubated with 100 ng/ml soluble IL-6R (sIL-6R) have a significantly lower [Cl−]i than untreated counterparts (20.6 ± 1.6 mm) (***p < 0.001; one-way ANOVA). C, Analysis of [Cl−]i among sensory neurons with a somatic diameter <30 μm shows that axotomy induced a significant increase in [Cl−]i from 24.9 ± 1.1 mm in controls to 66.9 ± 5.7 mm in axotomized neurons, and to 70.2 ± 5.1 mm in anti-IL-6R antibody-treated axotomized neurons, with no differences between treated and nontreated axotomized neurons (***p < 0.001; one-way ANOVA). D, Jak1-2 inhibitors, AG490 at 50 μm, cucurbitacin at 3 μm, and INCB 018424 at 300 nm significantly reduced [Cl−]i in medium to large axotomized neurons from 52.3 ± 2.8 mm to 39.9 ± 2.0 mm, 32.2 ± 5.7 mm, and 32.9 ± 1.6 mm, respectively (**p < 0.01, ***p < 0.001, one-way ANOVA; number of experiments in brackets).
Having determined that IL-6R is specifically expressed in medium- to large-sized axotomized neurons, we analyzed, among this subset of neurons, the effects of the blocking-function IL-6R antibody and of the soluble IL-6R on the [Cl−]i. Following overnight incubation with 10 μg/ml anti-IL-6R antibody, EGABAA was significantly less depolarized than in untreated neurons (−47.2 ± 2.6 mV, n = 15 versus −23.5 ± 1.9 mV, n = 8, p < 0.001, Fig. 4B). The soluble IL-6R competes with its membrane-bound counterpart for ligand binding and therefore could act as an antagonist in cells expressing IL-6R (März et al., 1999). Consistent with antagonistic effects, EGABAA was significantly less depolarized following soluble IL-6R treatment (−49.8 ± 2.0 mV, n = 6, p < 0.001). To further confirm the cell specificity of IL-6R expression, small axotomized neurons with a somatic diameter <30 μm were recorded in the presence of function-blocking anti-IL-6R antibody. Following axotomy, these neurons show chloride accumulation similar to medium to large neurons. However, anti-IL-6R antibody incubation did not significantly alter EGABAA, which averaged −20.2 ± 2.2 mV in axotomized neurons (n = 5) and −19.0 ± 1.8 mV in axotomized neurons treated with anti-IL-6R antibody (n = 7, Fig. 4C).
Finally, the functional implication for the regulation of chloride homeostasis of the downstream Janus kinase signaling initiated by IL-6R activation was investigated in medium to large neurons. We used three pharmacological inhibitors of these pathways, AG490, cucurbitacin and a specific Jak1-2 inhibitor, INCB 018424 (Quintás-Cardama et al., 2010). Following overnight incubation, these inhibitors shifted EGABAA among axotomized neurons toward hyperpolarized potentials from −17.5 ± 3.2 mV (n = 14) to −33.2 ± 1.3 mV (n = 11) for AG490; −41.9 ± 4.5 mV (n = 10) for cucurbitacin and −38.0 ± 1.3 mV (n = 11) for INCB (Fig. 4D). Our data support that signaling through neuronal IL-6R regulates [Cl−]i in medium- to large-sized sensory neurons following peripheral nerve injury.
IL-6 signaling promotes NKCC1 phosphorylation on plasma membrane in vivo
We previously reported that axotomy does not modify NKCC1 transcript expression (Pieraut et al., 2007). The contribution of the outwardly directed cation-Cl− cotransporters (KCC1-4) to the regulation of [Cl−]i in sensory neurons is unknown. We therefore used RT-qPCR to screen for the expression of KCC1-4 transcripts in control DRG and following nerve injury. Analysis of the relative expression of cation-Cl− cotransporters in DRG shows a preferential transcript expression of NKCC1, KCC1, and KCC3 (Fig. 5A). Three days after axotomy, no significant change in transcripts encoding the five Slc12a genes was observed (Fig. 5B). Gene expression analysis performed in DRG from IL-6−/− mice showed no significant changes in the relative expression of cotransporters (data not shown), and nerve injury did not significantly modify them (Fig. 5B). Our gene expression analysis thus confirms that transcriptional modification of cation-Cl− cotransporters cannot account for the effects of IL-6 on intracellular chloride concentration.
Figure 5.
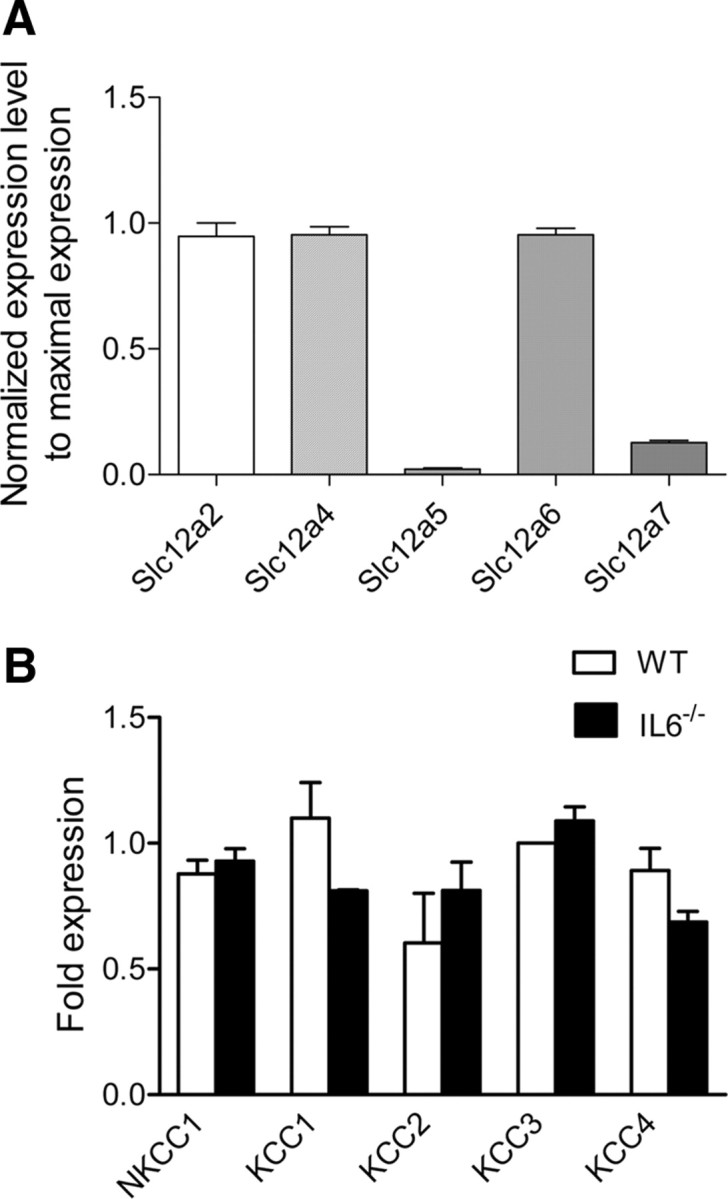
Expression and regulation of cation-chloride cotransporter transcripts in DRG following nerve injury. A, RT-qPCR was performed in control DRG to determine expression of cation-Cl− cotransporters. For each qPCR experiment, at least three different cation-Cl− cotransporter transcripts, including Slc12a2, were analyzed and their expression was normalized relative to the most expressed Slc12a family transcript (Slc12a2 in our experiments). Data are the mean of 4 experiments, with duplicate measures of each gene. In DRG, Slc12a2 (NKCC1), Slc12a4 (KCC1) and Slc12a6 (KCC3) are the most expressed transcripts. B, RT-qPCR shows no significant change in the expression of the Slc12a family in DRG 3 d after sciatic nerve injury, in wild-type or IL-6−/− mice. To compare between two different experimental conditions (control vs axotomy) each transcript expression is normalized to Polr2J, a control gene previously shown to keep a constant expression level. Data are the mean of 4 experiments (p > 0.05, Man–Whitney t test). Name of the corresponding transporter protein is used.
We previously shown that NKCC1 phosphorylation is a major contributor to [Cl−]i regulation following nerve injury (Pieraut et al., 2007). To assess whether IL-6 signaling is responsible for NKCC1 phosphorylation, we performed phospho-NKCC1 labeling in WT and IL-6−/− DRG. In control DRG from WT mice, no distinctive staining was observed among sensory neurons (Fig. 6A). Five days following nerve injury, an intense phospho-NKCC1 staining was observed close to the plasma membrane of a subset of sensory neurons (Fig. 6B). Analysis of phospho-NKCC1 expression in control IL-6−/− DRG was similar to WT, with no obvious staining (Fig. 6C). In contrast with WT DRG, phospho-NKCC1 staining was no longer detected in IL-6−/− sensory neurons after nerve injury (Fig. 6D). The neuronal expression of phospho-NKCC1 in axotomized sensory neurons was confirmed by colabeling with the glial marker glutamine synthetase (Fig. 6E). In addition, double labeling with IL-6R established its coexpression with phospho-NKCC1 in a subset of DRG sensory neurons (Fig. 6F). These data imply that IL-6 promotes expression of phospho-NKCC1 at or close to the plasma membrane of axotomized sensory neurons expressing IL-6R.
Figure 6.
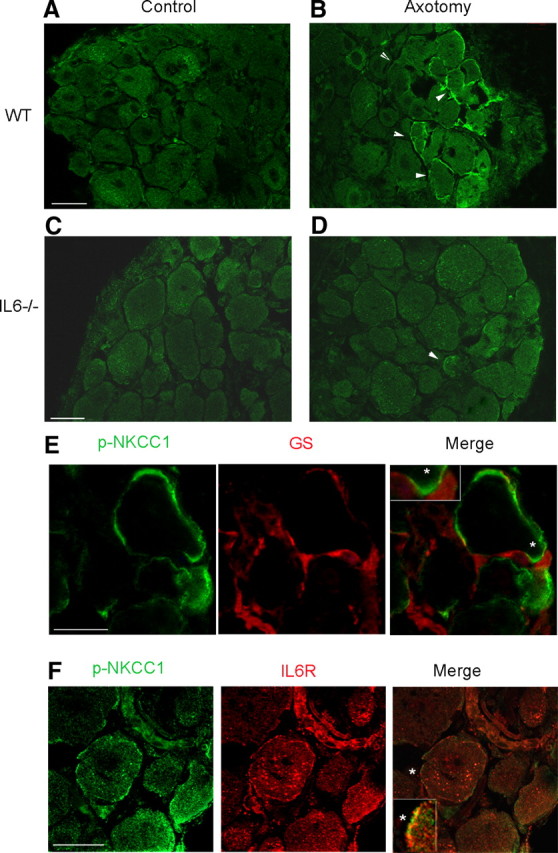
Il-6 regulates the expression of phospho-NKCC1 close to the plasma membrane of axotomized DRG neurons. A, Transverse section of L4–L6 DRG immunolabeled with an antibody generated against phospho-NKCC1; no signal is observed. B, Five days after nerve injury, phospho-NKCC1 staining is present at/over the plasma membrane in a subset of sensory neurons (arrows). C, Similar to WT DRG, phospho-NKCC1 staining was absent in transverse sections of L4–L6 DRGs from IL6−/− mice. D, Five days after nerve injury, only a few neurons have a plasma membrane showing phospho-NKCC1 staining in DRG from IL-6−/− mice (arrow). E, Confocal microscopy of double-immunofluorescent labeling for phospho-NKCC1 (p-NKCC1) and glutamine synthase (GS) shows no coexpression. F, Confocal microscopy of double-immunofluorescent labeling for p-NKCC1 and IL-6R shows expression in the same cell type. Merge image shows a co expression that appears as an alternate punctiform staining at the plasma level. Scale bars, 30 μm. Insets in merge images are 3× magnifications of the region marked by white asterisk. A–D, 20× objective; E, F, 40× objective.
IL-6 signaling contributes to neurite growth promoting effects of NKCC1
Previously, we reported that NKCC1 is involved in determining the velocity of elongated growth of axotomized neurons (Pieraut et al., 2007). To uncover whether regulation of NKCC1 through IL-6 signaling was involved in neurite growth of NF-200-positive large neurons, neuronal cultures from axotomized DRG were processed for NF-200 and β-III-tubulin immunocytochemistry. Somatic diameter and associated neurite growth were recorded for neurons that were distinguishable from their neighbors and analyzed according to their NF-200 expression and somatic size (Fig. 7A). For each neuron, the number of primary neurites expanding from the soma and the corresponding total maximal length were analyzed by using pan-neuronal β-III-tubulin staining. We show that large-sized sensory neurons (soma >30 μm) were NF-200 positive and characterized a higher number of primary neurites and a longer total length (3.3 ± 0.3 neurites/neuron and 1130 ± 119 μm, n = 26) compared with NF-200-positive or -negative small-sized neurons (soma <30 μm, 2.0 ± 0.2 neurites/neuron and 679 ± 78 μm, n = 26; p < 0.01, t tests) (Fig. 7E,F).
Figure 7.
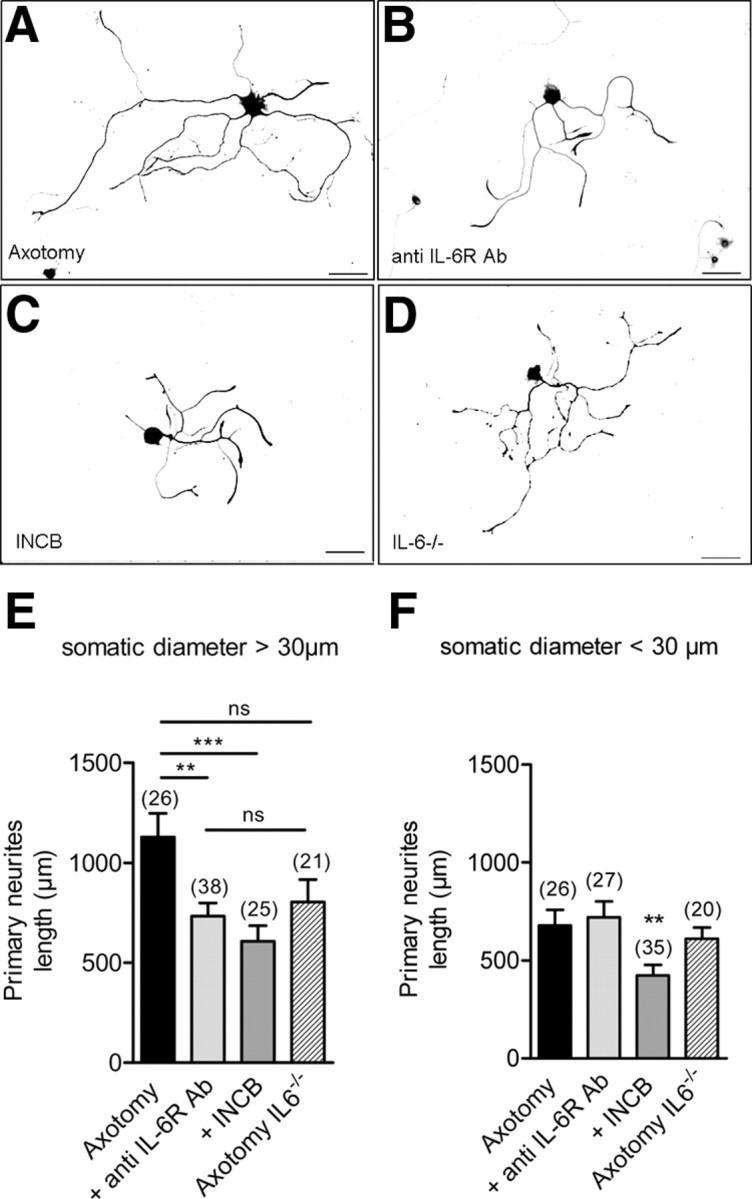
IL-6 signaling is involved in neurite growth of NF-200-positive medium- to large-sized sensory neurons. A–D, Photomicrographs of cultured medium- to large-sized neurons from axotomized DRG immunocytochemically stained with pan-neuronal marker β-III tubulin and NF-200 after 8 h in vitro (A), grown with 10 μg/ml blocking-function IL-6R antibody (B), or with 300 nm INCB 018424 (C), or from IL-6−/− axotomized DRG (D). Double labeling with β-III-tubulin and NF-200 allowed comparing neurite length of NF-200-positive and -negative neurons (scale bar, 100 μm; 10× objective). E, Analysis of total length of primary neurites in medium- to large-sized axotomized neurons (NF-200-positive neurons) shows a significant decrease in the presence of blocking-function IL-6R antibody or the Jak1-2 inhibitor INCB 018424, a specific Jak1-2 inhibitor. Il6 gene deletion did not significantly modify neurite length (**p < 0.01, ***p < 0.001; one-way ANOVA). F, Analysis of total length of primary neurites in small-sized neurons (NF-200-positive and -negative neurons) shows no significant effects of blocking-function IL-6R antibody or Il6 gene deletion. INCB 018424 was as efficient to decrease neurite length in small- than in large-sized neurons (**p < 0.01, ***p < 0.001; one-way ANOVA).
To ensure that, in our culture conditions, IL-6 signaling regulates neurite growth in a subset of neurons, 10 μg/ml IL-6R blocking-function antibody was added at the time of plating. Under this condition, there was a significant 40% decrease in the total length of primary neurites in large-sized neurons without effects on neurite number (733 ± 86 μm and 3.1 ± 0.5 neurites/neuron, n = 38; Fig. 7B,E). No significant effects of IL-6R blocking-function antibody were observed in the length and number of primary neurites in small-sized neurons (721 ± 81 μm and 2.3 ± 0.3 neurites/neuron, n = 27; Fig. 7F). Consistent with its inhibitory effects on Jak1-2, incubation with 300 nm INCB 018424 reduced by 50% total primary neurite length of large-sized neurons without effects on neurite number (607 ± 78 μm and 2.6 ± 0.4 neurites/neuron, n = 25; Fig. 7C,E). INCB did also significantly decreased neurite length of small-sized neurons without effects on neurite number (422 ± 55 μm and 1.8 ± 0.2 neurites/neuron, n = 35; Fig. 7F). Last, analysis of neurite growth from IL-6−/− DRG culture showed no significant effects of gene deletion on neurite length and number in large-sized neurons (30% decrease to 804 ± 113 μm and 2.6 ± 0.4 neurites/neuron, n = 21; Fig. 7D,E) and in small-sized neurons (609 ± 58 μm and 1.5 ± 0.1 neurites/neuron, n = 20; Fig. 7F). Our data demonstrate that inhibition of IL-6 signaling reduces neurite growth of large-sized NF200-positive axotomized sensory neurons, while Il-6 gene deletion leads to moderate changes in neurite phenotype.
Last, to uncover whether IL-6 signaling was contributing to neurite growth promoting effects of NKCC1, we analyzed IL-6 signaling under NKCC1 inhibition. Low external Cl− concentration or bumetanide were used to inhibit NKCC1 activity. Consistent with their effects on neurite growth velocity (Pieraut et al., 2007), low external Cl− concentration (12 mm instead of 150 mm) or 10 μm bumetanide significantly decreased primary neurite length from 1130 ± 119 μm (n = 26) to 605 ± 73 μm (n = 21) and 471 ± 46 μm (n = 17), respectively (Fig. 8A,C,E,F). Number of primary neurites was not significantly different from untreated axotomized neurons (3.3 ± 0.3 neurites/neuron, n = 26) and amounted 2.4 ± 0.2 neurites/neuron (n = 21) and 2.6 ± 0.3 neurites/neuron (n = 17), respectively (ANOVA). Addition of 10 μg/ml IL-6R blocking-function antibody to the low Cl− medium (Fig. 8B,E) or to bumetanide-treated cultures (Fig. 8D,F) did not lead to further decrease in neurite length (708 ± 71 μm, n = 29 and 486 ± 49 μm, n = 22, respectively) and did not modify the number of neurites (2.8 ± 0.2 neurites/neuron, n = 29 and 2.4 ± 0.2 neurites/neuron, n = 22, respectively).
Figure 8.
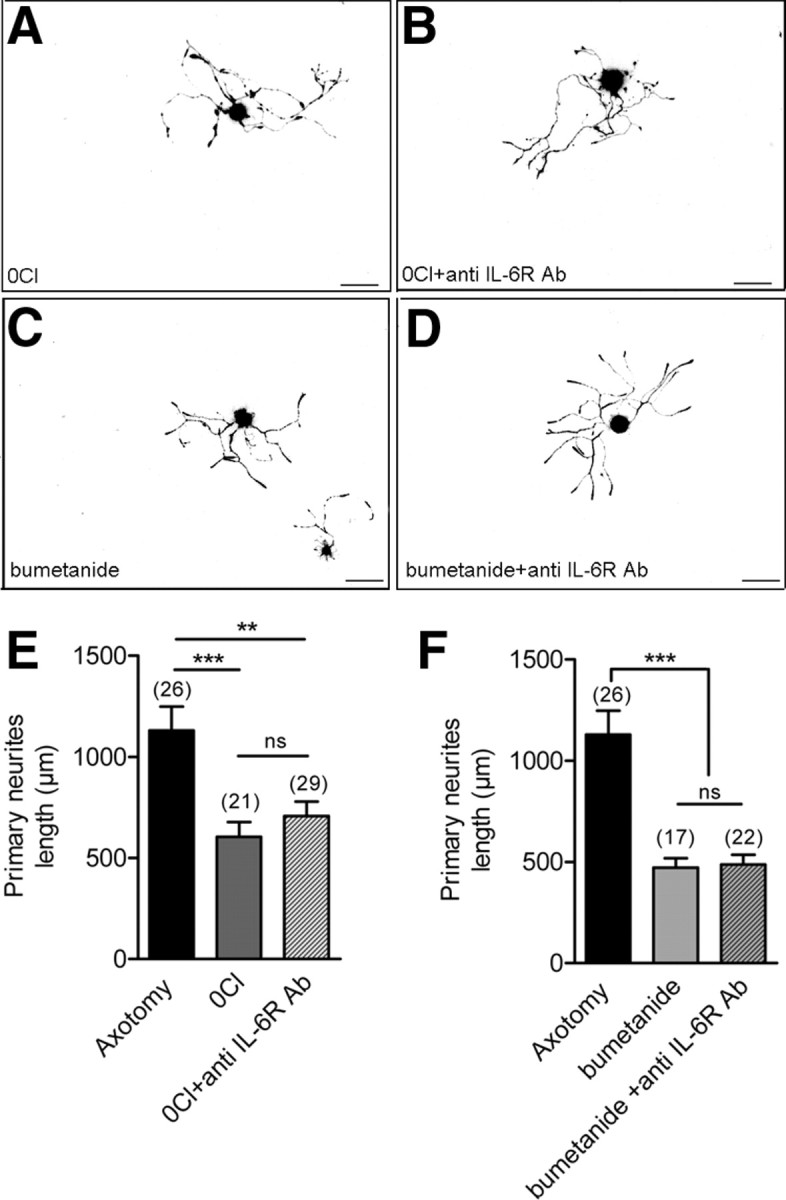
IL-6 signaling has no effects on neurite growth under NKCC1 inhibition. A–D, Photomicrographs of cultured medium- to large-sized neurons from axotomized DRG immunocytochemically stained with pan-neuronal marker β-III tubulin and NF-200 after 8 h in vitro grown in a low external Cl− concentration in the absence (A) and in the presence of 10 μg/ml anti-IL-6R blocking-function antibody (B), in control culture medium containing 10 μm bumetanide without (C), and in the presence of 10 μg/ml anti-IL-6R blocking-function antibody (D). Scale, bar 100 μm; 10× objective). E, Analysis of total length of primary neurites in medium- to large-sized axotomized neurons shows a significant decrease in a low external Cl− concentration (140 mm Na-methanesulfonate, 2 mm CaCl2, 1.5 mm MgCl2, 5 mm KCl, 10 mm glucose, and 10 mm HEPES). The addition of anti-IL-6R blocking-function antibody to the low external Cl− solution does not induce significant effects (**p < 0.01, ***p < 0.001; one-way ANOVA). F, Analysis of total length of primary neurites in medium- to large-sized axotomized neurons shows a significant decrease in axotomized neurons treated with bumetanide. The addition of anti-IL-6R blocking-function antibody to bumetanide-treated culture does not induce significant effects (***p < 0.001, one-way ANOVA).
Discussion
The present study reports that the cytokine, interleukin-6 and its nonsignaling receptor, IL-6R, are upregulated following peripheral nerve injury, in a subset of mechano- and proprioceptive DRG sensory neurons. IL-6 signaling among axotomized neurons expressing IL-6R is responsible for the increase in [Cl−]i and the expression of phospho-NKCC1 on the plasma membrane of axotomized sensory neurons. The contribution of NKCC1 to primary neurite growth in medium to large sensory neurons is mediated through IL-6 signaling.
Several functional and morphological studies demonstrated that DRG neurons do not express IL-6R under physiological conditions (Gadient and Otten, 1996; Thier et al., 1999) (Wang et al., 2009). These results are consistent with data showing that for many neuronal cells, particularly in nociception processing, IL-6 responses depend on trans-signaling due to the presence of soluble IL-6R (Jones et al., 2001; Obreja et al., 2005). The present study shows that DRGs express IL-6R mRNA and injury further increases its expression. Nerve injury induced expression of the protein at the plasma membrane of sensory neurons both in vivo and in vitro. Interestingly, we observed that under our experimental conditions, soluble IL-6R inhibited Cl− accumulation which could be attributed to competition for Il-6 binding with endogenously expressed IL-6R at plasma membrane (März et al., 1999).Consistent with a regulated expression of IL-6R, protein upregulation has also been reported in hypoglossal neurons and Schwann cells after injury (Hirota et al., 1996; Ito et al., 1998; Lara-Ramírez et al., 2008). In a model of pain induced by chronic nerve constriction, increased expression of IL-6R was localized in satellite cells surrounding sensory neurons (Dubový et al., 2010). Here, we show no colocalization between satellite cell markers and IL6-R. Moreover, in addition to studies showing that sensory neurons express IL-6 in situ and in vitro (Murphy et al., 1995; Thier et al., 1999), our in vitro data support that neurons are the main source of IL-6 secretion, confirming that nerve injury induces an autocrine neuronal IL-6 signaling.
Our data demonstrate that only a subpopulation of axotomized sensory neurons expresses IL-6R. This subpopulation belongs to the NF-200-positive DRG sensory neurons and expresses TrkB and parvalbumin, known markers of mechano- and proprioreceptors. In vitro experiments confirm IL-6R expression mainly among the medium and large somatic-diameter neurons, with, however, a somewhat higher percentage (34% versus 23%) of positively stained neurons than in in situ experiments. No IL-6R staining was observed in TrkA-positive neurons, suggesting that expression is not present in nociceptive neurons. However, correlations do exist between IL-6 signaling and pain processing, even in the absence of soluble IL-6R. It has been reported that IL-6 and NGF have synergistic effects on nociceptive plasticity (Cafferty et al., 2004; Melemedjian et al., 2010). It is therefore possible that NGF promotes expression of IL-6R in TrkA-expressing neurons, and long-term primary culture could also induce IL-6R expression in nociceptors (von Banchet et al., 2005). In these experiments, it is also possible that exogenous IL-6 exerted paracrine neuronal effects. Following axotomy, we report that small somatic-diameter neurons also increased their [Cl−]i, which is unrelated to IL-6R activation. The receptor for leukemia inhibitory factor is upregulated in a subset of nociceptive neurons following nerve injury. It is involved in nerve regeneration of small diameter sensory neurons and could be responsible for Cl− accumulation in nociceptive neurons (Cafferty et al., 2001; Gardiner et al., 2002). Cell-specific regulation of receptor expression appears to be a major event in the control cellular responses to nerve injury and repair.
We provide evidence that in DRG, the most expressed transcripts of cation-Cl− cotransporters from the Slc12a gene family code for NKCC1, KCC1, and KCC3. As previously shown, axotomy did not modify gene expression of NKCC1, and here we also shown that the expression of the four outward KCCs was unchanged. RT-qPCR in DRG does not discriminate between neuronal subpopulations. Therefore, we also performed transcriptional analysis of cotransporter expression in DRG from IL-6−/− mice and also found no significant modification. Actually, our data show that IL-6 promotes the expression of phospho-NKCC1 on the plasma membrane of DRG neurons expressing IL-6R. Unexpectedly, most small sensory neurons did not express phospho-NKCC1. This could be due to different cellular mechanisms leading to Cl− accumulation in nociceptors, such as an inhibition of the outwardly directed cation-Cl− cotransporters activity or a NKCC1 phosphorylation at different sites. The anti-phosphopeptide antibody that we used recognized two phosphoacceptor sites, Thr212 and Thr217, and available data imply that at least five phosphoacceptors are used in regulatory phosphorylation (Thr203, Thr207, Thr212, Thr217, and Thr230, in hNKCC1 and mNKCC1) (Flemmer et al., 2002). A phospho-peptide generated against three phosphorylated sites of hNKCC1 (Thr203, Thr207, and Thr212) was used to demonstrate NKCC1 phosphorylation in small nociceptive neurons exposed to inflammatory agents (Funk et al., 2008). This immediately questioned the identity of the kinases recognizing these threonines. The sequence of events leading to NKCC1 phosphorylation involves numerous kinases (Flatman, 2002). In particular the osmosensors Ste-20 related proline-alanine-rich kinase (SPAK) and the oxidative-stress response 1 kinase (OSR1) are involved (Delpire and Austin, 2010). Both of these kinases are under the control of upstream kinases such as the kinases with no lysine (WNK) (Kahle et al., 2010). In a recent study, Thr203 Thr207, and Thr212 were shown to interact with SPAK and OSR1, whereas Thr217 and Thr230 were not phosphorylated by SPAK and OSR1 in vitro (Vitari et al., 2006). As the anti-phosphopeptide antibody used in our study is generated against Thr212 and Thr217, this does not support a phosphorylation based exclusively on SPAK or OSR1 activity on NKCC1 following nerve injury. The sequence of intracellular events linking IL-6 signaling to the expression of phospho-NKCC1 is currently unknown. In addition to regulating activation or expression of specific kinase, IL-6 could also act by inhibiting the activity of phosphatase, leading to increased phosphorylation and NKCC1 activity (Darman et al., 2001; Pieraut et al., 2007).
The use of the selective Jak1-2 inhibitor provides evidence that neurite growth of conditioned axotomized neurons is under the dependence of cytokine receptors including the neuropoietic cytokines ciliary neurotrophic factor, IL-6, IL-10, IL-11, IL-22, LIF, oncostatin-M (Murray, 2007). In agreement with other studies, we show that IL-6 is one of the factors promoting neurite growth (Cafferty et al., 2004; Cao et al., 2006). In addition, we provide evidence that inhibition of IL-6R signaling specifically reduced total primary neurite length of NF-200-positive medium to large sensory neurons. Contradictory results concerning the effects of IL-6 on neurite growth of conditioned axotomized neurons have been reported using IL-6−/− mice (Cafferty et al., 2004; Cao et al., 2006). In our culture conditions, we did also get contradictory results as only mild changes in neurite length were obtained in IL-6−/− axotomized sensory neurons. This could be attributed to upregulation of overlapping functions with other neuropoietic cytokine receptors mediated by the common cytokine receptor subunit gp130. Indeed, IL-6 plays roles in neuronal survival and differentiation (März et al., 1997; Thier et al., 1999; Murphy et al., 2000) and Il6 gene deletion does not induce neuronal death (Cafferty et al., 2004). Consistent with redundancy, mice lacking individual members of IL-6 cytokine family display only mild phenotypes, as also observed in the present work, while animals lacking gp130 are not viable (Betz et al., 1998). The downstream targets of IL-6 leading to improved neurite growth are poorly known. Studies on gene expression profiling induced with exogenous application of IL-6/IL6R in embryonic DRG cells and PC12 cells point out the recruitment of many growth factors acting as autocrine or paracrine factors (Zhang et al., 2007; Kunz et al., 2009). Our results demonstrate that IL-6-induced Cl− accumulation through NKCC1 phosphorylation is a necessary downstream cellular step which promotes neurite growth in mechano- and proprioceptive neurons.
In conclusion, the present study provides new mechanistic insights into the regulation of chloride homeostasis, and establishes IL-6 signaling through neuronal IL-6R expression as a major regulator of chloride homeostasis among injured myelinated sensory neurons. Our study also opens new perspectives into the molecular partners involved in chloride regulation, especially within the nociceptive versus mechanoceptive sensory neurons.
Footnotes
This work was supported by the Institut National de la Santé et la Recherche Médicale (Inserm), the Association Française contre les Myopathies, the Fondation pour la Recherche Médicale (grant to S.M.), and the Ministère de la Recherche et la Technology (grant to O.L. and M.B.). We thank Dr. Julian Wong for critical reading of the manuscript; and the regional imaging platform RIO and the experimental histology platform RHEM for technical assistance.
References
- Andratsch M, Mair N, Constantin CE, Scherbakov N, Benetti C, Quarta S, Vogl C, Sailer CA, Uceyler N, Brockhaus J, Martini R, Sommer C, Zeilhofer HU, Müller W, Kuner R, Davis JB, Rose-John S, Kress M. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci. 2009;29:13473–13483. doi: 10.1523/JNEUROSCI.1822-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André S, Boukhaddaoui H, Campo B, Al-Jumaily M, Mayeux V, Greuet D, Valmier J, Scamps F. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. J Neurophysiol. 2003;90:3764–3773. doi: 10.1152/jn.00449.2003. [DOI] [PubMed] [Google Scholar]
- Betz UA, Bloch W, van den Broek M, Yoshida K, Taga T, Kishimoto T, Addicks K, Rajewsky K, Müller W. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J Exp Med. 1998;188:1955–1965. doi: 10.1084/jem.188.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bourane S, Garces A, Venteo S, Pattyn A, Hubert T, Fichard A, Puech S, Boukhaddaoui H, Baudet C, Takahashi S, Valmier J, Carroll P. Low-threshold mechanoreceptor subtypes selectively express MafA and are specified by Ret signaling. Neuron. 2009;64:857–870. doi: 10.1016/j.neuron.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J, Thompson SW. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci. 2001;21:7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Das P, Qiu J, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J Neurosci. 2004;24:4432–4443. doi: 10.1523/JNEUROSCI.2245-02.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Gao Y, Bryson JB, Hou J, Chaudhry N, Siddiq M, Martinez J, Spencer T, Carmel J, Hart RB, Filbin MT. The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J Neurosci. 2006;26:5565–5573. doi: 10.1523/JNEUROSCI.0815-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervero F, Laird JM, García-Nicas E. Secondary hyperalgesia and presynaptic inhibition: an update. Eur J Pain. 2003;7:345–351. doi: 10.1016/s1090-3801(03)00047-8. [DOI] [PubMed] [Google Scholar]
- Darman RB, Flemmer A, Forbush B. Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J Biol Chem. 2001;276:34359–34362. doi: 10.1074/jbc.C100368200. [DOI] [PubMed] [Google Scholar]
- Delpire E, Austin TM. Kinase regulation of Na+-K+-2Cl− cotransport in primary afferent neurons. J Physiol. 2010;588:3365–3373. doi: 10.1113/jphysiol.2010.190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouad F, Charbonnier LM, Bouffi C, Louis-Plence P, Bony C, Apparailly F, Cantos C, Jorgensen C, Noël D. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. 2007;25:2025–2032. doi: 10.1634/stemcells.2006-0548. [DOI] [PubMed] [Google Scholar]
- Dubový P, Klusáková I, Svízenská I, Brázda V. Satellite glial cells express IL-6 and corresponding signal-transducing receptors in the dorsal root ganglia of rat neuropathic pain model. Neuron Glia Biol. 2010;6:73–83. doi: 10.1017/S1740925X10000074. [DOI] [PubMed] [Google Scholar]
- Flatman PW. Regulation of Na-K-2Cl cotransport by phosphorylation and protein-protein interactions. Biochim Biophys Acta. 2002;1566:140–151. doi: 10.1016/s0005-2736(02)00586-2. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Funk K, Woitecki A, Franjic-Würtz C, Gensch T, Möhrlen F, Frings S. Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain. 2008;4:32. doi: 10.1186/1744-8069-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadient RA, Otten U. Postnatal expression of interleukin-6 (IL-6) and IL-6 receptor (IL-6R) mRNAs in rat sympathetic and sensory ganglia. Brain Res. 1996;724:41–46. doi: 10.1016/0006-8993(96)00264-8. [DOI] [PubMed] [Google Scholar]
- Galan A, Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 cotransporter. Neuroscience. 2005;133:245–252. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Gardiner NJ, Cafferty WB, Slack SE, Thompson SW. Expression of gp130 and leukaemia inhibitory factor receptor subunits in adult rat sensory neurones: regulation by nerve injury. J Neurochem. 2002;83:100–109. doi: 10.1046/j.1471-4159.2002.01101.x. [DOI] [PubMed] [Google Scholar]
- Geng Y, Hoke A, Delpire E. The Ste20 kinases Ste20-related proline-alanine-rich kinase and oxidative-stress response 1 regulate NKCC1 function in sensory neurons. J Biol Chem. 2009;284:14020–14028. doi: 10.1074/jbc.M900142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Kiyama H, Kishimoto T, Taga T. Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med. 1996;183:2627–2634. doi: 10.1084/jem.183.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Yamamoto M, Li M, Doyu M, Tanaka F, Mutch T, Mitsuma T, Sobue G. Differential temporal expression of mRNAs for ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), interleukin-6 (IL-6), and their receptors (CNTFR alpha, LIFR beta, IL-6R alpha and gp130) in injured peripheral nerves. Brain Res. 1998;793:321–327. doi: 10.1016/s0006-8993(98)00242-x. [DOI] [PubMed] [Google Scholar]
- Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 2001;15:43–58. doi: 10.1096/fj.99-1003rev. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Köhler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- Kunz D, Walker G, Bedoucha M, Certa U, März-Weiss P, Dimitriades-Schmutz B, Otten U. Expression profiling and Ingenuity biological function analyses of interleukin-6- versus nerve growth factor-stimulated PC12 cells. BMC Genomics. 2009;10:90. doi: 10.1186/1471-2164-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Ramírez R, Segura-Anaya E, Martínez-Gómez A, Dent MA. Expression of interleukin-6 receptor alpha in normal and injured rat sciatic nerve. Neuroscience. 2008;152:601–608. doi: 10.1016/j.neuroscience.2008.01.014. [DOI] [PubMed] [Google Scholar]
- März P, Herget T, Lang E, Otten U, Rose-John S. Activation of gp130 by IL-6/soluble IL-6 receptor induces neuronal differentiation. Eur J Neurosci. 1997;9:2765–2773. doi: 10.1111/j.1460-9568.1997.tb01705.x. [DOI] [PubMed] [Google Scholar]
- März P, Otten U, Rose-John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. Eur J Neurosci. 1999;11:2995–3004. doi: 10.1046/j.1460-9568.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30:15113–15123. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Aza BM, Chillingworth NL, Payne JA, Donaldson LF. Inflammation alters cation chloride cotransporter expression in sensory neurons. Neurobiol Dis. 2004;17:62–69. doi: 10.1016/j.nbd.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Murphy PG, Grondin J, Altares M, Richardson PM. Induction of interleukin-6 in axotomized sensory neurons. J Neurosci. 1995;15:5130–5138. doi: 10.1523/JNEUROSCI.15-07-05130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM. Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur J Neurosci. 2000;12:1891–1899. doi: 10.1046/j.1460-9568.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Miyazaki H, Niisato N, Marunaka Y. Essential role of NKCC1 in NGF-induced neurite outgrowth. Biochem Biophys Res Commun. 2007;359:604–610. doi: 10.1016/j.bbrc.2007.05.133. [DOI] [PubMed] [Google Scholar]
- Obreja O, Biasio W, Andratsch M, Lips KS, Rathee PK, Ludwig A, Rose-John S, Kress M. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128:1634–1641. doi: 10.1093/brain/awh490. [DOI] [PubMed] [Google Scholar]
- Pieraut S, Laurent-Matha V, Sar C, Hubert T, Méchaly I, Hilaire C, Mersel M, Delpire E, Valmier J, Scamps F. NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J Neurosci. 2007;27:6751–6759. doi: 10.1523/JNEUROSCI.1337-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–170. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, Rodgers JD, Haley P, Kantarjian H, Fridman JS, Verstovsek S. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita T, Totsuka T, Saito M, Yamasaki K, Taga T, Hirano T, Kishimoto T. Functional murine interleukin 6 receptor with the intracisternal A particle gene product at its cytoplasmic domain. Its possible role in plasmacytomagenesis. J Exp Med. 1990;171:2001–2009. doi: 10.1084/jem.171.6.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thier M, März P, Otten U, Weis J, Rose-John S. Interleukin-6 (IL-6) and its soluble receptor support survival of sensory neurons. J Neurosci Res. 1999;55:411–422. doi: 10.1002/(SICI)1097-4547(19990215)55:4<411::AID-JNR2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397:223–231. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Banchet GS, Kiehl M, Schaible HG. Acute and long-term effects of IL-6 on cultured dorsal root ganglion neurones from adult rat. J Neurochem. 2005;94:238–248. doi: 10.1111/j.1471-4159.2005.03185.x. [DOI] [PubMed] [Google Scholar]
- Wang L, Lee HK, Seo IA, Shin YK, Lee KY, Park HT. Cell type-specific STAT3 activation by gp130-related cytokines in the peripheral nerves. Neuroreport. 2009;20:663–668. doi: 10.1097/WNR.0b013e32832a09f8. [DOI] [PubMed] [Google Scholar]
- Zhang PL, Levy AM, Ben-Simchon L, Haggiag S, Chebath J, Revel M. Induction of neuronal and myelin-related gene expression by IL-6-receptor/IL-6: a study on embryonic dorsal root ganglia cells and isolated Schwann cells. Exp Neurol. 2007;208:285–296. doi: 10.1016/j.expneurol.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Zhong J, Dietzel ID, Wahle P, Kopf M, Heumann R. Sensory impairments and delayed regeneration of sensory axons in interleukin-6-deficient mice. J Neurosci. 1999;19:4305–4313. doi: 10.1523/JNEUROSCI.19-11-04305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]