

Abstract
Mononuclear phagocytes are important modulators of Alzheimer's disease (AD), but the specific functions of resident microglia, bone marrow-derived mononuclear cells, and perivascular macrophages have not been resolved. To elucidate the spatiotemporal roles of mononuclear phagocytes during disease, we targeted myeloid cell subsets from different compartments and examined disease pathogenesis in three different mouse models of AD (APPswe/PS1, APPswe, and APP23 mice). We identified chemokine receptor 2 (CCR2)-expressing myeloid cells as the population that was preferentially recruited to β-amyloid (Aβ) deposits. Unexpectedly, AD brains with dysfunctional microglia and devoid of parenchymal bone marrow-derived phagocytes did not show overt changes in plaque pathology and Aβ load. In contrast, restriction of CCR2 deficiency to perivascular myeloid cells drastically impaired β-amyloid clearance and amplified vascular Aβ deposition, while parenchymal plaque deposition remained unaffected. Together, our data advocate selective functions of CCR2-expressing myeloid subsets, which could be targeted specifically to modify disease burden in AD.
Introduction
A local activation of microglial cells is consistently detected in the brains of patients with Alzheimer's disease (AD), but the role of activated peripheral myeloid cells in the pathogenesis of AD has not been resolved (Akiyama et al., 2000; Prinz and Mildner, 2011). For example, microglial cells have been suggested to be responsible for pathological β-amyloid (Aβ) protein deposition (Wisniewski et al., 1989; Akiyama et al., 2000; Frackowiak et al., 2005), but at the same time, they have also been implicated in Aβ clearance (Chung et al., 1999; Bard et al., 2000; Wyss-Coray et al., 2001; Jantzen et al., 2002). Moreover, it has been shown recently that the interaction of CD40 with CD40 ligand on the surface of microglia is an important molecular signal for microglia activation in AD mouse models (Tan et al., 1999, 2002; Gate et al., 2010).
Recent work using GFP-labeled bone marrow (BM) chimeras and total-body irradiation increased the longstanding controversy over the issue of whether endogenous microglia or BM-derived phagocytes are beneficial or detrimental in neurodegeneration. One study concluded that BM-derived phagocytes are critical for restricting Aβ plaque formation (Simard et al., 2006). Other groups found that mononuclear phagocytes represent the minority of blood-borne cells, which engraft in the brains of APP23 transgenic mice (Stalder et al., 2005). A possible involvement of microglia in the pathogenesis of AD was suggested by a recent study showing that chemokine receptor 2 (CCR2) deficiency diminished the migratory potential of microglia and reduced the clearance of Aβ in transgenic APPswe mice (El Khoury et al., 2007). Moreover, stimulation of perivascular macrophage turnover reduced cerebral amyloid angiopathy (CAA) load independent of clearance by microglia (Hawkes and McLaurin, 2009). Blockade of innate immune responses by interruption of TGF-β-Smad 2/3 signaling in myeloid cells mitigated AD pathology and attenuated parenchymal and cerebrovascular Aβ deposits (Town et al., 2008).
One major caveat of all reports using BM chimeras, however, is the usage of total-body irradiation (including the brain) before BM transplantation to discriminate donor hematopoietic cells from resident microglia in the hosts (Malm et al., 2005; Stalder et al., 2005; Simard et al., 2006; Grathwohl et al., 2009), which potentially changes the brain microenvironment (Ajami et al., 2007; Mildner et al., 2007).
Furthermore, the direct circulating phagocyte or its progenitor immigrating into the diseased AD brain has not been identified yet. Ly-6Chi monocytes have been shown to be recruited into the inflamed CNS during autoimmune demyelination (Mildner et al., 2009), and bacterial (Mildner et al., 2008) and viral infection (Getts et al., 2008) with different roles for disease pathogenesis. Whether these monocyte subsets are involved in neurodegeneration of the CNS is not yet known. We therefore used a combinational approach of bone marrow transplantation and partial-body irradiation (head shielded, protected CNS) versus whole-body irradiation (unprotected CNS) to examine the engraftment of myeloid subsets and to circumvent the caveats of brain irradiation in mouse models of AD.
Materials and Methods
Mice and generation of BM chimeric mice.
Bone marrow chimeric mice were generated as described recently (Mildner et al., 2007). In brief, recipient mice were reconstituted with bone marrow cells (BMCs) derived from tibias and femurs from adult β-actin (ACTβ)-EGFP mice alone (CCR2+/+GFP+) or from double-mutant animals intercrossed with CCR2 ko mice (CCR2−/−GFP+). Specific body (protected) irradiation of mice was performed by a 6 MV X-ray Varian linear accelerator. Mice were put into a 20 × 20 × 3 cm cast Perspex-lined chamber (5 mm gauge, density 1.18 g/cm3) subdivided into eight single compartments and equipped with a removable cap. Spiracles were provided, and the heads of the mice were carefully adjusted to them. Mice were treated with parallel opposed fields with a maximum dose at a depth of 1.5 cm. The field size was adjusted to 14 × 20 cm, and therefore, the brains of the animals were outside the irradiation fields, and only the body was irradiated. A safety margin of 0.5 cm from the field border to the head was preserved, so that no dose could affect normal brain tissue. Dosimetry data were measured in a water phantom and corrected for the presence of tissue inhomogeneities and surrounding air. A total of 5 × 106 CCR2−/−GFP+ or CCR2+/+GFP+ BMCs were injected into the tail vein of recipients 24 h after irradiation. Since the skull bone marrow contributes to hematopoiesis, the reconstitution levels of chimeric mice were ∼60%, resulting in a total of 30–40% GFP+ cells. We therefore adapted the reconstitution level of total-irradiated (unprotected) mice by mixing CCR2−/−GFP+ or CCR2+/+GFP+ BMCs in a 1:3 ratio with CCR2+/+ BMCs, which resulted in similar reconstitution levels in all chimeric mice. Only protected and unprotected mice with a similar grade of myeloid chimerism were chosen for subsequent comparative analysis. All mice received an irradiation dosage of 1100 cGy and were injected with 5 × 106 BMCs into the tail vein, 24 h after the irradiation. APPswe/PS1 mice [Tg(APP695)3DboTg(PSEN1)5Dbo/J] were obtained from The Jackson Laboratory. APP23 mice were a kind gift from Matthias Staufenbiel, Novartis Institutes for BioMedical Research. Tg2576 APPswe mice were from Taconic. Mice were bred in-house under pathogen-free conditions. Females were used in all experiments.
Six to eight weeks after grafting, reconstitution was assessed by FACS analysis of peripheral blood. Blood samples were prepared at 4°C in buffer solution (PBS containing 2% FCS and 5 mm EDTA) and stained with CD11b, Ly-6C (eBioscience, BD PharMingen) as described previously (Mildner et al., 2009). For comparison of macrophages, adult microglia and Ly-6Chi monocytes of CCR2+/+, CCR2−/−, CCR2−/− GFP+, and CCR2+/+ GFP+ animals, cells were stained for α4-integrin, β1-integrin, PSGL-1 (P-selectin glycoprotein ligand-1), and CD11c (eBioscience, BD PharMingen). After lysis of erythrocytes with FACS lysis solution (Becton Dickinson) and washing, cell suspensions were analyzed on a FACS Calibur (Becton Dickinson). Data were acquired with WinMDI and FlowJo.
Quantification of phagocyte engraftment in the CNS.
After transcardial perfusion with PBS and subsequent 4% paraformaldehyde, 20 μm cryosections were obtained from the brains. Sections were incubated in PBS containing 5% fetal calf serum. Primary and preabsorbed antibodies were added overnight at a dilution of 1:100 for Iba-1 (Wako). Amyloid was stained with anti-human amyloid-β antibody (Signet antibodies; Covance), the antibody was added overnight at a dilution of 1:100. Cy3-conjugated secondary antibodies (Dianova) were added at a dilution of 1:100 for Iba-1 and 1:600 for other antibodies for 1 h. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI). GFP-expressing ramified cells and Iba-1+ microglia were counted in at least three sections each of individual animal. Quantification was performed independently by two scientists in a blinded manner. The results were compared with stereological analysis using a Leica DMRB/DIC fluorescence research microscope (100× magnification) and quantified using the optical fractionator method (Stereo Investigator system, MicroBrightField Bioscience). The number of engrafted cells was examined microscopically at a 200× microscopic magnification using a conventional fluorescence microscope (Olympus BX-61) equipped with a color camera (Olympus DP71).
Laser microdissection.
Microdissection of microglia was performed using a Zeiss PALM MicroBeam as described previously, with modification (Raasch et al., 2011). Fast immunochemistry of serial sections was performed with CD11b antibodies (Serotec). Immunostained sections were counterstained with DAPI to facilitate the identification of individual cells. RNA was isolated with the RNeasy Micro Plus Kit (Qiagen), and reverse transcription (RT), preamplification, and real-time PCR were performed using Applied Biosystems reagents according to the manufacturer's recommendations.
Real-time PCR.
RNA was extracted from brains at indicated time points after or without irradiation. The tissue was flushed with ice-cold HBSS and RNA was isolated using RNAeasy Mini kits (Qiagen) following the manufacturer's instructions. The samples were treated with DNase I (Roche), and 1 μg of RNA was transcribed into cDNA using oligo-dT primers and the SuperScript II RT kit (Invitrogen). cDNA (2.5 μl) was transferred into a 96-well Multiply PCR plate (Sarstedt), and 12.5 μl of ABsolute QPCR SYBR Green Master Mix (ABgene) plus 9.6 μl of ddH2O were added. The PCR was performed as described recently (Prinz et al., 2006). The following primer probe pairs were used: CCL3 (sense, TGCCCACGTCAAGGAGTATTT; antisense, TCTCTGGGTTGGCACACACTT), CXCL10 (sense, TGCTGGGTCTGAGTGGGACT; antisense, CCCTATGGCCCTCATTCTCAC), TNFα (sense, CATCTTCTCAAAATTCGAGTGACAA; antisense, TGGGAGTAGACAAGGTACAACCC), CCL2 (sense, TCTGGGCCTGCTGTTCACC; antisense, TTGGGATCATCTTGCTGGTG).
Aβ quantifications.
Quantification of Aβ was performed using human amyloid β1–40 and β1–42 ELISA kits (The Genetics Company) according to the manufacturer's protocol. Forebrains of mice were homogenized in PBS containing 1 mm EDTA and EGTA and protease inhibitor mixture, further extracted in RIPA buffer (containing, in mm: 25 Tris-HCl, pH 7.5, 150 NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS), and centrifuged at 20,000 × g for 30 min, and the pellet was solubilized in 2% SDS, 25 mm Tris-HCl, pH 7.5. Samples were separated by NuPAGE and immunoblotted using antibodies against APP, β-C-terminal fragment (β-CTF), and Aβ, and antibody E7 (Developmental Studies Hybridoma Bank), followed by incubation with appropriate secondary antibodies. Immunoreactivity was detected by enhanced chemiluminescence reaction (Millipore).
Statistical analysis.
Statistical differences of clinical scores were evaluated using a nonpaired Student‘s t test. Differences were considered significant when p < 0.05.
Results
Engraftment of BM-derived phagocytes in the brains of AD transgenic mice depends on CCR2
We generated double transgenic mice by intercrossing ACTβ-EGFP mice with CCR2−/− animals and obtained individuals that express GFP on CCR2+/+ (CCR2+/+GFP) or CCR2−/− (CCR2−/−GFP) background, respectively. To distinguish invading BM-derived mononuclear cells from brain endogenous microglia in transgenic mouse models of AD, we generated BM chimeras. Respective GFP-marked BM cells were transplanted into lethally total-body irradiated (including the brain) recipient APPswe/PS1 transgenic mice. CCR2−/− mice have a strong and specific reduction of the Ly-6Chi subpopulation of monocytes (Serbina and Pamer, 2006; Mildner et al., 2007). As expected, peripheral blood Ly-6Chi monocytes were strongly diminished in CCR2−/−GFP → APPswe/PS1 chimeric mice compared with CCR2+/+GFP → APPswe/PS1 mice 7 months after BM transplantation (Fig. 1A,B). Notably, the general reconstitution efficacy was similar in both chimeric groups, as indicated by comparable numbers of Ly-6CloGFP+ cells in the circulation.
Figure 1.
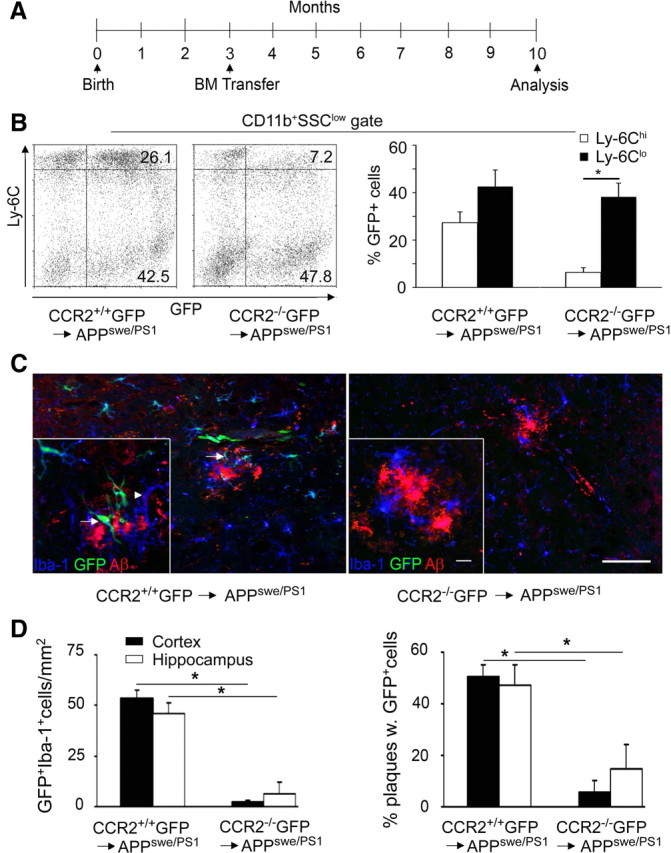
Engraftment of BM-derived phagocytes in the brains of AD transgenic mice depends on CCR2. A, Time scale of BM transfer experiments performed in APPswe/PS1 animals. Arrows mark the time points of generation and subsequent analysis of CCR2+/+GFP → APPswe/PS1 and CCR2−/−GFP → APPswe/PS1 chimeric mice. B, FACS analysis of peripheral blood 10 months after BM cell transfer reveals a lack of GFP+Ly-6Chi monocytes in CCR2−/−GFP → APPswe/PS1 chimeras compared with CCR2+/+ GFP donors. Percentages of the respective cell populations are indicated. Representative dot blots for the chimeric groups (left) and quantification of GFP+CD11b+Ly-6Chi and GFP+CD11b+Ly-6Clo cells (right) are shown. Data are expressed as means ± SEM. SSC, Side scatter. At least 5 mice per group were assessed. *p < 0.05 statistical significance. C, Immunohistochemistry of APPswe/PS1 BM chimeric brains. Fluorescence microscopy reveals a significant number of GFP-positive ramified cells in the hippocampus of CCR2+/+GFP → APPswe/PS1 mice (left), but very few in CCR2−/−GFP → APPswe/PS1 mice (right). Iba-1 immunoreactivity (blue) for phagocytes shows that some branched cells are GFP+ and therefore of donor origin (GFP, green, arrows), whereas others represent endogenous microglia expressing only Iba-1 (arrowheads). Immunostaining of β-amyloid is shown in red. Scale bars: 100 μm (overviews) and 25 μm (insets). D, Semiquantitative analysis of phagocyte engraftment (GFP+Iba-1+ cells) per area (left) and per β-amyloid plaque (right) in the hippocampus and cortex of BM chimeric animals. Bars show the means ± SEM from at least three sections per individual animal (n ≥ 5 per group). *p < 0.05 statistical significance.
To assess the ability of GFP-marked myeloid subsets derived from transplanted BM to differentiate into tissue macrophages in the AD brain, we histologically analyzed recipient mice 7 months after BM transplantation (Fig. 1C). Microscopic investigation of the hippocampus and the cortex revealed numerous parenchymal cells with ramified morphology. These donor-derived cells were positive for the microglia/macrophage marker, Iba-1, and partially decorated Aβ plaques. Notably, GFP+Iba-1+ cells were predominantly found in the CNS of CCR2+/+GFP → APPswe/PS1 mice, whereas the number of engrafted myeloid cells in CCR2−/−GFP → APPswe/PS1 chimeras was greatly reduced in the hippocampus (6.2 ± 5.8 cells/mm2 in CCR2−/−GFP → APPswe/PS1 compared with 45.8 ± 4.6 cells/mm2 in CCR2+/+GFP → APPswe/PS1) and in the cortex (2.3 ± 0.8 cells/mm2 in CCR2−/−GFP → APPswe/PS1 compared with 53.2 ± 4.0 cells/mm2 in CCR2+/+GFP → APPswe/PS1, Fig. 1D). Interestingly, only ∼50% of hippocampal and cortical Aβ plaques were surrounded by engrafted GFP+Iba-1+ cells. As expected, significantly fewer Aβ deposits were decorated by immigrated GFP+ cells in CCR2−/− GFP → APPswe/PS1 mice (Fig. 1D, right).
In conclusion, our data suggest that CCR2-expressing BM-derived cells are the main source of donor-derived Iba-1+ phagocytes in several brain regions with AD pathology after total-body irradiation and BM transplantation.
Irradiation conditions the brain for mononuclear phagocyte engraftment in AD transgenic mice
The possibility that total-body irradiation conditions the brain for phagocyte engraftment from the circulation has not been addressed in earlier studies on neurodegeneration in AD. We therefore adapted previous protocols using linear acceleration, which allow for selective irradiation that includes or excludes the brain before BM transplantation. These BM chimeras are subsequently referred to as “protected” mice, in contrast to the “unprotected” ones. As expected, the number of circulating donor-derived CD11b+ Ly-6ChiGFP+ and CD11b+Ly-6CloGFP+ monocytes was slightly decreased in protected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 protected) compared with the unprotected situation (APPswe/PS1 unprotected), because skull hematopoiesis was excluded from irradiation (Fig. 2A).
Figure 2.

CNS conditioning enhances the recruitment of BM-derived mononuclear phagocytes into the brains of AD transgenic mice. A, Assessment of blood chimerism in brain-protected and unprotected CCR2+/+GFP → APPswe/PS1 chimeras. Representative dot plots from individual mice are shown on the left, and the percentages of GFP+CD11b+Ly-6Clo and GFP+CD11b+Ly-6Chi cells are indicated. SSC, Side scatter. Quantification of Ly-6Clo- and Ly-6Chi-expressing GFP-positive cells in the peripheral blood of unprotected and protected CCR2+/+GFP → APPswe/PS1 BM chimeras is shown on the right. Data are means ± SEM. At least 5 mice per group were examined. B, Phagocyte engraftment from the hematopoietic compartment requires irradiation of the brains of AD transgenic mice. Ramified GFP- and Iba-1-expressing cells were examined 7 months after BMC transfer. Immunohistochemistry for Iba-1 (blue, left; red, right), Aβ deposits (red), GFP fluorescence (green), and DAPI staining of nuclei (blue) in cortical and hippocampal sections from unprotected and protected CCR2+/+GFP → APPswe/PS1 BM chimeras reveals that BM-derived (GFP+) Iba-1+ mononuclear phagocytes (arrows) are exclusively found in unprotected AD brains, whereas nonirradiated (protected) brains are devoid of parenchymal ramified GFP+ cells. Endogenous host microglia are Iba-1+GFP− (arrowheads). Scale bars, 30 μm. C–E, Semiquantitative analysis of regional myeloid cell engraftment (GFP+Iba-1+ cells) in the brain (cortex and hippocampus) (C) and spinal cord (D) of total-body irradiated (unprotected) and brain-shielded (protected) CCR2+/+GFP → APPswe/PS1 BM chimeras. Engraftment of BM-derived mononuclear cells strictly depends on irradiation of the nervous tissue in AD transgenic mice. The percentage of plaques surrounded by GFP+ mononuclear cells in the conditioned brains is shown in E. Data are means ± SEM from at least three sections per animal and at least 5 mice per group. n.d., Not detectable.
Importantly, histopathological analysis of the brains of APPswe/PS1 mice 7 months after BM transplantation revealed phagocyte engraftment only in regions of the brain that were conditioned by irradiation (Fig. 2B). In fact, amyloid plaques in irradiated cortex and hippocampus were partially surrounded by donor-derived GFP+Iba-1+ cells, whose cell bodies were distal from the edges of the deposits and whose cellular processes were directed toward the center of the plaques without apparently reaching the core of the deposits. Subsequent quantitative measurements revealed a comparable number of GFP-positive cells in hippocampal and cortical brain regions of unprotected CCR2+/+GFP → APPswe/PS1 mice (13.9 ± 4.3 cells/mm2 in the hippocampus and 12.7 ± 2.5 cells/mm2 in the cortex), whereas protected CCR2+/+GFP → APPswe/PS1 mice were completely devoid of any engrafted cells in the brain parenchyma and around amyloid plaques (Fig. 2C). However, engraftment of GFP-labeled cells in protected animals occurred in the nonshielded spinal cord of these animals, underscoring that conditioning by irradiation is a prerequisite for hematopoietic cell entry into the CNS parenchyma (Fig. 2D). To examine whether the turnover of perivascular macrophages (PVMs) also depends on irradiation, we examined the presence of GFP+ cells around blood vessels in the brains of AD transgenic mice. As expected from our previous work (Bechmann et al., 2001), donor-derived cells with elongated cell bodies and bipolar shape were detected around blood vessels in both protected and unprotected BM chimeras (data not shown). Quantitative examination revealed robust engraftment of donor-derived PVMs in the perivascular spaces that was not significantly decreased in the protected individuals (7.1 ± 1.9 cells/section compared with 8.9 ± 2.5 cells/section in unprotected samples). To assess the capacity of transferred BM cells to migrate to amyloid plaques, we determined the percentage of Aβ deposits surrounded by GFP+ cells in unprotected CCR2+/+GFP → APPswe/PS1 mice. Again, only a minority of amyloid plaques in the hippocampus were clearly associated with donor-derived cells, whereas most GFP+ cells were not located in proximity to Aβ deposits (Fig. 2E).
In summary, our data provide strong evidence that the engraftment of myeloid cells in the brain parenchyma of AD transgenic mice does not occur normally during disease progression, but requires prior CNS conditioning to sufficiently attract BM cells. Furthermore, engrafted phagocytes were not primarily attracted by amyloid plaques in the brain parenchyma. In contrast, turnover of PVMs around vessels did not depend on irradiation.
Brain conditioning alters the CNS milieu and mitigates Aβ deposition in AD transgenic mice
Since irradiation of the CNS induces a tremendous and rapid gene regulation (Linard et al., 2004; François et al., 2006), we examined the structural and molecular consequences of irradiation on the brains of APPswe/PS1 mice. Thorough histopathological examination of brain sections from protected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 protected) revealed a dense network of Iba-1-immunoreactive cells, which characterizes endogenous microglia under these experimental conditions (Fig. 3A). Microglia were densely packed and localized mainly around Aβ plaques. In sharp contrast to this situation, irradiation dramatically changed the morphology of Iba-1+ cellular networks and the association of Iba-1+ cells with amyloid plaques in the brains of unprotected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 unprotected) (Fig. 3A). BM-derived phagocytes expressing GFP and Iba-1 were now present in the brain, but endogenous GFP−Iba-1+ microglia appeared more separated and disintegrated from the local glial environment. In fact, quantitative measurements of Iba-1+ cells associated with amyloid plaques revealed a significant reduction in plaque-associated microglia/macrophages at 7 months after irradiation (Fig. 3B).
Figure 3.
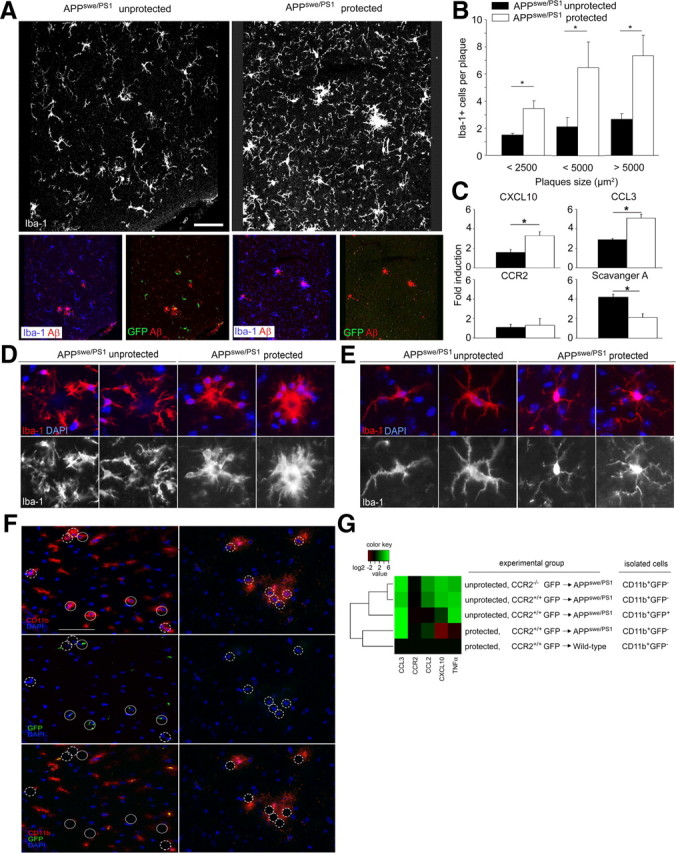
Irradiation changes the network of microglia, alters their morphology, and shapes the local inflammatory milieu. A, Iba-1 immunoreactivity (white) reveals dramatic changes of the microglia network upon irradiation in unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras compared with protected chimeras (top row). Only unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras (APPswe/PS1 unprotected) contain GFP+ (green) Iba-1-immunoreactive (blue) phagocytes, which partially surround Aβ plaques (red) (bottom row). B, Quantification of Iba-1+ cells per β-amyloid plaque indicates a significant reduction of plaque-associated Iba-1+ microglia/macrophages in unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras (APPswe/PS1 unprotected, black bars) compared with protected chimeras (APPswe/PS1 protected, white bars) independent of the plaque size. Data are means ± SEM from at least three sections per animal and at least 5 mice per group. *p < 0.05 statistical significance. C, Quantitative real-time PCR analysis of CXCL10, CCL3, CCR2, and scavenger A mRNA expression in the brains of unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras (black bars) compared with protected chimeras (white bars). Data are means ± SEM from at least 5 mice animals per group. *p < 0.05 statistical significance. D, Morphology of microglia/macrophages surrounding β-amyloid plaques in the brain. In unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras (APPswe/PS1 unprotected), Iba-1-immunoreactive microglia/macrophages (red) are dystrophic and dissociated from the plaque (left). In contrast, Iba-1+ microglia/macrophages in protected CCR2+/+GFP → APPswe/PS1 BM chimeras (APPswe/PS1 protected) cluster in and around the plaque and extend cellular protrusions into the core of the plaque (right). Nuclear DAPI staining in blue. E, Morphology of Iba-1-immunoreactive microglia/macrophages in the brain at sites distant from β-amyloid deposits. In unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras (left), Iba-1-immunoreactive microglia/macrophages (red) have enlarged cell bodies with short and spiny processes compared with the protected condition (right). Nuclear DAPI staining in blue. F, Laser microdissection of CD11b+GFP− endogenous microglia and CD11b+GFP+ engrafted macrophages from the hippocampus of unprotected and protected CCR2+/+GFP → APPswe/PS1 BM chimeras. Nuclear staining with DAPI is shown in blue, CD11b immunoreactivity in red (top), and GFP expression in green (middle). The bottom shows the overlay and indicates the results of microdissection. Closed circles indicate dissected endogenous microglia (CD11b+GFP−); dashed circles indicate BM-derived macrophages (CD11b+GFP+). Scale bar, 50 μm. G, Quantification of cytokine and chemokine mRNA expression in microdissected CD11b+GFP− and CD11b+GFP+ microglia/macrophages under different experimental conditions. Data are presented as a heat map with a log2 scale (brown, downregulated; green, upregulated). Rows indicate experimental groups; columns represent particular genes. Each data point reflects the median expression value of a particular gene resulting from three to four individual mice, normalized to the mean expression value of the respective gene in CD11b+GFP− cells from protected wild-type recipients.
We next analyzed the whole brain for expression of chemoattractant factors known to be involved in the recruitment of myeloid cells, and the phagocyte scavenger A as the key microglial factor required for the clearance of insoluble Aβ (Fig. 3C). Notably, we observed significantly less CXCL10 and CCL3 mRNAs in the brains of unprotected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 unprotected) compared with the protected chimeras. Interestingly, CCR2 mRNA expression was not altered, while gene expression of scavenger A was significantly higher in the brains of unprotected CCR2+/+ GFP → APPswe/PS1 (APPswe/PS1 unprotected) mice. These data clearly suggest robust long-term changes of the brain milieu, even several months after irradiation and BM transplantation.
Closer examination of microglia morphology by confocal microscopy uncovered striking changes of cellular organization (Fig. 3D). In protected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 protected), Iba-1+ parenchymal cells showed typical microglial morphology with round to spindle-shaped somata and a distinct arborization pattern. Cells were located at the border of the amyloid plaques, and their processes reached into the core of the plaques. In contrast, microglia in the brains of unprotected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 unprotected) were more dissociated from the Aβ deposits, with hardly any cellular processes reaching the center of the plaques. Similarly, parenchymal microglia that were not associated with amyloid plaques showed morphological signs of activation, namely retraction of cellular processes and rounding of the somata (Fig. 3E). Furthermore, activated microglia in unprotected CCR2+/+GFP → APPswe/PS1 mice (APPswe/PS1 unprotected) were covered by numerous spiny protrusions on the surface that could not be found in their counterparts in protected BM chimeras.
It has furthermore been proposed that BM-derived phagocytes are functionally different from endogenous microglia, e.g., by their increased immune gene expression (Simard et al., 2006). To determine whether endogenous microglia or BM-derived phagocytes are the cellular sources of potentially detrimental mediators in AD and whether the individual gene profiles might be influenced by irradiation, we generated BM chimeras and performed single-cell microdissections on brain sections in situ (Fig. 3F). Immunostained serial sections were used to dissect CD11b+GFP− endogenous microglia and donor-derived CD11b+GFP+ phagocytes from the brains of protected and unprotected CCR2+/+GFP → APPswe/PS1 BM chimeras by a pulsed UV laser beam. Sections were counterstained with DAPI to facilitate the identification of individual cells. Cytokine mRNA expression was subsequently analyzed by quantitative RT-PCR (Fig. 3G). We found a 24.06 ± 5.71-fold increase of CXCL10 mRNA expression in CD11b+GFP− endogenous microglia subjected to irradiation compared with CD11b+GFP− microglia from protected CCR2+/+ GFP → APPswe/PS1 mice, underscoring again the influence of prior brain conditioning. Note that CXCL10 mRNA expression was significantly decreased in whole brains of unprotected CCR2+/+GFP → APPswe/PS1 mice compared with the protected chimeras (Fig. 3C), suggesting differential gene expression in microglia versus other types of brain cells. In contrast, CXCL10 mRNA levels were slightly downregulated in microglia from protected CCR2+/+GFP → APPswe/PS1 mice compared with protected CCR2+/+GFP → wild-type mice (0.13 ± 0.05-fold compared with 1.74 ± 0.54-fold). CCL3 mRNA expression was strongly induced in CD11b+GFP− microglia from protected CCR2+/+GFP → APPswe/PS1 mice compared with protected CCR2+/+GFP → wild-type mice (67.81 ± 10.15-fold compared with 1.19 ± 0.28-fold), indicating a disease-associated induction of this chemokine. Whereas CCL3 gene expression was largely unchanged by irradiation, CCL2 and TNFα transcripts were always induced in CD11b+ cells from irradiated brains. In contrast, CCR2 mRNA expression in microglia/phagocytes was influenced neither by disease nor by brain irradiation. Interestingly, CD11b+GFP− microglia and CD11b+GFP+ BM-derived phagocytes from unprotected CCR2+/+GFP → APPswe/PS1 mice had different levels of CCL2 and CXCL10 mRNA expression (CCL2 mRNA, 6.70 ± 2.20 in CD11b+GFP− and 0.72 ± 0.24 in CD11b+GFP+ cells; CXCL10 mRNA, 20.89 ± 7.24 in CD11b+GFP− and 2.06 ± 0.30 in CD11b+GFP+ cells). No significant differences in cytokine and chemokine gene expression were observed between CD11b+GFP− microglia isolated from CCR2+/+GFP → APPswe/PS1 and CCR2−/−GFP → APPswe/PS1 mice. The chemokine and cytokine gene expression changes induced in microglia by irradiation coincide with the morphological changes observed in brain sections from unprotected BM chimeras, but they do not necessarily overlap with the pattern of gene regulation observed in whole-brain lysates. However, the results strongly suggest differential functional properties of endogenous microglia and BM-derived mononuclear cells.
To examine whether irradiation of the CNS induces cell death or proliferation of parenchymal cells, histopathological changes following irradiation were investigated in more detail (Fig. 4). Unprotected BM chimeras received intraperitoneal injections of 100 μg of BrdU every second day starting on day 0 after irradiation, and the brains were analyzed 0, 2, 4, 8, and 16 d post-irradiation. We did not observe any apoptotic TUNEL+ or proliferating Ki-67+ (TEC-3) or BrdU+ cells in the cerebellum, suggesting that these events are obviously not induced when cytokine mRNA levels peak in the brains following irradiation, as described previously (Mildner et al., 2007).
Figure 4.

Absence of cell death and proliferation in the brain following irradiation and bone marrow transplantation. Tissue sections from the CNS of chimeric mice were stained at indicated time points after irradiation and BM transfer with antibodies against Ki-67 (TEC-3, DAKO, 1:20) and BrdU (in situ Detection Kit, BD PharMingen) for proliferation and TUNEL (Roche) for apoptotic cells. Spleens of chimeric mice were used as positive controls (insets). Representative pictures (cerebellum) from each experimental group (n = 2–3 mice) are shown.
To determine whether irradiation-induced changes of brain homeostasis affect Aβ burden and APP processing, we examined β-amyloid content in brain lysates from protected and unprotected CCR2+/+GFP → APPswe/PS1 BM chimeric mice (Fig. 5A). Surprisingly, the amount of insoluble Aβ1–40 and Aβ1–42 was significantly reduced after brain irradiation, while soluble Aβ1–40 and Aβ1–42 remained unchanged. Importantly, the levels of full-length APP and β-CTF were comparable between both groups, suggesting that brain irradiation might inhibit the degradation/clearance of fibrillar insoluble Aβ1–40 and Aβ1–42 by mononuclear phagocytes, rather than altering the processing of APP (Fig. 5B).
Figure 5.

Aβ deposition is reduced in AD transgenic mice after brain irradiation and BMC transplantation. A, Determination of the amounts of soluble and insoluble Aβ1–40 and Aβ1–42 by sandwich ELISA in brain lysates from protected (white bars) and unprotected (black bars) CCR2+/+GFP → APPswe/PS1 BM chimeras 7 months after irradiation. Deposition of insoluble, but not soluble Aβ1–40 and Aβ1–42 is significantly reduced in unprotected (brain-irradiated) CCR2+/+GFP → APPswe/PS1 BM chimeras. Data are means ± SEM from at least 5 mice per group. *p < 0.05 statistical significance. B, Immunoblot analysis of brain lysates from unprotected and protected CCR2+/+GFP → APPswe/PS1 BM chimeras for APP, β-CTF, and Aβ. No differences in APP processing were found between protected and unprotected animals (n = 6 per group). Wild-type mice (wt) served as negative controls.
To confirm our findings in an alternative and less aggressive AD mouse model, we used APP23 mice (Fig. 6). In this transgenic line, APP gene expression is driven by the neuronal Thy-1 promoter, leading to a slower progression of AD pathology compared with APPswe/PS1 mice. When APP23 mice were used as recipients of BM cells from β-actin-EGFP mice, some invading ameboid-like GFP+ cells were found in the brains of unprotected CCR2+/+ GFP → APP23 mice, while none were detected in protected CCR2+/+GFP → APP23 BM chimeras (data not shown). These results are in line with the findings of Stalder et al. (2005), and indicate that hematopoietic cells also engraft in response to cerebral amyloidosis in this single transgenic AD model. In analogy to our results in APPswe/PS1 BM chimeric mice, unprotected CCR2+/+GFP → APP23 mice contained significantly less insoluble Aβ1–40 and Aβ1–42 in the brain compared with protected CCR2+/+GFP → APP23 mice, while the levels of soluble Aβ1–40 and Aβ1–42, and APP and β-CTF were unchanged (Fig. 6). Stereological analysis revealed no significant differences in β-amyloid plaque load in the neocortex and hippocampus of protected versus unprotected CCR2+/+GFP → APP23 mice (data not shown).
Figure 6.

Aβ deposition is reduced in AD transgenic mice after brain irradiation and BM transplantation. A, Determination of the amounts of soluble and insoluble Aβ1–40 and Aβ1–42 by sandwich ELISA in brain lysates from protected (white bars) and unprotected (black bars) CCR2+/+GFP → APP23 BM chimeras. Deposition of insoluble, but not soluble Aβ1–40 and Aβ1–42 is significantly reduced in unprotected (brain-irradiated) CCR2+/+GFP → APP23 BM chimeras. Data are means ± SEM from 5 mice per group. *p < 0.05 statistical significance. B, Immunoblot analysis of brain lysates from unprotected and protected CCR2+/+ GFP → APP23 BM chimeras for APP, β-CTF, and Aβ. No differences in APP processing were found between protected and unprotected animals (n = 5 per group).
Together, these results underscore the broad impact of brain irradiation before BM transplantation, which induces considerable and long-lasting morphological, genetic, and functional changes of brain cells, including microglia.
BM-derived mononuclear phagocytes do not modulate parenchymal Aβ load in CCR2-deficient chimeras
It has been proposed that BM-derived microglia are critical in restricting senile plaque formation in AD mouse models due to improved migration and increased phagocytosis compared with endogenous microglia (Simard et al., 2006). To establish whether BM-derived phagocytes are beneficial or detrimental to AD pathogenesis, we generated BM chimeras specifically lacking donor-derived cells in their brains. For this purpose, CCR2+/+GFP → APPswe/PS1 and CCR2−/−GFP → APPswe/PS1 mice were generated, which were total body irradiated before BM transplantation to condition the brains for potential myeloid cell entry. However, as we have shown before, unprotected CCR2−/−GFP → APPswe/PS1 mice lacked significant numbers of engrafted GFP+Iba-1+ cells compared with wild-type donors despite a comparable degree of blood chimerism (Fig. 1C). Surprisingly, determination of the levels of soluble and insoluble Aβ1–40 and Aβ1–42 in the brains of CCR2+/+ GFP → APPswe/PS1 and CCR2−/−GFP → APPswe/PS1 mice did not reveal any significant differences (Fig. 7A). Moreover, immunoblot analyses of brain lysates revealed similar quantities of APP, β-CTF, and Aβ (Fig. 7B). Thus, our results suggest that BM-derived phagocytes can be targeted to parenchymal β-amyloid plaques in a CCR2-dependent manner, but changes of Aβ load depend on irradiation-induced changes, such as activation of endogenous brain Aβ clearance or increased brain-to-blood efflux of Aβ, rather than reduced amyloidogenic APP metabolism or the recruitment of BM-derived phagocytes to parenchymal Aβ deposits.
Figure 7.

Parenchymal Aβ load in the brains of AD transgenic mice is independent from the presence of immigrating bone marrow-derived phagocytes. A, Determination of the amounts of soluble and insoluble Aβ1–40 and Aβ1–42 by sandwich ELISA in brain lysates from unprotected (brain-irradiated) CCR2+/+GFP → APPswe/PS1 (white bars) and CCR2−/−GFP → APPswe/PS1 (black bars) BM chimeric mice. Aβ deposition is independent of the engraftment of donor-derived phagocytes in the brain, which is significantly reduced in CCR2−/−GFP → APPswe/PS1 BM chimeras (see also Fig. 1C,D). B, Immunoblot analysis of brain lysates from CCR2+/+GFP → APPswe/PS1 and CCR2−/−GFP → APPswe/PS1 BM chimeric mice for APP, β-CTF, and Aβ. No differences in APP processing were found between the groups (n = 3–4 per group). Tubulin served as loading control.
Perivascular macrophages in AD transgenic mice clear Aβ in a CCR2-dependent manner
To further explore the role of hematopoietic cells in Aβ clearance, we took advantage of transgenic Tg2576 (APPswe) mice expressing the 695 aa isoform of human APP, a well established model for a nonaggressively progressing disease with established CCR2 dependency (El Khoury et al., 2007).
To generate APPswe mice deficient in CCR2, we bred APPswe mice with CCR2−/− mice and examined the survival times (Fig. 8A). As already described, APPswe mice deficient in CCR2 (APPsweCCR2−/−) had a marked increase in mortality compared with wild-type CCR2+/+, CCR2−/−, and APPswe mice (p < 0.05). By 120 d of age, 38% of APPsweCCR2−/− had died, compared with 100% survival in the other groups of mice. Interestingly, even APPswe mice had normal survival rates up to this time point. These data confirm that the presence of the chemokine receptor CCR2 modulates pathogenicity in this AD mouse model.
Figure 8.
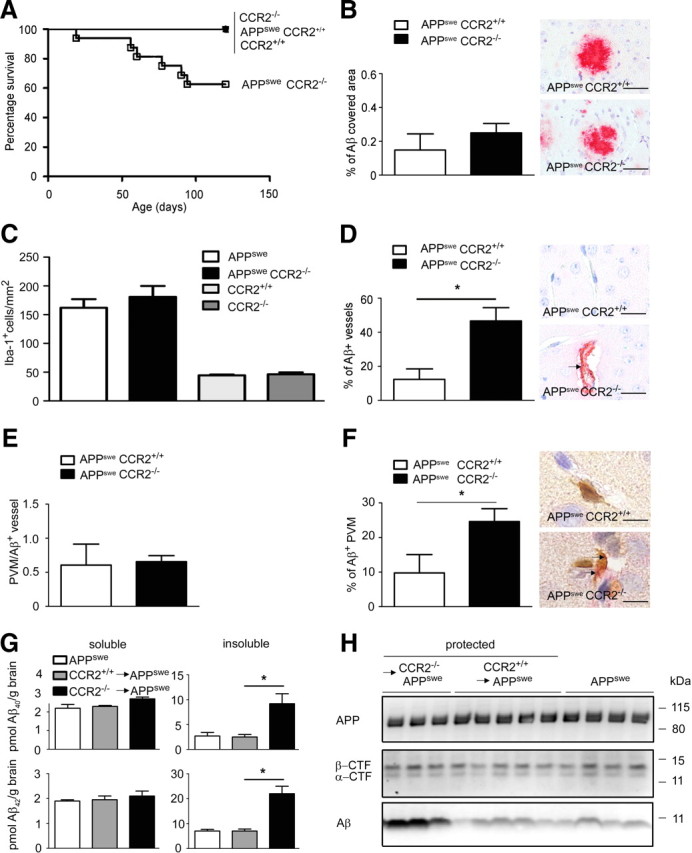
Perivascular macrophages in AD transgenic mice clear Aβ in a CCR2-dependent manner. A, Expression of CCR2 determines the survival of APPswe mice. Survival curves of wild-type CCR2+/+, CCR2−/−, APPswe CCR2+/+, and APPswe CCR2−/− mice are shown. In each experimental group at least 20 mice were used. B, Immunohistochemistry with an antibody against Aβ reveals no difference in the relative area covered by Aβ in the cortex of APPswe CCR2+/+ and APPswe CCR2−/− mice. Data are means ± SEM from at least 3 sections per animal and at least 3 mice per group. Representative immunostained sections are shown on the right. Scale bars, 100 μm. C, Semiquantitative analysis of Iba-1-immunoreactive cells in the brain reveals normal proliferation of microglia in the absence of CCR2. The number of Iba-1+ microglia/macrophages is significantly increased in the brains of APPswe CCR2+/+ (white bar, n = 4) and APPswe CCR2−/− mice (black bar, n = 4) compared with CCR2+/+ (light gray bar, n = 3) and CCR2−/− mice (dark gray bar, n = 4), respectively. The number of Iba-1+ cells is not different between APPswe CCR2+/+ and APPswe CCR2−/− mice, or between CCR2+/+ and CCR2−/− mice. Data are means ± SEM from at least 3 sections per animal and at least 3 mice per group. D, Immunohistochemistry with an antibody against Aβ reveals a significant increase in the percentage of blood vessels containing β-amyloid in the cortex of APPswe CCR2−/− compared with APPswe CCR2+/+ mice. Data are means ± SEM from at least 3 sections per animal and at least 3 mice per group. Asterisk indicates statistical significance (p < 0.05). Representative immunostained sections are shown on the right, and the arrow indicates vascular Aβ accumulation. Scale bars, 50 μm. E, Absence of CCR2 does not change the number of Iba-1+ PVMs per Aβ-containing blood vessel. Semiquantitative analysis reveals the same number of PVMs in APPswe CCR2+/+ as in APPswe CCR2−/− mice. Data are means ± SEM from at least 3 sections per animal and at least 3 mice per group. F, CCR2 deficiency increases the percentage of perivascular macrophages, which accumulate Aβ. Semiquantitative analysis was performed on at least 3 sections per animal and at least 3 mice per group (APPswe CCR2+/+ and APPswe CCR2−/− mice). Data are means ± SEM. *p < 0.05 statistical significance. Representative immunostained sections are shown on the right. Aβ+Iba-1+ PVMs were visualized by double immunohistochemistry (Aβ in red, Iba-1 in brown). The arrows point to aβ deposition in PVMs. Scale bars, 20 μm. G, Determination of the amounts of soluble and insoluble Aβ1–40 and Aβ1–42 by sandwich ELISA in brain lysates from nontransplanted APPswe mice (white bars), and from protected, i.e., brain shielded from irradiation, CCR2+/+GFP → APPswe (gray bars) and CCR2−/−GFP → APPswe (black bars) BM chimeric mice 7 months after transplantation. Deposition of insoluble, but not soluble Aβ1–40 and Aβ1–42 is significantly increased in APPswe mice transplanted with CCR2-deficient BMCs. Data are means ± SEM from at least 3 mice per group. *p < 0.05 statistical significance. H, Immunoblot analysis of brain lysates from APPswe mice, and from protected CCR2+/+GFP → APPswe and CCR2−/−GFP → APPswe BM chimeric mice for APP, α- and β-CTF, and Aβ. Aβ accumulation is enhanced in APPswe mice transplanted with CCR2-deficient BMCs, whereas APP processing is unchanged (n = 3–5 per group).
To determine whether the increased mortality in APPswe CCR2−/− mice was associated with elevated brain parenchymal β-amyloid deposits, we performed immunohistochemistry with antibodies against Aβ. Similar amounts of parenchymal Aβ deposition were found in the brains of aged APPsweCCR2−/− mice and age-matched APPsweCCR2+/+ control mice (Fig. 8B).
Both microglia and BM-derived phagocytes have been implicated in the clearance of Aβ from the brain (Bard et al., 2000). However, the mechanisms by which mononuclear phagocytes are recruited into AD brains are not known. CCR2 is the main chemokine receptor for CCL2 and mediates CCL2-induced leukocyte chemotaxis (Mildner et al., 2007; Prinz and Priller, 2010) Since El Khoury et al. (2007) suggested that CCR2 deficiency might affect microglia accumulation in APPswe mice, we performed immunohistochemical stainings for the well established macrophage/microglia marker, Iba-1, and quantified Iba-1-immunoreactive cells in the brains of APPsweCCR2−/−, APPsweCCR2+/+, CCR2−/−, and CCR2+/+ mice (Fig. 8C). As expected, diseased APPsweCCR2−/− and APPsweCCR2+/+ animals exhibited more Iba-1+ cells per square millimeter than did CCR2−/− and CCR2+/+ mice. Surprisingly, both APPsweCCR2−/− and APPsweCCR2+/+ mice had comparable numbers of Iba-1+ cells in the brain (APPsweCCR2−/−, 181.0 ± 18.9 cells/mm2; APPsweCCR2+/+, 162.1 ± 15.0 cells/mm2), clearly pointing to a redundant role of CCR2 for microglia recruitment and accumulation in AD. This finding is in contrast with the findings of El Khoury et al. (2007). However, CCR2−/− and CCR2+/+ mice also contained similar numbers of Iba-1+ cells in the brain (CCR2−/−, 44.0 ± 1.4 cells/mm2; CCR2+/+, 46.1 ± 3.9 cells/mm2).
APPsweCCR2−/− mice show enhanced β-amyloid deposition around blood vessels, a situation known as CAA (El Khoury et al., 2007). We confirmed these findings by Aβ immunohistochemistry (Fig. 8D). In APPsweCCR2−/− mice, 46.6 ± 7.8% of all cortical vessels showed Aβ deposits compared with 12.3 ± 6.2% in APPsweCCR2+/+ mice. We therefore postulated that the absence of CCR2 leads to increased Aβ accumulation in small blood vessels, possibly leading to intracerebral hemorrhages and premature death. In search of a potential mechanism, we asked whether the homeostasis and turnover of PVMs might be dependent on the presence of CCR2. To this end, we performed double immunohistochemistry for Iba-1 as macrophage marker and for GFAP to visualize the astrocyte end feet lining the glia limitans on brain sections from CCR2−/− and CCR2+/+ mice (data not shown). Semiquantitative evaluation revealed a similar amount of PVMs regardless of the genotype, namely 14.1 ± 2.8 Iba-1+ PVMs/section localized below the glia limitans in CCR2+/+ mice and 13.6 ± 1.7 Iba-1+ PVMs/section in normal CCR2−/− mice. To determine whether CCR2−/− mononuclear phagocytes might have a defect in their ability to migrate to cerebrovascular Aβ deposits, we next compared the number of Iba-1+ PVMs around Aβ+ vessels in 120-d-old APPsweCCR2−/− and APPsweCCR2+/+ mice (Fig. 8E). The number of Iba-1+ PVMs per Aβ-containing vessel was independent of CCR2 expression, suggesting that CCR2 plays a redundant role in PVM recruitment to cerebrovascular Aβ deposits.
Since cerebrovascular Aβ levels are mainly regulated by transport processes that shuttle Aβ across the blood–brain barrier (BBB) (Mucke, 2009), CCR2 may play an important role in Aβ clearance by PVMs. We determined the percentage of PVMs that incorporated Aβ by double immunohistochemistry for Iba-1 and Aβ (Fig. 8F). Aβ-containing Iba-1+ PVMs were significantly more frequent in APPsweCCR2−/− mice compared with APPsweCCR2+/+ mice (APPsweCCR2−/−, 24.6 ± 3.8 cells/mm2; APPsweCCR2+/+, 9.8 ± 5.3 cells/mm2). Thus, our data suggest that CCR2 deficiency affects Aβ clearance/transport by mononuclear phagocytes associated with blood vessels, but not by parenchymal microglia in AD transgenic mice.
To restrict CCR2 expression in APPswe mice to the radioresistant vascular compartment, we generated protected BM chimeric mice. Brain shielding in these mice still allows for sufficient exchange of PVMs, while the entry of mononuclear phagocytes into the brain parenchyma is prevented. To test whether CCR2 deficiency in the periphery, including PVMs, induces a change in cerebral Aβ accumulation, we analyzed protected CCR2−/− → APPswe and CCR2+/+ → APPswe mice at 7 months after BM transplantation (Fig. 8G,H). Notably, amyloid burden was dramatically increased in CCR2−/− → APPswe mice compared with CCR2+/+ → APPswe and APPswe mice. Biochemical analysis revealed an increase of both insoluble Aβ1–40 and Aβ1–42 in the brains of CCR2−/− → APPswe mice, whereas soluble Aβ1–40 and Aβ1–42 remained unchanged (Fig. 8H). To address steady-state APP metabolism, we also analyzed β-CTF and nonamyloidogenic CTF (α-CTF), but we did not detect any differences between the genotypes (Fig. 8H).
To examine whether CCR2−/− microglia and myeloid cells have a defect in their ability to phagocytose Aβ, we exposed the cells to FAM-labeled Aβ1–42 in vitro and performed flow cytometry (Fig. 9). We found that phagocytosis by adult microglia and bone marrow-derived macrophages was unchanged in the absence of CCR2. Accordingly, application of the CCR2 ligand, CCL2, did not modulate Aβ uptake. Next, we examined whether CCR2 deficiency or synthesis of GFP might affect the expression of immune-related molecules and adhesion factors on adult microglia, circulating Ly-6Chi monocytes, and macrophages using flow cytometry (Fig. 10). Baseline expression levels of CD11c, PSGL-1, and integrins α4 and β1 were similar in all myeloid cell populations derived from CCR2+/+, CCR2+/+GFP, CCR2−/−, and CCR2−/−GFP mice, demonstrating that expression of neither CCR2 nor GFP influences the homeostatic baseline expression of immune molecules on the surface of microglia, monocytes, and macrophages.
Figure 9.
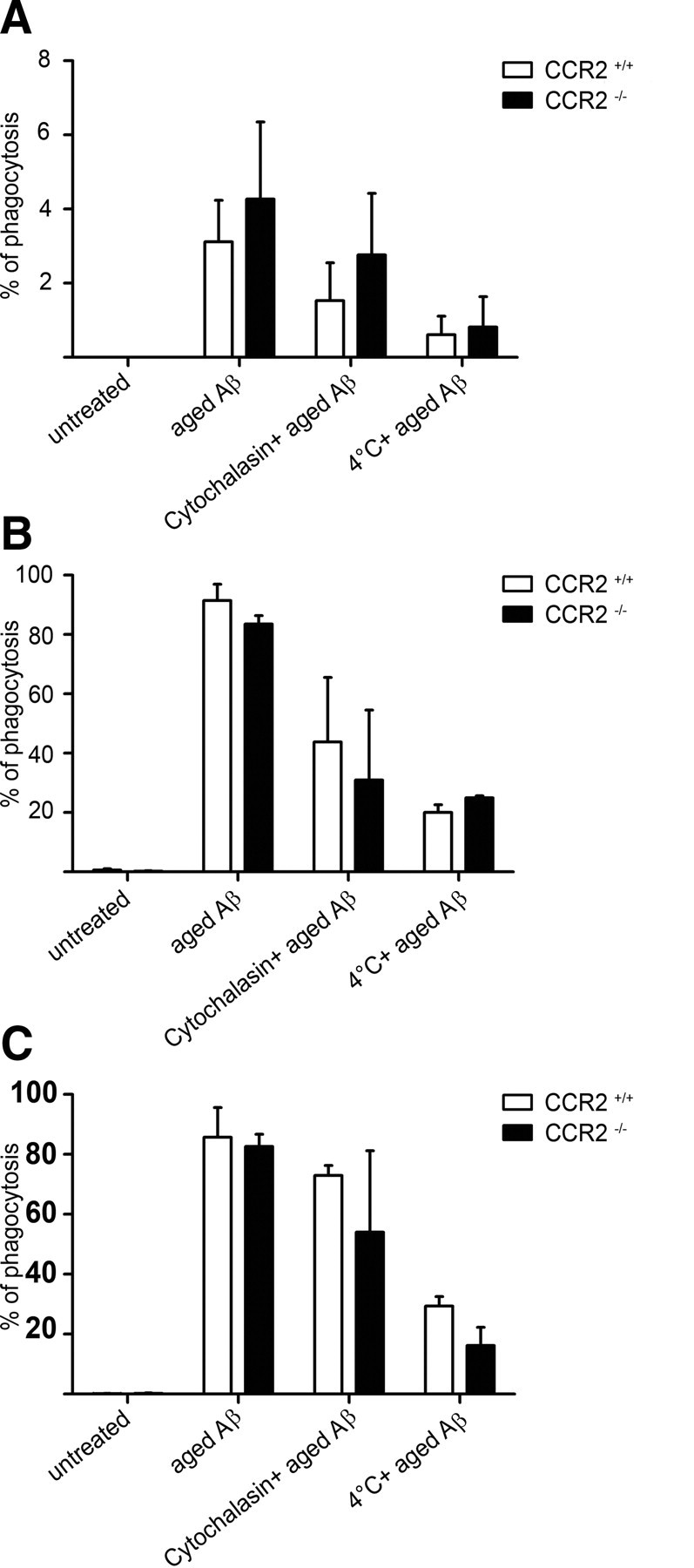
In vitro phagocytosis of Aβ1–42 is unchanged in the absence of CCR2. A, B, Phagocytosis of aged 5′-FAM Aβ1–42 by ex vivo isolated adult microglia (A) and bone marrow-derived macrophages (BMDM) (B) from CCR2−/− mice (black bars) and CCR2+/+ mice (white bars) as measured by flow cytometry 6 h after aged 5′-FAM amyloid β1–42 challenge. C, Addition of recombinant murine CCL2 (10 ng/ml) has no influence on Aβ phagocytosis in BMDM. Data are means ± SEM from 3 independent experiments with at least 3 mice per group.
Figure 10.

Expression of immune and adhesion molecules on myeloid cells is independent of CCR2 or GFP expression. Flow cytometric comparison of immune surface markers on myeloid cell subsets, including adult microglia, blood Ly-6Chi monocytes, and macrophages derived from CCR2+/+, CCR2+/+GFP, CCR2−/−, and CCR2−/−GFP mice. All cells were gated on CD11b, and expression of CD11c, PSGL-1, integrin α4, and integrin β1 was assessed by FACS under baseline conditions. Isotype antibodies were used as negative controls.
Our results imply that peripheral macrophages, e.g., PVMs, rather than parenchymal microglia modulate β-amyloid deposition in the brains of AD transgenic mice by clearing Aβ in a CCR2-dependent fashion.
Discussion
Here, we describe and characterize a specific myeloid subpopulation of CCR2-expressing cells as the precursors of newly recruited tissue phagocytes in the brains of irradiated AD transgenic mice. Our findings are in line with the dominant role that MCP-1/CCL2, the ligand for CCR2, plays in chronic inflammation in the human AD brain (Janelsins et al., 2005; Sokolova et al., 2009). In mouse models of AD chimeras, CCR2+ myeloid cells are partially recruited to regions of Aβ deposition, namely to senile plaques and the cerebrovascular compartment, where they exert differential functions. The recruitment of mononuclear phagocytes from the periphery to parenchymal β-amyloid plaques depended on CCR2 expression and conditioning of the brain (for example, irradiation), whereas Iba-1+ PVMs were recruited to vascular β-amyloid deposits in the absence of CCR2, but they needed this receptor for Aβ clearance. Thus, our data offer new insights into the mechanisms leading to the engraftment of BM/blood-derived mononuclear phagocytes in the brains of chimeric AD transgenic mice, and suggest distinct spatiotemporal roles for specific myeloid subpopulations in disease pathogenesis.
Monocytes and tissue macrophages, such as microglia, are highly mobile immune effector cell populations with distinct functional features. Microglia already colonize the CNS during early embryogenesis, when they arise from hemangioblastic mesoderm and populate the developing neuroectoderm in rodents after embryonic day 8.5 (Alliot et al., 1999), whereas monocytes circulate in the blood during adulthood and cannot normally enter the brain, which is anatomically separated from the periphery by the BBB (Ransohoff and Perry, 2009). Early studies in irradiated rats using MHC mismatch as a marker of donor BM cells found only limited turnover of microglia with their peripheral myeloid counterparts (Hickey et al., 1992). However, more recent studies documented an early and rapid engraftment of BM-derived phagocytes in several models of neurodegenerative diseases, such as facial nerve axotomy (Priller et al., 2001), scrapie (Priller et al., 2006), the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of Parkinson's disease (Kokovay and Cunningham, 2005), and Alzheimer's disease models (Malm et al., 2005; Stalder et al., 2005; Simard et al., 2006).
It is quite essential to note that the vast majority of BM chimera data addressing the functional impact and kinetics of newly recruited phagocytes versus endogenous microglia were obtained using total-body irradiation, without selective marrow-specific targeting of the myeloablative treatment. As we have demonstrated here, the involvement of the head during the irradiation procedure has a substantial impact on the local CNS environment. First, we observed substantial changes of the cellular networks of glial cells, as well as the morphology and localization of microglia in relation to β-amyloid plaques in AD transgenic mice. It was surprising to find that these alterations persist for several months after irradiation, indicating long-term effects of this conditioning regimen. Indeed, as we could show by single-cell microdissection followed by quantitative real-time PCR, TNFα, CCL2, and CXCL10 mRNA levels in microglia were greatly affected by irradiation. Subtle changes of BBB integrity or inactivation of repulsory signals following irradiation might also promote myeloid cell engraftment (Diserbo et al., 2002; Yuan et al., 2003). Finally, we cannot exclude any irradiation-induced neuronal damage that might cause distraction of microglia processes directly or indirectly.
CCL2 is induced not only by irradiation, but also during brain diseases, including AD (Ishizuka et al., 1997; Janelsins et al., 2005; Yamamoto et al., 2005; Prinz and Priller, 2010). Our results suggest that this chemokine might be involved in the recruitment of CCR2-expressing myeloid cells into the brains of AD transgenic mice. In line with this, transgenic overexpression of CCL2 in astrocytes of APP transgenic mice was associated with increased myeloid cell accumulation around plaques (Yamamoto et al., 2005). Here, we show for the first time that CCR2-expressing myeloid cells are the source of newly immigrated phagocytes in the brains of chimeric AD transgenic mice. However, in contrast to a previous report (Simard et al., 2006), we did not detect any effects of BM-derived phagocytes on parenchymal Aβ plaque load.
It has further been reported that CCR2 deficiency may hasten disease progression in AD transgenic mice, most likely as a result of impaired amyloid degradation by brain endogenous phagocytes, e.g., microglia (El Khoury et al., 2007). However, our data indicate that microglia proliferation and accumulation were unaltered in the absence of CCR2. Two other recent studies also question the involvement of microglia in the pathogenesis of AD. In vivo multiphoton imaging revealed that resident microglia are not important in the de novo formation of β-amyloid deposits in AD transgenic mice (Meyer-Luehmann et al., 2008). Moreover, ablation of the majority of parenchymal microglia using an inducible suicide gene approach in two mouse models of AD suggested that neither amyloid plaque formation and maintenance, nor amyloid-associated neuritic dystrophy depended on the presence of microglia (Grathwohl et al., 2009). Our findings are in line with a recent finding that CCR2 deficiency in APPswe/PS1 mice provokes a rapid cognitive decline closely correlated with brain accumulation of soluble Aβ oligomers and a robust mRNA expression of TGF-β1, TGF-β-R2, and CX3CR1 in microglia (Naert and Rivest, 2011). Similar to our study, the authors further found no impaired microglia proliferation around AD plaques in the absence of CCR2. In sum, CCR2 has certainly complex functions in AD that go well beyond the control of the CNS immigration of defined myeloid subsets to a role in remodeling of Aβ deposition.
Our data further indicate the pathogenic role of another specialized subset of mononuclear phagocytes, namely PVMs, for Aβ clearance in the brains of AD transgenic mice. These phagocytes are located in the perivascular spaces, where they are important for immune surveillance and usually involved in the transport of Aβ across the BBB (Mucke, 2009). Recent experimental evidence highlighted the critical role of PVMs in the regulation of cerebral amyloid angiopathy (Hawkes and McLaurin, 2009). We found that the recruitment of PVMs from the BM to cerebrovascular Aβ deposits in AD transgenic mice occurred independent of CCR2. However, PVMs cleared Aβ from the brain in a CCR2-dependent manner. Previous results suggested that CCR2 deficiency abolished the chemotaxis of peritoneal macrophages to supernatants from Aβ-stimulated macrophages (El Khoury et al., 2007). However, our data suggest that increased Aβ levels in the brains of APPsweCCR2−/− mice were neither due to impaired recruitment of CCR2-deficient phagocytes from the blood into the brain, nor due to defects in the activation or proliferation of CCR2-deficient microglia. Instead, our results imply a role for CCR2 in the clearance of Aβ by PVMs. We speculate that PVMs from CCR2−/− mice might be inefficient in shuttling Aβ outside the brain along a CCL2-mediated vascular gradient. Thus, our study could provide the missing link between the landmark findings on the role of CCR2 and PVMs in mouse models of AD (El Khoury et al., 2007; Hawkes and McLaurin, 2009). Our results are also in line with earlier observations that microglia do not have the ability to clear Aβ from the extracellular brain milieu, whereas peripheral macrophages can remove Aβ in vivo by phagocytosis (Wisniewski et al., 1989, 1991).
What is the relevance of our findings for AD patients? There are several reports indicating that ∼60% of patients with AD have a disturbed BBB (Algotsson and Winblad, 2007; Bowman et al., 2007). Moreover, AD patients are generally aged and often have a history of cerebrovascular events caused by CAA, including ischemic insults (Koistinaho and Koistinaho, 2005). Therefore, it can be assumed that circulating mononuclear phagocytes may engraft in the brains in AD patients without conditioning by irradiation. This is relevant for myeloid cell-based gene therapy in AD. In fact, infusion of millions of genetically engineered BM-derived CD11b+cells into APPswe/PS1 transgenic mice resulted in some parenchymal myeloid cell engraftment and modulation of amyloid deposition (Lebson et al., 2010). The impact of gene therapeutic approaches using myeloid precursors for CNS diseases has been highlighted in a mouse model of metachromatic leukodystrophy (Biffi et al., 2004), and more recently, in children with X-linked adrenoleukodystrophy (Cartier et al., 2009).
In conclusion, our results identified CCR2+ mononuclear cells as the source of immigrating phagocytes in the brains of chimeric AD transgenic mice. Myeloid cells are recruited to sites of cerebral amyloidosis, namely parenchymal β-amyloid plaques and cerebrovascular Aβ deposits. Moreover, we defined the differential roles of these BM/blood-derived mononuclear phagocytes in the pathophysiology of AD, and point to a dialectic function of myeloid cells inside and outside of the brain. It appears that the spatiotemporal role of phagocytes, i.e., when and where they are present and active, is a crucial issue.
Footnotes
This work was supported by the German Research Council (DFG, PR 577/5-1 and FOR1336 to M.P., I.B., and J.P.). M.P. was supported by the Bundesministerium fuer Bildung und Forschung-funded competence network of neurodegenerative disorders (DZNE). A.M. was a fellow of the Gertrud Reemtsma Foundation. We thank Olga Kowatsch, Doris Bode, and Tina El Gaz for excellent technical assistance; Dr. Matthias Staufenbiel for providing APP23 mice; and Drs. Bettina Bert and Heidrun Fink for behavioral analysis.
The authors declare no conflict of interest.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algotsson A, Winblad B. The integrity of the blood–brain barrier in Alzheimer's disease. Acta Neurol Scand. 2007;115:403–408. doi: 10.1111/j.1600-0404.2007.00823.x. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Kwidzinski E, Kovac AD, Simbürger E, Horvath T, Gimsa U, Dirnagl U, Priller J, Nitsch R. Turnover of rat brain perivascular cells. Exp Neurol. 2001;168:242–249. doi: 10.1006/exnr.2000.7618. [DOI] [PubMed] [Google Scholar]
- Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, Bordignon C, Naldini L. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–1129. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood–brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology. 2007;68:1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrère F, Blanche S, Audit M, Payen E, Leboulch P, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E, Amourette C. Blood–brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol. 2002;80:670–678. doi: 10.1139/y02-070. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Potempska A, LeVine H, Haske T, Dickson D, Mazur-Kolecka B. Extracellular deposits of A beta produced in cultures of Alzheimer disease brain vascular smooth muscle cells. J Neuropathol Exp Neurol. 2005;64:82–90. doi: 10.1093/jnen/64.1.82. [DOI] [PubMed] [Google Scholar]
- François S, Bensidhoum M, Mouiseddine M, Mazurier C, Allenet B, Semont A, Frick J, Saché A, Bouchet S, Thierry D, Gourmelon P, Gorin NC, Chapel A. Local irradiation not only induces homing of human mesenchymal stem cells at exposed sites but promotes their widespread engraftment to multiple organs: a study of their quantitative distribution after irradiation damage. Stem Cells. 2006;24:1020–1029. doi: 10.1634/stemcells.2005-0260. [DOI] [PubMed] [Google Scholar]
- Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer's disease: the blood-borne identity. J Neural Transm. 2010;117:961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF, Vass K, Lassmann H. Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol. 1992;51:246–256. doi: 10.1097/00005072-199205000-00002. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer's disease mice. J Neuroinflammation. 2005;2:23. doi: 10.1186/1742-2094-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Interactions between Alzheimer's disease and cerebral ischemia—focus on inflammation. Brain Res Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Cunningham LA. Bone marrow-derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson's disease. Neurobiol Dis. 2005;19:471–478. doi: 10.1016/j.nbd.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Lebson L, Nash K, Kamath S, Herber D, Carty N, Lee DC, Li Q, Szekeres K, Jinwal U, Koren J, Dickey CA, Gottschall PE, Morgan D, Gordon MN. Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J Neurosci. 2010;30:9651–9658. doi: 10.1523/JNEUROSCI.0329-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linard C, Marquette C, Mathieu J, Pennequin A, Clarençon D, Mathé D. Acute induction of inflammatory cytokine expression after gamma-irradiation in the rat: effect of an NF-kappaB inhibitor. Int J Radiat Oncol Biol Phys. 2004;58:427–434. doi: 10.1016/j.ijrobp.2003.09.039. [DOI] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6C(hi)CCR2(+) monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mildner A, Djukic M, Garbe D, Wellmer A, Kuziel WA, Mack M, Nau R, Prinz M. Ly-6G+CCR2− myeloid cells rather than Ly-6ChighCCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J Immunol. 2008;181:2713–2722. doi: 10.4049/jimmunol.181.4.2713. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Mucke L. Neuroscience: Alzheimer's disease. Nature. 2009;461:895–897. doi: 10.1038/461895a. [DOI] [PubMed] [Google Scholar]
- Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2011;31:6208–6220. doi: 10.1523/JNEUROSCI.0299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller J, Flügel A, Wehner T, Boentert M, Haas CA, Prinz M, Fernández-Klett F, Prass K, Bechmann I, de Boer BA, Frotscher M, Kreutzberg GW, Persons DA, Dirnagl U. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- Priller J, Prinz M, Heikenwalder M, Zeller N, Schwarz P, Heppner FL, Aguzzi A. Early and rapid engraftment of bone marrow-derived microglia in scrapie. J Neurosci. 2006;26:11753–11762. doi: 10.1523/JNEUROSCI.2275-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Mildner A. Microglia in the CNS: immigrants from another world. Glia. 2011;59:177–187. doi: 10.1002/glia.21104. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX(3)CR1 in myeloid cell entry in the CNS. J Neuroimmunol. 2010;224:80–84. doi: 10.1016/j.jneuroim.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Brück W, Becher B. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, Knobeloch KP, Bethea JR, Waisman A, Knust M, Del Turco D, Deller T, Blank T, Priller J, Brück W, Pasparakis M, Prinz M. I{kappa}B kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-{kappa}B in the central nervous system. Brain. 2011;134:1184–1198. doi: 10.1093/brain/awq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer's disease. Brain Pathol. 2009;19:392–398. doi: 10.1111/j.1750-3639.2008.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci. 1989;16:535–542. doi: 10.1017/s0317167100029887. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Barcikowska M, Kida E. Phagocytosis of beta/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol. 1991;81:588–590. doi: 10.1007/BF00310142. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Horiba M, Buescher JL, Huang D, Gendelman HE, Ransohoff RM, Ikezu T. Overexpression of monocyte chemotactic protein-1/CCL2 in beta-amyloid precursor protein transgenic mice show accelerated diffuse beta-amyloid deposition. Am J Pathol. 2005;166:1475–1485. doi: 10.1016/s0002-9440(10)62364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Gaber MW, McColgan T, Naimark MD, Kiani MF, Merchant TE. Radiation-induced permeability and leukocyte adhesion in the rat blood–brain barrier: modulation with anti-ICAM-1 antibodies. Brain Res. 2003;969:59–69. doi: 10.1016/s0006-8993(03)02278-9. [DOI] [PubMed] [Google Scholar]