

Abstract
Infantile neuroaxonal dystrophy (INAD) is a fatal neurodegenerative disease characterized by the widespread presence of axonal swellings (spheroids) in the CNS and PNS and is caused by gene abnormality in PLA2G6 [calcium-independent phospholipase A2β (iPLA2β)], which is essential for remodeling of membrane phospholipids. To clarify the pathomechanism of INAD, we pathologically analyzed the spinal cords and sciatic nerves of iPLA2β knock-out (KO) mice, a model of INAD. At 15 weeks (preclinical stage), periodic acid-Schiff (PAS)-positive granules were frequently observed in proximal axons and the perinuclear space of large neurons, and these were strongly positive for a marker of the mitochondrial outer membrane and negative for a marker of the inner membrane. By 100 weeks (late clinical stage), PAS-positive granules and spheroids had increased significantly in the distal parts of axons, and ultrastructural examination revealed that these granules were, in fact, mitochondria with degenerative inner membranes. Collapse of mitochondria in axons was accompanied by focal disappearance of the cytoskeleton. Partial membrane loss at axon terminals was also evident, accompanied by degenerative membranes in the same areas. Imaging mass spectrometry showed a prominent increase of docosahexaenoic acid-containing phosphatidylcholine in the gray matter, suggesting insufficient membrane remodeling in the presence of iPLA2β deficiency. Prominent axonal degeneration in neuroaxonal dystrophy might be explained by the collapse of abnormal mitochondria after axonal transportation. Insufficient remodeling and degeneration of mitochondrial inner membranes and presynaptic membranes appear to be the cause of the neuroaxonal dystrophy in iPLA2β-KO mice.
Introduction
Infantile neuroaxonal dystrophy (INAD) is a fatal neurodegenerative disease with various neurological symptoms (Gregory et al., 2008b). Widespread formation of axonal swellings, referred to as spheroids, and tubulovesicular structures are observed in the CNS and PNS (Cowen and Olmstead, 1963; Khateeb et al., 2006). Gene abnormality in the PLA2G6 [calcium-independent phospholipase A2β (iPLA2β)] gene is associated with 80% of INAD cases (Morgan et al., 2006) and is sometimes found in patients with dystonia–parkinsonism (Paisan-Ruiz et al., 2009). The enzyme activity is impaired by the mutation associated with INAD but not in dystonia–parkinsonism (Engel et al., 2010). Recently, iPLA2β knock-out (KO) mice (Malik et al., 2008; Shinzawa et al., 2008) and iPLA2β gene-mutated mice (Wada et al., 2009) have been reported to show progressive motor deficits, and their neuropathological changes are very similar to those of INAD, although the pathomechanism remains unknown.
iPLA2β is a phospholipase A2 family member that hydrolyzes the sn-2 ester bond in phospholipids, including glycerophospholipids such as phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and cardiolipin, to yield free fatty acids and lysophospholipids (Burke and Dennis, 2009). The functions of iPLA2β include remodeling of membrane phospholipids (Balsinde et al., 1997), fatty acid oxidation (Strokin et al., 2003), release of docosahexaenoic acid (DHA) and arachidonic acid (AA) (Green et al., 2008), cell growth and signaling (Hooks and Cumming, 2008), and cell death (Shinzawa and Tsujimoto, 2003). In the brain of iPLA2β-deficient mice, DHA metabolism is reduced at 4 months without overt neuropathology (Basselin et al., 2010), and an iPLA2β inhibitor has been reported to attenuate linoleic acid (LA) incorporation of cardiolipin (CL) in rat heart (Zachman et al., 2010). In monkey brain, iPLA2β is localized in axon terminals and dendritic spines of neurons (Ong et al., 2005). Although iPLA2β also exists in various organs (Song et al., 2010), no known non-neurological dysfunction has been reported in INAD (Gregory and Hayflick, 2008a).
Mitochondria are subcellular micro-organelles that are integral to all eukaryotic cells, being responsible for metabolic and respiratory functions. Their characteristic feature is a double-membranous architecture that separates four distinct compartments: the outer membrane, intermembrane space, inner membrane, and matrix (McBride et al., 2006). The inner membrane is highly folded into invaginations known as cristae, in which oxidative phosphorylation is catalyzed, and the phospholipids of which it is composed contain a high proportion of CL relative to that in the other membranes. The outer membrane has numerous pores for uptake and exchange of specific metabolites (McBride et al., 2006). iPLA2β has also been reported to exist in, and to protect, mitochondria (Seleznev et al., 2006a).
Previously, in iPLA2β-KO mice, we demonstrated the presence of characteristic periodic acid-Schiff (PAS)-positive granules, which appeared early in apparently normal axons and later in spheroids. To clarify the pathomechanism of neuroaxonal dystrophy, we analyzed the spinal cords and sciatic nerves of iPLA2β-KO mice, especially in the context of PAS-positive granules and spheroids. First, quantitative pathological analysis, immunohistochemistry, and ultrastructural analysis were performed, and second, imaging mass spectrometry (IMS) was performed to visualize the distribution of phospholipid in membranes.
Materials and Methods
Animals.
Mice with homozygous disruption of the iPLA2β gene on a C57BL/6 background (Shinzawa et al., 2008), aged 15 weeks (n = 2, preclinical stage, one male and one female), 56 weeks (n = 4, early clinical stage, four females), and 95–103 weeks (n = 5, late clinical stage, two males and three females), and wild-type (WT) mice, aged 56 weeks (two males) and 95–103 weeks (one male and four females), were used. After being given an overdose of isoflurane, each animal was perfused with PBS and then 4% paraformaldehyde (PFA), followed by removal of the spinal cord and sciatic nerves. Spinal cords were immersed in the same fixative overnight at 4°C and then dehydrated and embedded in paraffin blocks. Four-micrometer-thick paraffin sections were prepared and stained with PAS. Some of the spinal cords fixed in 4% PFA were cryoprotected, and 10-μm-thick frozen sections were prepared for immunohistochemistry. Small pieces of the spinal cord and sciatic nerve were fixed with 2.5% glutaraldehyde and processed to Epon blocks as described previously (Sumi et al., 2006). Epon sections, 1 μm thick, were stained with thionin and PAS. Sciatic nerves were analyzed in transverse and longitudinal views. For analysis using liquid chromatography/electrospray ionization tandem mass spectrometry (LC/ESI-MS/MS) and imaging mass spectrometry (IMS), quickly frozen spinal cords without fixation from WT and iPLA2β-KO mice aged 56 and 102 weeks were stored at −80°C. All animals were handled in accordance with the Guidelines for Animal Experimentation of Osaka University and those of the Japanese Government.
Immunohistochemistry.
Deparaffinized sections were incubated for 30 min with 0.3% H2O2 to quench endogenous peroxidase activity and then washed with PBS. The primary antibodies used were a mouse monoclonal antibody against cytochrome c (cyt c) oxidase subunit I (CCO) (component of complex IV, which is the terminal enzyme in the respiratory chain on the inner membrane; 1:50; Invitrogen), a rabbit polyclonal antibody against the 20 kDa translocase of the outer mitochondrial membrane (TOM20) (one of the import receptors of mitochondrial outer membrane pores; 1:100; Dako), and a mouse monoclonal antibody against 4-hydroxy-2-nonenal (4-HNE) (an oxidized secondary product that is formed when organic lipids consisting of polyunsaturated fatty acid (PUFA) receive oxidization stress; 1:100; NOF Corporation). Autoclave treatment was performed for 9 min before incubation with the antibody against 4-HNE. Fixed frozen sections were dried and washed in PBS and incubated with a mouse monoclonal antibody against cyt c (1:100; BD Pharmingen). Goat anti-rabbit and anti-mouse Ig conjugated to peroxidase-labeled dextran polymer (Dako Envision+; Dako) were used as secondary antibodies. Reaction products were visualized with 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories), and hematoxylin was used to counterstain the cell nuclei. The immunostaining patterns were compared with the PAS staining pattern, and some of the specimens were stained with PAS after the immunohistochemical staining procedures.
Quantitative pathological analysis.
To study the distal parts of axons, 1-μm-thick transverse Epon sections of the lumbar cord and sciatic nerves of iPLA2β-KO mice (15, 56, and 95–103 weeks), which had been stained with thionin and PAS, were examined. Video images were obtained using a digital camera connected to a microscope (oil immersion, 100× objective). In the lumbar cord, five fields (100 × 100 μm × 5) in the most posterior part of the posterior horns were examined. The numbers of PAS-positive granules, spheroids, and vacuoles exceeding 5 μm in diameter were counted in each mouse. In sciatic nerves, four fields (100 × 100 μm × 4) in the endoneurium were examined. The numbers of PAS-positive granules, myelin ovoids, and large fibers, with a diameter exceeding 7 μm, were counted in each mouse. The diameters of axons were measured with the aid of image analysis software (VH-H1A5; Keyence), and the data were compared statistically between iPLA2β-KO mice aged 56 and 100 weeks by the Wilcoxon's rank sum test (Excel Toukei version 6.0; Esumi).
Ultrastructural analysis.
Ultrathin sections of the spinal cord from iPLA2β-KO mice aged 15, 56, and 100 weeks were cut and stained with uranyl acetate and lead citrate and examined using a transmission electron microscope (H-7650; Hitachi High-Technologies).
Liquid chromatography/electrospray ionization tandem mass spectrometry.
For LC/ESI-MS/MS, spinal cord tissue blocks were collected into glass vials for lipid extraction (∼10 mg for each), and total lipids were extracted by the Folch method (Folch et al., 1957). The ESI-MS/MS analysis was performed using a 4000Q-TRAP quadrupole linear ion trap hybrid mass spectrometer (Applied Biosystems/MDS Sciex) with an ACQUITY Ultra Performance Liquid Chromatography (Waters). A chromatographic method was developed using an ACQUITY UPLCTM BEH C18 column (2.1 × 50 mm inner diameter, 0.17 μm particle), fitted with an identically packed guard column (2.1 × 5 mm) (Waters). The column oven was maintained at 40°C. The following gradient elution with mobile phase A (acetonitrile/methanol/water at 19:19:2 v/v/v, containing 0.1% formic acid and 0.028% ammonium) and mobile phase B (isopropanol, 0.1% formic acid, and 0.028% ammonium) was used at a flow rate of 0.4 ml/min: 0–10 min, 5% B → 5% B; 10–15 min, 5% B → 50% B; 15–20 min, 50% B → 50% B; and 20–25 min, 5% B.
Mass spectrometry conditions.
Tandem mass spectrometry analysis was performed in both positive, for phosphatidylcholines (PCs) and phosphatidylethanolamines (PEs), and negative, for CLs, electrospray ionization mode with the following settings: for PCs and PEs, ion spray voltage, 5500 V; curtain (nitrogen), 10 arbitrary units; and collision gas (nitrogen), “high.” Specific detection was performed by multiple reaction monitoring (MRM) with the following settings: dwell time, 50 ms; declustering potential, 60 V; and resolutions of Q1 and Q3, “unit.” For CLs, the following settings were used: ion spray voltage, −4500 V; curtain (nitrogen), 10 arbitrary units; and collision gas (nitrogen), “high.” For MRM, the following settings were used: dwell time, 50 ms; declustering potential, −80 V; and resolutions of Q1 and Q3, “unit.” The characteristic fragmentation patterns of individual lipid species were determined by enhanced product ion scanning. The results were expressed as logarithmic values of the detected intensity ratio (KO/WT), i.e., log(2, Intensity KO/Intensity WT).
Imaging mass spectrometry.
Tissues blocks were sectioned at a thickness of 8 μm at −18°C using a cryostat (CM 3050; Leica Microsystems), as described previously (Schwartz et al., 2003; Sugiura et al., 2006). A 2,5-dihydroxybenzoic acid (DHB) solution (40 mg/ml DHB, 20 mm potassium acetate, 70% MetOH, and 0.1% trifluoroacetic acid) was used as the matrix solution for imaging of PC in positive ion detection mode (Sugiura and Setou, 2009). 9-Aminoacridine dissolved in 70% ethanol (50 mg/ml) was used for imaging of PE. For detection of PE in negative ion detection mode, before matrix application, tissue sections were washed with 50 mm ammonium formate for 30 s to remove endogenous salts. The matrix solution was sprayed over the tissue surface using a 0.2 mm caliber nozzle airbrush (Procon Boy FWA Platinum; Mr. Hobby). Matrices were applied simultaneously to the tissue sections that were to be compared, to equalize the analyte extraction and cocrystallization conditions. IMS measurements were performed using a matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI TOF/TOF)-type instrument (Ultraflex 2 TOF/TOF; Bruker Daltonics) equipped with a 355-nm Nd:YAG laser and a modified laser focusing system. Signals between mass-to-charge ratio (m/z) of 400 and 1000 were collected. Raster scans on tissue surfaces were performed automatically using FlexControl and FlexImaging 2.0 software (Bruker Daltonics). The number of laser irradiations was 200 shots for each data point. Image reconstruction was performed using FlexImaging 2.0 software.
Results
Histochemical analysis of the spinal cord
In the spinal cord of iPLA2β-KO mice at 15 weeks (presymptomatic stage), small PAS-positive granules, 0.5–1.5 μm in diameter, were frequently observed in the perinuclear space and proximal axons of anterior horn cells (Figs. 1A,C, 2A) but were rarely seen in the white matter. Most of the anterior horn cells contained several PAS-positive granules but appeared normal in shape. Some of the anterior horn cells were swollen and filled with the granules (Fig. 1C). Many PAS-positive granules were also observed in the cytoplasm of dorsal root ganglion cells (Fig. 1G). In the posterior horn, few PAS-positive granules were found (Fig. 2H). At 56 weeks (early clinical stage), swollen axons (spheroids) containing PAS-positive granules became frequent, and some of the PAS-positive granules were large and irregularly shaped (Fig. 2B). In the posterior horn, PAS-positive granules were increased in number, and spheroids filled with PAS-positive granules and small vacuoles became apparent (Fig. 2I). At 100 weeks (late clinical stage), large spheroids with or without PAS-positive granules and large vacuoles were frequently observed in the spinal cord (Figs. 1H, 2J). Severe degeneration of proximal axons was apparent, and many PAS-positive granules were found in remaining large fibers (Fig. 2C).
Figure 1.
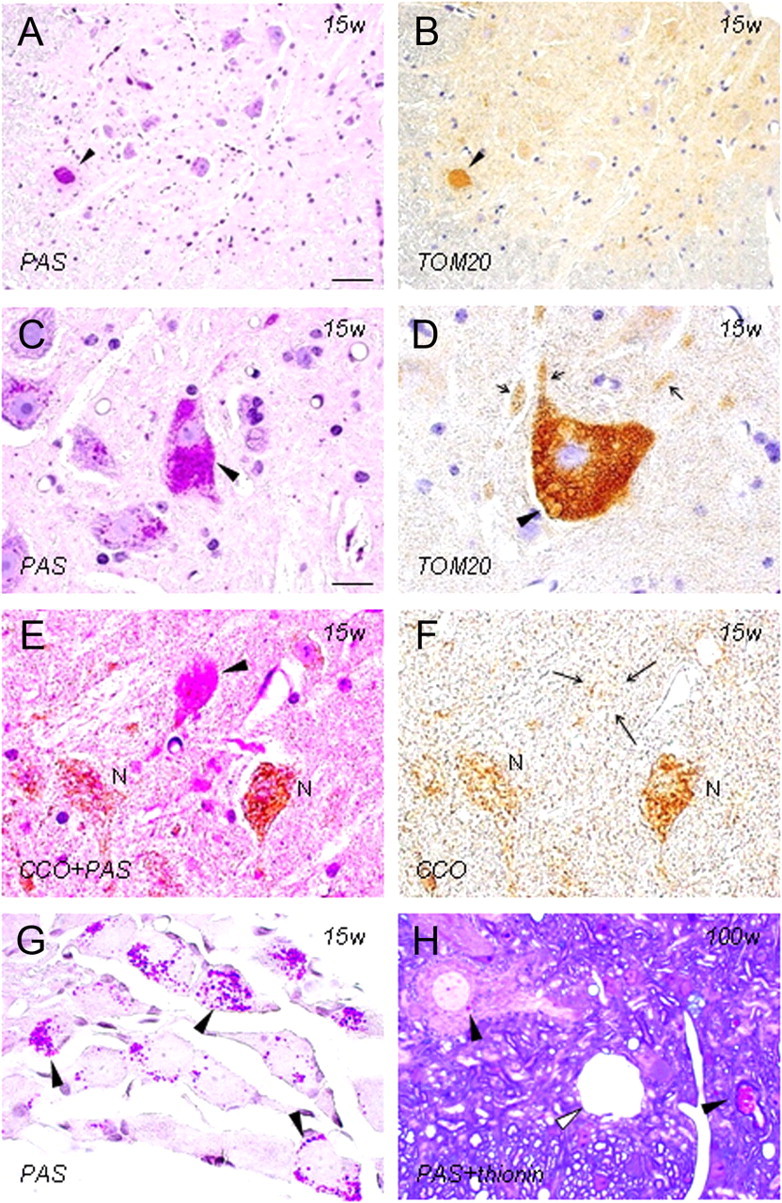
Immunohistochemistry of PAS-positive granules as components of mitochondrial membranes in iPLA2β-KO mice. A–G, 15 weeks; H, 100 weeks; A–F, H, anterior horn; G, dorsal root ganglia; A, C, G, PAS staining; B, D, immunohistochemistry for TOM20; E, double staining with PAS and immunohistochemistry for CCO; F, immunohistochemistry for CCO; H, double staining with PAS and thionin. A and B are serial sections, and E and F are the same section. A, B, A strongly PAS-positive anterior horn cell (arrowhead in A) shows strong positivity for TOM20 (arrowhead in B). C, An anterior horn cell (arrowhead) is filled with PAS-positive granules. Apparently normal anterior horn cells also contain some PAS-positive granules. D, There are many vesicles whose rims are positive for TOM20 (arrowhead) in the anterior horn cell and in neurites (arrows). E, F, An anterior horn cell (arrowhead in E) filled with PAS-positive granules is almost negative for CCO (arrows in F). The cytoplasm of other anterior horn cells (N), which contain a few PAS-positive granules, is stained for CCO. G, There are many PAS-positive granules in the cytoplasm of dorsal root ganglion cells (arrowheads). H, PAS-positive granules are evident in the perinuclear space of anterior horn cells and myelinated axons (arrowheads). A large vacuole (white arrowhead) is present in the neuropil. Scale bars: (in A) A, B, 40 μm; (in C) C–H, 20 μm.
Figure 2.
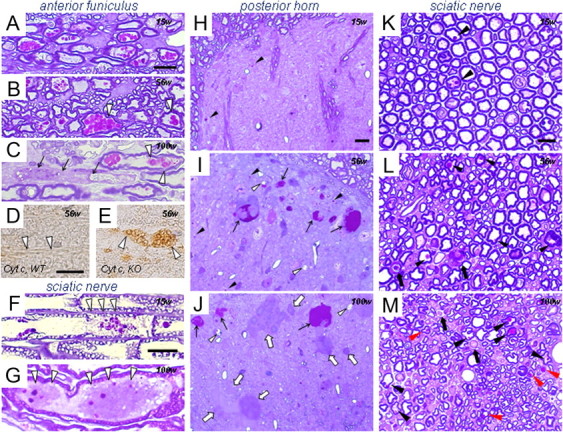
PAS-positive granules in proximal and distal parts of axons in iPLA2β-KO mice. A, F, H, K, 15 weeks; B, D, E, I, L, 56 weeks; C, G, J, M, 100 weeks. A–E, Anterior funiculus; F, G, K–M, sciatic nerve; H–J, posterior horn. Semithin Epon sections stained with PAS and thionin (A–C, F–M) and frozen sections immunostained for cyt c (D, E). A, Strongly PAS-positive granules are evident in proximal axons. B, Large PAS-positive granules are frequently present in swollen axons (white arrowheads). C, Degeneration of a proximal axon (arrows), with PAS-positive granules still evident in remaining axons. Some of these granules are irregular in shape and color (white arrowheads). D, E, Many vesicles strongly immunopositive for cyt c (white arrowheads) are evident in the proximal axons in iPLA2β-KO mice (E) but are not observed in WT mice (white arrowheads in D). F, Focal axonal degeneration (white arrowheads) around vacuolated or irregular PAS-positive granules. G, Two spheroids (white arrowheads) containing PAS-positive granules. H, Only a few PAS-positive granules (arrowheads) are evident. I, Large numbers of PAS-positive granules (arrowheads) and PAS-positive spheroids (arrows). Small vacuoles (white arrowheads) are also evident. J, PAS-negative spheroids (white arrows) are frequent, in addition to PAS-positive spheroids (arrows). Vacuoles (white arrowheads) are larger than those at 56 weeks in I. K, A few large fibers contain PAS-positive granules (arrowheads) in the axon. L, The number of large fibers is apparently reduced, and myelin ovoids (arrows) and dark axons are frequently evident. PAS-positive granules (arrowheads) are increased in number compared with the situation at 15 weeks (K). M, Reduction of large fibers is severe. Myelin ovoids are present (arrows). PAS-positive granules of various sizes are evident in both large (black arrowheads) and small (red arrowheads) fibers. Scale bars: (in A) A–C, 10 μm; (in D) D, E, 10 μm; (in F) F, G, 10 μm; (in H) H–J, 10 μm; (in K) K–M, 10 μm.
Histochemistry of the sciatic nerve
In transverse sections from control mice at 100 weeks, few myelin ovoids were found, and myelin of large fibers was often redundant. In iPLA2β-KO mice at 15 weeks, myelin splitting was frequently evident, and myelin ovoids were rare. A few PAS-positive granules were observed in large fibers (Fig. 2K). In longitudinal sections, focal axonal degeneration was detected around the vacuolated PAS-positive granules (Fig. 2F). In transverse sections of iPLA2β-KO mice at 56 weeks, large fibers were apparently decreased in number, and myelin ovoids and dark axons were evident (Fig. 2L). Large fibers with redundant, split, or thin myelin were frequent. PAS-positive granules were increased in both number and size. At 100 weeks, axonal degeneration had become more severe (Fig. 2M). PAS-positive granules were observed in both large and small fibers and also in spheroids (Fig. 2G). In longitudinal view, nodal demyelination was not evident in iPLA2β-KO mice at any age.
Progressive increase of PAS-positive granules and axonal degeneration in the distal parts of axons
In both posterior horns of the lumbar cord and sciatic nerves of iPLA2β-KO mice, the number of PAS-positive granules was very small at 15 weeks (n = 2), second largest at 56 weeks (n = 4), and largest at 100 weeks (n = 5), with statistical significance (p < 0.05, Wilcoxon's rank-sum test) (Fig. 3A,D). Spheroids in the posterior horns were significantly more frequent at 100 weeks than at 56 weeks (p < 0.05, Wilcoxon's rank-sum test) (Fig. 3B). In iPLA2β-KO mice, there was no significant difference in the number of vacuoles in the posterior horns at 56 and 100 weeks (Fig. 3C). Myelin ovoids in sciatic nerves were very few at 15 weeks and were observed more frequently at 100 weeks than those at 56 weeks (Fig. 3E). Large fibers were apparently reduced in number at 56 and 100 weeks, and their number was significantly smaller at 100 weeks than at 56 weeks (p < 0.05, Wilcoxon's rank-sum test) (Fig. 3F).
Figure 3.
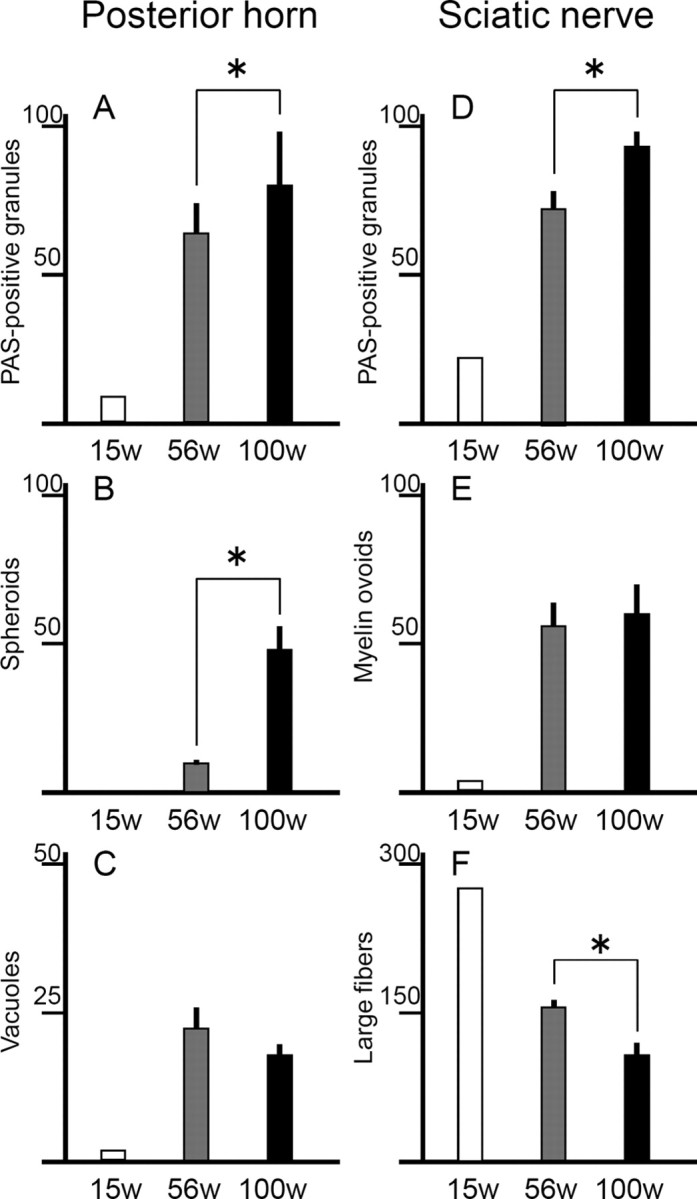
Progressive increase of PAS-positive granules and axonal degeneration in distal parts of axons in iPLA2β-KO mice. A–C, Posterior horns; D–F, sciatic nerves. For iPLA2β-KO mice, white bars represent data at 15 weeks (n = 2, mean), gray bars data at 56 weeks (n = 4, mean ± SD), and black bars data at 100 weeks (n = 5, mean ± SD). *p < 0.05, Wilcoxon's rank-sum test. A, D, In both posterior horns and sciatic nerves, PAS-positive granules are significantly more frequent at 100 weeks than at 56 weeks (p < 0.05, Wilcoxon's rank-sum test). B, In the posterior horns, spheroids are significantly more frequent at 100 weeks than at 56 weeks (p < 0.05, Wilcoxon's rank-sum test). C, The number of vacuoles (>5 μm) in the posterior horn of iPLA2β-KO mice does not differ significantly between 56 and 100 weeks. E, The number of myelin ovoids in sciatic nerves of iPLA2β-KO mice does not differ significantly between 56 and 100 weeks. F, Large fibers in sciatic nerves are significantly fewer at 100 weeks than at 56 weeks (p < 0.05, Wilcoxon's rank-sum test).
Immunohistochemical analysis of mitochondrial markers
At 15 weeks, the cytoplasm of swollen anterior horn cells filled with PAS-positive granules (Fig. 1A,C) was strongly positive for TOM20 in serial sections (Fig. 1B). There were many vesicles whose rims were strongly positive for TOM20 (Fig. 1D). PAS and immunohistochemical double staining showed that most of the PAS-positive granules in swollen anterior horn cells were strongly stained for TOM20 but negative for CCO (Fig. 1E,F). Overall, the immunoreactivities for TOM20 and CCO in the spinal cord were not apparently reduced at 15, 56, or 100 weeks (data not shown). In iPLA2β-KO mice at 15, 56 (Fig. 2E), and 100 weeks, many vesicles were strongly immunopositive for cytochrome c in proximal axons, but no such vesicles were observed in WT mice (Fig. 2D).
Increase of 4-HNE in spinal cords from iPLA2β-KO mice
The increase of 4-HNE was observed mainly in the white matter of the spinal cords of iPLA2β-KO mice from 15 weeks (Fig. 4C) and became prominent with age (Fig. 4D–G), whereas no staining was observed in WT mice (Fig. 4A,B). Some of the distal parts of axons were immunopositive for 4-HNE (Fig. 4C,E,G), and some spheroids and vacuoles were also immunostained in the anterior horns (Fig. 4H). However, most of the large spheroids in the posterior horns (Fig. 4G) and axons in the anterior root (Fig. 4I) were negative for 4-HNE. The distributions of PAS-positive granules (Fig. 1A,C,H, 2A–C,H–J) were not correlated with those of 4-HNE.
Figure 4.
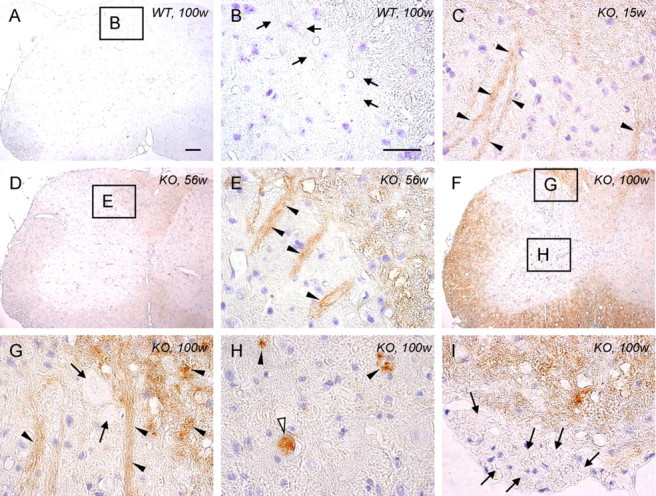
High expression of 4-HNE in the spinal cord of iPLA2β-KO mice. A, B, WT mice at 100 weeks; C, KO mice at 15 weeks; D, E, KO mice at 56 weeks; F–I, KO mice at 100 weeks. A, B, No staining is evident in the control. B is a high-magnification view of the square in A. Axons in the posterior horn are negative for 4-HNE (arrows). C, Some of the axons are immunopositive for 4-HNE (arrowheads). D, E, The white matter and the axons in the posterior horn (arrowheads in E) are more strongly immunopositive for 4-HNE than at 15 weeks (C). E is a high-magnification view of the square in D. F–H, The white matter is more strongly immunopositive for 4-HNE than at 56 weeks (D, E). G and H are high-magnification views of the top and bottom squares in F, respectively. The axons are strongly immunopositive for 4-HNE (arrowheads in G), but large spheroids in the posterior horn are negative (arrows in G). The vacuoles (arrowheads in H) and the spheroid (white arrowhead in H) are strongly positive for 4-HNE. I, Axons in the anterior root are negative for 4-HNE (arrows). Scale bars: (in A) A, D, F, 100 μm; (in B), C, E, G–I, 10 μm.
Ultrastructural analysis of the anterior horn and anterior funiculus
In iPLA2β-KO mice at 15 weeks, abnormal mitochondria were frequently found in the perinuclear space of large neurons and large myelinated axons in the anterior horns (Fig. 5A,B) but were rare in the posterior horns. The cristae were partly or almost wholly diminished, and dense granules occupied the spaces created as a result (Fig. 5A). The structure of the cristae surrounding the granules appeared normal. Also there were many severely degenerated mitochondrion-like structures containing numerous dense granules, which were enclosed by a single membrane (Fig. 5B). The abnormal mitochondria were 0.5–1.5 μm in diameter and, in terms of size and distribution, appeared very similar to PAS-positive granules, which were observed microscopically after PAS staining. At 56 weeks, many dense granules were scattered around the collapsed abnormal mitochondria with degenerated cristae, and focal loss of axonal cytoskeletal elements was also evident in the vicinity (Fig. 5C). Another type of mitochondrial abnormality, characterized by diffusely degenerated, branching, and tubular cristae, was frequently evident (Fig. 6A,B). This type of degenerated mitochondrion was observed also in the axons of the white matter. Tubulovesicular structures, similar in size to abnormal mitochondria, were also present near them in spheroids (Fig. 6C). Collapse of the abnormal mitochondria without dense granules was also accompanied by focal disappearance of the axonal cytoskeleton (Fig. 6D). At 100 weeks, degenerated axons and abnormal mitochondria became more frequent. Some abnormal mitochondrion-like structures, with many dense granules enclosed by a single membrane, were accumulated on one side of the axon and appeared static (Fig. 5D). Most of the myelinated or unmyelinated spheroids were filled with tubulovesicular structures and abnormal mitochondria with degenerated inner membranes (Fig. 6E,F).
Figure 5.
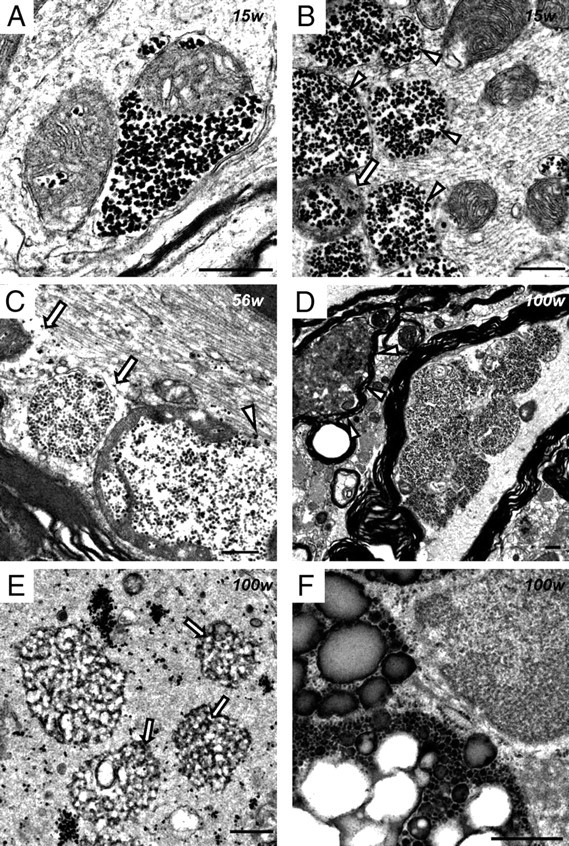
Electron microscopic observation of abnormal mitochondria with dense granules in iPLA2β-KO mice. A, B, 15 weeks; C, 56 weeks; D–F, 100 weeks; A–D, anterior horn; E, F, posterior horn. A, A proportion of the mitochondrial cristae are diminished, and dense granules occupy the resulting space. The outer membrane is morphologically preserved. B, An abnormal mitochondrion containing dense granules (arrow) and abnormal mitochondrion-like structures (arrowheads) with a single membrane enclosing many dense granules. Neurofilaments are mostly preserved. C, Dense granules are released from abnormal mitochondria with broken membranes (white arrowhead). Focal loss of the axonal cytoskeleton is evident around abnormal mitochondria with degenerated cristae (white arrows). D, Several abnormal mitochondrion-like structures have accumulated on one side of the axon. A highly degenerated axon is also evident (white arrowheads). E, Dense granules are gathered or scattered in spheroids. Small and round tubulovesicular structures contain dense granules (arrows). F, Spheroid containing three abnormal aggregates consisting of various-sized dense granules. The smallest one resembles an abnormal mitochondrion-like structure (B–D) but has no membrane around it. Scale bars, 500 nm.
Figure 6.
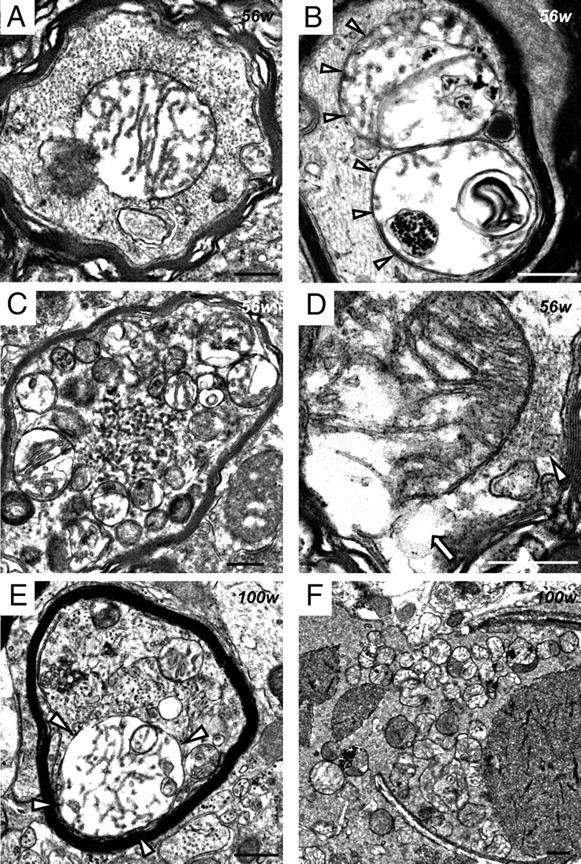
Abnormal mitochondria with tubular and branching cristae and tubulovesicular structures in iPLA2β-KO mice. A–D, 56 weeks; E, F, 100 weeks. A–F, Anterior horn. A, A myelinated fiber contains abnormal mitochondria, whose cristae are diffusely degenerated, tubular, and branching. Neurofilaments are mostly preserved. B, Dense granules, degenerative membranes, and tubular and branching cristae are evident in the severely degenerated mitochondria (white arrowheads). C, A swollen axon is filled with round tubulovesicular structures and similar-sized abnormal mitochondria. D, There is focal loss (white arrow) of the axonal cytoskeleton (white arrowhead) near the broken membrane of an abnormal mitochondrion. E, A myelinated axon is entirely filled with tubulovesicular structures. Abnormal mitochondria with tubular and branching cristae are also evident (white arrowheads). F, Many abnormal mitochondria and various-sized round tubulovesicular structures are evident in a large spheroid. Scale bars, 500 nm.
Ultrastructural analysis of the posterior horn
At 15 weeks, abnormally expanded and loose presynaptic membranes containing synaptic vesicles were observed (Fig. 7A). In some axon terminals, parts of the plasma membranes and synaptic vesicles had disappeared, accompanied by degenerative membranous structures and apparently normal mitochondria (Fig. 7B). Tubulovesicular structures sometimes followed degenerative axons of myelinated small fibers (Fig. 7C). After 56 weeks, vacuoles containing degenerative membranes and mitochondria were frequently evident. Irregularly shaped spheroids 3–5 μm in diameter were a common feature, and these contained various amounts of dark mitochondria, numerous dense granules, degenerative membranes, and tubulovesicular structures (Fig. 5E). Abnormal aggregations, which contained many dense granules of various sizes without limiting membranes, were also found in spheroids (Fig. 5F). Irregularly shaped and large spheroids were also evident at the ends of degenerated small fibers (Fig. 7D). Occasionally, large spheroids containing differing densities of tubulovesicular structures were attached to each other (Fig. 7E,F). Spheroids with a lower density of tubulovesicular structures contained degenerative axoplasm and dark mitochondria (Fig. 7F). Newly formed abnormal mitochondria were rare in the posterior horn.
Figure 7.
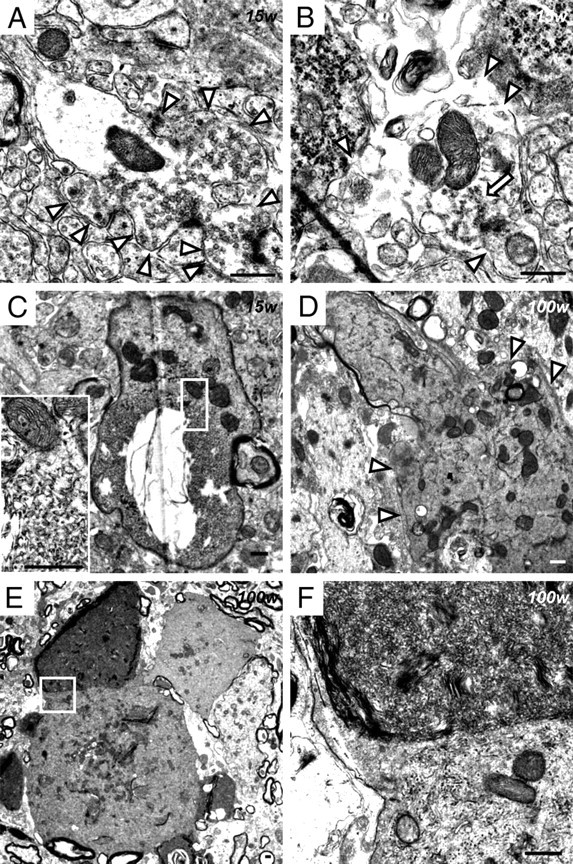
Electron microscopic observation of presynaptic membranes and tubulovesicular structures at the axon terminals of iPLA2β-KO mice. A–C, 15 weeks; D–F, 100 weeks. A–F, Posterior horn. A, An abnormally expanded and loose presynaptic membrane (white arrowheads). Synaptic vesicles, apparently normal mitochondria, and postsynaptic densities are evident. B, Most of the presynaptic membranes have disappeared (white arrowheads). Degenerative membranous or vesicular structures (white arrow) and apparently normal mitochondria are evident. C, The tubulovesicular structures are followed by a degenerative axon containing dark mitochondria in a myelinated small fiber. The inset is a high-magnification view of the white square, which shows tubulovesicular structures and mitochondria containing dense granules. D, A large and irregular spheroid (white arrowheads), containing tubulovesicular structures, degenerative membranes, and mitochondria, has formed at the axon terminal of a degenerated small fiber. E, A large spheroid contains tubulovesicular structures of various densities. F, High-magnification view of the white square in E. Two types of tubulovesicular structures are attached to each other. The top one shows a high density of tubulovesicular structures, whereas the bottom one has a low density of tubulovesicular structures and contains mitochondria. Scale bars, 500 nm.
Differences in phospholipids and fatty acids between WT and KO mice demonstrated by LC/ESI-MS/MS
Among PC species, signals for one containing DHA (16:0/22:6) and one containing AA (16:0/20:4) were prominently increased, whereas one containing oleic acid (OA) (18:0/18:1) was decreased, in the spinal cords of iPLA2β-KO mice (Fig. 8A). Other PC species, such as PC (16:0/16:0), PC (16:0/16:1), PC (18:0/20:4), PC (18:0/22:6), and PC (18:1/22:6), were also increased in iPLA2β-KO mice (Fig. 8A). Moreover, all five PE species analyzed, including PE (1-alkenyl-18:1/18:1), PE (1-alkenyl-18:0/18:1), PE (1-alkenyl-18:0/20:4), PE (18:0/20:4), and PE (1-alkenyl-18:0p/22:6), were increased in iPLA2β-KO mice (Fig. 8B). PE (18:0/20:4) was the most heavily accumulated species in analyzed PEs (Fig. 8B). Moreover, increases in the six known major molecular species of CLs (Sparagna et al., 2005) were detected in iPLA2β-KO mice (Fig. 8C). Among them, CL (18:2/18:2/20:4/22:6), CL (18:0/18:2/22:6/22:6), and CL (18:2/18:2/18:2/22:6) showed large increases, and the remaining three species, CL (18:1/18:2/20:4/20:4), CL (18:1/18:2/18:2/22:6), and CL (18:2/20:4/20:4/20:4), showed mild increases (Fig. 8C).
Figure 8.
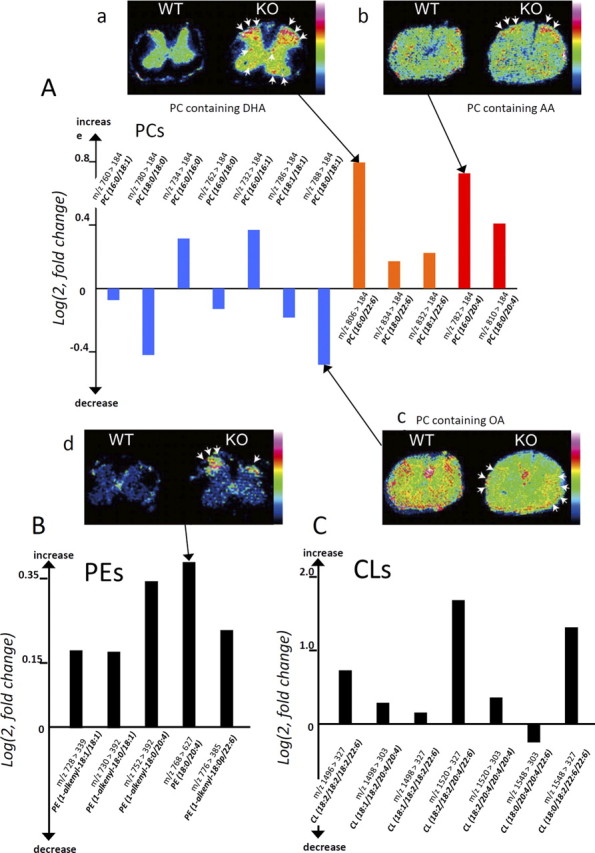
Quantitative lipid profiling of spinal cords from iPLA2β-KO mice and wild-type mice. In the graphs (A–C), relative abundances of PCs (A), PEs (B), and CLs (C) in iPLA2β-KO mice at 56 weeks, which were quantified by liquid chromatography/electrospray ionization tandem mass spectrometry in multiple reaction monitoring mode, are shown by bars, respectively. The vertical axis represents the logarithmic value of the detected intensity ratio (KO/WT), i.e., log(2, Intensity KO/Intensity WT). The bar with a positive value indicates an increase in the KO, whereas one with a negative value indicates a reduction. Blue bars represent PC species containing saturated or monounsaturated fatty acids, orange bars those containing DHA, and red bars those containing AA. In the figures (a–d), the distributions of lipid species that showed notable differences between KO mice and WT mice at 102 weeks are demonstrated by matrix-assisted laser desorption/ionization-imaging mass spectrometry. A-a, Among PC species, one containing DHA is prominently increased in the gray matter of a KO mouse, especially in the posterior horn (arrows), compared with a WT mouse. A-b, A PC species containing AA is accumulated in the posterior horn of a KO mouse (arrows). A-c, A myelin constituting PC species, one containing OA, is decreased in the white matter region of a KO mouse (arrows). B, All five analyzed PE species were increased in the KO spinal cord, and the most accumulated species was prominently found in the posterior horn of a KO mouse (arrows in d). C, Six of the seven CL molecular species analyzed were increased.
IMS of the spinal cords of iPLA2β-KO mice
By using mass spectrometry-based molecular detection, MALDI IMS can distinctly visualize the tissue distributions of various species of phospholipids that have different fatty acid compositions (Sugiura and Setou, 2009). In the positive ion mode, an ion signal at m/z 844 was detected and its distribution was visualized. This signal was identified as a DHA-containing PC (diacyl-16:0/22:6), which was clearly increased in the gray matter of the spinal cord of iPLA2β-KO mice, especially in the posterior horn (Fig. 8A-a). IMS analyses also demonstrated an increase of PC containing AA (diacyl-16:0/20:4) in the posterior horn (Fig. 8A-b) and a decrease of PC containing OA (diacyl-18:0/18:1) (Fig. 8A-c), a component of myelin (Sugiura et al., 2009), in the white matter. In the negative ion mode, PE containing PUFAs (diacyl-18:0/20:4) was shown to be increased in the posterior horn of iPLA2β-KO mice (Fig. 8B-d). These results were compatible with those of LC/ESI-MS/MS, but CLs were not detected by IMS, possibly because they were present in excessively small amounts.
Discussion
In the present study, we detected the presence of abnormal mitochondria with degenerated inner membranes from a young age in mice with KO of the group VIA phospholipase A2 (iPLA2β) gene. The collapsed and degenerated mitochondria were accompanied by focal axonal degeneration, and the plasma membranes were also degenerated at the axon terminals. Thus, two kinds of degenerative membranes in axons appeared to be intrinsically associated with the pathomechanism of neuroaxonal dystrophy in iPLA2β-KO mice.
In INAD, spheroids often contain PAS-positive granular material as well as membranous material (Cowen and Olmstead, 1963; Mahadevan et al., 2000), similar to those in iPLA2β-KO mice. The PAS-positive granules in iPLA2β-KO mice were identified ultrastructurally as abnormal mitochondria with degenerated inner membranes. Absence of immunoreactivity for CCO would have been attributable to degeneration of cristae, as reported previously in mice (Kirkinezos et al., 2005). Because PAS stains structures containing a high proportion of carbohydrate macromolecules and the dense granules in abnormal mitochondria resembled glycogen granules ultrastructurally, PAS staining of abnormal mitochondria might be attributable to glycosylated metabolites. Abnormal mitochondria with tubular and branching cristae, similar to those reported in INAD patients (Itoh et al., 1993; Mahadevan et al., 2000), were also observed in iPLA2β-KO mice after 56 weeks. The difference in the structure of abnormal mitochondria might be explained by differences in the transport of metabolites attributable to membrane dysfunction (Sagun et al., 2005). Abnormal mitochondria and similar-sized tubulovesicular structures were frequently colocalized in axons, suggesting that tubulovesicular structures, a pathological hallmark of INAD, might partly originate from the abnormal mitochondria.
The vulnerability of mitochondrial inner membranes in iPLA2β-KO mice might be attributable to rich production of reactive oxygen species (ROS) (McBride et al., 2006) and a rich content of PUFAs that can be readily peroxidized, such as LA in CL (Zhao et al., 2010). Mitochondrial inner membranes would be easily affected because of deficiency of iPLA2β, which can hydrolyze peroxidized fatty acids to repair the membrane phospholipids oxidized by ROS, as reported previously (Zachman et al., 2010; Zhao et al., 2010). Although the evidence of lipid peroxidation in PAS-positive granules was not obtained, the increase of CL demonstrated by LC/ESI-MS/MS suggested insufficient remodeling of mitochondrial inner membranes in iPLA2β-KO mice. Oxidation of CL leads to subsequent loss of mitochondrial membrane potential and the release of cytochrome c and other apoptotic proteins (Seleznev et al., 2006). In fact, liver mitochondria isolated from iPLA2β-KO mice showed increased sensitivity to Ca2+ overload, resulting in easy loss of membrane potential compared with those from control mice (our unpublished data). Such mitochondrial dysfunction in the cytoplasm would finally trigger cell death as a result (Tsujimoto and Shimizu, 2007; Kagan et al., 2009). Many swollen mitochondria, which were strongly immunopositive for cytochrome c in the proximal axons of iPLA2β-KO mice, might release cytochrome c when their membranes are broken. Ultrastructually, the axonal cytoskeleton showed focal disappearance in the proximity of collapsed abnormal mitochondria, suggesting that massive release of cytochrome c and other stress inducers such as ROS and lipid peroxide from the disrupted mitochondrial membranes would injure axons and impair neuronal function.
There have been no reported descriptions of non-neurological symptoms, laboratory findings, or pathological changes in internal organs in INAD (Cowen and Olmstead, 1963; Nardocci et al., 1999), although dysfunction of spermatozoa (Bao et al., 2004), a reduced insulin secretory response (Zhao et al., 2010), and acceleration of age-related changes in bone morphology (Ramanadham et al., 2008) have been reported in adult iPLA2β-KO mice. The relatively mild phenotypes of iPLA2β deficiency in non-neurological tissues suggest that iPLA2β plays an especially important role in the nervous system, although it is widely distributed in various organs (Bao et al., 2004). Mitochondria are micro-organelles integral to all types of eukaryotic cell (McBride et al., 2006), although the lipid and protein compositions of mitochondrial membranes differ among various organs (Stepien et al., 1992). In anterior horn cells, which have marked polarity, long axons, and extensive dendritic networks (Bäumer et al., 2010), mitochondria must meet the high energy demands of neuronal function. Conversely, even in the same neuron, synaptic and nonsynaptic mitochondria differ in their functions and lipid components, the level of CL being higher in the latter than in the former (Kiebish et al., 2008). Selective degeneration of nonsynaptic mitochondria in large neurons of the spinal cord in iPLA2β-KO mice might be associated with the individual characteristics of mitochondria unique to each functional type of cell.
Neuronal mitochondria are transported by axonal flow, connecting to kinesin motor protein with mitofustin 2 on the outer membrane (Misko et al., 2010). Because abnormal mitochondria with degenerated inner membranes were evident in the cytoplasm and proximal axons from an early stage and became significantly more frequent in the distal parts of axons later, they appeared to move to the distal portions very slowly. Prominent degeneration of axons and less marked degeneration of the neuronal cytoplasm in neuroaxonal dystrophy (Cowen and Olmstead, 1963) could be explained by the collapse of abnormal mitochondria in axons after being transported within the cell. However, apparently normal mitochondria were observed at degenerated axon terminals, suggesting that the membranes of axon terminals would also be affected by iPLA2β deficiency, regardless of any mitochondrial dysfunction.
iPLA2β shows differences in enzymatic specificity for certain fatty acids. The specific activity of iPLA2β with LA (18:2n-6), palmitic acid (16:0), OA (18:1n-9), and AA (20:4n-6) esterified at the sn-2 position is 10.0, 4.3, 3.0, and 2.0 μmol · min−1 · mg−1 protein, respectively (Green et al., 2008), suggesting iPLA2β selectivity for LA. Conversely, the net rate of LA release from brain phospholipids, including CLs, is at least five times lower than that of DHA (22:6n-3) from PC (Green et al., 2008), suggesting the importance of iPLA2β for enzymatic release of DHA from PC within the brain. IMS demonstrated a prominent increase of DHA-containing PC in the gray matter, especially the posterior horn, which would have resulted from compensatory production of phospholipids in a background of membrane degeneration and lack of acyl decomposition in PC attributable to iPLA2β deficiency (Basselin et al., 2010). DHA is one of the important PUFAs (Green et al., 2008), possessing double- or triple-bond carbon chains and accounting for the water solubility and liquefaction of membranes (Stillwell and Wassell, 2003). Therefore, an excessive DHA content resulting from iPLA2β deficiency might cause abnormal expansion and degeneration of membranes. Ultrastructurally, the mitochondrial and presynaptic membranes, both of which contain PC with DHA (Omoi et al., 2006; Mitchell et al., 2007), were affected, and tubulovesicular structures were formed in the middle and ends of axons. The vulnerability of mitochondrial and presynaptic membranes might be explained by the selective localization of iPLA2β in mitochondria (Seleznev et al., 2006) and synapses (Ong et al., 2005).
An age-dependent increase of 4-HNE was demonstrated in the axons and white matter of the spinal cord in iPLA2β-KO mice. 4-HNE is considered to be an oxidized secondary product of lipids including CL (Liu et al., 2011) and would reflect mitochondrial dysfunction (Roede and Jones, 2010). Because CL was increased approximately threefold in iPLA2β-KO mice and the distribution of 4-HNE was not correlated with increased PCs with DHA or AA, the accumulation of 4-HNE would be related to insufficient remodeling and degeneration of the mitochondrial membrane, which was ultrastructurally prominent in axons of the white matter. The fact that the distribution of 4-HNE was inconsistent with that of PAS-positive granules, which were identical to abnormal mitochondria containing dense granules, and the distal parts of axons were positive for 4-HNE suggested that 4-HNE might be accumulated in the distal part of degenerated axons that lacked axonal flow for transport of PAS-positive granules and that 4-HNE might not be massively produced until the mitochondrial double membranes had been disrupted. The presence of 4-HNE in axons might also aggravate axonal degeneration by attacking targets within the cell and expanding into the extracellular space (Roede and Jones, 2010).
In conclusion, neuroaxonal dystrophy in iPLA2β deficiency is thought to be caused by two types of pathomechanism, which are associated with insufficient remodeling of the mitochondrial inner membrane and presynaptic membrane of axon terminals. The former mechanism results in insufficient remodeling of the mitochondrial inner membrane, anterograde transport (Fig. 9A1), and collapse of abnormal mitochondria with membrane degeneration in the middle of axons (Fig. 9A2). The latter mechanism results in insufficient remodeling and degeneration of presynaptic membranes at axon terminals (Fig. 9B). Although mitochondrial inner membranes and presynaptic membranes differ from each other, the degeneration of both finally results in the appearance of tubulovesicular structures. Spheroids containing different densities of tubulovesicular structures might originate from the two different types of membrane (Fig. 9B4,B5), i.e., those in transported mitochondria and those in presynapses. Our findings provide new insight into the pathomechanisms of neuroaxonal dystrophy in iPLA2β deficiency and confirm the pathological importance of tubulovesicular structures as a marker of INAD.
Figure 9.
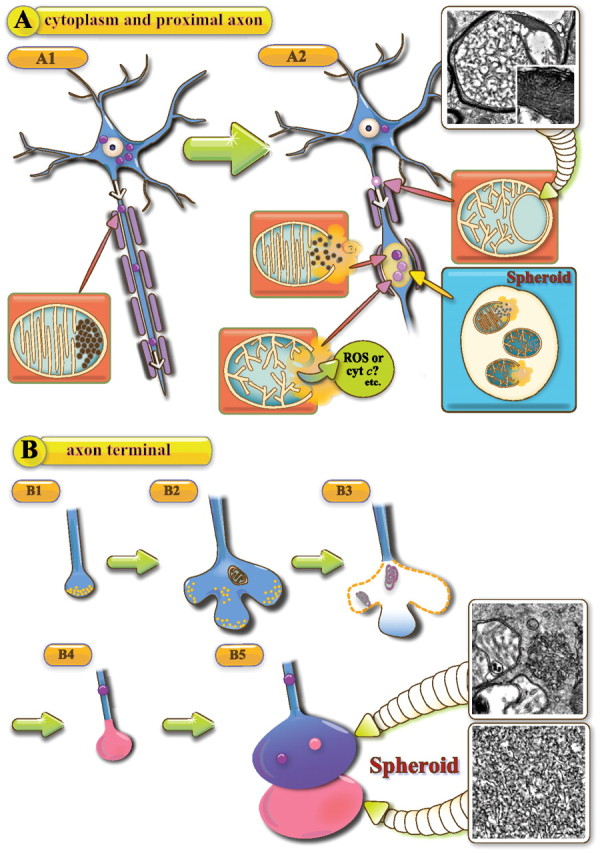
Schematic representation of the hypothetical pathomechanism of neuroaxonal dystrophy in iPLA2β-KO mice. A, Neuronal cytoplasm and proximal axon; B, axon terminal. A1, PAS-positive granules (purple color), which are indistinguishable from abnormal mitochondria, are anterogradely transported from the cytoplasm to the axon (blue color). A2, When abnormal mitochondria collapse, release of cyt c and ROS induces spheroid formation in the middle of the axon. The photograph shows an abnormal mitochondrion containing tubulovesicular and normal cristae at 56 weeks. The square in the photo is a high-magnification view of the white square. B1, Normal axon terminal with synaptic vesicles (yellow color). B2, The presynaptic membrane is abnormally expanded. B3, The presynaptic membrane and synaptic vesicles have disappeared, and degenerative membranes (purple color) are evident in the corresponding area. B4, A spheroid (pink color) with tubulovesicular structures has formed at the axon terminal. B5, Two types of tubulovesicular structures are attached to each other at the axon terminal. The distal one (pink color) shows a high density and the proximal one (blue color) a low density of tubulovesicular structures and PAS-positive granules. The photographs in the squares show two types of tubulovesicular structures observed at 100 weeks. The proximal tubulovesicular structure is followed by a swollen axon.
Footnotes
This study was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) (H.S.-A.), a Grant-in-Aid for Young Scientists (B) (K.S.) from JSPS, Grant S0801035 from the Ministry of Education, Science, Sports and Culture of Japan (S.K.), a Grant-in-Aid for Young Scientists (S) from JSPS (M.S.), a Grant-in-Aid for Creative Scientific Research (Y.T.), and a Health and Labor Sciences Research on Measures for Incurable Disease, Ministry on Health, Labor. and Welfare of Japan (S.S. and S.K.). We thank all members of our laboratory (particularly Chieko Tada and Rie Yoshida) for their technical assistance, discussion, and comments.
The authors declare no competing financial interests.
References
- Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basselin M, Rosa AO, Ramadan E, Cheon Y, Chang L, Chen M, Greenstein D, Wohltmann M, Turk J, Rapoport SI. Imaging decreased brain docosahexaenoic acid metabolism and signaling in iPLA(2) β (VIA)-deficient mice. J Lipid Res. 2010;51:3166–3173. doi: 10.1194/jlr.M008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D, Ansorge O, Almeida M, Talbot K. The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev Mol Med. 2010;12:e21. doi: 10.1017/S1462399410001523. [DOI] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen D, Olmstead EV. Infantile neuroaxonal dystrophy. J Neuropathol Exp Neurol. 1963;22:175–236. doi: 10.1097/00005072-196304000-00001. [DOI] [PubMed] [Google Scholar]
- Engel LA, Jing Z, O'Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One. 2010;5:e12897. doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Green JT, Orr SK, Bazinet RP. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J Lipid Res. 2008;49:939–944. doi: 10.1194/jlr.R700017-JLR200. [DOI] [PubMed] [Google Scholar]
- Gregory A, Hayflick SJ. GeneReviews. Seattle: University of Washington; 2008a. Infantile neuroaxonal dystrophy. [Google Scholar]
- Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, et al. Neurodegeneration associated with genetic defects in phspholipase A2. Neurology. 2008b;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks SB, Cumming BS. Role of Ca2+-independent phospholipase A2 in cell growth and signaling. Biochem Pharm. 2008;76:1059–1067. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Negishi H, Obayashi C, Hayashi Y, Hanioka K, Imai Y, Itoh H. Infantile neuroaxonal dystrophy: immunohistochemical and ultrastructural studies on the central and peripheral nervous systems in infantile neuroaxonal dystrophy. Kobe J Med Sci. 1993;39:133–146. [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, Birk OS. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. 2006;79:942–948. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Han X, Cheng H, Lunceford A, Clarke CF, Moon H, Chuang JH, Seyfried TN. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J Neurochem. 2008;106:299–312. doi: 10.1111/j.1471-4159.2008.05383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A–SOD1 mice. J Neurosci. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Porter NA, Schneider C, Brash AR, Yin H. Formation of 4-hydroxynonenal from cardiolipin oxidation: intramolecular peroxyl radical addition and decomposition. Free Radic Biol Med. 2011;50:166–178. doi: 10.1016/j.freeradbiomed.2010.10.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan A, Santosh V, Gayatri N, Ratnavalli E, NandaGopal R, Vasanth A, Roy AK, Shankar SK. Infantile neuroaxonal dystrophy and giant axonal neuropathy: overlap diseases of neuronal cytoskeletal elements in childhood? Clin Neuropathol. 2000;19:221–229. [PubMed] [Google Scholar]
- Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol. 2008;172:406–416. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a power house. Curr Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TW, Buffenstein R, Hulbert AJ. Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): a comparative study using shotgun lipidomics. Exp Gerontol. 2007;42:1053–1062. doi: 10.1016/j.exger.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, et al. PLA2G6, encoding a phospholipase A2, is mutated neurodegenerative disorders with high brain iron. Nat Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, Verga L, Angelini L, Savoiardo M, Bugiani O. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology. 1999;52:1472–1478. doi: 10.1212/wnl.52.7.1472. [DOI] [PubMed] [Google Scholar]
- Omoi NO, Arai M, Saito M, Takatsu H, Shibata A, Fukuzawa K, Sato K, Abe K, Fukui K, Urano S. Influence of oxidative stress on fusion of pre-synaptic plasma membranes of the rat brain with phosphatidyl choline liposomes, and protective effect of vitamin E. J Nutr Sci Vitaminol (Tokyo) 2006;52:248–255. doi: 10.3177/jnsv.52.248. [DOI] [PubMed] [Google Scholar]
- Ong WY, Yeo JF, Ling SF, Farooqui AA. Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J Neurocytol. 2005;34:447–458. doi: 10.1007/s11068-006-8730-4. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanadham S, Yarasheski KE, Silva MJ, Wohltmann M, Novack DV, Christiansen B, Tu X, Zhang S, Lei X, Turk J. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)-null mice. Am J Pathol. 2008;172:868–881. doi: 10.2353/ajpath.2008.070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roede JR, Jones DP. Reactive species and mitochondrial dysfunction; mechanistic significance of 4-hydroxynonenal. Environ Mol Mutagen. 2010;51:380–390. doi: 10.1002/em.20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagun KC, Cárcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005;19:1657–1667. doi: 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
- Schwartz SA, Reyzer ML, Caprioli RM. Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: practical aspects of sample preparation. J Mass Spectrom. 2003;38:699–708. doi: 10.1002/jms.505. [DOI] [PubMed] [Google Scholar]
- Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa K, Tsujimoto Y. PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J Cell Biol. 2003;163:1219–1230. doi: 10.1083/jcb.200306159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase deficiency in mice: a model of human neurodegenerative disease. J Neurosci. 2008;28:2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Bao S, Lei X, Jin C, Zhang S, Turk J, Ramanadham S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA(2)beta. Biochim Biophys Acta. 2010;1801:547–558. doi: 10.1016/j.bbalip.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparagna GC, Johnson CA, McCune SA, Moore RL, Murphy RC. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res. 2005;46:1196–1204. doi: 10.1194/jlr.M500031-JLR200. [DOI] [PubMed] [Google Scholar]
- Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- Stillwell W, Wassall SR. Docosahexaenoic acid: membrane properties of a unique fatty acid. Chem Phys Lipids. 2003;126:1–27. doi: 10.1016/s0009-3084(03)00101-4. [DOI] [PubMed] [Google Scholar]
- Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+ Br J Pharmacol. 2003;139:1014–1022. doi: 10.1038/sj.bjp.0705326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Setou M. Selective imaging of positively charged polar and nonpolar lipids by optimizing matrix solution composition. Rapid Commun Mass Spectrom. 2009;23:3269–3278. doi: 10.1002/rcm.4242. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Shimma S, Setou M. Thin sectioning improves the peak intensity and signal-to-noise ratio in direct tissue mass spectrometry. J Mass Spectrom Soc Jpn. 2006;54:45–48. [Google Scholar]
- Sugiura Y, Konishi Y, Zaima N, Kajihara S, Nakanishi H, Taguchi R, Setou M. Visualization of the cell-selective distribution of PUFA-containing phosphatidylcholines in mouse brain by imaging mass spectrometry. J Lipid Res. 2009;50:1776–1788. doi: 10.1194/jlr.M900047-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi H, Nagano S, Fujimura H, Kato S, Sakoda S. Inverse correlation between the formation of mitochondria-derived vacuoles and Lewy-body-like hyaline inclusions in G93A superoxide-dismutase-transgenic mice. Acta Neuropathol (Berl) 2006;112:52–63. doi: 10.1007/s00401-006-0056-x. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- Wada H, Yasuda T, Miura I, Watabe K, Sawa C, Kamijuku H, Kojo S, Taniguchi M, Nishino I, Wakana S, Yoshida H, Seino K. Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in Pla2g6. Am J Pathol. 2009;175:2257–2263. doi: 10.2353/ajpath.2009.090343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachman DK, Chicco AJ, McCune SA, Murphy RC, Moore RL, Sparagna GC. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J Lipid Res. 2010;51:525–534. doi: 10.1194/jlr.M000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K, Turk J, Ma ZA. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151:3038–3048. doi: 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]