

Abstract
A large body of evidence indicates that nitric oxide (NO) and cGMP contribute to central sensitization of pain pathways during inflammatory pain. Here, we investigated the distribution of cyclic nucleotide-gated (CNG) channels in the spinal cord, and identified the CNG channel subunit CNGA3 as a putative cGMP target in nociceptive processing. In situ hybridization revealed that CNGA3 is localized to inhibitory neurons of the dorsal horn of the spinal cord, whereas its distribution in dorsal root ganglia is restricted to non-neuronal cells. CNGA3 expression is upregulated in the superficial dorsal horn of the mouse spinal cord and in dorsal root ganglia following hindpaw inflammation evoked by zymosan. Mice lacking CNGA3 (CNGA3−/− mice) exhibited an increased nociceptive behavior in models of inflammatory pain, whereas their behavior in models of acute or neuropathic pain was normal. Moreover, CNGA3−/− mice developed an exaggerated pain hypersensitivity induced by intrathecal administration of cGMP analogs or NO donors. Our results provide evidence that CNGA3 contributes in an inhibitory manner to the central sensitization of pain pathways during inflammatory pain as a target of NO/cGMP signaling.
Introduction
Pain caused by tissue injury and inflammation is accompanied by sensitization of the nociceptive system. This sensitization is driven by peripheral and central mechanisms and manifests as pain in response to normally innocuous stimuli (allodynia), increased responses to noxious stimuli (hyperalgesia), or spontaneous pain, and it can persist long after the initial injury is resolved (Woolf, 2004; Basbaum et al., 2009). Recent evidence indicates that various cGMP signaling pathways in the superficial dorsal horn of the spinal cord essentially contribute to the mechanisms underlying pain sensitization (Schmidtko et al., 2009). One “pain-relevant” cGMP-dependent signaling pathway is initiated by release of nitric oxide (NO) and subsequent cGMP production by NO-sensitive guanylyl cyclase (NO-GC; also called soluble guanylyl cyclase) in lamina I projection neurons and in inhibitory interneurons of the dorsal horn (Ding and Weinberg, 2006; Ruscheweyh et al., 2006; Schmidtko et al., 2008a). Studies with intrathecally (i.t.) administered inhibitors of NO synthases or NO-GC (Luo and Cizkova, 2000) and studies in knock-out mice devoid of NO-GC activity (Schmidtko et al., 2008a) confirmed that NO-mediated cGMP production plays a key role in pain sensitization. However, cGMP can also be synthesized by particulate guanylyl cyclases, and recent studies indicate that in primary afferent neurons cGMP is produced by particulate guanylyl cyclase types A and B upon activation by natriuretic peptides (Kishimoto et al., 2008; Schmidtko et al., 2008a; Schmidt et al., 2009; Zhang et al., 2010).
A major target of cGMP in nociceptive processing is cGMP-dependent protein kinase I (cGKI, synonym PKG-I). The contribution of cGKI to pain sensitization is reflected by the reduced inflammatory hyperalgesia in cGKI−/− mice (Tegeder et al., 2004), and by antinociceptive effects of cGKI inhibitors (Tao et al., 2000; Schmidtko et al., 2003). However, there is accumulating evidence that cGMP may also exert antinociceptive effects within the spinal cord independently from cGKI activation (Iwamoto and Marion, 1994; Tegeder et al., 2002, 2004; Schmidtko et al., 2008b). This indicates that, in addition to cGKI, other cGMP targets might be involved in nociceptive signaling.
In addition to cGMP-dependent protein kinases, classical cGMP targets include phosphodiesterases, which hydrolyze cyclic nucleotides, and ion channels. The best characterized class of ion channels that are modulated by cGMP are the cyclic nucleotide-gated cation (CNG) channels, which are opened by direct binding of cyclic nucleotides. Activation of CNG channels results in membrane depolarization and cytosolic Ca2+ influx, and plays a fundamental role in visual and olfactory signal transduction (Kaupp and Seifert, 2002; Craven and Zagotta, 2006; Biel and Michalakis, 2009). In the present study we hypothesized that CNG channels might contribute to the cGKI-independent effects of cGMP during nociceptive signaling. We analyzed the expression of CNG channel subunits in the spinal cord and in dorsal root ganglia (DRGs) and thereby identified the subunit CNGA3 as a putative contributor to the processing of inflammatory pain. In addition, we characterized the nociceptive behavior of mice lacking CNGA3 to further elucidate the role of cGMP in pain sensitization.
Materials and Methods
Animals
Experiments were performed in 6- to 12-week-old mice lacking CNGA3 (CNGA3−/−) and littermate wild-type (WT) control mice of either sex backcrossed onto C57BL/6N background for >10 generations (Biel et al., 1999). Male C57BL/6N mice (Harlan) were additionally used for reverse transcriptase (RT)-PCR analyses. Animals were housed on a 12 h light/dark cycle with standard rodent chow and water available ad libitum. All experiments were approved by the local Ethics Committee for Animal Research.
Real-time RT-PCR
Lumbar spinal cords and DRGs (L4–L6) were rapidly dissected and snap frozen in liquid nitrogen. Total RNA was isolated under RNase-free conditions using RNA isolation kits (for spinal cord, RNeasy Lipid Tissue Mini Kit, Qiagen; for DRGs, RNAqueous Micro Kit, Ambion); DNase was treated for 15 min to minimize genomic DNA contamination and quantified with a NanoDrop ND-1000 spectrophotometer. cDNA was synthesized from 200 ng of RNA using random hexamers of the Verso cDNA kit (Thermo Fisher Scientific). Real-time RT-PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) using TaqMan gene expression assays for CNGA1 (catalog # Mm00833234_m1), CNGA2 (catalog # Mm00432614_m1), CNGA3 (catalog # Mm00802288_m1), CNGA4 (catalog # Mm01278645_m1), CNGB1 (catalog # Mm01249077_m1), CNGB3 (catalog # Mm00489232_m1), and 18S rRNA (catalog # 4310893E), purchased from Applied Biosystems. Reactions (total volume, 10 μl) were performed in triplicate by incubating at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Water controls were included to ensure specificity. Relative expression of target gene levels was determined using the comparative ΔΔCt method, with Ct indicating the cycle number at which the signal of the PCR product crosses an arbitrary threshold set within the exponential phase of the PCR. The amount of target gene mRNA was normalized to 18S rRNA (Chen et al., 2008). Control experiments revealed stable 18S rRNA expression levels in the spinal cord during hindpaw inflammation normalized to GAPDH or β-actin.
Laser microdissection
Lumbar spinal cords L4–L6 were rapidly dissected, frozen in tissue freezing medium on dry ice, and stored at −80°C until sectioning. Serial transverse cryostat sections (16 μm) were cut and collected on PEN (polyethylene-naphthalene, RNase-free) slides (Leica Microsystems). The sections were briefly stained in 1% cresyl violet solution (Sigma-Aldrich) and dehydrated rapidly through 75–100% ethanol. A Leica DM6000B LMD system was used to collect the superficial dorsal horn (laminae I–III) and ventral horn (laminae VII–IX) separately. Total RNA was isolated from the microdissected tissue by using the RNeasy Plus Micro Kit (Qiagen) according to the manufacturer's instructions. Integrity of the isolated RNA was determined using the Agilent 2100 bioanalyzer and RNA 6000 Pico LabChip Kit (Agilent Technologies). Electropherograms demonstrated clear 18S and 28S rRNA peaks and no significant shift of RNA fragments to shorter migration times, indicating high RNA quality [RIN (RNA integrity number) >7]. Real-time RT-PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) as described above using TaqMan gene expression assays for CNGA3 and GAPDH (catalog # Mm99999915_g1; Applied Biosystems). Control experiments revealed stable GAPDH expression levels in the dorsal and ventral horn during hindpaw inflammation.
In situ hybridization
Total RNA from mouse lumbar spinal cord was extracted and transcribed into cDNA using the One Step RT-PCR Kit (Qiagen). The primers 5′-ccaagaggctgtggaaacat-3′ and 5′-ggagcgttcatgttggaaat-3′ were used to synthesize a fragment corresponding to nucleotides 844–1363 of CNGA3 mRNA (NCBI accession number NM_009918) by PCR. For control experiments, a second fragment corresponding to nucleotides 1410–2176 of CNGA3 mRNA was produced using primers 5′-cagttccggaaggtaaccaa-3′ and 5′-ctgtcaggtgagaagccaca-3′. The primers 5′-ggccaaggtgagcggcatgt-3′ and 5′-acttctgcgacacgggcagc-3′ were used to synthesize a fragment corresponding to nucleotides 547–1208 of GABA vesicular transporter (VGAT) mRNA (NCBI accession number NM_009508). Agarose gel electrophoresis of the PCR products presented a single band with the expected size. The PCR products were eluted from the agarose gel using the QIAEX II Agarose Gel Extraction Kit (Qiagen), cloned into the pCR4-TOPO plasmid vector (Invitrogen), and amplified in One Shot TOP10 Chemically Competent E. coli cells (Invitrogen). Then the plasmid-DNA was purified using the QIAprep Spin Miniprep Kit (Qiagen), quantified with a NanoDrop spectrophotometer, and sequenced for verification (LGC Genomics). Restriction digestions of the plasmid DNA were performed by incubation with PmeI and NotI (for CNGA3) or Pvu2 (for VGAT) restriction endonucleases. Finally, sense and antisense probes labeled with digoxigenin (for CNGA3) or fluorescein (for VGAT) were transcribed in vitro using T3 or T7 RNA polymerases (Roche Diagnostics).
Mice were intracardially perfused with 4% paraformaldehyde (PFA) in 0.1 m: PBS, pH 7.4, under deep anesthesia. The lumbar spinal cords and DRGs were dissected, postfixed in 4% PFA for 10 min, cryoprotected into 30% sucrose overnight, and frozen in tissue freezing medium on dry ice. Cryostat sections were cut at a thickness of 16 μm and stored at −80°C until use. Tissue sections were incubated with 3% H2O2 for 5 min, fixed in 4% PFA for 5 min, and prehybridized in hybridization buffer (50% formamide, 5× SSC, 5× Denhardt's solution, 500 μg/ml herring sperm DNA, 250 μg/ml yeast tRNA in nuclease-free water) at 65°C for 1.5–2 h. Digoxigenin-labeled CNGA3 sense or antisense probes (600 ng/ml in hybridization buffer) were incubated at 65°C over 2 nights. In double in situ hybridization experiments, fluorescein-labeled VGAT probes (400 ng/ml) were simultaneously incubated. After hybridization, the slides were washed in 0.2× SSC and PBS, blocked for 1–2 h in blocking buffer (0.12 m maleic acid, 0.15 m NaCl, pH 7.5, 1% Blocking Reagent; Roche Diagnostics), and incubated with alkaline phosphatase-conjugated anti-digoxigenin antibody (1:1000; Roche Diagnostics) at 4°C overnight. Sections were permeabilized in PBS containing 0.1% Tween (PBST) and equilibrated in alkaline phosphatase buffer (0.1 m Tris-HCl, 0.1 m NaCl, 0.05 m MgCl2, pH 9.5, 0.2 mm levamisole, 0.2% Tween). Fluorescent detection of digoxigenin-labeled probes using 2-hydroxy-3-naphthoic acid-2′-phenylanilide phosphate (HNPP; Roche Diagnostics) was performed at room temperature for 15–30 min. In double in situ hybridization experiments, the slides were then blocked for 2 h in blocking buffer and incubated with biotin-conjugated anti-FITC antibody (1:500; AbD Serotec) at 4°C overnight. Then sections were incubated with horseradish peroxidase-conjugated streptavidin for 30 min at room temperature and detected with TSA fluorescein tyramide reagent (1:200 in amplification diluent; PerkinElmer) for 2–6 min. Sections were finally rinsed in PBS and coverslipped.
In in situ hybridization experiments combined with immunohistochemistry, hybridization of CNGA3 was performed as described above. After washing and blocking, sections were incubated with biotin-conjugated anti-digoxigenin antibody (1:500; Abcam) at 4°C overnight, horseradish peroxidase-conjugated streptavidin (1:100; PerkinElmer) for 30 min, and the fluorescent HRP substrate tyramide (1:100 in amplification diluent; TSA Cyanine 3 System, PerkinElmer) for 3–10 min. Then the slides were incubated with mouse anti-NeuN (clone A60; 1:500; Millipore), rabbit anti-glial fibrillary acidic protein (GFAP; 1:500; Millipore), rabbit anti-stathmin-like 2 (STMN2; 1:500; Proteintech), or rabbit anti-glutamine synthetase (1:300; Sigma Aldrich) dissolved in PBST at 4°C for 24–72 h. Finally, sections were incubated for 2 h with Alexa Fluor 488-conjugated secondary antibodies (1:800; Invitrogen), rinsed in PBS, and coverslipped.
Images were taken using an Axio Observer.Z1 microscope (Zeiss) and a monochrome CCD camera (AxioCam Mrm; Zeiss). The microscope was equipped with the Zeiss Apotome oscillating grating in the epifluorescence beam, which resulted in a significant reduction of out-of-focus stray light. In double staining experiments, images taken with different filters were pseudocolored and superimposed using the Zeiss AxioVision 4.7.2 software. Brightness and contrast of the final images were adjusted using Adobe Photoshop CS software.
Cell counting
The number of neurons in the dorsal horn (laminae I–IV) was quantified by an observer blinded to the animal treatment using in situ hybridization for CNGA3 mRNA and VGAT mRNA or immunohistochemistry for NeuN as described above. Lumbar (L4–L5) spinal cord sections at intervals of ∼300 μm (3–4 sections per mouse, 3 mice per group) were stained and images of the dorsal horn were captured under a 10× objective. Using NIH ImageJ software equipped with the MacBiophotonics MBF plugin (McMaster Biophotonics Facility, McMaster University, Hamilton, ON, Canada), the image contrast was adjusted such that the background level just disappeared (command: image > adjust > color threshold). Images were then converted into 8-bit (command: image > type >8-bit), and the cell bodies were selected and counted according to their size using nucleus counter plugin (command: plugins > particle analysis > nucleus counter; smallest particle size, 50; threshold method: otsu). All parameters were validated in control experiments by manual counting. A total of 7106 cells were counted for quantification.
Behavioral testing
Littermate WT and CNGA3−/− mice were used in all behavioral tests. Animals were habituated to the experimental room and were investigated by observers blinded for the genotype and treatment of the animals.
Rotarod test.
Motor coordination was assessed with a Rotarod Treadmill for mice (Ugo Basile) at a constant rotating speed of 32 rpm. All mice had three training sessions before the day of the experiment. The fall-off latency was averaged from five tests and the cutoff time was 120 s.
Hot-plate test.
Mice were placed into a Plexiglas cylinder (diameter, 20 cm; height, 18 cm) on a metal surface maintained at 50, 52, or 54°C (Hot Plate, Ugo Basile). Cutoff times were 60, 40, and 20 s, respectively, to prevent tissue damage. The time between placement and shaking or licking of the hindpaws or jumping off the plate was recorded. Only one test per animal was performed, since repeated measures might cause profound latency changes (Mogil et al., 1999).
Tail-flick test.
Mice were loosely wrapped in a cotton towel, with the tail extended. A radiant heat stimulus (Plantar test, Ugo Basile) was applied to the middle of the tail. The cutoff time was 10 s to prevent tissue injury. The tail-flick latency was calculated as the mean of three measurements with at least 5 min in between.
Dynamic-plantar test.
The mechanical sensitivity of the plantar side of a hindpaw was assessed with an automated von Frey-type testing device (Dynamic Plantar Aesthesiometer, Ugo Basile). The steel rod (2 mm diameter) was pushed against the paw with ascending force until a strong and immediate withdrawal occurred. The force went from 0 to 5 g over a 10 s period and then remained constant at 5 g for a further 10 s (cutoff time 20 s). The paw withdrawal latency was calculated as the mean of five to six consecutive trials with at least 20 s in between.
Formalin test.
Formalin (20 μl of a 0.5% formaldehyde solution) was injected subcutaneously into the dorsal surface of one hindpaw (Hunskaar et al., 1985; Bráz and Basbaum, 2010). The time spent licking the formalin-injected paw was recorded in 5 min intervals up to 60 min after formalin injection.
Zymosan-induced mechanical hyperalgesia.
A zymosan A suspension (15 μl, 5 mg/ml in 0.1 m PBS, pH 7.4; Sigma-Aldrich) was injected into the plantar subcutaneous space of a hindpaw (Meller and Gebhart, 1997), and the paw withdrawal latencies upon mechanical stimulation were measured using the dynamic-plantar test.
Mechanical allodynia after intrathecal drug administration.
For intrathecal drug delivery, a catheter was implanted onto the lumbosacral spinal cord (L4–L5) as described previously (Schmidtko et al., 2008b). The mechanical sensitivity of both hindpaws was assessed using the dynamic-plantar test. Following baseline measurements, 8-(4-chlorophenylthio)guanosine-3′,5′-cyclic monophosphate (8-pCPT-cGMP; 20 nmol; Biolog), 8-(4-chlorophenylthio)guanosine-3′,5′-cyclic monophosphorothioate (Rp-8-pCPT-cGMPS; 20 nmol; Biolog), C-type natriuretic peptide (CNP; 200 ng; Merck), or 1-hydroxy-2-oxo-3-(3-aminopropyl)-3-isopropyl-1-triazene (NOC-5; 10 μg; Axxora) dissolved in 2 μl of 0.9% saline was intrathecally injected, followed by 4 μl of artificial CSF (containing, in mm: 141.7 Na+, 2.6 K+, 0.9 Mg2+, 1.3 Ca2+, 122.7 Cl−, 21.0 HCO3−, 2.5 HPO42−, 3.5 dextrose, bubbled with 5% CO2 in 95% O2 to adjust pH to 7.2). After the experiment, mice were killed and the catheter position was verified by injection of 5 μl of trypan blue. Only animals in which trypan blue marked the lumbar spinal cord were included.
Spared nerve injury model.
Mice were anesthetized with isoflurane, and the tibial and common peroneal branches of the sciatic nerve were ligated and sectioned distally, while the sural nerve was left intact (Decosterd and Woolf, 2000). Mechanical allodynia was determined using the dynamic-plantar test. Cold allodynia was measured on a 10°C cold plate (AHP-1200CPHC; Teca). The time between placement and shaking or flinching the spared nerve injury (SNI)-operated hindpaw was recorded. A 60 s cutoff was used.
Statistics
Statistical analysis was performed with SPSS software using the Student's t test for paired comparisons, or one-way ANOVA for multiple comparisons followed by a Fisher post hoc test. When mice were tested at different time points, we used a repeated-measures ANOVA and differences between groups at each time point were analyzed with a Fisher post hoc test. Rotarod fall-off latencies were analyzed with the Mann–Whitney U test and are expressed as median and interquartile range. All other data are presented as the mean ± SEM. For all tests, a probability value p < 0.05 was considered as statistically significant.
Results
Expression of CNG channel subunits in the spinal cord
Native mammalian CNG channels are tetramers composed of A and B subunits (CNGA1-4, CNGB1, and CNGB3). The main properties of CNG channels are determined by the principal subunits CNGA1-3, whereas the subunits CNGA4, CNGB1, and CNGB3 have modulatory functions (Craven and Zagotta, 2006; Biel and Michalakis, 2009). To assess a potential contribution of CNG channels to nociceptive processing, we first examined the expression levels of CNG channel subunits in the mouse spinal cord by real-time RT-PCR. Two principal CNG channel subunits (CNGA2 and CNGA3) and all three modulatory subunits (CNGA4, CNGB1 and CNGB3) were detected in the spinal cord of naive mice. The relative mRNA expression was 1.6 ± 0.4 (CNGA2), 1.5 ± 0.2 (CNGA3), 3.4 ± 0.3 (CNGA4), 8.6 ± 1.7 (CNGB1), and 1.1 ± 0.2 (CNGB3). To further identify pain-relevant CNG channel subunits, we investigated the mRNA expression in the spinal cord following hindpaw inflammation evoked by the proinflammatory yeast extract zymosan. Interestingly, mRNA levels of CNGA3 were significantly increased in the spinal cord 4–8 h after intraplantar zymosan injection, whereas the expression of other CNG channel subunits was not affected (Fig. 1). These data point to CNGA3 as the most likely candidate in nociceptive processing in the spinal cord.
Figure 1.

Expression of CNG channel subunits in the mouse spinal cord after intraplantar zymosan injection. Data are expressed as mRNA ratio of zymosan-injected mice (tissue extracted 4, 8, and 24 h after injection) relative to untreated control mice whose mRNA expression levels were set to 1 (indicated as a dotted line). CNGA3 mRNA was significantly upregulated at 4 and 8 h after zymosan injection, whereas mRNA of CNGA2, CNGA4, CNGB1, and CNGB3 was not affected. CNGA1 mRNA was not detected. The quantity of CNG channel subunit mRNA relative to 18S mRNA was calculated by the 2−ΔΔCT method. n = 4–6 per group. All data are presented as mean ± SEM. *p < 0.05.
CNGA3 distribution in the spinal cord
We then examined the distribution of CNGA3 in the mouse spinal cord in more detail. We used laser microdissection (Burbach et al., 2003) to isolate superficial dorsal horn (laminae I–III) and ventral horn (laminae VII–IX) spinal cord tissues (Fig. 2A,B), and analyzed the CNGA3 mRNA levels in the isolated tissues by subsequent real-time RT-PCR. As shown in Figure 2C, in naive animals CNGA3 mRNA was expressed to a higher extent in the superficial dorsal horn compared with the ventral horn (relative expression levels 1.0 ± 0.1 and 0.3 ± 0.1, respectively). Of note, induction of hindpaw inflammation by intraplantar zymosan injection significantly increased the CNGA3 mRNA expression in the superficial dorsal horn but not in the ventral horn (Fig. 2C), further suggesting a role of CNGA3 in spinal nociceptive processing.
Figure 2.

CNGA3 expression in the ipsilateral dorsal and ventral horn of the spinal cord after intraplantar zymosan injection. A–C, Spinal cord sections were stained with cresyl violet (A), and tissues of the superficial dorsal horn (laminae I–III) and ventral horn (laminae VII–IX) were collected using laser microdissection (B). Total RNA was isolated from collected tissues and subjected to real-time RT-PCR analysis (C). The quantity of CNGA3 mRNA relative to GAPDH mRNA was calculated by the 2−ΔΔCT method. n = 3 per group. Note that in naive animals, more CNGA3 mRNA was detected in the superficial dorsal horn compared with the ventral horn, and that zymosan-induced CNGA3 upregulation occurs only in the superficial dorsal horn. Scale bar, 250 μm.
We next analyzed the CNGA3 distribution in the spinal cord using nonradioactive in situ hybridization experiments. CNGA3 mRNA was seen throughout the spinal cord, with the highest expression in the superficial dorsal horn (Fig. 3A) and moderate expression in the intermediate spinal cord and the ventral horn. As expected, no hybridization signals were detected using the control sense probe (Fig. 3B). A similar distribution pattern was observed with an alternative antisense probe (see Materials and Methods) targeting a nonoverlapping sequence position of CNGA3 mRNA (data not shown). To characterize the distribution of CNGA3 in the superficial dorsal horn more precisely, we combined in situ hybridization of CNGA3 with immunohistochemistry or in situ hybridization of standard markers. Notably, double in situ hybridization experiments revealed that most CNGA3 mRNA-positive neurons in the superficial dorsal horn also expressed VGAT, a marker of inhibitory neurons (Fig. 3C–E). In contrast, the staining pattern of the astroglial marker GFAP essentially differed from CNGA3 mRNA distribution (Fig. 3F–H), suggesting that CNGA3 is not expressed in astroglial cells.
Figure 3.

Cellular localization of CNGA3 in the spinal cord. A, In situ hybridization using an antisense probe for CNGA3 reveals high levels of CNGA3 mRNA expression within the superficial dorsal horn. B, Specificity of the CNGA3 antisense probe was confirmed by incubating with the respective sense probe directed against the complementary mRNA sequence. Dotted lines delineate gray matter. C–E, Double in situ hybridization of CNGA3 mRNA and VGAT mRNA. F–H, Double labeling by in situ hybridization of CNGA3 mRNA and immunohistochemistry for GFAP. CNGA3 mRNA was detected using HNPP (A, C) or TSA cyanine 3 (F) and appears in red. Markers were detected using TSA fluorescein (D) or Alexa Fluor 488 (G) and appear in green. Pictures C–H were taken from lamina II. Data indicate that CNGA3 is mainly expressed in inhibitory neurons of the superficial dorsal horn. Scale bars: A, 100 μm; C, 10 μm.
We then compared the CNGA3 mRNA distribution in the dorsal horn between naive mice and mice 8 h after intraplantar zymosan injection, i.e., at a time point of increased CNGA3 mRNA levels in the spinal cord. The gross distribution pattern of CNGA3 mRNA in the dorsal horn was similar between naive and zymosan-treated mice. In situ hybridization of CNGA3 combined with immunohistochemistry for NeuN revealed that in both groups the CNGA3 hybridization signals were exclusively found in NeuN-immunoreactive cells, indicating that the zymosan injection did not induce CNGA3 in non-neuronal cells. The proportion of NeuN-immunoreactive cells that coexpress CNGA3 mRNA in the dorsal horn was 42 ± 4% and 45 ± 7% in naive and zymosan-treated mice, respectively. Moreover, the proportion of CNGA3 mRNA-positive cells that coexpress VGAT mRNA was 92 ± 1% in naive mice and 90 ± 1% in zymosan-treated mice. These data suggest that the zymosan-induced upregulation of CNGA3 mRNA mainly occurred in inhibitory neurons, which already express CNGA3 under basic conditions. In summary, our findings demonstrate a neuronal expression pattern of CNGA3 in the spinal cord with high levels in the superficial dorsal horn, and imply that CNGA3 is mainly expressed in inhibitory neurons and upregulated in these neurons after intraplantar zymosan injection.
CNGA3 distribution in DRGs
Real-time RT-PCR analyses revealed that CNGA3 mRNA is also expressed in mouse DRGs, with a relative expression level of 0.4 ± 0.1 compared with the spinal cord. Using in situ hybridization experiments combined with immunohistochemistry of the neuronal marker STMN2, we observed that CNGA3 mRNA expression in DRGs was confined to non-neuronal cells (Fig. 4A,B), in contrast to the spinal cord. No hybridization signals were obtained using the control sense probe (Fig. 4C). Notably, most CNGA3 hybridization signals were detected in close proximity to, and surrounded by, immunoreactivity for the satellite glial cell marker glutamine synthetase (Fig. 4D–F), indicating that CNGA3 mRNA is likely to be expressed in satellite glial cells. Interestingly, mRNA levels of CNGA3 were significantly increased in ipsilateral DRGs 4–24 h after intraplantar zymosan injection (Fig. 4G), similar to the zymosan-induced CNGA3 upregulation in the dorsal horn. Using in situ hybridization experiments in DRGs of mice 8 h after intraplantar zymosan injection, we could not observe an altered staining pattern compared with naive animals or an induction of CNGA3 mRNA in dorsal root ganglion (DRG) neurons. Hence, we conclude that in DRGs, CNGA3 mRNA is constitutively expressed in satellite glial cells and upregulated in these cells during paw inflammation.
Figure 4.

Cellular localization of CNGA3 in dorsal root ganglia. A, B, In situ hybridization experiments detected CNGA3 mRNA (red) in DRGs (A), and double labeling with immunohistochemistry for the neuronal marker stathmin-like 2 (STMN2; green) indicates that CNGA3 mRNA is not expressed in DRG neurons (B). C, Specificity of the CNGA3 antisense probe was confirmed by incubating with the respective sense probe directed against the complementary mRNA sequence. D–F, Double labeling by in situ hybridization of CNGA3 mRNA and immunohistochemistry for the satellite glial cell marker glutamine synthetase (GS; green) suggests CNGA3 to be mainly expressed in satellite glial cells. G, Real-time RT-PCR analyses demonstrating that CNGA3 mRNA was significantly upregulated in DRGs 4–24 h after intraplantar zymosan injection. The quantity of CNGA3 mRNA relative to GAPDH mRNA was calculated by the 2−ΔΔCT method. n = 4 per group. Data are presented as mean ± SEM. *p < 0.05. Scale bar, 20 μm.
Nociceptive behavior in CNGA3−/− mice
To assess the role of CNGA3 in nociceptive processing in vivo, we compared the nociceptive behavior of CNGA3−/− mice with that of littermate WT mice in models of acute, inflammatory, and neuropathic pain. CNGA3−/− mice are viable, fertile, and do not show reduced life expectancy (Biel et al., 1999). The macroscopic morphology of DRGs and the spinal cord, and the distribution of terminals of nociceptive and thermoreceptive primary afferents in the superficial dorsal horn appeared normal in CNGA3−/− mice (data not shown). The mRNA expression of other CNG channel subunits in the spinal cord of CNGA3−/− mice did not significantly differ from that in WT mice (relative mRNA expression in CNGA3−/− mice compared with WT mice: CNGA2, 98.8%; CNGA4, 104.3%; CNGB1, 110.5%; CNGB3, 94.2%, respectively; CNGA1, not detected in CNGA3−/− and WT mice; n = 3 mice per genotype). Furthermore, the motor coordination and balance were not impaired in CNGA3−/− mice, as analyzed in the rotarod test [median fall-off latencies: CNGA3−/− mice, 120.0 s (interquartile range 96.7–120.0 s); WT mice, 120.0 s (interquartile range 120.0–120.0 s); p = 0.301; n = 8 per group).
Acute pain
To determine whether or not acute nociception is altered in CNGA3−/− mice, the latency to acute thermal stimuli (50, 52, and 54°C hot-plate test, tail-flick test) and mechanical stimuli (dynamic-plantar test) was measured. No significant differences in latency times were found between CNGA3−/− and WT mice (Table 1), indicating an intact immediate response to acute noxious thermal and mechanical stimulation in CNGA3−/− mice.
Table 1.
Acute nociception in CNGA3−/− mice
| Model | CNGA3−/− mice | WT mice | p | n |
|---|---|---|---|---|
| Hot-plate test (50°C) | 35.1 ± 5.6 s | 33.6 ± 4.0 s | 0.831 | 8 |
| Hot-plate test (52°C) | 24.5 ± 1.2 s | 25.2 ± 1.5 s | 0.701 | 16 |
| Hot-plate test (54°C) | 11.3 ± 1.1 s | 11.8 ± 1.0 s | 0.738 | 16 |
| Tail-flick test | 4.1 ± 0.1 s | 4.0 ± 0.2 s | 0.664 | 8 |
| Dynamic-plantar test | 8.3 ± 0.3 s | 8.1 ± 0.3 s | 0.620 | 16 |
Inflammatory pain
We then analyzed the behavior of CNGA3−/− mice in models of inflammatory pain. In the formalin test, injection of 0.5% formalin into a hindpaw resulted in the typical phasic nociceptive response consisting of licking the injected paw. In the first phase (1–10 min), which results from direct activation of TRPA1-expressing nociceptors (McNamara et al., 2007), a tendency toward increased paw licking in CNGA3−/− mice compared with WT mice was detected (Fig. 5A), which was, however, not significant (Fig. 5B; p = 0.079). Interestingly, in the second phase of paw licking (11–60 min), which reflects a combination of ongoing sensory input and central sensitization (Vardeh et al., 2009), the licking behavior was increased in CNGA3−/− mice compared with WT mice (Fig. 5A). Statistical analyses revealed a significant difference in phase 2 between groups (Fig. 5B).
Figure 5.
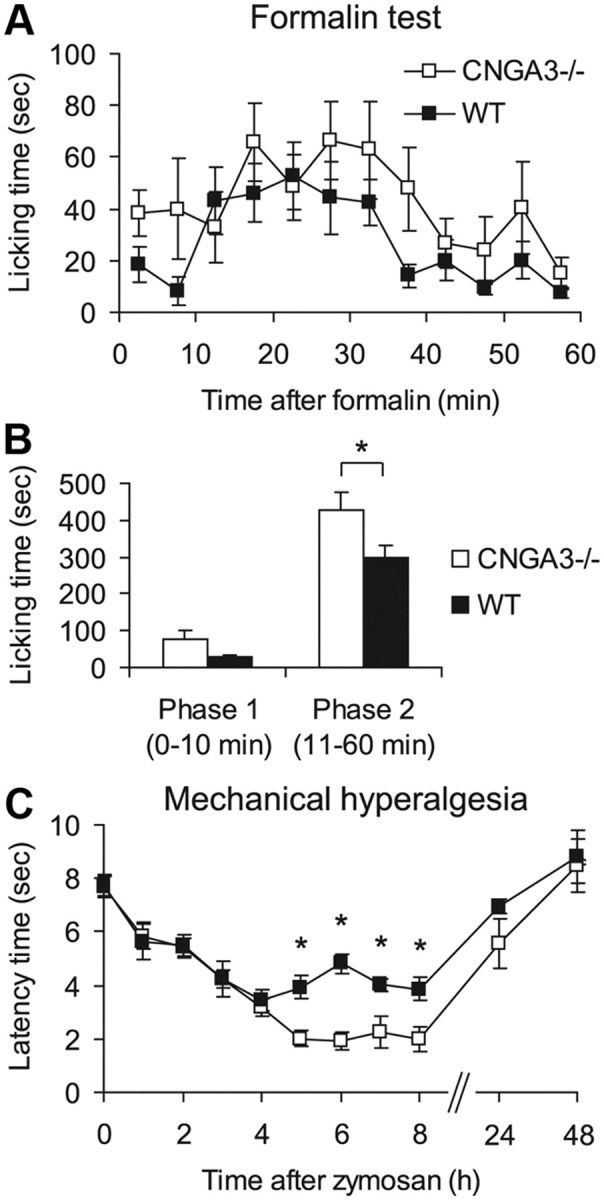
Inflammatory pain in CNGA3−/− mice. A, B, Formalin test. A, Time course of licking behavior of the formalin-injected hindpaw. B, Sum of paw-licking time in phase 1 (0–10 min) and phase 2 (11–60 min) demonstrating increased licking behavior in CNGA3−/− mice. C, Zymosan-induced mechanical hyperalgesia. Time course of paw withdrawal latency time after mechanical stimulation demonstrates increased mechanical hyperalgesia in CNGA3−/− mice after zymosan injection. All data are presented as mean ± SEM. *p < 0.05, comparing CNGA3−/− and WT mice. n = 7–9 per group.
In a second model of inflammatory pain, we analyzed the extent of mechanical hyperalgesia after zymosan injection into a hindpaw. As shown in Figure 5C, hyperalgesia developed similarly in CNGA3−/− and WT mice during the first 4 h after zymosan injection. However, between 4 and 8 h after injection, the extent of hyperalgesia remained nearly constant in WT mice, whereas it was significantly increased in CNGA3−/− mice (Fig. 5C). Together, these data suggest an inhibitory role of CNGA3 during the processing of persistent inflammatory pain.
cGMP-induced pain
To assess the impact of CNGA3 for cGMP signaling in spinal nociceptive processing, we determined the nociceptive behavior of CNGA3−/− and WT mice following intrathecal administration of CNG channel-activating cGMP analogs. Currently available CNG channel-activating cGMP analogs also modulate the activity of cGKI, which essentially contributes to spinal nociceptive processing. Therefore, we assessed two compounds with different cGKI-modulating properties: (1) 8-pCPT-cGMP, an activator of CNG channels and of cGKI, and (2) Rp-8-pCPT-cGMPS, which activates CNG channels but inhibits cGKI (Kramer and Tibbs, 1996). As shown in Figure 6A, injection of 8-pCPT-cGMP (20 nmol, i.t.) evoked mechanical allodynia in both CNGA3−/− and WT mice. However, in CNGA3−/− mice, the extent of mechanical allodynia was significantly increased and lasted for a longer time compared with WT mice. As expected, injection of Rp-8-pCPT-cGMPS (20 nmol, i.t.) did not modulate the paw withdrawal latency time in WT mice (Fig. 6B), confirming earlier reports (Schmidtko et al., 2008a). However, intrathecal Rp-8-pCPT-cGMPS induced mechanical allodynia in CNGA3−/− mice (Fig. 6B). Mechanical allodynia was not observed in both genotypes after intrathecal administration of saline, which was used as negative control (Fig. 6C). These data indicate that CNGA3 contributes in an inhibitory manner to cGMP-mediated nociceptive signaling.
Figure 6.
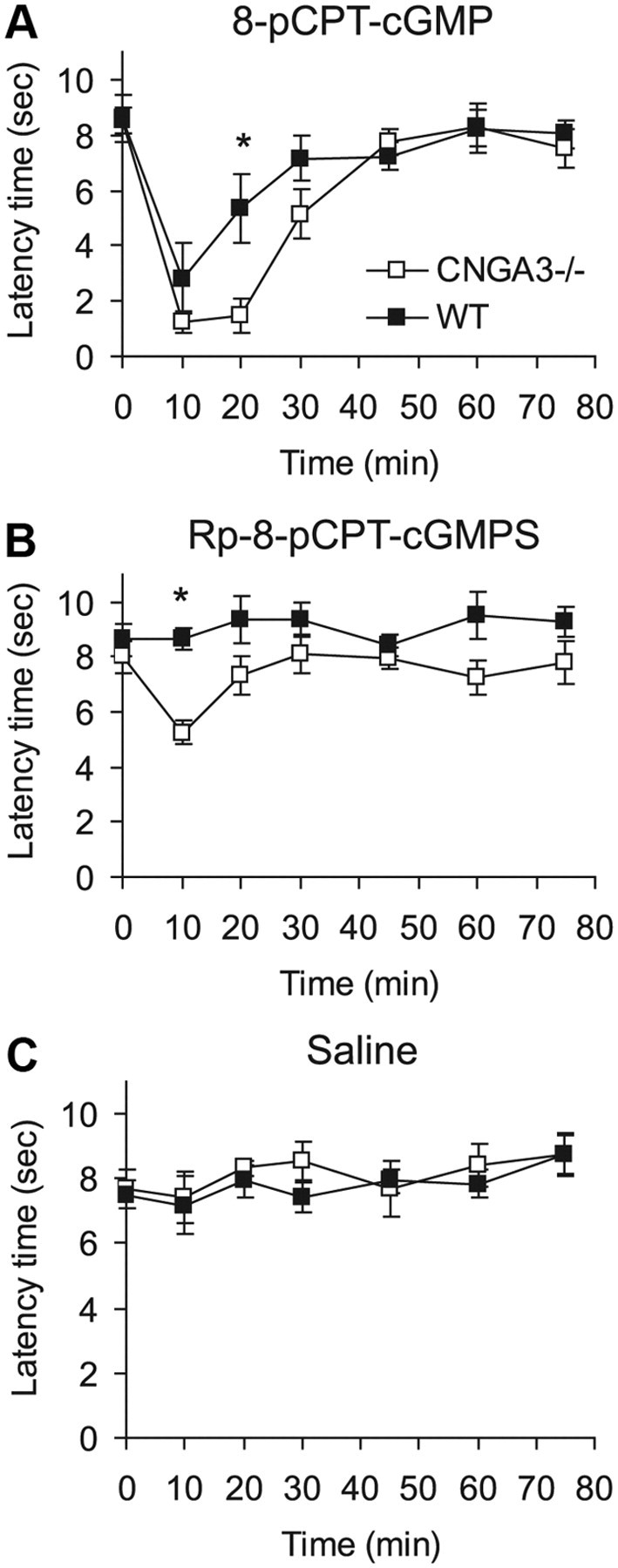
cGMP-induced nociceptive behavior in CNGA3−/− mice. A–C, Time course of mechanical allodynia induced by intrathecal administration of the cGMP analogs 8-pCPT-cGMP (20 nmol; A) and Rp-8-pCPT-cGMPS (20 nmol; B), or of saline (C). Data indicate that the extent of cGMP-induced allodynia is increased in CNGA3−/− mice. All data are presented as mean ± SEM. *p < 0.05, comparing CNGA3−/− and WT mice. n = 6–8 per group.
Natriuretic peptide- and NO-induced pain
During spinal nociceptive processing, cGMP exerts pronociceptive effects after production by at least two different guanylyl cyclases, NPR-B and NO-GC (Schmidtko et al., 2009). To identify the guanylyl cyclase mediating CNGA3 activation in the spinal cord, we assessed the nociceptive behavior of CNGA3−/− and WT mice after intrathecal administration of a NPR-B ligand (CNP) and a NO-GC activator (NOC-5). As shown in Figure 7A, CNP (200 ng, i.t.) evoked mechanical allodynia to a similar extent in CNGA3−/− and WT mice. By contrast, after administration of NOC-5 (10 μg, i.t.), the mechanical allodynia was significantly increased in CNGA3−/− mice compared with WT mice (Fig. 7B). Hence, CNGA3 obviously contributes in an inhibitory manner to spinal pain sensitization evoked by NO donors.
Figure 7.
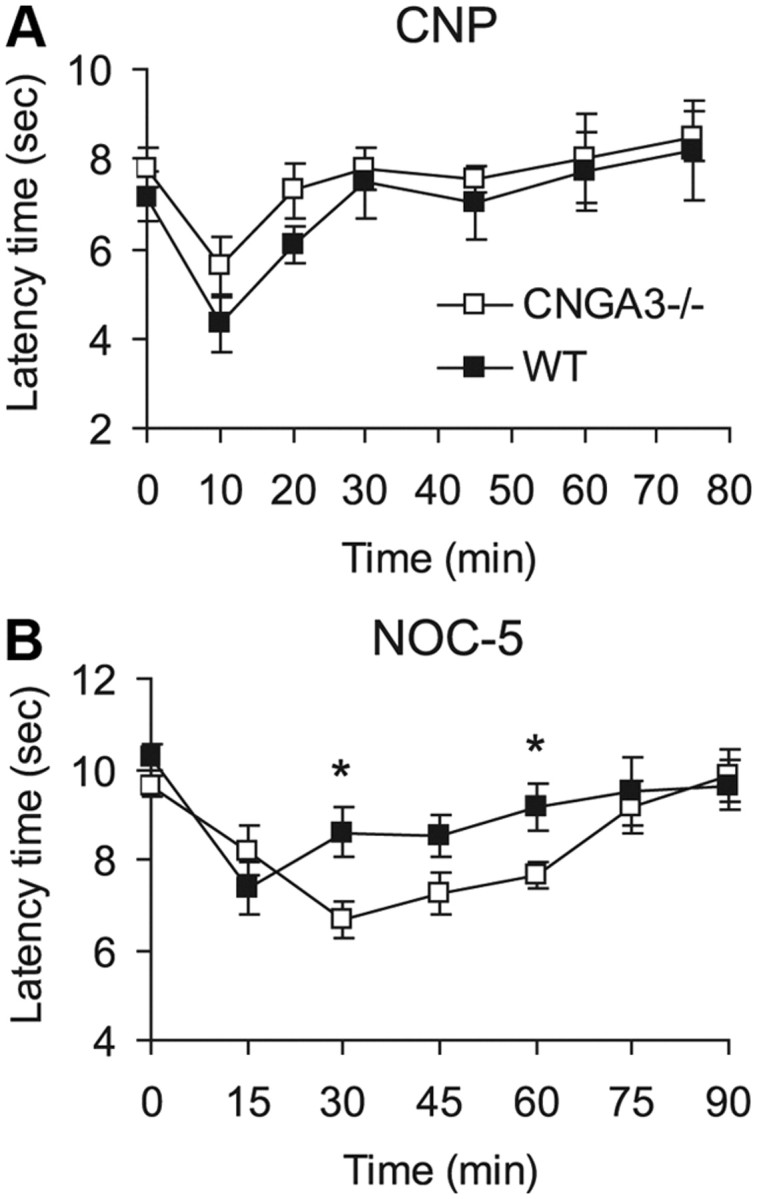
Natriuretic peptide and NO-induced nociceptive behavior in CNGA3−/− mice. A, B, Time course of mechanical allodynia induced by intrathecal administration of CNP (200 ng; A) or NOC-5 (NO donor, 10 μg; B). Data indicate that the extent of NO-induced allodynia, but not that of CNP-induced allodynia, is increased in CNGA3−/− mice. All data are presented as mean ± SEM. *p < 0.05, comparing CNGA3−/− and WT mice. n = 7–9 per group.
Neuropathic pain
The role of CNGA3 in neuropathic pain was tested in the SNI model by analyzing SNI-induced mechanical and cold allodynia. During the entire 28 d observation period, mechanical allodynia of the affected hindpaw developed similarly in CNGA3−/− and WT mice (Fig. 8A). SNI-evoked cold allodynia was also similar in CNGA3−/− and WT (Fig. 8B). Thus, CNGA3 seems not to be critically involved in the development of neuropathic pain after peripheral nerve injury.
Figure 8.
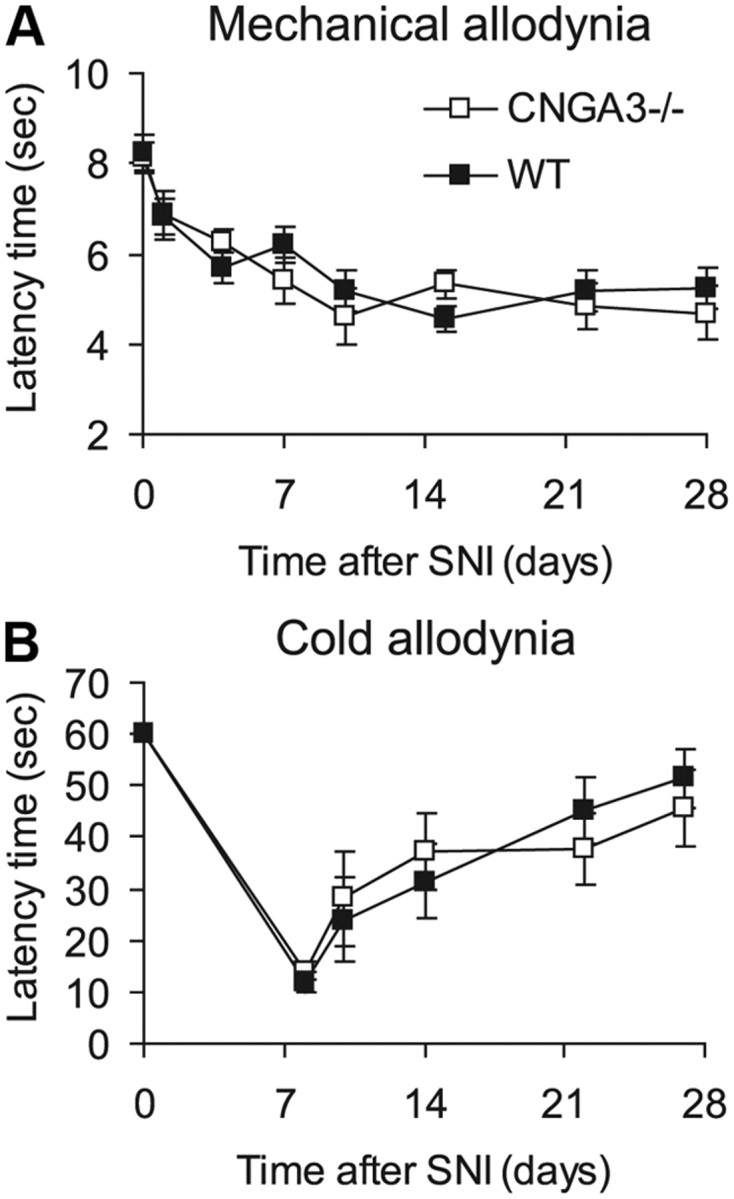
SNI-induced neuropathic pain in CNGA3−/− mice. A, Time course of paw withdrawal latency time after mechanical stimulation. B, Time course of reaction latency time after exposure to a cold plate at 10°C (cutoff time, 60 s). Statistical analyses revealed no significant difference between CNGA3−/− and WT mice. All data are presented as mean ± SEM. n = 8 per group.
Discussion
In the present study, we identified the CNG channel subunit CNGA3 as a novel effector of NO/cGMP signaling in nociceptive processing. CNGA3 is expressed in inhibitory dorsal horn neurons of the spinal cord and in non-neuronal DRG cells. The nociceptive behavior of CNGA3-deficient mice suggests that CNGA3 mediates inhibitory effects during inflammatory pain.
CNG channels are key players in sensory transduction in photoreceptors and olfactory neurons. In addition, CNG channels were detected in other tissues including kidney, heart, sperm cells, endocrine tissues, and brain. However, the functional role of CNG channels in these tissues remains mostly elusive (Kaupp and Seifert, 2002; Biel and Michalakis, 2009). Here, we detected five of six known mammalian CNG channel subunits in the mouse spinal cord based on their mRNA expression levels. Interestingly, induction of hindpaw inflammation by zymosan injection induced an upregulation of the principal CNG channel subunit CNGA3 but not of other subunits in the superficial dorsal horn of the spinal cord. We also observed an upregulation of CNGA3 mRNA in the spinal cord 3–6 d after intrathecal injections of PBS, which presumably induces local transient inflammation (data not shown). These data indicate that CNGA3 might play a particular role in nociceptive processing and during inflammation. In general, CNG channels are composed of four subunits forming a tetramer with a central pore. This raises the question whether homomeric CNGA3 channels may operate in nociceptive processing. It has been shown that in heterologous expression systems, CNGA3 can form functional channels in the absence of other CNG subunits (Weyand et al., 1994; Biel et al., 1996). However, in photoreceptors and olfactory neurons, CNG channels are heteromers composed of different subunits (Biel and Michalakis, 2009). Hence, it remains to be determined whether CNGA3 forms homomeric or heteromeric channels, and whether or not other subunits that have been detected in our study (i.e., CNGA2, CNGA4, CNGB1, and CNGB3) contribute to nociceptive processing.
We used in situ hybridization for CNGA3 mRNA to identify its distribution and cellular localization, because we could not detect a specific CNGA3 protein signal using four different CNGA3 antibodies and various staining protocols (data not shown). Based on immunohistochemical and Western blot experiments in WT and CNGA3−/− mice, the tested commercially available CNGA3 antibodies turned out to be unspecific. Another antibody that specifically detects CNGA3 in the retina (Biel et al., 1999) did not detect CNGA3 in the spinal cord, nor in DRGs. Lower CNGA3 expression levels or steric effects that impair the antibody binding may account for the lack of CNGA3 protein detection. In the spinal cord, we observed that most CNGA3 mRNA-positive cells also expressed mRNA of the vesicular GABA transporter VGAT, indicating that CNGA3 is localized to inhibitory interneurons. Thus, it is tempting to speculate that cGMP produced in inhibitory interneurons of the dorsal horn might stimulate CNG channels expressing CNGA3. In contrast to spinal cord, the expression of CNGA3 in DRGs is restricted to non-neuronal cells, most likely satellite glial cells. These cells are thought to exert regulatory functions on DRG neurons after peripheral nerve injury (Hanani, 2005; Ohara et al., 2009). However, the functions of satellite glial cells in inflammatory pain conditions are incompletely resolved. Coupling by gap junctions among satellite glial cells seems to increase after injection of Complete Freund's Adjuvant into a hindpaw (Dublin and Hanani, 2007), but the contribution of satellite glial cells to nociceptive signaling during inflammatory pain will need to be worked out in future studies.
Acute nociceptive responses were unaffected in CNGA3−/− mice, confirming earlier reports that NO/cGMP signaling is not required for acute pain reflexes (Schmidtko et al., 2009). Moreover, the neuropathic pain behavior was similar in CNGA3−/− and WT mice, suggesting that CNGA3 does not essentially contribute to the development of hypersensitivity evoked by nerve injury. In contrast to acute and neuropathic pain, the inflammatory pain behavior of CNGA3−/− mice differed from that of WT mice after injection of formalin or zymosan into a hindpaw. In particular, the formalin-induced paw licking and the zymosan-induced mechanical hypersensitivity were increased in the mutants compared with that in WT mice, suggesting that CNGA3 normally attenuates the hypersensitivity associated with inflammatory pain. This hypothesis is supported by the observation that naive CNGA3−/− mice developed an increased allodynia after intrathecal injection of the cGKI- and CNG channel-activating cGMP analog, 8-pCPT-cGMP, because the attenuating effect of CNGA3 is not present in CNGA3−/− mice. Moreover, naive CNGA3−/− but not WT mice developed allodynia after intrathecal injection of Rp-8-pCPT-cGMPS, which inhibits cGKI and activates CNG channels. It is likely that the cGKI inhibitory effect of Rp-8-pCPT-cGMPS does not affect the behavior in naive mice because cGKI is not, or is only moderately, activated, consistent with findings that cGKI inhibitors induced antinociceptive effects in inflammatory pain models but not in naive animals (Tao et al., 2000; Schmidtko et al., 2003, 2008a). Therefore, the induction of allodynia in naive CNGA3−/− mice by Rp-8-pCPT-cGMPS seems to be mediated by cGKI-independent mechanisms. Although highly speculative, activation of CNGA3-negative CNG channel subunits in excitatory neurons might mediate this effect. In this hypothetical scenario, the simultaneous activation of CNGA3-negative CNG channels in excitatory neurons and of CNGA3-positive CNG channels in inhibitory neurons could explain why Rp-8-pCPT-cGMPS induced allodynia in CNGA3−/− mice but not in WT mice.
Given the fact that various cGMP-producing guanylyl cyclases and cGMP effectors are present in different cells of the nociceptive system (Schmidtko et al., 2009), one has to consider that cGMP may generally induce both pronociceptive and antinociceptive effects. A major pronociceptive mechanism of cGMP is mediated by activation of cGKI in primary afferent neurons, which may result in an increased glutamate release from central terminals to activate second-order nociceptive neurons in the spinal cord. Notably, pronociceptive cGKI activation in primary afferent neurons occurs in a NO-independent manner by activation of the particulate guanylyl cyclase NPR-B (Kishimoto et al., 2008; Schmidtko et al., 2008a; Schmidt et al., 2009). Pronociceptive effects of cGMP are also mediated by NO-dependent activation of NO-GC, as indicated by the considerably reduced nociceptive behavior in mice lacking NO-GC (Schmidtko et al., 2008a). However, NO-GC is expressed in different areas of the spinal cord, i.e., in NK1 receptor-positive projection neurons in lamina I and in inhibitory interneurons of the dorsal horn, as well as in DRG satellite glial cells (Ding and Weinberg, 2006; Ruscheweyh et al., 2006; Schmidtko et al., 2008a). It is currently not known whether or not NO-GC-mediated cGMP production in all these cells results in pronociceptive effects. Several lines of evidence indicate that cGMP can exert pronociceptive effects in NK1-R-positive lamina I projection neurons. In particular, previous studies revealed that long-term potentiation of synapses between C-fibers and lamina I projection neurons is NO dependent (Ikeda et al., 2006), and that selective ablation of these neurons considerably attenuated inflammatory and neuropathic pain (Mantyh et al., 1997; Nichols et al., 1999; Khasabov et al., 2002). However, our data indicate that cGMP produced by NO-GC in inhibitory neurons exerts antinociceptive effects that are mediated by CNGA3. This assumption is supported by the observation that CNGA3−/− mice developed an exaggerated mechanical allodynia after intrathecal injection of the NO donor NOC-5, but not of the NPR-B ligand CNP. Interestingly, in a recent study, it was shown that cGMP may also exert antinociceptive effects in DRG neurons after activation of the particulate guanylyl cyclase NPR-A by B-type natriuretic peptide (Zhang et al., 2010).
The complexity of the distribution of cGMP generators and effectors in the nociceptive system may also explain why both pronociceptive and antinociceptive effects have been observed after intrathecal injection of NO donors and cGMP analogs. In fact, several studies revealed that NO donors and cGMP analogs, in addition to their well characterized pronociceptive effects, may also inhibit pain (Iwamoto and Marion, 1994; Pehl and Schmid, 1997; Tegeder et al., 2002, 2004; Schmidtko et al., 2008b). Interestingly, the antinociceptive effects of cGMP analogs were not antagonized by coadministration with a cGKI inhibitor and were also observed in cGKI−/− mice, suggesting that cGKI-independent mechanisms are involved. Our data indicate that CNGA3 contributes to these inhibitory effects of cGMP. Considering that (1) CNG channels containing CNGA3 in retinal cones are activated by low micromolar concentrations of cGMP, (2) activation of CNG channels causes an influx of Na+ and Ca2+ that leads to depolarization of the plasma membrane (Kaupp and Seifert, 2002; Biel, 2009), and (3) the evidence we here provide that CNGA3 is expressed in inhibitory neurons, it is likely that cGMP-mediated activation of CNG channels in inhibitory interneurons results in GABA release that, in turn, inhibits inflammatory pain processing. Moreover, activation of CNG channels in satellite glial cells might modulate the inflammatory pain processing by so far unidentified mechanisms. Hence, if NO donors or cGMP analogs are administered, several factors including dose, route of administration, and selectivity toward cGKI and/or CNG channel activation determine whether pro- or antinociceptive effects are being observed.
In conclusion, we have presented evidence for an inhibitory role of CNGA3 in the processing of hyperalgesia during inflammatory pain. It remains to be determined whether other CNG channel subunits are also involved in nociceptive processing and mediate pro- and/or antinociceptive effects of cGMP.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (SFB815-A14), the Interdisciplinary Center for Neuroscience Frankfurt (ICNF), the Paul and Ursula Klein-Stiftung, and the Novartis-Stiftung. We thank Karin Schilling, Christine Manderscheid, Annett Häussler, Judith Fuchs, and Heike Korff for excellent technical assistance.
References
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M. Cyclic nucleotide-regulated cation channels. J Biol Chem. 2009;284:9017–9021. doi: 10.1074/jbc.R800075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Michalakis S. Cyclic nucleotide-gated channels. Handb Exp Pharmacol. 2009;191:111–136. doi: 10.1007/978-3-540-68964-5_7. [DOI] [PubMed] [Google Scholar]
- Biel M, Zong X, Ludwig A, Sautter A, Hofmann F. Molecular cloning and expression of the Modulatory subunit of the cyclic nucleotide-gated cation channel. J Biol Chem. 1996;271:6349–6355. doi: 10.1074/jbc.271.11.6349. [DOI] [PubMed] [Google Scholar]
- Biel M, Seeliger M, Pfeifer A, Kohler K, Gerstner A, Ludwig A, Jaissle G, Fauser S, Zrenner E, Hofmann F. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc Natl Acad Sci U S A. 1999;96:7553–7557. doi: 10.1073/pnas.96.13.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bráz JM, Basbaum AI. Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain. 2010;150:290–301. doi: 10.1016/j.pain.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach GJ, Dehn D, Del Turco D, Deller T. Quantification of layer-specific gene expression in the hippocampus: effective use of laser microdissection in combination with quantitative RT-PCR. J Neurosci Methods. 2003;131:83–91. doi: 10.1016/s0165-0270(03)00232-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Geis C, Sommer C. Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J Neurosci. 2008;28:5836–5845. doi: 10.1523/JNEUROSCI.4170-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Ding JD, Weinberg RJ. Localization of soluble guanylyl cyclase in the superficial dorsal horn. J Comp Neurol. 2006;495:668–678. doi: 10.1002/cne.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007;21:592–598. doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Iwamoto ET, Marion L. Pharmacologic evidence that spinal muscarinic analgesia is mediated by an l-arginine/nitric oxide/cyclic GMP cascade in rats. J Pharmacol Exp Ther. 1994;271:601–608. [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22:9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto I, Tokudome T, Horio T, Soeki T, Chusho H, Nakao K, Kangawa K. C-type natriuretic peptide is a Schwann cell-derived factor for development and function of sensory neurones. J Neuroendocrinol. 2008;20:1213–1223. doi: 10.1111/j.1365-2826.2008.01778.x. [DOI] [PubMed] [Google Scholar]
- Kramer RH, Tibbs GR. Antagonists of cyclic nucleotide-gated channels and molecular mapping of their site of action. J Neurosci. 1996;16:1285–1293. doi: 10.1523/JNEUROSCI.16-04-01285.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Cizkova D. The role of nitric oxide in nociception. Curr Rev Pain. 2000;4:459–466. doi: 10.1007/s11916-000-0070-y. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/s1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80:67–82. doi: 10.1016/s0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Ohara PT, Vit JP, Bhargava A, Romero M, Sundberg C, Charles AC, Jasmin L. Gliopathic pain: when satellite glial cells go bad. Neuroscientist. 2009;15:450–463. doi: 10.1177/1073858409336094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehl U, Schmid HA. Electrophysiological responses of neurons in the rat spinal cord to nitric oxide. Neuroscience. 1997;77:563–573. doi: 10.1016/s0306-4522(96)00495-2. [DOI] [PubMed] [Google Scholar]
- Ruscheweyh R, Goralczyk A, Wunderbaldinger G, Schober A, Sandkühler J. Possible sources and sites of action of the nitric oxide involved in synaptic plasticity at spinal lamina I projection neurons. Neuroscience. 2006;141:977–988. doi: 10.1016/j.neuroscience.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Stonkute A, Jüttner R, Koesling D, Friebe A, Rathjen FG. C-type natriuretic peptide (CNP) is a bifurcation factor for sensory neurons. Proc Natl Acad Sci U S A. 2009;106:16847–16852. doi: 10.1073/pnas.0906571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Ruth P, Geisslinger G, Tegeder I. Inhibition of cyclic guanosine 5′-monophosphate-dependent protein kinase I (PKG-I) in lumbar spinal cord reduces formalin-induced hyperalgesia and PKG upregulation. Nitric Oxide. 2003;8:89–94. doi: 10.1016/s1089-8603(02)00165-9. [DOI] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, König P, Heine S, Motterlini R, Ruth P, Schlossmann J, Koesling D, Niederberger E, Tegeder I, Friebe A, Geisslinger G. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neurosci. 2008a;28:8568–8576. doi: 10.1523/JNEUROSCI.2128-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008b;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32:339–346. doi: 10.1016/j.tins.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Tao YX, Hassan A, Haddad E, Johns RA. Expression and action of cyclic GMP-dependent protein kinase Ialpha in inflammatory hyperalgesia in rat spinal cord. Neuroscience. 2000;95:525–533. doi: 10.1016/s0306-4522(99)00438-8. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Schmidtko A, Niederberger E, Ruth P, Geisslinger G. Dual effects of spinally delivered 8-bromo-cyclic guanosine mono-phosphate (8-bromo-cGMP) in formalin-induced nociception in rats. Neurosci Lett. 2002;332:146–150. doi: 10.1016/s0304-3940(02)00938-2. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Del Turco D, Schmidtko A, Sausbier M, Feil R, Hofmann F, Deller T, Ruth P, Geisslinger G. Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc Natl Acad Sci U S A. 2004;101:3253–3257. doi: 10.1073/pnas.0304076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119:287–294. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand I, Godde M, Frings S, Weiner J, Müller F, Altenhofen W, Hatt H, Kaupp UB. Cloning and functional expression of a cyclic-nucleotide-gated channel from mammalian sperm. Nature. 1994;368:859–863. doi: 10.1038/368859a0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140:441–451. doi: 10.7326/0003-4819-140-8-200404200-00010. [DOI] [PubMed] [Google Scholar]
- Zhang FX, Liu XJ, Gong LQ, Yao JR, Li KC, Li ZY, Lin LB, Lu YJ, Xiao HS, Bao L, Zhang XH, Zhang X. Inhibition of inflammatory pain by activating B-type natriuretic peptide signal pathway in nociceptive sensory neurons. J Neurosci. 2010;30:10927–10938. doi: 10.1523/JNEUROSCI.0657-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]