

Abstract
Extracellular deposition of the amyloid-β peptide (Aβ) in the brain parenchyma is a hallmark lesion of Alzheimer's disease (AD) and a predictive marker for the progression of preclinical to symptomatic AD. Here, we used multiphoton in vivo imaging to study Aβ plaque formation in the brains of 3- to 4-month-old APPPS1 transgenic mice over a period of 6 months. A novel head fixation system provided robust and efficient long-term tracking of single plaques over time. Results revealed an estimated rate of 35 newly formed plaques per cubic millimeter of neocortical volume per week at 4–5 months of age. At later time points (i.e., in the presence of increasing cerebral β-amyloidosis), the number of newly formed plaques decreased. On average, both newly formed and existing plaques grew at a similar growth rate of 0.3 μm (radius) per week. A solid knowledge of the dynamics of cerebral β-amyloidosis in mouse models provides a powerful tool to monitor preclinical Aβ targeting therapeutic strategies and eases the interpretation of diagnostic amyloid imaging in humans.
Introduction
Extracellular deposition of the amyloid-β peptide (Aβ) in the brain parenchyma is a hallmark lesion of Alzheimer's disease (AD) (Hardy and Selkoe, 2002). Position emission tomography (PET) imaging using Pittsburgh compound B (PIB) has shown that cerebral β-amyloid deposition is an early and predictive marker for the progression of preclinical to symptomatic AD (Morris et al., 2009; Weiner et al., 2010).
Cerebral β-amyloidosis and its impact on neuronal connectivity and function can be studied in transgenic mice, although PIB binding in mouse models of AD is poor compared with that in the human brain (Klunk et al., 2005; Jucker, 2010). Thus, the kinetics of cerebral β-amyloidosis in mice has primarily been studied by multiphoton microscopy using methoxy-X04, an amyloid-binding Congo red derivative (Dong et al., 2010). Multiphoton microscopy, compared with PET imaging, has the advantage of high spatial resolution and allows observation of individual amyloid lesions and the associated cellular structures over time.
The most intriguing research questions of the present study were whether β-amyloid plaques steadily increase in size, stay constant once they developed, or grow and shrink dynamically over time. Furthermore, do newly formed plaques differ in their fate from already existing ones and does the size of the plaques affect their growth kinetics? A solid characterization of the development and progression of β-amyloidosis is not only important for the understanding of AD pathogenesis but is also instrumental for the development of Aβ and/or amyloid-based treatment strategies.
The few studies that have addressed this topic have yielded somewhat inconsistent results (Bolmont et al., 2008; Meyer-Luehmann et al., 2008; Yan et al., 2009). Whereas some authors reported that plaques appear rapidly within 24 h and stay stable in size within the following 14 d of imaging (Meyer-Luehmann et al., 2008), others found that small- and medium-sized plaques grow over a time period of 4 weeks with few plaques also shrinking in size (Bolmont et al., 2008). Finally, it was shown that plaques grow in size within 7, 28, and 90 d intervals, but only if using the thinned skull imaging technique. No growth was found with the cranial window technique because of a reported surgery-induced activation of microglia (Yan et al., 2009).
Here, we report a long-term in vivo imaging approach in APPPS1 transgenic mice (Radde et al., 2006) over a period of 6 months. We have developed an improved setup that allows imaging with automatic rapid relocation to previously marked areas. Surgery-induced gliosis was controlled by combined imaging of microglia and β-amyloid plaques in a subset of mice.
Materials and Methods
Mice.
Hemizygous APPPS1 mice (Radde et al., 2006) that express human APPKM670/671NL and PS1L166P under the control of the Thy-1 promoter (Andrä et al., 1996) were crossed with hemizygous Iba1-EGFP mice (Hirasawa et al., 2005). Both lines were generated on a C57BL/6 background. For this study, APPPS1 littermates were analyzed, either with (GFP+) or without (GFP−) microglia expressing green fluorescent protein; male and female mice were used (APPPS1×GFP+: male, n = 2, and female, n = 2; APPPS1×GFP−: male, n = 2, and female, n = 2). One female APPPS1×GFP+ mouse showed obscuring dural thickening in response to the surgery; this mouse was excluded from the analysis (final n = 7). Mice were group housed under pathogen-free conditions. After surgery, mice were single housed. All procedures were in accordance with animal protocols approved by local Animal Care and Use Committees.
Surgery.
A round cranial window (4 mm diameter) was installed under general anesthesia (fentanyl, 0.05 mg/kg; midazolam, 5 mg/kg; medetomidine, 0.50 mg/kg). The anesthetized animals were secured on a modified stereotactic frame while placed on a warming pad. The skin and the periosteum were removed to expose the skull. Lines forming a circle (from the midline ±2 mm and at ∼0.0 and −4.0 bregma) were gently drilled onto the skull surface. Using a pair of angled forceps, the respective portion of the skull was removed. Care was given not to disturb or puncture the dura mater. A sterile, custom-made glass coverslip (thickness, 0.13 mm) was slowly lowered until it was parallel and flush with the edge of the skull by the use of a modified micromanipulator. The coverslip was then sealed to the skull with light-curing dental cement (Flowline; Heraeus Kulzer). A custom-made titanium ring (14 mm diameter) was then sealed onto the skull above the cranial window with light-curing dental cement to secure the mouse to a custom-made head fixation plate during imaging (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). After surgery, the animals were allowed to recover for 1 week before starting the imaging process.
Imaging.
Twenty-four hours before each imaging session, mice were injected intraperitoneally with 350 μl of amyloid staining solution [4% vol of 10 mg/ml methoxy-X04 (Klunk et al., 2002) in DMSO, and 7.7% vol CremophorEL (Sigma-Aldrich) in 88.3% vol PBS]. The systemic injection 24 h before imaging ensured a homogenous distribution of the methoxy-X04 in the entire imaged area (neocortex). Before each imaging session, mice were anesthetized with isoflurane (induction at 3%, subsequently reduced and held constant at 1–1.5%). Texas Red dextran (70,000 Da molecular weight; 12.5 mg/ml in sterile PBS; Invitrogen) was injected intravenously, providing a fluorescent angiogram (Spires et al., 2005), which served demonstration purposes in shown images. The cranial window was cleaned with ddH2O, and the mouse was secured under the microscope by fitting the titanium ring in a custom-built head fixation apparatus connected to a motorized XY stage (Luigs & Neumann). The head fixation system in combination with the Mark and Find function in the Leica confocal software allowed for relocating the previously imaged locations with a precision of a few micrometers. If necessary, fine-tuning was performed manually with respect to the characteristic three-dimensional plaque pattern.
All imagery was taken using a 40× HCX APO water-immersion objective (0.8 numerical aperture; Leica Microsystems). The Leica SP2 confocal microscope was equipped with a Spectra Physics Mai-Tai laser (tunable 710–990 nm), which provided multiphoton excitation at 810 nm for methoxy-X04 and 910 nm for GFP (Texas Red was simultaneously excited at 810 nm). Signals were detected via non-descanned detectors and the following filters: BP 525/50 for methoxy-X04, BP 515/30 for GFP, and BP 590/60 for Texas Red. Settings were optimized for the first imaging session and kept constant for each xyz location.
For each mouse, six different xyz locations were chosen, which were imaged for methoxy-X04/Texas Red on a weekly basis (for 7–9 weeks), followed by biweekly imaging and finally monthly imaging periods (up to 25 weeks). In the double-transgenic APPPS1×GFP+ mice, two of the six locations were additionally imaged for GFP. From the initial 42 locations (7 mice; 6 locations), 2 locations had to be excluded because of local dural thickening. At each xyz location, z-stacks of 150–180 μm were taken, starting at a depth of ∼20 μm from the cortical surface at a z-spacing of 1.0033 μm.
Data analysis.
Before additional processing, images were deblurred using AutoquantX (Media Cybernetics; for APPPS1×GFP− mice) or Huygens (SVI; for APPPS1×GFP+ mice). Stacks of different time points were registered for subsequent analysis using customized algorithms in Amira (Amira 4.1.1; Mercury Computer Systems). The final analysis was done in Imaris (Imaris 7.0; Bitplane) using surface detection and tracking. Time point 1 was excluded from the analysis as some of the plaques were stained by methoxy-X04 but were too weak to be detected by the software (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). For the plaque analysis, the surfaces of individual plaques were automatically tracked over time and the volume for each time point was measured. Care was taken to only include plaques that did not extend over the edges of the imaged volume. All detected plaques were double-checked manually after the automatic tracking over time, categorized into (pre-) “existing” or “new” plaques or removed if considered unreliable. The definition of new plaque was used if a plaque was not present in the previous imaging time point. The term existing was used if the plaque was detected from the first included imaging time point (week 2) and could be tracked until the end of the observation period. To analyze plaque growth and compare the growth rate of existing (=larger) and new (=smaller) plaques, measured volumes were considered as spheres and categorized according to their radii.
Statistics.
Linear regression lines were calculated for the growth of the radii of each individual plaque. The linear model was chosen because of the general high coefficient of determination (mean R2 = 0.69 ± 0.011) indicating a good fit with our original data set. To characterize the general trend of growth, averaged regression lines (medians of intercept and slope calculated from all individual regression lines of the respective group) or the mean growth rate (with 95% confidence interval) were reported. Slopes of individual plaques were compared for existing and new plaques by using nonparametric statistics (Mann–Whitney U test). Mean values are given ± SEM.
Results
Visualization of amyloid plaques and microglia over time
Simultaneous imaging of β-amyloid plaques, microglia, and blood vessels was performed as described in Materials and Methods. A total of 40 amyloid-containing volumes from 7 APPPS1 tg mice (four without and three with additional GFP expression under the Iba-1 promoter) were imaged starting at an age of 3.7 months over a total of up to 25 weeks (Fig. 1A). The first 7–9 imaging sessions were performed on a weekly basis followed by intervals of 2 or 4 weeks. A coordinate system with a newly designed head fixation plate was used, allowing automatic and highly precise relocation to previously marked positions even in absence of rhodamine dextran labeling of the vasculature as a landmark (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Thus, image sequences of disparate timescales could be reliably generated for quantitative analysis.
Figure 1.

β-Amyloid plaques grow over time. A, Timeline in weeks after surgery (marked with “S”). APPPS1 transgenic mice were 3.7 months of age at the time of surgery and were imaged for up to 25 weeks. Imaging time points (blue) are exemplarily shown for one mouse and numbered time points correspond to the images shown in B. B, Long-term imaging of the same region of interest. β-Amyloid deposits (green) are stained with methoxy-X04, and blood vessels (red) were made visible by Texas Red dextran. A maximum z-projection is shown for the imaged volume including truncated plaques located at the rim of the volume. These show fluctuations in signal strength (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material). C, β-Amyloid plaque surfaces were reconstructed and tracked to obtain changes in plaque size over time. Truncated plaques were generally excluded from analysis (D). Calculated plaque radii are shown over time for the position depicted in B and C. Different colors indicate individual plaques tracked over time. Individual plaque radii fluctuate between the weekly imaging sessions but showed a robust increase over the entire observation period. Indicated are the individual regression lines. E, When all mice and all measured volumes are considered (7 mice, 40 volumes, and 388 tracked plaques), an overall increase in radius is observed over time, indicated by the “overall regression line” (medians of intercept and slope calculated from all individual regression lines; mean slope, 0.315 ± 0.008 μm/week). The dots represent all plaque radii at the respective imaging sessions, whereby imaging week 1 corresponds to the first appearance of each plaque during the course of the study. F, Size distribution of plaques in mice at 4, 7, and 9 months of age (corresponding to imaging weeks 4/5, 16/17, and 24/25, average of 3 mice, 16 volumes). A shift from smaller to bigger plaques is observed over time [plaques were categorized in 11 subgroups from 1 (small) to 11 (large) according to their radii]. Scale bars: B, C, 30 μm.
Amyloid plaques grow over time
An example of β-amyloid plaque development over time is shown in Figure 1B. As a first analysis, all detected plaques (preexisting and newly appearing ones) were measured by a surface detection algorithm to obtain their volumes (Fig. 1C). Over the entire observation time, nearly all plaques increased constantly in size (only 4 of 388 plaques showed a negative growth rate). The applied linear model best described the constant increase in radius, which was calculated from the respective volume of each plaque (for details, see Materials and Methods). Individual plaques exhibited different positive growth rates over the observation period as exemplarily shown in Figure 1D. When considering the growth curves of all individual plaques (n = 388), an overall (median) radius growth rate of 0.3 μm/week was observed (Fig. 1E). A gradual shift from smaller plaque radii at 4 months of age to larger radii at 7 and 9 months of age was observed (Fig. 1F) consistent with an overall increase in plaque size over time.
Appearance and growth of newly developed amyloid plaques
During the 25 week imaging interval, a total of 132 newly developed β-amyloid plaques was detected (Fig. 2). The rate at which they appeared differed widely over time [i.e., with increasing β-amyloid deposition (Fig. 2C)]. Within the first 6 imaging weeks (corresponding to 4–5 months of age of the mice), the number of new plaques peaked and decreased thereafter (Fig. 2C). New plaques were mainly small in size, with 87% having a radius of <4 μm (Fig. 2D). Only 5 of the 132 plaques started with a radius of >6 μm (Fig. 2D) and were detected within the weekly imaging intervals. Visual verification of the few larger newly appearing plaques revealed that these plaques developed from weakly stained diffuse β-amyloid deposits. The size distribution of preexisting plaques was much broader (radii ranging from 0.9 to 16 μm), and only 20% of the plaques had radii <4 μm (Fig. 2D).
Figure 2.

The appearance of newly formed β-amyloid plaques declines over time. A, Example of a newly formed plaque (arrow), which appeared at imaging week 9. B, Reconstructed surfaces of the same region of interest with the appearing plaque indicated in yellow. C, The rate of newly formed plaques declines over time with the highest rate of newly appearing plaques between imaging weeks 3 and 6 (corresponding to 4–5 months of age of the mice; plaque appearance is shown per region of interest with a mean volume of 0.015 mm3). Error bars indicate SEM. D, Size distribution of preexisting and newly appearing plaques. New plaques were mainly small in size with 87% having a radius of < 4 μm. Scale bars: A, B, 10 μm.
Do newly appearing plaques grow differently than existing ones?
The radii of newly formed plaques and preexisting plaques grew at very similar rates as their median growth rates did not differ significantly (0.286 μm/week for existing plaques compared with 0.306 μm/week for new ones; Mann–Whitney U test, p = 0.4) (Fig. 3A,B). The underlying physiological model suggests that new amyloid “layers” are constantly added to the surface of existing plaques—despite the difference in initial size. However, such a model implies that, if the plaque volume is considered, the observed linear growth of the radii will lead to a nonlinear growth of the volumes. Figure 3C reflects this fact by depicting the volume of each plaque instead of its radius, whereby the prediction curves for the volumes were calculated from the median radii (shown as lines in Fig. 3A). As new layers add more volume to large plaques compared with small plaques, large plaques will show a steeper increase in their volume compared with the small plaques. The apparent discrepancy between the underlying linear growth of the radius and the nonlinear growth of the volume becomes even more striking when looking at the factor by which the volume increases over time: Figure 3D reveals that the volume of newly developed plaques increases >50-fold in 10 weeks, whereas the volume of preexisting ones increases only 5-fold, a fact that is completely consistent with equal linear growth rates of the radii of new and preexisting plaques (see also supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 3.

Existing and newly formed plaques show on average the same increase in radius over time. A, Single measurements for existing (blue) and newly appearing (red) plaques shown as dots together with the resulting mean regression lines (median of intercept and slope calculated from all individual regression lines for the two groups). Imaging week 1 corresponds to the first appearance of each plaque during the course of the study. A similar growth rate was observed for new and preexisting plaques (0.286 μm/week for existing plaques compared with 0.306 μm/week for new ones). B, Comparison of the slopes of the individual regression lines plotted for existing (blue) and newly formed (red) plaques. No significant difference is observed for the slopes between new and existing plaques (box plot whiskers denote the 10th and 90th percentiles; Mann–Whitney U test, p = 0.4). C, The according increase in plaque volume was calculated by using the radii and shown with the respective lines. This prediction shows that existing (and therefore initially larger) plaques gain more volume than newly formed ones. Our data (shown as dots) support this prediction. Imaging week 1 corresponds to the first appearance of each plaque during the course of the study. D, When comparing the volume at the first appearance of a plaque to its volume 10 weeks later, newly formed plaques showed a much higher “fold increase” than existing ones. This is in line and predicted by our model based on linearly growing radii (see also supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Error bars indicate SEM.
No surgery- or imaging-induced gliosis
Microglia morphology and changes were studied in the APPPS1×GFP+ mice. No signs of an imaging- or surgery-induced activation of microglia was observed at any time point within the 6 month imaging period (Fig. 4A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In contrast, the expected and previously described local increase in the number of microglia cells surrounding the growing β-amyloid plaques was observed (Bolmont et al., 2008). Histopathological analysis of microglia and astrocytes 1 week after surgery also revealed no difference between animals that underwent cranial window surgery and control mice without (Fig. 4B).
Figure 4.
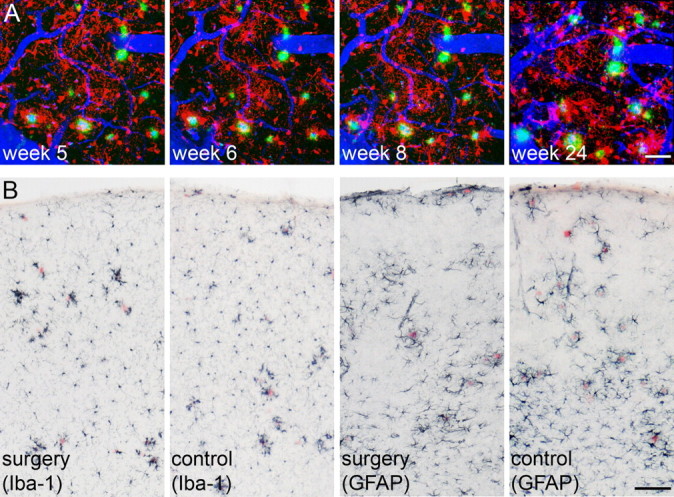
No indication for long-term imaging- or surgery-induced gliosis. A, Time series of a double-transgenic APPPS1×GFP+ mouse reveals no overall, but only local amyloid-associated increase in activated microglia over time. β-Amyloid deposits are shown in green (methoxy-X04), blood vessels in blue (Texas Red dextran), and microglia in red (endogenous GFP expression). For illustration of β-amyloid together with microglia, the two channels were subtracted from each other, using a channel arithmetic Matlab script (The MathWorks) embedded in Imaris. B, Additional mice received either open-skull cranial window surgery or served as controls (n = 5/group). Immunohistochemical staining for Iba-1 and GFAP 1 week later did not indicate any differences in morphology and number of microglia (left) or astroglia (right) between the two groups. Images are taken from area of surgery or respective areas in control mice. β-Amyloid was stained with Congo red. Scale bars: A, 40 μm; B, 200 μm.
Discussion
In the present study, we used in vivo multiphoton microscopy to examine the appearance and development of β-amyloid plaques over a 6 month time period. We selected 3- to 4-month-old APPPS1 transgenic mice because of their rapid and robust β-amyloid plaque development at that age (Radde et al., 2006). Through the use of a novel head fixation system, we could reliably and with high precision locate previously imaged positions without the necessity of rhodamine dextran-labeled vasculature as a landmark.
We found the highest rate of new plaque appearance in animals between 4 and 5 months of age with a striking decrease at later time points (i.e., older mice with a higher plaque load form fewer new plaques). Moreover, our results show that the radii of newly appearing and existing plaques grew similarly and constantly over the 6 month observation period despite the difference in initial size. These results favor a physiological model whereby the amount of soluble Aβ is the primary and limiting factor in plaque growth. In accordance with these findings, a critical relationship between Aβ concentration in the interstitial fluid and β-amyloid plaque growth was reported (Yan et al., 2009). During the presence of excessive soluble Aβ, Aβ would be continually added in layers to the existing deposits. Until a certain limit is reached, continued plaque growth could be explained by the larger plaques being exposed to a larger area of the neuropil compared with small plaques.
A constant increase in the radii of plaques naturally leads to an exponential increase in plaque volume, consistent with previously reported quantitative morphology (Radde et al., 2006). The increase in total β-amyloid load over time is in line with the present finding of an age-related change in the average size of plaques: In young animals (i.e., animals with a comparably lower overall β-amyloid load), the majority of plaques had a radius of <8 μm. With increasing age, there is a shift of the plaque size distribution toward larger plaques (up to 22 μm in radius).
With additional aging and β-amyloid load, the exponential demand for soluble Aβ to incorporate into plaques might exceed the production, and hence one would expect that plaque growth slows down. This is consistent with a lack of plaque growth previously described in very old mice (Christie et al., 2001; Yan et al., 2009). However, no obvious saturation could be detected in APPPS1 mice within our imaging time period (4–9 months of age).
Nevertheless, we have noticed a decrease of newly formed plaques in the period of sustained plaque growth. This observation may be explained that, with a given constant Aβ production, fewer new plaques will form in a “competitive” β-amyloid-laden environment because of diffusion kinetics. In this situation, soluble Aβ is more likely to be sequestered by existing plaques than forming a new seed. The latter would require a critical local concentration that cannot be reached above a certain plaque load.
The appearance and development of β-amyloid plaques has been previously addressed and yielded inconsistent results (Bolmont et al., 2008; Meyer-Luehmann et al., 2008; Yan et al., 2009). Several factors might contribute to the diverging findings: (1) As described above, the age and therefore the β-amyloid load of the respective mouse models are likely to play a fundamental role. (2) The duration of the imaging period is essential as imaging over short periods of time might be under the limit of detection for small increases in size (Meyer-Luehmann et al., 2008). (3) As plaques are amorphous three-dimensional structures, a three-dimensional analysis will be most sensitive to detect changes in size. Moreover, the detection stringency (i.e., the set threshold) might further affect the respective outcome in whether the diffuse periphery of a plaque is included in the analysis or not. As this is the interface of microglia plaque interaction, fluctuations are most likely to happen here and might explain the shrinkage described previously between imaging time points in a subset of plaques (Bolmont et al., 2008). (4) Finally, the role of microglia activation attributable to cranial window surgery and/or multiphoton imaging was discussed to influence the kinetics of β-amyloid deposition (Yan et al., 2009). In the course of this study, however, we could not detect surgery- or imaging-induced changes in morphology or number of microglia cells [only the expected pathology-related changes were observed, as previously described (Bolmont et al., 2008)]. Thus, similarly to the thinned-skull technique, the cranial window appears to be an appropriate technique for long-term imaging of amyloid lesions. In addition, it provides greater imaging depth: cranial window, 600–800 μm (Helmchen and Denk, 2005), versus thinned skull, 200 μm (Xu et al., 2007) to 300–400 μm (Yang et al., 2010). Moreover, it allows the observation of more imaging sites (4 mm diameter cranial window vs 1–2 mm for thinned skull). It was also reported that open-skull preparation influences dendritic spine turnover (Xu et al., 2007). However, a surgery-induced gliosis was also described in this study. Therefore, additional experiments are needed to investigate whether there is a direct link between cranial window technique and spine turnover under optimized surgery conditions.
In summary, the current study delivers new insights into the development of β-amyloid plaques and especially the time course of newly forming deposits. Future therapeutic experiments (e.g., immunization) can use the time course of plaque appearance to evaluate efficacy. The direct effect of a compound on β-amyloid deposits delivered in a time-dependent manner can then be observed in our mouse model using multiphoton imaging.
Footnotes
This work was supported by European Union FP7 HEALTH (Project LUPAS), the Competence Network on Degenerative Dementias Grant BMBF-01GI0705, Alzheimer's Association Grant IIRG-05-13464, and a PhD grant from the charitable Hertie Foundation (J.K.H.). We thank M. Meyer-Luehmann, T. Bolmont, and S. Grathwohl for experimental help. We also thank R. Barbizan Sühs, M. Hruscha, C. Krüger, and A. Bosch for assistance with data analysis and technical support, and B. Schmidt for providing methoxy-X04.
The authors declare no competing financial interests.
References
- Andrä K, Abramowski D, Duke M, Probst A, Wiederhold KH, Bürki K, Goedert M, Sommer B, Staufenbiel M. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging. 1996;17:183–190. doi: 10.1016/0197-4580(95)02066-7. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Revilla-Sanchez R, Moss S, Haydon PG. Multiphoton in vivo imaging of amyloid in animal models of Alzheimer‘s disease. Neuropharmacology. 2010;59:268–275. doi: 10.1016/j.neuropharm.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J Neurosci Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-β in Alzheimer's disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner MW, Aisen PS, Jack CR, Jr, Jagust WJ, Trojanowski JQ, Shaw L, Saykin AJ, Morris JC, Cairns N, Beckett LA, Toga A, Green R, Walter S, Soares H, Snyder P, Siemers E, Potter W, Cole PE, Schmidt M. The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202–211.e7. doi: 10.1016/j.jalz.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009;29:10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc. 2010;5:201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]