

Abstract
Duchenne muscular dystrophy is caused by mutations in the Dystrophin gene and is characterized by muscle degeneration and the occurrence of mental deficits in a significant number of patients. Although Dystrophin and its closely related ortholog Utrophin are present at a variety of synapses, little is known about their roles in the nervous system. Previously, we reported that absence of postsynaptic Dystrophin from the Drosophila neuromuscular junction (NMJ) disrupts synaptic homeostasis, resulting in increased stimulus-evoked neurotransmitter release. Here, we show that RhoGAP crossveinless-c (cv-c), a negative regulator of Rho GTPase signaling pathways, genetically interacts with Dystrophin. Electrophysiological characterization of the cv-c-deficient NMJ and the use of presynaptic- and postsynaptic-specific transgenic rescue versus RNA interference reveal that the absence of postsynaptic cv-c results in elevated evoked neurotransmitter release. The cv-c mutant NMJ exhibits an increased number of presynaptic neurotransmitter release sites and higher probability of vesicle release without apparent changes in postsynaptic glutamate receptor numbers or function. Moreover, we find that decreasing expression of the Rho GTPase Cdc42 suppresses the high neurotransmitter release in the cv-c and Dystrophin mutants, suggesting that Cdc42 is a substrate of Cv-c. These results indicate that Dystrophin and the Rho GTPase signaling pathway likely interact at the postsynaptic side of the NMJ to maintain synaptic homeostasis. The absence of this postsynaptic pathway results in presynaptic structural and functional alterations, suggesting that retrograde signaling mechanisms are affected.
Introduction
Duchenne muscular dystrophy (DMD), a common inherited muscle degenerative disease, is caused by the absence of Dystrophin (Hoffman et al., 1987; Koenig et al., 1987). With its interacting partners in the Dystrophin glycoprotein complex (DGC), the best studied functions of Dystrophin are at the sarcolemma, in which it protects the muscle against contraction-induced injury and acts as a scaffold to localize a variety of signaling pathway components (for review, see Rando, 2001). The observations that a significant number of DMD patients display mental deficits (Rapaport et al., 1991; Anderson et al., 2002) and that Dystrophin, its ortholog Utrophin, and other members of the DGC are present at a variety of synapses indicate that Dystrophin also plays as yet poorly understood roles in the nervous system (for review, see Waite et al., 2009; Pilgram et al., 2010).
The remarkable evolutionary conservation of the domain structure of Dystrophin suggests that at least some of its interactions and functions are conserved. Recent studies have demonstrated the utility of the fruit fly Drosophila melanogaster, with its single Dystrophin (Dys) gene (Roberts and Bobrow, 1998), as a model for exploration of the roles of Dys at both extrasynaptic (Shcherbata et al., 2007; van der Plas et al., 2007; Christoforou et al., 2008; Kucherenko et al., 2008; Taghli-Lamallem et al., 2008) and synaptic (van der Plas et al., 2006; Fradkin et al., 2008) sites. We reported previously that the Dys DLP2 isoform is enriched at the postsynaptic side of the larval neuromuscular junction (NMJ) (van der Plas et al., 2006). Its absence from the muscle results in increased evoked neurotransmitter release by the presynaptic motoneuron. This led us to speculate that retrograde signaling from the muscle to motoneuron is altered in the Dys mutant. Retrograde signaling pathways influencing neurotransmitter release have also been inferred in previous murine and Drosophila studies (Plomp et al., 1992; Davis and Bezprozvanny, 2001; Turrigiano and Nelson, 2004), including those focused on the Drosophila larval NMJ (Marqués and Zhang, 2006). Little is known, however, about the components of these pathways.
To identify genes encoding Dys-interacting proteins, including possibly those involved in retrograde signaling, we performed a candidate-based genetic screen in the Drosophila wing. Here, we show that a Rho GTPase activating protein (RhoGAP) encoding gene crossveinless-c (cv-c) interacts with Dys during formation of the wing posterior crossvein. RhoGAPs counteract the Rho GTPase exchange factors (RhoGEFs) to inhibit Rho signaling pathways in a variety of biological processes (for review, see Hall, 1998).
We demonstrate in transgenic RNA interference (RNAi) and rescue studies that, as in the Dys-deficient mutant, evoked presynaptic neurotransmitter release is significantly enhanced at the NMJ when Cv-c is lacking postsynaptically. Our analyses indicate that this is likely attributable to the observed increase in neurotransmitter release sites, leading to an elevated probability of vesicle release. Heterozygosity for a null allele of the Rho GTPase Cdc42 can suppress the increased neurotransmitter release observed in cv-c and Dys mutants, supporting our hypothesis that the interaction of Dys with Rho signaling pathways is required for appropriate synaptic homeostasis at the NMJ.
Materials and Methods
Fly stocks.
The following mutant alleles were used in the genetic enhancer screen and obtained from the Bloomington Drosophila Stock Center: sogU2, sax4, tkv7, cv-21, witB11, witA12, cv-d1, egfrf2, cv-c1, dpphr92, mysts2, mysolfC-x3, dNOSC, h1, det1, ab1, ast1, cg1, kniri-1, tt1, rhove-1, ciw, ci1, and H1. gbb4 and gbb1 were obtained from M. O'Connor (University of Minnesota, Minneapolis, MN). w1118, the genetic background in which the Dys mutation was generated, served as the wild-type control genotype for stainings, electrophysiology, and transmission electron microscopy analyses. The Dys DLP2 isoform null mutant allele DysE6 was described previously (van der Plas et al., 2006). The cv-c mutant alleles cv-c1, cv-cC524, UAS–cv-c–RNA interference flies (Billuart et al., 2001) and Cdc424 were obtained from the Bloomington Drosophila Stock Center; cv-cM62 and UAS–cv-c–RA (Denholm et al., 2005) were gifts from R. Ray (University of Sussex, Brighton, UK). All cv-c genetic lesions have been described previously (Denholm et al., 2005). The cv-c alleles were derived from distinct genetic backgrounds. To minimize possible genetic background effects, we backcrossed each mutant allele used against wild-type control flies for at least five generations before using them in electrophysiological analyses.
The following regulatory protein GAL4 (GAL4) driver lines were used: G14–GAL4 and OK6–GAL4 (Aberle et al., 2002). The upstream activating sequence (UAS)-bearing GS12472 P-element insertion (Toba et al., 1999), 1.9 kb upstream of the DLP2 ATG initiator codon, was used to overexpress DLP2 (van der Plas et al., 2006).
Genetic enhancer screen.
The DysE6 wing crossvein phenotype described below was used as the basis for a genetic enhancer screen. Stocks bearing mutant alleles of candidate genes (supplemental Table 1, available at www.jneurosci.org as supplemental material) reported previously to be involved in wing vein morphogenesis were crossed with DysE6 at 25°C to generate transheterozygous progeny (e.g., DysE6/+; mutant gene X/+ or DysE6/mutant gene X). Adult fly wing posterior crossveins were scored 5 d after eclosion.
Quantification of bouton number and muscle size.
Body walls of third-instar larvae were stained with anti-FasII (1D4) antibody (Vactor et al., 1993). Photographs of body wall muscle number 4 in segments A2–A5 from five larvae (40 muscles in total) for each genotype were made, and the muscle area was determined using NIH ImageJ. In addition, the numbers of boutons from at least 40 muscle number 4 fibers were manually counted in these same preparations.
Immunohistochemistry and RNA in situ hybridization.
Third-instar larvae were dissected in cold PBS, and their body walls were fixed in 4% formaldehyde in PBS and then incubated overnight at 4°C with mouse anti-Bruchpilot NC82 monoclonal antibody (Wagh et al., 2006), followed by application of goat anti-mouse Alexa Fluor 488 antibody (Invitrogen). Body wall synapses were visualized by confocal microscopy (Leica TCS SL; Leica Microsystems), the number of NC82-positive domains at synaptic termini on muscles 6 and 7 in 5 segments (A2–A6) were counted, and total bouton area was measured and analyzed using the Leica Application Suite software.
RNA in situ hybridizations were performed as described previously (Tautz and Pfeifle, 1989). The cv-c antisense RNA probe was generated by in vitro transcription of a linearized template plasmid containing sequences of the chromosome 3R cv-c gene (base pairs 10219833–10221479 of GenBank accession number AE014297.2). The sense control probe was generated by transcribing the other strand of the same plasmid template.
Wing vein imaging.
Whole adult flies were immersed in 100% EtOH and stored at −20°C for 1 d. Wings were then dissected in 100% EtOH, equilibrated in 50% EtOH/50% glycerol, and then mounted in 50% EtOH/50% glycerol on a microscope slide. Photographs were made using a light microscope (Axio Scope; Carl Zeiss) equipped with a digital camera (Axio Cam, Carl Zeiss).
Transmission electron microscopy.
Larval dissection, fixation, embedding, and sectioning were performed as described previously (Lin et al., 1994). Semi-serial sections of body walls from w1118, DysE6, and cv-c1 mutants were prepared, and electron micrographs were made of type Ib boutons on muscles 6 and 7 using a transmission electron microscope (Tecnai 12 Biotwin; FEI).
Electrophysiology.
Electrophysiological recordings were performed as described previously (van der Plas et al., 2006). Briefly, a microelectrode filled with 3 m KCl was inserted into muscle 6 (segments A3–A4) of dissected third-instar female larvae bathed in HL3 containing 0.6 mm Ca2+ unless described otherwise (Stewart et al., 1994). The intracellular measurements were recorded using a Geneclamp 500B amplifier (Molecular Devices), low-pass filtered at 10 kHz, high-pass filtered at 0.5 Hz, and digitized using a Digidata 1322A and pClamp9 software (Molecular Devices). Miniature excitatory junction potentials (mEJPs) were recorded continuously for 1 min. Thirty EJPs were recorded at 0.3 Hz stimulation after the appropriate axon was stimulated by a pulse generator (Master-8; AMPI) via a suction electrode. Electrical input resistance of all muscle fibers recorded was above 4 MΩ. The mean mEJP amplitude and frequency were analyzed by using the peak detection feature of Mini-Analysis 6.0 (Synaptosoft); all events were confirmed by eye. EJP amplitudes were analyzed using Clampfit 9.0, and amplitudes were normalized to a membrane potential of −60 mV. NMJ quantal content (QC) was calculated by dividing the mean EJP amplitude (calculated from 30 events) corrected for nonlinear summation (B. A. Stewart, personal communication) by the mean mEJP amplitude (calculated from 100 events). Failure analyses were performed in HL3 with 0.15 mm Ca2+. Paired-pulse facilitation (PPF) was assessed at 0.6 and 0.25 mm Ca2+ using 50 ms interstimulus intervals.
Statistical analyses.
One-way ANOVA was performed with least significant differences or Bonferroni's for post hoc multiple comparisons (PASW statistics 17.0; SPSS) for statistic analyses. Differences were considered significant when p < 0.05. Occasionally, p < 0.001 is indicated separately to make a distinction between the levels of significance (see figure legends for details).
Results
Dystrophin interacts genetically with RhoGAP cv-c during wing crossvein formation in Drosophila
To identify genes that interact with the Drosophila Dys DLP2 isoform, we performed a screen for genes that interact dominantly with a single copy of the DysE6 allele during formation of the wing crossveins. The Drosophila wing exhibits a stereotypic pattern of five longitudinal and two crossveins, the anterior crossvein and the posterior crossvein (pcv). These veins lie between the dorsal and ventral wing surfaces. The pcv is usually partially missing in the homozygous Dys mutant (Fig. 1D), whereas the pcv appears wild type in the heterozygous Dys mutant (Fig. 1A). This phenotype serves as an excellent basis for the identification of genes interacting with Dys because the absence of a crossvein does not affect viability and can be readily scored in the adult fly. The wing vein phenotype and its frequency of occurrence are described in detail in the legend of supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Figure 1.

Dystrophin interacts genetically with cv-c to stabilize posterior crossvein formation in Drosophila wings. Wings of heterozygous DysE6/+ (A) and cv-c1/+ (B) mutants exhibit a wild-type pcv as in w1118 (C). The double-heterozygous mutant DysE6/cv-c1 (F) only has a remnant of the pcv (arrow) between the fourth and fifth longitudinal veins remaining. This detached pcv phenotype is also observed in homozygous DysE6 (D) and cv-c1 (E) mutants. This result indicates a genetic interaction between Dys and cv-c in the wing.
The Drosophila Dys gene locus contains multiple promotors that regulate the transcription of at least six distinct isoforms: the three large isoforms DLP1–DLP3 and the smaller isoforms Dp186, Dp117, and Dp205 (for review, see Pilgram et al., 2010). The pcv phenotype is caused by the homozygous loss of the large isoforms; the phenotype is observed in the mutant allele DysE6 that lacks only these isoforms (van der Plas et al., 2006). The observation that the large isoforms are expressed in the developing wing disk (data not shown) is consistent with their roles in wing vein formation.
We selected a number of Drosophila mutants reported previously to display abnormal wing vein formation as candidates to test for interaction with DysE6 (supplemental Table 1, available at www.jneurosci.org as supplemental material) (see Materials and Methods). We generated flies heterozygous for the DysE6 allele and one allele of the selected mutant alleles. Importantly, the pcv phenotype was not observed in individuals heterozygous for any of the candidate genes tested (data not shown). Two of the 22 mutant genes display transheterozygous interactions with DysE6. One is detached (det), which subsequently was demonstrated to be an allele of Dys (Christoforou et al., 2008). The other is cv-c1, a hypomorphic mutant allele of the RhoGAP cv-c gene (Denholm et al., 2005). Two additional alleles of cv-c, cv-cM62 and cv-cC524, were found to display similar transheterozygous interactions with DysE6 (data not shown).
RhoGAP cv-c regulates neurotransmitter release at the Drosophila larval NMJ
Previously, we described a role for the large Dys isoform DLP2 in maintaining synaptic homeostasis at the Drosophila third-instar larval NMJ (van der Plas et al., 2006). We therefore evaluated whether cv-c also plays roles at this synapse. First, we determined where the cv-c gene is expressed in third-instar larvae by RNA in situ hybridization (see Materials and Methods). cv-c is expressed in the brain lobes but absent from the ventral nerve cord in which the motoneuron cell bodies are located (Fig. 2A). mRNA is also present in the eye, wing, and antennal imaginal disks (Fig. 2A,B), the primordia for the corresponding adult structures. Furthermore, mRNA expression is observed throughout the larval body wall muscle fibers (Fig. 2, compare C, D). cv-c mRNA was reported previously to be expressed in the embryonic visceral and somatic mesoderm (Denholm et al., 2005). The Dys DLP2 isoform is also expressed in the larval body wall musculature, eye, antennal, and wing disks and absent from the ventral nerve cord (van der Plas et al., 2006).
Figure 2.

The RhoGAP cv-c mRNA is expressed in the third-instar larval brain, wing disc, and muscle fibers but not in the motoneurons. cv-c mRNA is expressed in the brain lobes (arrowheads) and the eye-antennal discs (black arrows) but not in the larval neuropil in which the motoneuron cell bodies are located (white arrow) (A). cv-c mRNA is expressed in the wing disc (B) and throughout the muscle fiber (C). A sense strand cv-c probe was used as a control to determine background staining levels in muscle fibers (D).
To investigate the roles of Cv-c in synaptic function at the larval NMJ, we performed intracellular electrophysiological recordings at an identified third-instar larval muscle fiber in cv-c mutants and controls (see Materials and Methods). We recorded three cv-c mutant genotypes, the hypomorphic homozygous viable cv-c1 allele and the embryonic lethal alleles cv-cC524 and cv-cM62, which were present in combination with a wild-type chromosome or the cv-c1 allele to permit collection of third-instar larvae.
mEJP amplitudes from the various cv-c mutant allelic combinations were not significantly different from control larvae (Fig. 3A,B), which suggests that the postsynaptic response to the quantal release of glutamate from a single synaptic vesicle is unaltered. Frequencies of these spontaneous mEJPs were somewhat variable between the mutants but not significantly different from wild type (Fig. 3D). However, EJP amplitudes evoked by stimulation at 0.3 Hz were significantly increased above control levels, ranging from 1.4-fold to 1.6-fold, in all allelic combinations (p < 0.001) (Fig. 3A,C) (measured values are given in supplemental Table 2, available at www.jneurosci.org as supplemental material). As a result, QC in the cv-c mutant allelic combinations was, on average, ∼70% higher than the QC in the wild-type control (Fig. 3E). The QC is a measure of the number of neurotransmitter-containing vesicles released during stimulation and is defined by the mean EJP value divided by the mean mEJP amplitude (see Materials and Methods). Interestingly, the presence of one mutant allele also exhibits increased QC, indicating that cv-c is haploinsufficient for NMJ synaptic function (Fig. 3E). The increased QC in the cv-c mutants was confirmed by failure analysis (Boyd and Martin, 1956), an alternate method to determine neurotransmitter release that is independent of the mEJP amplitude (Fig. 3F). Together, these results indicate that cv-c is required at the larval NMJ to maintain normal levels of neurotransmitter release.
Figure 3.

Electrophysiological recordings reveal that cv-c mutants have a Ca2+ independent increased QC compared with wild-type controls. A, Representative traces of EJPs and mEJPs recorded from w1118 controls and cv-c1 mutants. B–E, Bar graph representations of mean ± SEM values of mEJP amplitudes (B), EJP amplitudes (C), mEJP frequencies (D), and QC (E) for the following genotypes: w1118, cv-c1, cv-c1/+, cv-c1/cv-cC524, cv-cC524/+, cv-c1/cv-cM62, and cv-cM62/+. Although the mEJPs and their frequencies are not significantly different among these genotypes, the EJPs and QC of all cv-c mutant alleles are significantly increased compared with wild-type animals. All measured values are given in supplemental Table 2 (available at www.jneurosci.org as supplemental material). F, Using failure analyses, the increased QC in cv-c1 mutants compared with that in w1118 control larvae was confirmed (QCs for w1118 controls vs cv-c1, failure analysis, 2.2 ± 0.5 vs 4.7 ± 0.6; direct method, 2.6 ± 1.0 vs 4.4 ± 0.7). G, Paired-pulse facilitation is presented in the graph by the ratio of EJP2/EJP1 with an interstimulus interval of 50 ms measured at 0.25 and 0.6 mm Ca2+. Although facilitation occurs at both Ca2+ concentrations in wild-type larvae (n = 3 and n = 6, respectively), it does not in cv-c1 mutants (n = 3 and n = 12), indicating an increased probability of synaptic vesicle release in the latter. *p < 0.05. H, Graph of the QC, depicted as log10 QC, as a function of the external Ca2+ concentration showing that the elevated QC in cv-c1 mutants compared with wild-type controls is Ca2+ independent. n represents the number of larvae that were analyzed.
We determined whether an increase in the probability of vesicle release underlies the increased QC at the cv-c1 mutant NMJ by performing PPF. PPF is a form of short-term adaptation of synaptic transmission (Zucker and Regehr, 2002) and is quantified by calculation of the ratio of the amplitudes of two consecutive EJPs evoked at short interstimuli intervals. When the probability of release of EJP1 decreases, as is observed at lower external Ca2+ concentrations, PPF increases (Rohrbough et al., 1999; Sandstrom, 2004). Indeed, facilitation in wild-type control larvae is higher (∼180%) at 0.25 mm external Ca2+ than at a Ca2+ concentration of 0.6 mm (∼115%) (Fig. 3G). In contrast, virtually no facilitation was observed at the cv-c1 mutant NMJ. Instead, a depression occurs at both Ca2+ concentrations, as if transmission during the initial EJP is already in a facilitated state (Fig. 3G). Therefore, we conclude that the probability of synaptic release is significantly increased at the cv-c mutant NMJ.
We then investigated whether the increased neurotransmitter release in the mutants was dependent on Ca2+ levels by comparing the QC of the mutants with the controls at different external Ca2+ concentrations ranging from 0.15 to 0.6 mm. The mutant QC was significantly higher than the control QC at all concentrations tested, although the slopes of the regression lines did not differ (Fig. 3H). These results indicate that the Ca2+ cooperativity is not altered in the mutant animals. Therefore, the properties of the Ca2+ sensors that regulate Ca2+-dependent vesicle fusion are not likely to be altered when cv-c levels are reduced.
RhoGAP cv-c is required at the postsynaptic side of the NMJ to modulate presynaptic neurotransmitter release
We used two different strategies to determine whether cv-c acts presynaptically or postsynaptically at the NMJ. First, we used transgenic RNA interference to reduce cv-c expression levels at individual sides of the synapse. Second, we investigated whether QC could be restored to wild-type levels in the cv-c mutant background by expressing cv-c presynaptically versus postsynaptically. We used the UAS–GAL4 transcriptional activation system (Brand and Perrimon, 1993) to effect tissue-specific expression of either double-stranded (ds) RNA (Billuart et al., 2001) or a cv-c cDNA (Denholm et al., 2005). The G14–GAL4 and OK6–GAL4 drivers (Aberle et al., 2002) were used for pan-muscle and pan-motoneuron expression, respectively.
Expression of cv-c dsRNA in the motoneuron (OK6/RNAi–cv-c) increased QC somewhat, but the increase was not significant when compared with OK6–GAL4 driver only control (Fig. 4). In contrast, reduction of cv-c expression in the muscle (G14/RNAi–cv-c) resulted in significantly increased QC, phenocopying the cv-c mutant phenotype. Moreover, rescue of the cv-c1 phenotype was only observed when cv-c expression was restored in the muscle in the heterozygous mutant background (QC = 47.9 ± 7.1 in G14/+;cvc1/UAS–cv-c–RA larvae; QC = 62.9 ± 3.5 in G14/+;cvc1/+ control larvae) and not in the motoneuron (QC = 65.2 ± 8.8 in OK6/+;cv-c1/UAS–cv-c–RA larvae). These results indicate that cv-c acts postsynaptically to maintain wild-type levels of neurotransmitter release at the NMJ, a finding consistent with our observations that cv-c is expressed in muscle but not detected in motoneuron cell bodies.
Figure 4.

RNA interference and rescue experiments reveal that RhoGAP cv-c is required postsynaptically at the larval NMJ. Bar graph representations are shown of the mean ± SEM values of the QC (left y-axis, dark bars) and the mEJP amplitudes (right y-axis, light bars) normalized to wild-type controls recorded from w1118 third-instar larvae, cv-c1/+, G14–GAL4/+, OK6–GAL4/+, G14–GAL4/RNAi–cv-c, OK6–GAL4/RNAi–cv-c, G14–GAL4/+;cv-c1/+, OK6–GAL4/+;cv-c1/+, G14–GAL4/+;cv-c1/UAS–cv-c–RA, and OK6–GAL4/+;cv-c1/UAS–cv-c–RA. Transgenic RNA interference targeting cv-c in muscle phenocopies the heterozygous cv-c1 elevated QC mutant phenotype, i.e., QC is 72.0% higher than w1118 controls. Presynaptic RNA interference targeting cv-c in motoneurons results in a moderate increase of ∼37.0% relative to w1118 controls (p < 0.05), but this increase is not statistically significantly different from the OK6–GAL4/+ driver control. Postsynaptic expression of cv-c in a heterozygous cv-c1 mutant background reduces QC to wild-type levels in contrast to presynaptic expression. All measured values are presented in supplemental Table 2 (available at www.jneurosci.org as supplemental material). Asterisks mark significant differences relative to w1118 controls: **p < 0.001.
The number of presynaptic neurotransmitter release sites is increased in the cv-c mutant
The increased neurotransmitter release observed in cv-c mutants might be caused by changes in either the presynaptic or postsynaptic apparatus, such as an increase in (1) presynaptic motoneuron terminal size or bouton numbers, (2) the number of active zones (AZs) with T-bars (dense bodies), which are presumptive presynaptic neurotransmitter release sites, or (3) the number or the sensitivity of the postsynaptic neurotransmitter receptors. The Drosophila NMJ harbors glutamatergic synapses, and the best characterized postsynaptic neurotransmitter receptors subunits are GluRIIA and GluRIIB (Schuster et al., 1991; Petersen et al., 1997; DiAntonio et al., 1999; Sigrist et al., 2002; Featherstone et al., 2005). We therefore investigated whether there were apparent differences in the glutamate receptor field in the cv-c1 mutants relative to controls. No changes in the numbers and intensities of the glutamate receptor subunit GluRIIA- and GluRIIB-positive domains were observed (data not shown). This observation is consistent with the mEJPs recordings, mentioned above, which indicated that postsynaptic sensitivity to glutamate is unchanged in the mutant. We also counted the number of boutons and measured muscle size in cv-c1 mutants but did not observe any differences from controls (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Overall, cv-c1 mutant NMJ ultrastructure appeared wild type, but we observed an increase in the number of T-bars at the mutant NMJ relative to controls (Fig. 5A vs C). Increased numbers of T-bars were also observed at the DysE6 mutant NMJ (Fig. 5B) (van der Plas et al., 2006). We performed immunohistochemistry using an antibody that recognizes the Bruchpilot (Brp) protein, which is associated with T-bar structures to quantify these apparent increases in T-bar number (Fig. 5D–F) (see Materials and Methods) (Kittel et al., 2006; Wagh et al., 2006). We found that there is a statistically significant 1.23-fold increase in the number of Brp+ zones in cv-c1 and DysE6 boutons relative to the control (Fig. 5G). This result was confirmed by morphometric analyses of EM micrographs of boutons of cv-c1 mutants versus controls showing a significant increase in the number of T-bars per AZ in the cv-c mutant (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), whereas the total length of the AZs per bouton perimeter was unchanged (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Thus, increased numbers of vesicle release sites in the cv-c mutant likely account for the higher QC observed.
Figure 5.

The number of active zones with T-bars per bouton is increased in the absence of RhoGAP cv-c. No major change of the bouton morphology of NMJs of muscle 6 and 7 at the ultrastructural level was observed among w1118 (A), DysE6 (B), and cv-c1 (C) larvae, except for an increase in the numbers of T-bars (arrows). Scale bars, 1 μm. This increase in T-bar numbers was confirmed using the NC82 antibody that recognizes Bruchpilot, a T-bar associated protein. NC82 antibody stainings of w1118 (D), DysE6 (E), and cv-c1 (F) larval body walls are shown. Scale bars, 5 μm. G, Bar graph of the number of NC82-positive AZs per bouton area is shown for each genotype (N = 5), showing a significant increase for DysE6 (1.31 ± 0.03; n = 98) and cv-c1 (1.28 ± 0.04; n = 97) compared with the wild-type control w1118 (1.06 ± 0.02; n = 78). *p < 0.001. N represents the number of larvae and n the number of boutons that were analyzed.
Dystrophin and cv-c likely act in the same or parallel pathways at the NMJ
Thus far, we have shown a genetic interaction of cv-c1 and DysE6 during the formation of the pcv and found that these genes display similar third-larval instar mutant NMJ phenotypes with respect to increased levels of neurotransmitter release and increased numbers of presynaptic neurotransmitter release sites. Both genes are expressed in muscle and not apparently in the motoneuron and are required at the postsynaptic side of the synapse. Next, we set out to investigate whether the two genes interact at the NMJ. However, in contrast to their roles in wing vein formation, Dys and cv-c are haploinsufficient for their roles in regulating neurotransmitter release (Fig. 3) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (van der Plas et al., 2006), preventing us from performing epistatic analyses of their function in that tissue. Moreover, the single heterozygous alleles show a similar increase in QC as observed at the transheterozygous cv-c1/DysE6 NMJ, possibly indicating that the maximum level of QC is already reached in the single heterozygous alleles (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Therefore, we used three alternative approaches to determine whether cv-c and Dys genetically interact at the NMJ.
First, we investigated whether Dys expression is altered at the cv-c1 mutant NMJ but did not observe changes in the levels or the localization of the Dys protein (data not shown) compared with the wild-type controls. Second, we attempted to restore wild-type neurotransmitter levels in the cv-c1 mutant by increasing expression levels of DLP2 in muscle or, conversely, by increasing expression levels of cv-c postsynaptically at the DysE6 mutant NMJ. We used a UAS-binding site containing P-element, GS12472, inserted upstream of DLP2 (Toba et al., 1999; van der Plas et al., 2006), and a UAS–cv-c–RA line (Denholm et al., 2005) driven by G14–GAL4 to increase expression of DLP2 and Cv-c, respectively, in the muscle. We observed significantly reduced QC values when DLP2 was expressed in the cv-c1 background but not when Cv-c was expressed in the DysE6 mutant background (Fig. 6A). This result suggests that Dys and Cv-c function in the same or parallel pathways.
Figure 6.
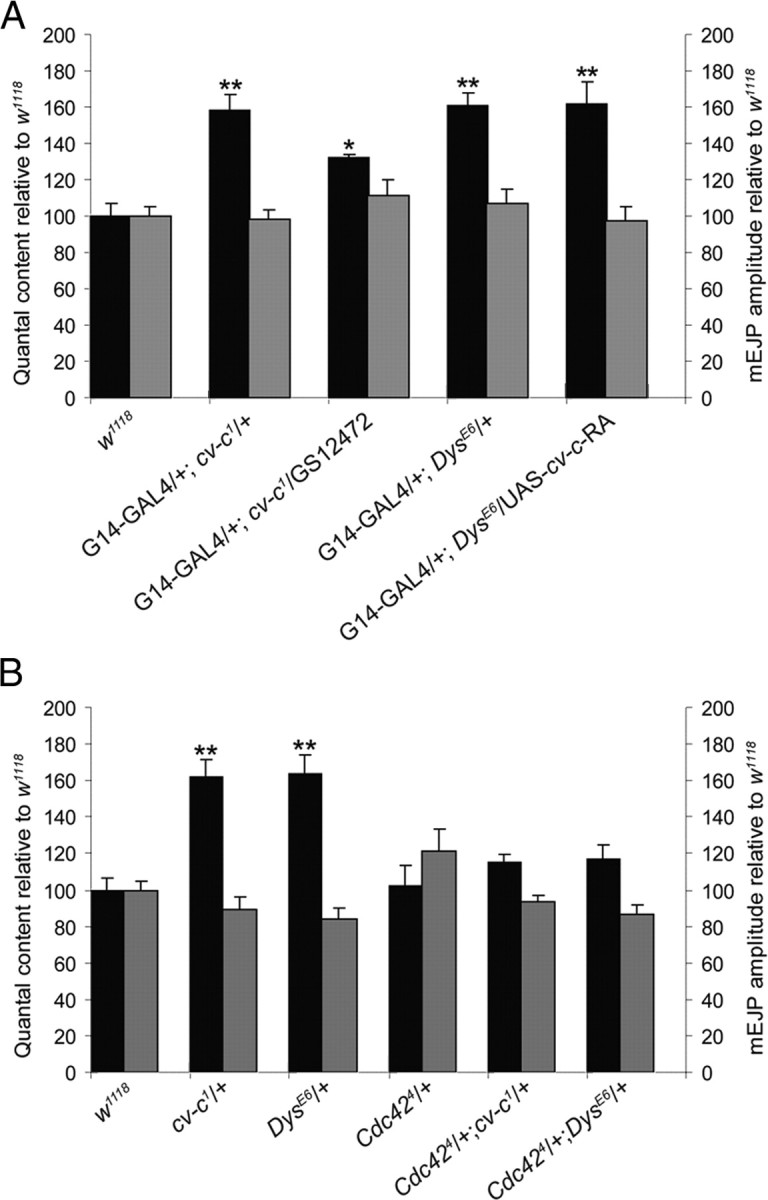
Dys interacts with a Rho GTPase signaling pathway at the larval NMJ. Bar graph representations are shown of the mean ± SEM values of the QC (left y-axis, dark bars) and the mEJP amplitudes (right y-axis, light bars) normalized to wild-type controls recorded from w1118 third-instar larvae. A, Overexpression of RhoGAP cv-c in the heterozygous DysE6 mutant muscle fibers does not reduce the increased QC level, whereas overexpression of Dys in the muscle in the heterozygous cv-c1 mutant partially reduces the otherwise increased QC toward wild-type levels. B, Heterozygosity for the Cdc424 mutant allele in either the heterozygous cv-c1 or DysE6 mutant background restores QC to wild-type levels. This suggests that Cdc42 is a substrate of Cv-c in a homeostatic signaling pathway operating at the NMJ and that Dystrophin genetically interacts with the Rho GTPase signaling pathway. All measured values are presented in supplemental Table 2 (available at www.jneurosci.org as supplemental material).
Third, we tried to rescue the effect on transmitter release observed in DysE6 mutants by simultaneously removing the possible target of Cv-c. The rationale for this experiment is that, if Dys and Cv-c act to negatively regulate Rho GTPase signaling, then removing potential Rho substrates in either mutant background would suppress their increased neurotransmitter release. Therefore, we first determined which Rho GTPase might suppress the cv-c synaptic phenotype. In other studies, it had been shown that Rho1, Rac1, Rac2, and Cdc42 were tissue-specific targets of Cv-c in the Malpighian tubuli and the epidermis and in cell-free assays (Denholm et al., 2005; Sato et al., 2010). We find that removing one copy of Cdc42 in the background of cv-c1 restored neurotransmitter release to wild-type levels (Fig. 6B). No interaction was found with rho1 or rac1 (data not shown). Interestingly, reduced levels of Cdc42 also rescue the increased QC observed in the DysE6 mutant (Fig. 6B), further suggesting that Dystrophin DLP2 functions to regulate a Rho GTPase signaling pathway required for homeostasis of the Drosophila third-instar larval NMJ.
Discussion
During development, homeostatic mechanisms ensure that levels of neurotransmitter release are adjusted to compensate for the dramatic increases in the size of synaptic terminals and their targets. At the NMJ, achieving appropriate synaptic homeostatic endpoints requires retrograde signaling from the muscle to the motoneuron, a process that has been studied extensively in Drosophila (for review, see Marqués and Zhang, 2006). Some of these retrograde molecular pathways, such as the bone morphogenetic protein pathway, are also involved in synaptic growth (Aberle et al., 2002; McCabe et al., 2003, Ball et al., 2010), whereas mutations in other genes can prevent appropriate synaptic homeostasis without visible effect on synapse size or overall morphology (Haghighi et al., 2003; Frank et al., 2006; Dickman and Davis, 2009). The challenges ahead include understanding how the latter types of pathways regulate synapse function.
We reported previously that the absence of the postsynaptic Dys DLP2 isoform results in elevated presynaptic neurotransmitter release at the Drosophila NMJ (van der Plas et al., 2006). Here, we made use of a wing crossvein phenotype exhibited by the DysE6 DLP2 mutant to perform a genetic screen for Dys-interacting genes whose possible functions at the NMJ could be subsequently determined. We identified the RhoGAP cv-c as a Dys mutant modifier. While this screen was in progress, other genes that interact with Dys during wing vein formation were reported (Christoforou et al., 2008; Kucherenko et al., 2008). cv-c was not found as an enhancer of Dys in these studies, but the genes mutated were not identified for the majority of the ethane methyl sulfonate-induced modifiers (Kucherenko et al., 2008), leaving open the possibility that cv-c was among them.
We determined that Cv-c is expressed postsynaptically at the NMJ and found that overall synapse morphology and postsynaptic responsiveness to neurotransmitter are unaltered in the cv-c mutant. Reduced expression of Cv-c results in significantly increased QC as a result of elevation of EJP amplitudes similar to that observed in the Dys mutant. Rescue and transgenic RNA interference experiments indicate that Cv-c is required postsynaptically to maintain appropriate levels of presynaptic vesicle release.
Our results indicate that the elevated QC is attributable to an increased probability of release, associated with higher numbers of T-bars per AZ, as was reported previously for the DysE6 DLP2 mutant (van der Plas et al., 2006) and for animals with decreased postsynaptic calcium/calmodulin-dependent kinase II (CaMKII) activity (Haghighi et al., 2003). The brp (Kittel et al., 2006) and unc-51 (Wairkar et al., 2009) mutant NMJs lack T-bars, display low QC, and, at least for brp, are not in a facilitated state as determined by paired-pulse assays. Conversely, the few remaining active synaptic sites at the rab3 mutant NMJ display elevated neurotransmitter release attributable to increased T-bar density (Graf et al., 2009). Together with our findings that dys and cv-c mutant NMJs are in a facilitated state and have increased numbers of T-bars, Brp+ punctate, and elevated QC, these results provide additional support for the hypothesis that increased numbers of T-bars correlate with increased QC and synaptic facilitation.
Insight into the interaction of Dys and cv-c came from our observation that reduced levels of the Rho GTPase Cdc42 suppress both the cv-c and DysE6 mutant synaptic phenotype. Cv-c is a member of the RhoGAP family, which together with the RhoGEFs and Rho GDP dissociation inhibitors, controls the nucleotide state of the Rho GTPases that cycle between an inactive GDP-bound and an active GTP-bound state (for review, see Hall, 1998). RhoGAPs promote the GTP-hydrolyzing activity of the Rho GTPases, thereby increasing the level of the GDP-bound, inactive form of the GTPases. Rho signaling pathways have been shown to regulate actin cytoskeleton organization, vesicle trafficking, and many other biological processes. Rho1, Rac1, Rac2, and Cdc42 have been found to be Cv-c targets both in vivo and in cell-free enzyme assays (Denholm et al., 2005; Sato et al., 2010).
We found that removal of one copy of Cdc42 in the cv-c background completely restores wild-type levels of neurotransmitter release. Heterozygosity for null alleles of rho1 or rac1 does not suppress the cv-c synaptic phenotype, but we cannot rule out the possibility that their expression levels are sufficiently high that halving them does not bring them below the threshold necessary to see suppression. The observation that heterozygosity for Cdc42 essentially completely suppresses the cv-c phenotype indicates, however, that it is likely the principle Rho GTPase gene interacting with cv-c to regulate presynaptic neurotransmitter release. Moreover, reduction of Cdc42 levels in the DysE6 mutant background also suppresses the increase in neurotransmitter release, providing additional evidence that Dys and Cv-c act together via Cdc42 to maintain normal synaptic function.
Cv-c and Dys are both required postsynaptically to maintain wild-type levels of neurotransmitter release. When we overexpressed Dys postsynaptically in the heterozygous cv-c1 mutant, we observed a limited, but significant, decrease in QC. This apparent partial rescue might be attributable to the increased recruitment of other RhoGAPs by overexpressed Dys. Postsynaptic overexpression of cv-c did not rescue the heterozygous DysE6 mutant phenotype, possibly as a result of the failure of Cv-c to localize properly in the absence of Dys. Although it remains unclear how Cv-c and Dys interact, we hypothesize that decreased Dys levels result in a reduction of the activity of Cv-c leading to increased activity of Cdc42.
The genetic interaction of cv-c and Cdc42 may indicate that Cdc42 is a direct target of Cv-c. However, it is also possible that the two genes act in either parallel pathways or in a single trans-synaptic pathway in which other RhoGAPs act on presynaptic Cdc42. The former possibility is consistent with proposed models for the regulation of synaptic homeostasis in which multiple feedback mechanisms are predicted to act simultaneously in parallel pathways (Davis, 2006). With respect to the possibility of a single trans-synaptic pathway, Cdc42 has been shown to interact genetically with the presynaptic RhoGEF gene ephexin (exn) whose protein product functions downstream of the Ephrin receptor to regulate the Cacophony (Cac) calcium channel (Frank et al., 2009). This pathway mediates rapid NMJ homeostasis and is also likely involved in slower long-term homeostatic responses because Cac was shown previously to be required for the compensatory increases in QC observed in the GluRIIA glutamate receptor mutant (Frank et al., 2006). Although exn (Frank et al., 2009) and cv-c (this work) were shown to act presynaptically and postsynaptically, respectively, neither study formally demonstrated where Cdc42 is required for homeostasis. Thus, it remains possible that Cdc42 plays roles in separate presynaptic and postsynaptic homeostatic pathways or in a single trans-synaptic pathway at one or both sides of the synapse.
The interactions of dys and Rho GTPase signaling during synaptic homeostasis could converge on, among other postsynaptic processes, cytoskeletal remodeling, clustering of neurotransmitter receptors, or Ca2+ handling. Synaptic actin networks play dynamic roles in modulating presynaptic and postsynaptic function (Cingolani and Goda, 2008). The postsynaptically localized DLP2 isoform bears the conserved Dystrophin N-terminal actin-binding domain. However, although the importance of this domain for the sarcolemmal interactions of Dystrophin has been established (Judge et al., 2006), its role at the synapse has not been reported. Additional cytoskeletal interactions of Dystrophin at the NMJ were revealed by the analysis of Ankyrin-deficient mice (Ayalon et al., 2008). Ankyrin stabilizes membrane-associated microtubule networks and is required for the localization of Dystrophin and other DGC proteins to the NMJ.
There have been several reports of Rho pathway involvements in postsynaptic receptor clustering. The Drosophila RhoGEF RtGEF (dPix) regulates the postsynaptic localization of GluRIIA through the Cdc42 effector kinase dPak (Parnas et al., 2001). Absence of dPix also compromises retrograde signaling, as reflected by reduced mEJPs and EJP amplitudes (Parnas et al., 2001). Similarly, the extracellular Dystroglycan-binding proteins Laminin and Agrin both signal through Rho GTPase-dependent pathways to initiate ACh receptor clustering (Weston et al., 2007). Finally, there is evidence for a RhoA-dependent postsynaptic role of ephexin-1 in regulation of clustering of acetylcholine neurotransmitter receptors at the murine NMJ (Shi et al., 2010). However, a role for Dys and/or cv-c in postsynaptic receptor clustering at the NMJ is unlikely because we observed no apparent changes in the number or location of the glutamate receptors in their respective mutant animals.
Last, changes in Ca2+ flux in response to the Dys/Cv-c/Cdc42 pathway may underlie the aberrant homeostatic endpoints observed at the Dys and cv-c mutant NMJs. Mammalian Dystrophin-deficient muscle fibers have extensively studied deficits in Ca2+ handling (for review, see Hopf et al., 2007). Interestingly, postsynaptic inhibition of CaMKII results in increased QC at the Drosophila NMJ (Haghighi et al., 2003). Although Dystrophin and CaMKII are known to interact physically via evolutionarily conserved motifs (for review, see Michalak et al., 1996), they have yet to be functionally linked at the NMJ.
At present, therefore, there are a number of potential targets of the Rho GTPase pathway that is disturbed in the DysE6 mutant. Identifying the targets of the Cv-c/Cdc42 pathway at the NMJ will increase our understanding of the role of Dystrophin at the synapse and likely lead to the identification of more general aspects of the pathways contributing to synaptic homeostasis.
Footnotes
This work was funded by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek. We thank Renée Bakker for help with the initial experiments, the Bloomington Drosophila Stock Center, R. Ray, and M. O'Connor for fly stocks, and J. Plomp and R. Ray for their comments on this manuscript.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Anderson JL, Head SI, Rae C, Morley JW. Brain function in Duchenne muscular dystrophy. Brain. 2002;125:4–13. doi: 10.1093/brain/awf012. [DOI] [PubMed] [Google Scholar]
- Ayalon G, Davis JQ, Scotland PB, Bennett V. An Ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell. 2008;135:1189–1200. doi: 10.1016/j.cell.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, Haghighi AP. Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron. 2010;66:536–549. doi: 10.1016/j.neuron.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Billuart P, Winter CG, Maresh A, Zhao X, Luo L. Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell. 2001;107:195–207. doi: 10.1016/s0092-8674(01)00522-0. [DOI] [PubMed] [Google Scholar]
- Boyd IA, Martin AR. The end-plate potential in mammalian muscle. J Physiol. 1956;132:74–91. doi: 10.1113/jphysiol.1956.sp005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Christoforou CP, Greer CE, Challoner BR, Charizanos D, Ray RP. The detached locus encodes Drosophila Dystrophin, which acts with other components of the Dystrophin associated protein complex to influence intercellular signalling in developing wing veins. Dev Biol. 2008;313:519–532. doi: 10.1016/j.ydbio.2007.09.044. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Denholm B, Brown S, Ray RP, Ruiz-Gómez M, Skaer H, Hombría JC. crossveinless-c is a RhoGAP required for actin reorganisation during morphogenesis. Development. 2005;132:2389–2400. doi: 10.1242/dev.01829. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci. 1999;19:3023–3032. doi: 10.1523/JNEUROSCI.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene Dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Rushton E, Rohrbough J, Liebl F, Karr J, Sheng Q, Rodesch CK, Broadie K. An essential Drosophila glutamate receptor subunit that functions in both central neuropil and neuromuscular junction. J Neurosci. 2005;25:3199–3208. doi: 10.1523/JNEUROSCI.4201-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradkin LG, Baines RA, van der Plas MC, Noordermeer JN. The Dystrophin Dp186 isoform regulates neurotransmitter release at a central synapse in Drosophila. J Neurosci. 2008;28:5105–5114. doi: 10.1523/JNEUROSCI.4950-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Daniels RW, Burgess RW, Schwarz TL, DiAntonio A. Rab3 dynamically controls protein composition at active zones. Neuron. 2009;64:663–677. doi: 10.1016/j.neuron.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Turner PR, Steinhardt RA. Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem. 2007;45:429–464. doi: 10.1007/978-1-4020-6191-2_16. [DOI] [PubMed] [Google Scholar]
- Judge LM, Haraguchiln M, Chamberlain JS. Dissecting the signaling and mechanical functions of the Dystrophin-glycoprotein complex. J Cell Sci. 2006;119:1537–1546. doi: 10.1242/jcs.02857. [DOI] [PubMed] [Google Scholar]
- Kittel RJ, Hallermann S, Thomsen S, Wichmann C, Sigrist SJ, Heckmann M. Active zone assembly and synaptic release. Biochem Soc Trans. 2006;34:939–941. doi: 10.1042/BST0340939. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Kucherenko MM, Pantoja M, Yatsenko AS, Shcherbata HR, Fischer KA, Maksymiv DV, Chernyk YI, Ruohola-Baker H. Genetic modifier screens reveal new components that interact with the Drosophila Dystroglycan-Dystrophin complex. PLoS One. 2008;3:e2418. doi: 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Fetter RD, Kopczynski C, Grenningloh G, Goodman CS. Genetic analysis of Fasciclin II in Drosophila: defasciculation, refasciculation, and altered fasciculation. Neuron. 1994;13:1055–1069. doi: 10.1016/0896-6273(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Marqués G, Zhang B. Retrograde signaling that regulates synaptic development and function at the Drosophila neuromuscular junction. Int Rev Neurobiol. 2006;75:267–285. doi: 10.1016/S0074-7742(06)75012-7. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Michalak M, Fu SY, Milner RE, Busaan JL, Hance JE. Phosphorylation of the carboxyl-terminal region of Dystrophin. Biochem Cell Biol. 1996;74:431–437. doi: 10.1139/o96-047. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the rho-type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Pilgram GS, Potikanond S, Baines RA, Fradkin LG, Noordermeer JN. The roles of the Dystrophin-associated glycoprotein complex at the synapse. Mol Neurobiol. 2010;41:1–21. doi: 10.1007/s12035-009-8089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. The Dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- Rapaport D, Passos-Bueno MR, Brandão L, Love D, Vainzof M, Zatz M. Apparent association of mental retardation and specific patterns of deletions screened with probes cf56a and cf23a in Duchenne muscular dystrophy. Am J Med Genet. 1991;39:437–441. doi: 10.1002/ajmg.1320390414. [DOI] [PubMed] [Google Scholar]
- Roberts RG, Bobrow M. Dystrophins in vertebrates and invertebrates. Hum Mol Genet. 1998;7:589–595. doi: 10.1093/hmg/7.4.589. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, Pinto S, Mihalek RM, Tully T, Broadie K. latheo, a Drosophila gene involved in learning, regulates functional synaptic plasticity. Neuron. 1999;23:55–70. doi: 10.1016/s0896-6273(00)80753-9. [DOI] [PubMed] [Google Scholar]
- Sandstrom DJ. Isoflurane depresses glutamate release by reducing neuronal excitability at the Drosophila neuromuscular junction. J Physiol. 2004;558:489–502. doi: 10.1113/jphysiol.2004.065748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Sugimura K, Satoh D, Uemura T. Crossveinless-c, the Drosophila homolog of tumor suppressor DLC1, regulates directional elongation of dendritic branches via down-regulating Rho1 activity. Genes Cells. 2010;15:485–500. doi: 10.1111/j.1365-2443.2010.01399.x. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H. Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science. 1991;254:112–114. doi: 10.1126/science.1681587. [DOI] [PubMed] [Google Scholar]
- Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, Baker D, Ruohola-Baker H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007;26:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Butt B, Ip FC, Dai Y, Jiang L, Yung WH, Greenberg ME, Fu AK, Ip NY. Ephexin1 is required for structural maturation and neurotransmission at the neuromuscular junction. Neuron. 2010;65:204–216. doi: 10.1016/j.neuron.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Schuster CM. The postsynaptic glutamate receptor subunit DGluR-IIA mediates long-term plasticity in Drosophila. J Neurosci. 2002;22:7362–7372. doi: 10.1523/JNEUROSCI.22-17-07362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- Taghli-Lamallem O, Akasaka T, Hogg G, Nudel U, Yaffe D, Chamberlain JS, Ocorr K, Bodmer R. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell. 2008;7:237–249. doi: 10.1111/j.1474-9726.2008.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Toba G, Ohsako T, Miyata N, Ohtsuka T, Seong KH, Aigaki T. The gene search system. A method for efficient detection and rapid molecular identification of genes in Drosophila melanogaster. Genetics. 1999;151:725–737. doi: 10.1093/genetics/151.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- van der Plas MC, Pilgram GS, Plomp JJ, de Jong A, Fradkin LG, Noordermeer JN. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci. 2006;26:333–344. doi: 10.1523/JNEUROSCI.4069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Plas MC, Pilgram GS, de Jong AW, Bansraj MR, Fradkin LG, Noordermeer JN. Drosophila Dystrophin is required for integrity of the musculature. Mech Dev. 2007;124:617–630. doi: 10.1016/j.mod.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Vactor DV, Sink H, Fambrough D, Tsoo R, Goodman CS. Genes that control neuromuscular specificity in Drosophila. Cell. 1993;73:1137–1153. doi: 10.1016/0092-8674(93)90643-5. [DOI] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wairkar YP, Toda H, Mochizuki H, Furukubo-Tokunaga K, Tomoda T, Diantonio A. Unc-51 controls active zone density and protein composition by downregulating ERK signaling. J Neurosci. 2009;29:517–528. doi: 10.1523/JNEUROSCI.3848-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the Dystrophin-associated glycoprotein complex. Ann Med. 2009;41:344–359. doi: 10.1080/07853890802668522. [DOI] [PubMed] [Google Scholar]
- Weston CA, Teressa G, Weeks BS, Prives J. Agrin and laminin induce acetylcholine receptor clustering by convergent, Rho GTPase-dependent signaling pathways. J Cell Sci. 2007;120:868–875. doi: 10.1242/jcs.03367. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]