

Abstract
Extracellular matrix metalloproteinase inducer (EMMPRIN, CD147) is a member of the Ig superfamily, with various physiological roles including the induction of matrix metalloproteinases (MMPs), leukocyte activation, and tumor progression. In this study, we illustrate a novel involvement of EMMPRIN in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). We found EMMPRIN levels to be upregulated on peripheral leukocytes before onset of EAE clinical signs and on infiltrating leukocytes and resident cells within the CNS in symptomatic mice. In EAE brain sections, EMMPRIN expression was localized with MMP-9 protein and activity. The increased EMMPRIN level was also characteristic of brain samples from MS subjects, particularly in plaque-containing areas. To evaluate the implications of elevated EMMPRIN levels, we treated EAE mice with an EMMPRIN function-blocking antibody and found reduced EAE clinical severity accompanied by decreased CNS parenchymal infiltration of leukocytes. Amelioration of EAE clinical signs by the anti-EMMPRIN antibody was critically dependent on its administration around the period of onset of clinical signs, which is typically associated with significant influx of leukocytes into the CNS. Moreover, the reduction in disease severity in anti-EMMPRIN-treated mice was associated with diminished MMP proteolytic activity at the glia limitans, the final barrier before parenchymal infiltration of leukocytes. Together, our results are the first to emphasize a role for EMMPRIN in MS and EAE, whereby EMMPRIN regulates leukocyte trafficking through increasing MMP activity. These results identify EMMPRIN as a novel therapeutic target in MS.
Introduction
Extracellular matrix metalloproteinase inducer (EMMPRIN, CD147) is a cell-surface glycoprotein investigated extensively in tumor biology (Biswas et al., 1995). The mechanism of EMMPRIN in tumor invasiveness is, in part, through inducing the expression of several matrix metalloproteinases (MMPs), including MMP-1, -2, -3, -9, and -11 (Biswas et al., 1995; Guo et al., 1997). EMMPRIN-expressing cells induce MMP production in an autocrine and paracrine manner (Tang et al., 2004).
In normal CNS tissue, EMMPRIN is found extensively at birth, whereas expression in adulthood is limited to brain endothelial cells (Fan et al., 1998). However, EMMPRIN must have important CNS functions, as EMMPRIN null mice display various sensory deficits (Igakura et al., 1996; Naruhashi et al., 1997; Chen et al., 2004).
Multiple sclerosis (MS) is an immune-mediated disease of the CNS with prominent demyelination and axonal degeneration. An animal model, experimental autoimmune encephalomyelitis (EAE), mimics several immunological features of MS. Many leukocyte subsets infiltrate into the CNS in MS and EAE through a multistep process. This involves the initial rolling and adhesion of leukocytes onto endothelial cells mediated through specific adhesion molecules and integrins (Engelhardt, 2008). After their diapedesis across the endothelial cell layer, leukocytes encounter two basement membranes: the first is the endothelial basement membrane, and the second is the parenchymal basement membrane or glia limitans (Agrawal et al., 2006).
Leukocytes have been shown to readily cross the endothelial basement membrane but require proteases to transmigrate the glia limitans and enter the CNS parenchyma. Available evidence indicates this proteolytic activity is provided principally by MMPs (Mun-Bryce and Rosenberg, 1998; Agrawal et al., 2006; Toft-Hansen et al., 2006). The use of MMP inhibitors (Toft-Hansen et al., 2006) and mutant mice (Agrawal et al., 2006) resulted in leukocytes being trapped in the perivascular space between the endothelial basement membrane and the glia limitans, and an attenuation of EAE disease.
There are 24 MMP members (Yong et al., 2001; Parks et al., 2004), and several of these are elevated simultaneously in the CNS of MS subjects (Anthony et al., 1997; Lindberg et al., 2001) and EAE-afflicted animals (Clements et al., 1997; Toft-Hansen et al., 2004; Weaver et al., 2005). Besides contributing to the trafficking of leukocytes into the CNS, MMPs have roles in regulating leukocyte activation, demyelination, and neurotoxicity (Yong et al., 2001). The simultaneous elevation of multiple MMPs in MS and EAE implis that targeting these with broad-spectrum inhibitors, or by affecting an upstream inducer of the expression of multiple MMPs such as EMMPRIN, would likely be more effective in resolving the disease.
As there is no existent association of EMMPRIN with MS, we have addressed whether EMMPRIN is upregulated in MS and EAE and whether EMMPRIN lies upstream to induce MMPs to promote leukocyte transmigration into the CNS. Our results highlight a role for EMMPRIN as a prominent upstream on-switch for MMP activity and a crucial regulator of leukocyte migration into the CNS to produce pathology.
Materials and Methods
Animals and EAE induction.
Six- to eight-week-old female C57BL/6 mice were used for EAE immunization. All procedures are in accordance with guidelines of the Canadian Council of Animal Care and have received approval by the local ethics committee. For immunization, 50 μg of MOG35–55 peptide in complete Freund's adjuvant containing 10 mg/ml of heat-inactivated Mycobacterium tuberculosis H37RA (Difco) was injected subcutaneously, 50 μl on either side of the tail base. Animals were supplemented with 300 ng of pertussis toxin injected intraperitoneally on days 0 and 2 after myelin oligodendrocyte glycoprotein (MOG) immunization. The animals were monitored daily for weight loss and changes of EAE disease score using a scale of 1–15 described previously (Weaver et al., 2005).
Human tissue samples.
Fresh, frozen adult human CNS tissue samples for Western blots were obtained from the Human Brain and Spinal Fluid Resource Center in Los Angeles, California. Fixed and paraffin-embedded MS tissues for immunohistochemical localization of EMMPRIN were from archival specimens at the University of Calgary.
Western blots.
Mouse and human CNS tissue were sonicated in protein lysis buffer containing 1% Triton X-100 and a protease inhibitor tablet (Roche Diagnostics). Samples were separated by SDS-PAGE on 12% gels, transferred to polyvinylidene difluoride membranes, and probed using antibodies to rat anti-mouse EMMPRIN (OX-114; Serotec) or mouse anti-human EMMPRIN (109403; R&D Systems). EMMPRIN appears around 50 kDa in Western blots. To determine whether similar amounts of total protein were loaded per well, immunoblots were stripped (Restore Western blot stripping buffer; Pierce) and reprobed with an anti-actin antibody (mouse anti-actin; Millipore).
With regard to anti-EMMPRIN antibodies for Western blots or other applications such as flow cytometry and immunohistochemistry (see below), we determined in preliminary optimization experiments that specific anti-EMMPRIN antibodies worked best for particular techniques; therefore, we had to use several EMMPRIN antibodies in this study, and they are all listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material). The specificity of the EMMPRIN antibodies was based on reduced signal when recombinant EMMPRIN was preincubated with antibodies and by the use of secondary antibody controls.
Gelatin zymography.
Gelatin gel zymography was used to identify MMP-2 and -9 present in mouse CNS tissue as described previously (Agrawal et al., 2006). Briefly, gelatin-binding proteins were first enriched by treatment of samples with gelatin Sepharose and then separated on a 10% polyacrylamide gel containing 1 mg/ml gelatin under nonreducing conditions. Gels were washed in a Triton X-100 renaturing buffer, followed by incubation in developing buffer containing 0.02% Brij 35, and stained with Coomassie blue, followed by destaining.
In situ zymography and immunofluorescence staining.
Gelatinolyic activity was localized in CNS sections by in situ zymography as described previously (Agrawal et al., 2006). Briefly, 10 μg/ml DQ-gelatin-FITC (EnzCheck; Invitrogen) in 50 mm Tris-HCl, pH 7.4, plus 1 mm CaCl2 was applied to unfixed, frozen cerebellar cryosections. These were incubated for 4 h in a humid chamber at 37°C. Digestion of the gelatin-FITC substrate results in unquenching of fluorescein and the localization of sites of proteolytic activity in the cerebellar samples. Each section was then washed in PBS and fixed in −20°C methanol before immunofluorescence staining as described previously (Agrawal et al., 2006). Primary antibodies used in immunofluorescence staining included anti-pan laminin as a marker of basement membranes (a kind gift from Dr. L. Sorokin, Institute of Physiological Chemistry and Pathobiochemistry, Westfälische Wilhelms-Universität, Münster, Germany), anti-CD45 (30F11) as a pan leukocyte marker (BD Bioscience), anti-CD3 as a T-cell marker (RM-153; Serotec), anti-mouse EMMPRIN (OX-114; Serotec) (supplemental Table 1, available at www.jneurosci.org as supplemental material), anti-glial fibrillary acidic protein (GFAP) as a marker of astrocytes (Sigma), anti-Iba-1 as a marker of macrophages and microglia (Wako), and anti-MMP-9 as a marker of active and inactive protease (Millipore Bioscience Research Reagents). Bound antibodies were visualized using Alexa 488-conjugated anti-rabbit or Alexa 546-conjugated anti-rat secondary antibody (Jackson ImmunoResearch Laboratories) or Pacific blue goat anti-rabbit secondary antibody (Invitrogen). Sections were examined and photographed using a BX51 fluorescence microscope (Olympus) and a Retiga 2000R camera from Q imaging.
Areas of the CNS examined included the lumbar sacral spinal cord, as this is a region that is commonly and rapidly affected in EAE, as well as the cerebellum, which presents with prominent perivascular inflammatory cuffs.
Fluorescence-activated cell sorting.
Mice were first perfused with physiological saline, and then spleens, lymph nodes, and CNS were dissected out in HBSS. Single-cell suspensions were isolated from these tissues by sieving through a 70 μm nylon filter. Cells from the lumbar sacral spinal cords were further separated into neural and leukocyte populations by discontinuous density gradient centrifugation using isotonic Percoll (GE Healthcare). Flow cytometry was performed using fluorescence-conjugated antibodies against CD45.2 (leukocytes), CD3 (T-cells), CD11b (monocytoid cells), or CD11c (dendritic cells) (all from BD PharMingen), GFAP (astrocytes), or EMMPRIN (E-Bioscience or Serotec) (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Anti-EMMPRIN antibody administration to EAE mice.
An anti-mouse EMMPRIN (clone RL73.2) function-blocking antibody (E-Bioscience) (supplemental Table 1, available at www.jneurosci.org as supplemental material) or rat IgG2a isotype control (E-Bioscience) was injected intraperitoneally (100 μg per mouse) in a volume of 100 μl into mice at specific time points (see Results) after EAE induction.
Statistical analyses.
We used Instat version 3 software (GraphPad) for our statistical analyses. A two-tailed unpaired Student's t test was used where two experimental groups with parametric scores were compared. In the case of EAE clinical scores, the ordinal scale required a nonparametric Mann–Whitney U test.
Results
EMMPRIN levels are increased in mouse CNS during EAE
In nonimmunized CNS sections, low EMMPRIN immunofluorescence was found restricted to endothelial cells of blood vessels (Fig. 1 A), as has been reported previously (Fan et al., 1998). In EAE mice, however, high EMMPRIN levels were evident not only around inflamed vessels but also on other cellular profiles in the vicinity of inflammation (Fig. 1 B). Consistent with these results, EMMPRIN levels detected using Western blots were found to be substantially increased in the CNS at the onset of clinical signs of EAE (day 10 after immunization) and at peak disease (day 15); indeed, elevated EMMPRIN levels were already evident at the presymptomatic stage at day 5 (Fig. 1 C).
Figure 1.
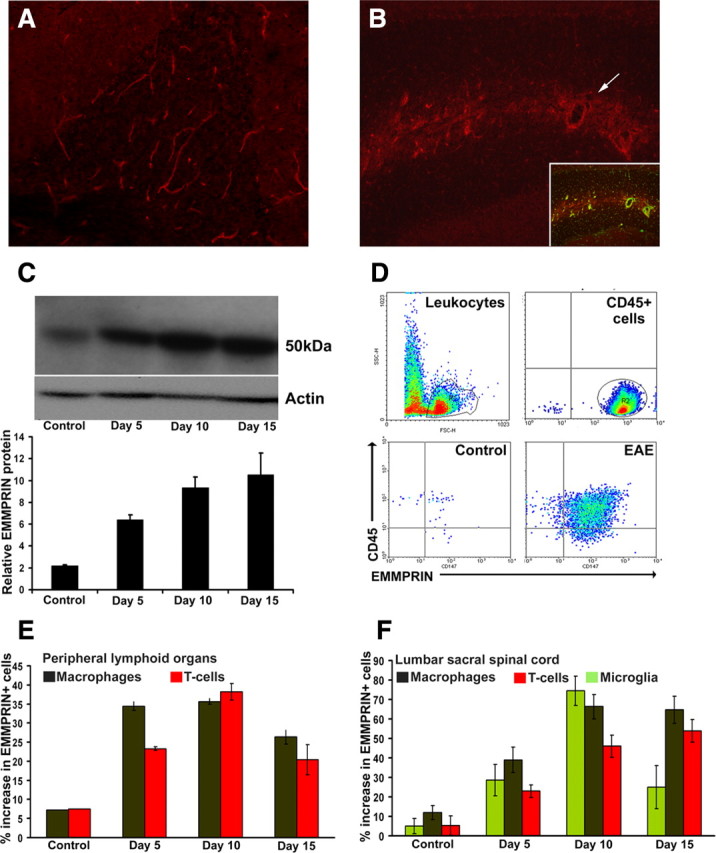
EMMPRIN expression is upregulated in EAE. Immunofluorescence staining (A, B), Western blot analysis (C), and flow cytometry (D–F) revealed higher EMMPRIN protein expression in mice immunized for EAE compared with naive controls. A, B, EMMPRIN expression was restricted to blood vessels in control cerebellum (A; 100× original magnification) and dramatically upregulated in EAE around inflamed blood vessels [B; 200× original magnification; the inset is double-immunofluorescence staining for pan laminin in green (vessels) and EMMPRIN in red] and also on other cellular profiles not associated with vessels. C, EMMPRIN, detected at 50 kDa in mouse spinal cord, increased (mean ± SEM; n = 3) in mice immunized for EAE at the presymptomatic stage (day 5), at the time of appearance of clinical signs (day 10), and at the peak clinical severity (day 15) compared with naive controls. Actin was used as a loading control. D, FACS analyses gating for CD45+ cells found increased numbers of EMMPRIN+ cells in lumbar sacral spinal cord isolates from EAE mice compared with controls. E, F, A time-point analysis on CD45+ gated cells from peripheral lymphoid organ (E) and lumbar sacral spinal cord (F) revealed increase in EMMPRIN in the T-cell (CD3+, red), macrophage (CD45highCD11bhigh, brown), and microglia (CD45low, CD11bhigh, green) populations with EAE progression. Data for fluorescence-activated cell sorting are mean ± SEM. pooled from nine mice each.
Various leukocyte subsets elevate EMMPRIN expression during EAE
To determine the cellular populations in EAE that expressed EMMPRIN, we first used flow cytometry of various live cell populations isolated from the lumbar sacral spinal cord and peripheral lymphoid organs (spleen and lymph nodes) gated on CD45+ (Fig. 1 D). A higher percentage of EMMPRIN-positive cells was detected in peripheral lymphoid organs on CD11b+ monocyte/macrophages and CD3+ T-cell populations at the presymptomatic period (day 5), at the onset of clinical signs (day 10), and at later periods (day 15) (Fig. 1 E) compared to naive control mice. In the lumbar sacral spinal cord, increased EMMPRIN expression was found in infiltrating T-cells (CD3+), macrophages (CD45high CD11bhigh), as well as microglia (CD45lowCD11bhigh) (Fig. 1 F, supplemental Fig. 1, available at www.jneurosci.org as supplemental material) at days 10 and 15, with a small increase already apparent at presymptomatic EAE (day 5).
We next used immunofluorescence staining on 4% paraformaldehyde-fixed cryostat sections from the lumbar sacral spinal cord. Consistent with fluorescence-activated cell-sorting (FACS) results, at peak EAE disease, EMMPRIN was highly expressed in infiltrating CD3+ T-cells (Fig. 2 A–C), in CNS resident GFAP+ astrocytes (Fig. 2 D–F) and in Iba-1+ macrophage/microglia (Fig. 2 G–I). Indeed, quantitation by flow cytometry shows that EMMPRIN+ astrocytes increased from 1% in normal brain to 92% in EAE mice, whereas the proportion of EMMPRIN+ macrophage/microglia was elevated from 9.4% in normal to 32% in the CNS in EAE mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These findings show that in EAE, EMMPRIN levels increase not only in CNS infiltrating cells but also in CNS resident cells.
Figure 2.

EMMPRIN is expressed by both CNS resident and infiltrating cells in EAE. At peak EAE disease (day 15), various cell types in the spinal cord were examined for EMMPRIN expression. A–C, Using immunofluorescence staining, we show that a large number of CD3+ infiltrating T-cells are also EMMPRIN positive; a high-magnification inset in C shows CD3 colocalization with EMMPRIN. D–F, GFAP+ astrocytes also expressed high amounts of EMMPRIN protein. G–I, Finally, Iba-1-positive macrophage/microglial cells stained positive for EMMPRIN. Pictures are representatives from similar data from nine mice each. All micrographs are 200× original magnification.
Increased EMMPRIN levels colocalize with increased MMP activity in the mouse CNS during EAE
Immunofluorescence staining of the cerebellum using a pan laminin (pan LM) antibody to demarcate the endothelial basement membrane and glia limitans, and a CD45+ antibody to stain leukocytes, revealed in EAE-afflicted mice the accumulation of leukocytes in perivascular cuffs as well as in the parenchyma (Fig. 3 A–C). High EMMPRIN immunofluorescence was found to coincide with increased MMP-9 fluorescence in and around the perivascular cuffs (Fig. 3 D–F). To determine whether EMMPRIN levels coincide with active MMP-2/9 levels, we used in situ gelatin zymography that detects signals selective for MMP activity, particularly those for active MMP-2 and/or MMP-9 (Oh et al., 1999; Agrawal et al., 2006). EMMPRIN expression was found to be coincident with MMP-2/9 activity (Fig. 3 G–I, arrows). We were unable to measure the activity of other members of the MMP family known to be elevated in EAE, such as MMP-3 (Weaver et al., 2005), because of insensitivity of techniques for non-MMP-2/9 in situ.
Figure 3.
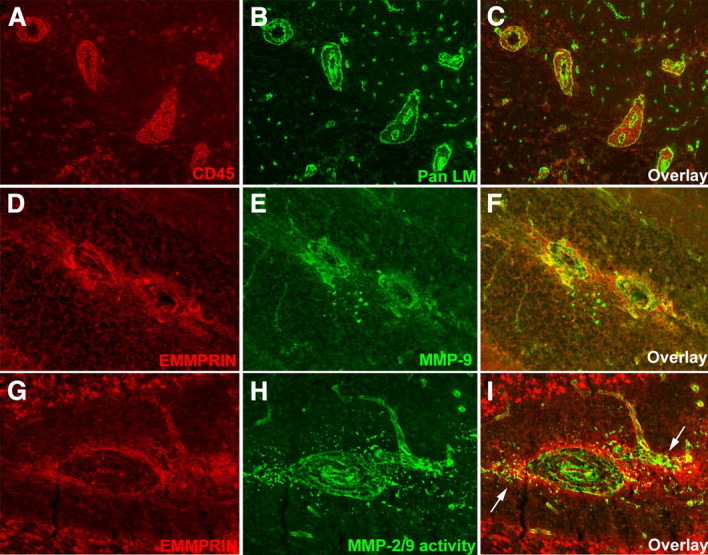
In EAE, EMMPRIN staining around inflammatory cuffs coincide with MMP-9 expression and MMP-2/9 activity. A–C, In the white matter of cerebellum of mice with peak EAE clinical severity, perivascular inflammatory cuffs typified by CD45+ leukocytes (red; A, C) sandwiched between the two laminin-containing (Pan LM; green; B, C) basement membranes of vessels are common (200× original magnification). D–F, High EMMPRIN levels (red; D, F) are found in and around the inflammatory cuffs coincident with high levels of MMP-9 protein (green; E, F) (200× original magnification). G–I, Similarly, EMMPRIN staining (red; G, I) colocalizes (I, arrows) with MMP-2/9 activity detected by in situ zymography (green; H, I) at sites of perivascular cuffs (400× magnification); in situ zymography cannot differentiate between the activity of MMP-2 and -9.
Elevation of EMMPRIN in MS
Having determined that EMMPRIN is upregulated in EAE, we examined whether this similarly occurred in MS. Western blot determinations showed that EMMPRIN levels in brain samples were higher in all four MS cases compared with healthy controls (Fig. 4 A), with higher EMMPRIN levels in active (MS1 and MS2) compared with inactive (MS3 and MS4) MS plaques. Within the MS cases, EMMPRIN levels were highest in plaques compared to adjacent normal appearing gray and white matter (Fig. 4 B, one case shown).
Figure 4.
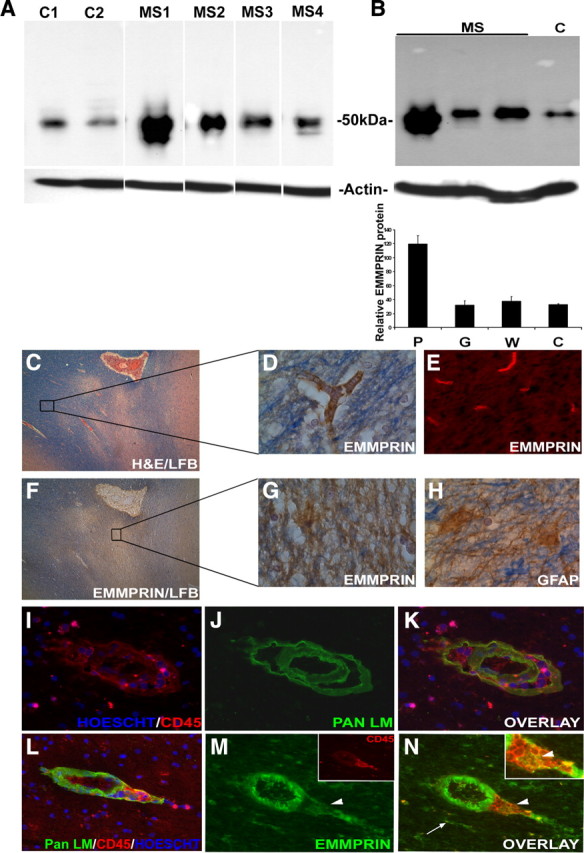
High EMMPRIN levels detected in MS plaques. A, Western blots revealed high EMMPRIN levels (50 kDa) in CNS samples from individual MS patients (MS1–2, with active lesions; MS3–4, inactive lesions) versus healthy donors (C1 and C2). Results are obtained from the same blot, and the white lines are attributable to irrelevant samples that have been spliced out. Similar levels of actin as the loading control in all samples are indicated. Samples C2 and MS4 have extra bands above and below the EMMPRIN band, respectively. The extra band in C2 is observed in a blot stained with the secondary antibody only (data not shown) and likely represents nonspecific binding. The lower bands in sample MS4 were not detected with the secondary antibody only and may be lower-molecular-weight glycosylated forms of EMMPRIN. B, Among the MS cases (where an example from one MS case is displayed), EMMPRIN was highly expressed in the MS plaque (P) compared to adjacent normal appearing white (W) and gray (G) matter. C, Hematoxylin and eosin with luxol fast blue staining of an MS plaque-containing specimen (40× original magnification) was used to examine EMMPRIN expression in both plaque and adjacent nonaffected white matter. D, E, G, Both immunohistochemistry (D) using 3,3′-diaminobenzidine tetrahydrochloride (DAB) and immunofluorescence staining (E) revealed EMMPRIN staining in normal-appearing areas to be restricted to blood vessels, whereas in plaques, EMMPRIN immunoreactivity was widespread (G). H, Immunohistochemistry for GFAP-positive astrocytes showed a similar pattern of expression as EMMPRIN. I–K, Immunofluorescence staining of an active MS plaque revealed the presence of inflammatory cuffs marked with pan laminin (Pan LM) staining for basement membranes surrounding the blood vessel and CD45+ leukocyte accumulation and infiltration into the CNS parenchyma. L–N, EMMPRIN, found to be predominantly expressed on endothelial cells of the inflammatory cuffs with this particular antibody, also colocalized with infiltrating CD45+ cells (M, inset; N, inset, arrowhead), and CNS resident cells (N, arrow). Hoescht dye was used to stain the nucleus of cells blue (I, K, L).
Immunohistochemical analysis of paraffin-embedded sections for EMMPRIN shows that in normal appearing white matter areas of MS CNS (Fig. 4 C, box), EMMPRIN was restricted to endothelial cells of blood vessels (Fig. 4 D,E). However, in the plaque-containing samples evident by extensive demyelination (loss of luxol fast blue staining) (Fig. 4 F, box), EMMPRIN immunohistochemistry was widespread (Fig. 4 G) and was similar to the staining pattern of GFAP+-reactive astrocytes in the same area (Fig. 4 H). We next performed immunofluorescence staining, on frozen sections, of MS CNS plaques, choosing the same MS cases used in the Western blots described previously in Figure 4, A and B. The results revealed the presence of typical inflammatory cuffs marked by pan laminin (Fig. 4 J,K) staining of the endothelial basement membrane and glia limitans, which surround blood vessels in the CNS, as well as an accumulation of CD45+ leukocytes (Fig. 4 I,L) in the perivascular cuff and within the CNS parenchyma. Staining with a mouse anti-human EMMPRIN antibody that was optimal for frozen tissue (but not for the paraffin-embedded sections) revealed prominent EMMPRIN expression (Fig. 4 M,N), not only on endothelial cells of blood vessels but also colocalized with infiltrating leukocytes (Fig. 4 M, inset, N, inset, arrowhead) and CNS resident cells (Fig. 4 N, arrow). These findings suggest an upregulation of EMMPRIN in MS and support a role for EMMPRIN in MS.
The blockade of EMMPRIN in EAE attenuates disease activity
To determine a role for EMMPRIN in EAE, a function-blocking antibody for EMMPRIN or an isotype control was administered to mice at days 8, 11, and 15 after EAE induction (Fig. 5 A), and EAE disease (onset, severity, and progress) was monitored for each group. Anti-EMMPRIN treatment reduced EAE severity, with a peak clinical disease score of 4 (involvement of tail and hindlimbs) versus 9 (involvement of tail, hindlimbs and forelimbs) in the isotype group. Disease scores between groups were significantly different (*p = 0.01–0.05, Mann–Whitney U test).
Figure 5.
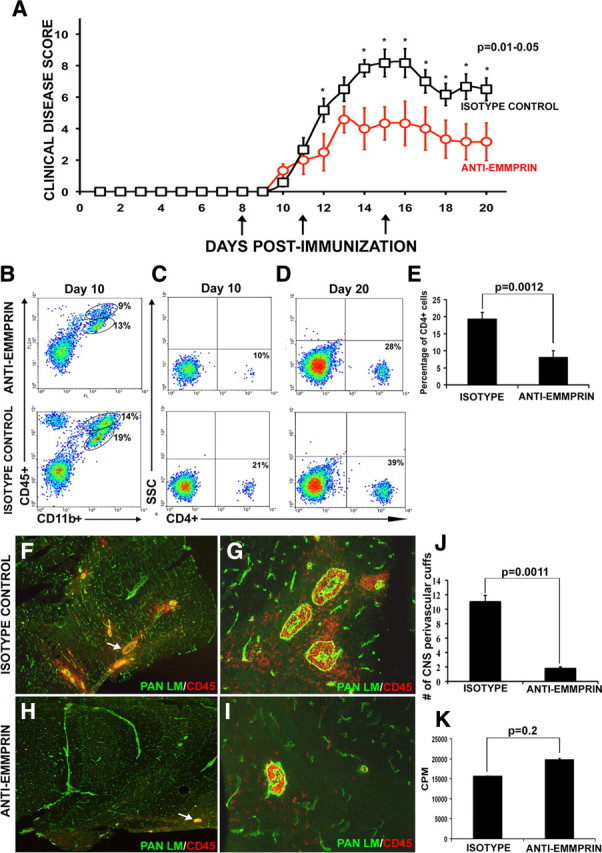
Treatment with anti-EMMPRIN function-blocking antibody attenuates EAE disease severity. A, Mice immunized for EAE were treated with an anti-EMMPRIN function-blocking antibody at days 8, 11, and 15 (arrows) after MOG immunization, and EAE disease severity was found to be significantly reduced compared with mice treated with an isotype antibody control. The data points that are statistically different are indicated (*p = 0.01–0.05, Mann–Whitney U test). Results are mean ± SEM. from six mice each; this was reproduced in another experiment. B–D, Using fluorescence-activated cell sorting, the percentage of CD45+CD11b+ macrophage/microglia (B; the ellipse with the higher CD45 represents macrophage, whereas the lower ellipse depicts microglia) and CD4+ T-cells at days 10 (C) and 20 (D) in the CNS were observed to be lower in mice treated with anti-EMMPRIN compared with those treated with isotype control. E, Percentage of CD4+ T-cells was lower in the CNS of EAE mice treated with anti-EMMPRIN compared with treatment with isotype control. F–J, Immunofluorescence staining for pan laminin (green) and CD45+ (red) cells of mouse cerebellum reveal a significantly higher number of perivascular cuffs (arrows) and CNS parenchymal infiltrates in isotype-treated [40× (F) and 200× (G) original magnification, respectively] animals compared with those treated with anti-EMMPRIN [40× (H) and 200× (I) original magnification, respectively]; the quantified results of the total number of perivascular cuffs observed in both groups is displayed in J. Results are representative from six mice in each group. K, Importantly, the recall response of T-cells to MOG35–55 was not significantly different between the anti-EMMPRIN- and isotype control-treated groups. CPM, Counts per minute in a proliferation assay.
CD45+ populations of cells (presumably T-cells) that are CD11b− (Fig. 5 B, top left corner) were reduced in the anti-EMMPRIN-treated group compared with isotype controls. Fewer CD45+CD11b+ macrophage/microglia were found in anti-EMMPRIN-treated EAE mice compared with isotype antibody controls (macrophage, 9 vs 14%; microglia, 13 vs 19%, respectively) (Fig. 5 B). Moreover, less CD4+ T-cells were found in the lumbar sacral spinal cord of anti-EMMPRIN-treated animals versus isotype controls at days 10 (10 vs 21%) and 20 (28 vs 39%) of EAE mice (Fig. 5 C,D, respectively). Overall, a lowered percentage of CD4+ cells were detected in anti-EMMPRIN-treated compared with isotype-treated mice (Fig. 5 E). Similarly, 14 d after immunization, immunofluorescence staining of the cerebellum using a pan LM antibody to demarcate the endothelial basement membrane and glia limitans and a CD45+ antibody to stain leukocytes revealed a higher density of perivascular cuffs and parenchymal infiltrates in the isotype-treated EAE mice (Fig. 5 F,G) compared with anti-EMMPRIN animals (Fig. 5 H,I). Although we expected to observe larger perivascular cuffs in the anti-EMMPRIN-treated animals, our results showed the presence of smaller and fewer inflammatory cuffs. EMMPRIN is expressed on endothelial cells of blood vessels in the CNS (Fan et al., 1998), and it is possible that treatment with the function-blocking anti-EMMPRIN antibody impedes the binding of leukocytes to endothelial cells. If so, this suggests a possible role for EMMPRIN in leukocyte adhesion to endothelial cells, although this would have to be investigated further.
The number of total cerebellar perivascular cuffs was counted in three mice in each group. Briefly, each cerebellum was completely sectioned into 10-μm-thick coronal sections and placed onto glass slides. Antibodies against pan laminin and CD45 were used to stain components of the basement membranes and infiltrating cells, respectively, to illustrate a perivascular cuff. Every perivascular cuff in all sections was counted, taking care that the same cuff was not counted twice. Significantly fewer perivascular cuffs were observed in anti-EMMPRIN-treated mice compared with isotype controls (Fig. 5 J). Fewer of the perivascular cuffs were found to be associated with CNS parenchymal infiltration of CD45+ cells in anti-EMMPRIN-treated EAE mice compared with isotype controls (42 vs 95%).
We addressed whether the decrease in leukocyte infiltration into the CNS parenchyma in the anti-EMMPRIN group was caused by lowered peripheral generation of antigen-specific T-cells. Splenocytes and lymph node cells were harvested at day 10 after MOG35–55 immunization and were restimulated with MOG35–55 in vitro for 3[H]-thymidine incorporation. No significant difference in T-cell proliferation was found between anti-EMMPRIN and isotype antibody treatments (Fig. 5 K), indicating that the lowered cellular infiltration into the CNS in anti-EMMPRIN animals was not contributed by reduced generation of antigen-specific T-cells.
Mechanism of action for EMMPRIN in EAE
To support the possible contention that anti-EMMPRIN antibody reduces EAE severity through a mechanism after activation of MOG-specific T-cells, we determined whether treatment could still affect EAE clinical scores if administered on days 0, 4, and 8 after MOG immunization, time points when antigen-reactive cells and other disease-promoting leukocytes are generated and propagated. This treatment paradigm did not result in modulation of EAE disease (supplemental Fig. 2, group I, available at www.jneurosci.org as supplemental material). Another group of mice treated with anti-EMMPRIN antibody on days 4, 8, and 11 after immunization showed a separation of initial course of disease compared with isotype antibody-treated mice (supplemental Fig. 2, group II, available at www.jneurosci.org as supplemental material). Collectively, these results of anti-EMMPRIN antibody administered at various times suggest that anti-EMMPRIN treatment was critically important between days 8 and 11 and that the continuation of treatment with anti-EMMPRIN after day 11 helps to contain disease activity.
How then is anti-EMMPRIN reducing the trafficking of leukocytes into the CNS parenchyma? We suggest that it may be the result of reduced MMP activity required for penetration of cells across the glia limitans. To address this, we subjected cerebellar cryosections to double-immunofluorescence staining for pan laminin and/or CD45+ cells, combined with gelatin in situ zymography. We found MMP activity in close proximity to CD45+ cells within the cuff or at the advancing front of leukocytes that infiltrated into the CNS parenchyma (Fig. 6 A,B). Importantly, while MMP activity could be detected in the perivascular cuff and advancing front of leukocytes within cerebellar slices of isotype antibody controls (Fig. 6 C–E), these were barely detectable or absent in sections from anti-EMMPRIN-treated animals at day 15 of EAE (Fig. 6 F–H). These data are compatible with reduced MMP-2 and -9 levels in anti-EMMPRIN-treated mice compared with isotype antibody controls examined through gel zymography of tissue homogenates (Fig. 6 I). Thus, together with the coincident expression of EMMPRIN with MMP-2/9 activity described previously (Fig. 3), the results of anti-EMMPRIN treatment validate that EMMPRIN acts as a prominent upstream on-switch for MMP activity at the blood–brain barrier in EAE, and this particularly promotes the entry of leukocytes into the CNS parenchyma.
Figure 6.
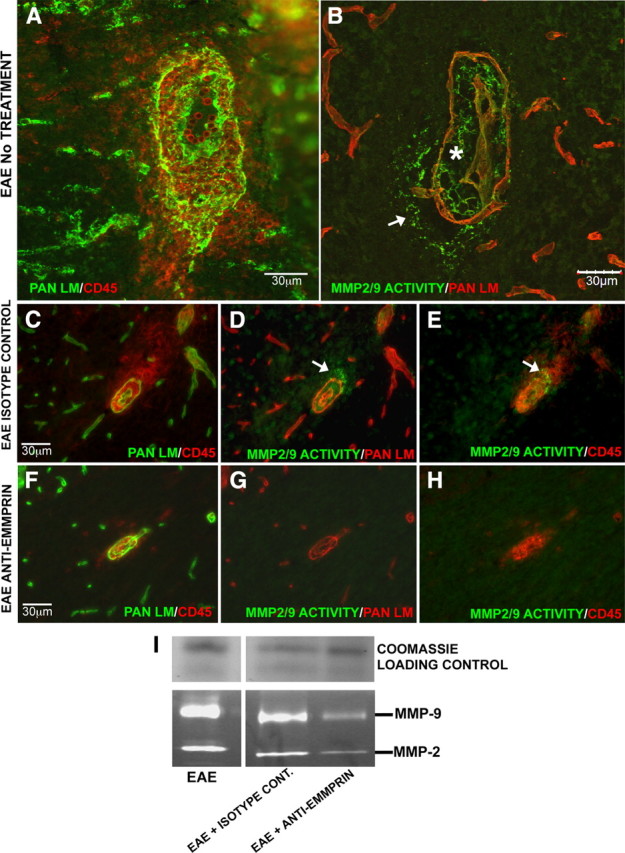
Anti-EMMPRIN antibody treatment reduces MMP activity in the CNS. A–C, Inflamed vessels with numerous CD45+ cells both within the perivascular cuff and invading the CNS parenchyma (B; asterisk and arrow, respectively) were observed in cerebellum cryosections of EAE mice with no treatment or isotype control antibody treatment (A, C), double-immunofluorescence stained for pan laminin (green) and CD45+ (red). B, D, E, Immunofluorescence staining for pan laminin (red) coupled with in situ zymography (green) showed MMP activity corresponding to the sites where leukocytes infiltrate the glia limitans to enter the CNS parenchyma. F, In EAE mice treated with anti-EMMPRIN antibody, double-immunofluorescence staining showed CD45+ cells to be restricted to the perivascular cuff, with very little to no infiltration into the CNS parenchyma. G–I, Immunofluorescence coupled with in situ zymography for MMP activity and gelatin gel zymography showed a reduction in MMP-2 and MMP-9 activity and levels with anti-EMMPRIN treatment. Coomassie blue was used to stain a nongelatin gel to verify equal sample loading on gel zymograms (I). In all cases, results are representatives from two independent experiments with at least three animals per treatment in each group.
Discussion
This study is the first to investigate a role for EMMPRIN in MS and EAE. We found an increase in EMMPRIN levels in both active MS plaques and in the CNS of EAE-afflicted mice. Indeed, in the latter model where a temporal study was possible, EMMPRIN expression in circulating leukocytes and within the CNS in infiltrated immune cells and CNS resident cells was elevated before onset of clinical signs; the further elevation of EMMPRIN coincided with increasing disease severity. Examined at peak clinical disease, EMMRPIN expression correlated with MMP-9 protein expression and MMP-2/9 enzymatic activity. When a function-blocking antibody to EMMPRIN was injected into EAE mice, we observed not only a reduction in disease severity correspondent with reduced cellular infiltrates, but also a marked decrease in MMP activity at the glia limitans. Together, our data implicate EMMPRIN as an upstream regulator of MMP activity-mediated transmigration of leukocytes into the CNS.
MMPs are implicated in the pathology of MS and EAE as marked by several lines of evidence (Hartung and Kieseier, 2000; Yong et al., 2001, 2007; Opdenakker et al., 2003). Many members are elevated in the serum, CSF, and brain tissue of patients with MS (for review, see Yong et al., 2007) and in the CNS of mice afflicted with EAE (Weaver et al., 2005; Toft-Hansen et al., 2006). In human subjects, the period between the first and second clinical attacks of MS is accompanied by an increase in levels of MMP-9 in the serum compared with controls (Correale and Bassani Molinas Mde, 2003). The expression of MMPs has consequences, as these proteases influence the activation of T-cells in the periphery (Li et al., 2007), the transmigration of leukocytes into the CNS (Rosenberg, 2002; Toft-Hansen et al., 2004; Agrawal et al., 2006), and demyelination and axonal/neuronal loss within the CNS (Matyszak and Perry, 1996; Newman et al., 2001). In support, pharmacological inhibitors (e.g., GM6001, Ro31-9790, and BB1101) of MMPs alleviate the clinical course and neuropathology of EAE (for review, see Yong et al., 2007). Moreover, EAE is reduced in mice that are genetically deficient for MMP-2, -7, -8, and -9 (Dubois et al., 1999; Agrawal et al., 2006; Folgueras et al., 2008; Buhler et al., 2009). As well, a first-line therapy used in MS, interferon-β, reduces the expression of several MMPs in leukocytes of MS patients (Stuve et al., 1996; Bernal et al., 2009). Despite these results, it has not been clear why there is the extensive upregulation of the multiple MMPs in MS and EAE. Our data render EMMPRIN a likely candidate upstream of these elevated MMPs.
As noted previously, leukocytes readily cross the endothelial basement membrane but require MMPs to transmigrate the glia limitans and enter the CNS parenchyma (Mun-Bryce and Rosenberg, 1998; Agrawal et al., 2006; Toft-Hansen et al., 2006). Thus, while the reduction of proteolytic activity with anti-EMMPRIN treatment helps account for the attenuation of parenchymal infiltration of leukocytes, it is surprising that we did not observe large perivascular cuffs that could represent immune cells that were prevented from penetrating across the glia limitans into the CNS parenchyma; instead, we observed fewer and smaller cuffs in the anti-EMMPRIN mice. This finding suggests that EMMPRIN may also affect leukocyte recruitment or their adhesion onto cerebral vessels, thereby generally reducing the number of inflammatory cells that attach onto cerebral vessels. In this regard, EMMPRIN is known to interact with the cyclophilins that can alter leukocyte chemotaxis (Damsker et al., 2009). Moreover, EMMPRIN colocalizes with integrins (Berditchevski et al., 1997) expressed on endothelial cells of blood vessels and infiltrating T-cells, and it is possible that anti-EMMPRIN treatment may alter integrin function and thereby affect the adhesion of leukocytes onto endothelial cells.
The period of treatment with anti-EMMPRIN that results in amelioration of EAE symptoms is instructive. Treatment at a time interval when leukocytes are being activated (days 0, 4, and 8) did not affect EAE, indicating that although EMMPRIN has been reported to promote leukocyte maturation and activation (Chiampanichayakul et al., 2006), the efficacy of the anti-EMMPRIN antibody was likely not related to affecting this process. This contention is supported by our data that anti-EMMPRIN administration did not change the antigen recall response of peripheral leukocytes. Importantly, treatment with anti-EMMPRIN at days 8–11 ameliorates EAE; this is a time window when leukocytes are known to migrate into the CNS in EAE. When mice with anti-EMMPRIN treatment at days 8 and 11 received another injection at day 15, the reduction in EAE symptoms was sustained compared with mice that did not receive EMMPRIN antibody at this point. Whereas this indicates a continual role for EMMPRIN in leukocyte migration into the CNS at the later time point, we cannot exclude the possibility that EMMPRIN has other functions within the CNS, as suggested by the high and persistent (data not shown) expression of EMMPIN on astrocytes in EAE and MS lesions. It is probable that the damaged blood–brain barrier in EAE permits the entry of the anti-EMMPRIN antibody into the CNS to neutralize EMMPRIN-dependent activity on CNS resident cells that is distinct from regulating leukocyte migration. The recent reports of EMMPRIN being elevated within the CNS in stroke (Burggraf et al., 2007; Zhu et al., 2008) and Alzheimer's disease (Zhou et al., 2005) support the idea that there could be other functions of EMMPRIN in the affected adult CNS that is unrelated to large-scale leukocyte migration. Indeed, in early life, EMMPRIN has roles in neural development unrelated to immunity (Naruhashi et al., 1997; Curtin et al., 2007).
Recently, it was shown that when mice null for the cyclophilin D gene were induced with EAE, they recovered earlier than their wild-type counterparts (Forte et al., 2007). Although these null mice had similar inflammatory scores as wild-type controls, they had more spared axons, which were attributed to the neurons in cyclophilin D null mice being more resistant to the reactive oxygen and nitrogen species encountered in EAE (Forte et al., 2007). Cyclophilins are implicated in the cell-surface expression of EMMPRIN, and EMMPRIN has been identified as being essential for cyclophilin-mediated signaling (Yurchenko et al., 2005), suggesting a role for EMMPRIN in cyclophilin associated effects in EAE.
EMMPRIN null mice do exist, wherein both male and female homozygotes are sterile. Impaired spermatogenesis (arrest at the metaphase of the first meiosis) is observed in EMMPRIN null male mice (Igakura et al., 1998), whereas the female sterility is thought to be caused by the failure of fertilization and implantation (Igakura et al., 1998). EMMPRIN null mice also have several neurological and cognitive defects, reduced blood–brain barrier integrity (Igakura et al., 1996), and cycling defects in lymphocytes (Renno et al., 2002). Together, this has made EMMPRIN null mice a poor candidate to investigate EAE in our opinion.
In summary, this study is the first to describe a role for EMMPRIN in MS and EAE. Our results highlight EMMPRIN in the regulation of MMP expression and activity, with resultant impact on the transmigration of leukocytes across the glia limitans into the CNS. Our findings emphasize EMMPRIN to be a novel therapeutic target to ameliorate MS.
Footnotes
This work was supported by an operating grant from the Canadian Institutes of Health Research. We thank Yan Fan, Hollie Mowbray, and Jackie Williamson for skilled technical assistance. S.M.A. was supported by fellowships from the Multiple Sclerosis Society of Canada and the Alberta Heritage Foundation for Medical Research. V.W.Y. is a Canada Research Chair (Tier 1) in Neuroimmunology. W.W.T. was sponsored by the National Institute of Neurological Disorders and Stroke–National Institute of Mental Health, the National Multiple Sclerosis Society, and the Department of Veteran Affairs.
References
- Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin L. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23:406–415. [PubMed] [Google Scholar]
- Berditchevski F, Chang S, Bodorova J, Hemler ME. Generation of monoclonal antibodies to integrin-associated proteins. Evidence that alpha3beta1 complexes with EMMPRIN/basigin/OX47/M6. J Biol Chem. 1997;272:29174–29180. doi: 10.1074/jbc.272.46.29174. [DOI] [PubMed] [Google Scholar]
- Bernal F, Elias B, Hartung HP, Kieseier BC. Regulation of matrix metalloproteinases and their inhibitors by interferon-beta: a longitudinal study in multiple sclerosis patients. Mult Scler. 2009;15:721–727. doi: 10.1177/1352458509102920. [DOI] [PubMed] [Google Scholar]
- Biswas C, Zhang Y, DeCastro R, Guo H, Nakamura T, Kataoka H, Nabeshima K. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995;55:434–439. [PubMed] [Google Scholar]
- Buhler LA, Samara R, Guzman E, Wilson CL, Krizanac-Bengez L, Janigro D, Ethell DW. Matrix metalloproteinase-7 facilitates immune access to the CNS in experimental autoimmune encephalomyelitis. BMC Neurosci. 2009;10:17. doi: 10.1186/1471-2202-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burggraf D, Martens HK, Dichgans M, Hamann GF. Matrix metalloproteinase (MMP) induction and inhibition at different doses of recombinant tissue plasminogen activator following experimental stroke. Thromb Haemost. 2007;98:963–969. doi: 10.1160/th07-03-0194. [DOI] [PubMed] [Google Scholar]
- Chen S, Kadomatsu K, Kondo M, Toyama Y, Toshimori K, Ueno S, Miyake Y, Muramatsu T. Effects of flanking genes on the phenotypes of mice deficient in basigin/CD147. Biochem Biophys Res Commun. 2004;324:147–153. doi: 10.1016/j.bbrc.2004.08.232. [DOI] [PubMed] [Google Scholar]
- Chiampanichayakul S, Peng-in P, Khunkaewla P, Stockinger H, Kasinrerk W. CD147 contains different bioactive epitopes involving the regulation of cell adhesion and lymphocyte activation. Immunobiology. 2006;211:167–178. doi: 10.1016/j.imbio.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Clements JM, Cossins JA, Wells GM, Corkill DJ, Helfrich K, Wood LM, Pigott R, Stabler G, Ward GA, Gearing AJ, Miller KM. Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumour necrosis factor-alpha inhibitor. J Neuroimmunol. 1997;74:85–94. doi: 10.1016/s0165-5728(96)00210-x. [DOI] [PubMed] [Google Scholar]
- Correale J, Bassani Molinas Mde L. Temporal variations of adhesion molecules and matrix metalloproteinases in the course of MS. J Neuroimmunol. 2003;140:198–209. doi: 10.1016/s0165-5728(03)00204-2. [DOI] [PubMed] [Google Scholar]
- Curtin KD, Wyman RJ, Meinertzhagen IA. Basigin/EMMPRIN/CD147 mediates neuron-glia interactions in the optic lamina of Drosophila . Glia. 2007;55:1542–1553. doi: 10.1002/glia.20568. [DOI] [PubMed] [Google Scholar]
- Damsker JM, Okwumabua I, Pushkarsky T, Arora K, Bukrinsky MI, Constant SL. Targeting the chemotactic function of CD147 reduces collagen-induced arthritis. Immunology. 2009;126:55–62. doi: 10.1111/j.1365-2567.2008.02877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Masure S, Hurtenbach U, Paemen L, Heremans H, van den Oord J, Sciot R, Meinhardt T, Hammerling G, Opdenakker G, Arnold B. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest. 1999;104:1507–1515. doi: 10.1172/JCI6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des. 2008;14:1555–1565. doi: 10.2174/138161208784705432. [DOI] [PubMed] [Google Scholar]
- Fan QW, Yuasa S, Kuno N, Senda T, Kobayashi M, Muramatsu T, Kadomatsu K. Expression of basigin, a member of the immunoglobulin superfamily, in the mouse central nervous system. Neurosci Res. 1998;30:53–63. doi: 10.1016/s0168-0102(97)00119-3. [DOI] [PubMed] [Google Scholar]
- Folgueras AR, Fueyo A, Garcia-Suarez O, Cox J, Astudillo A, Tortorella P, Campestre C, Gutierrez-Fernandez A, Fanjul-Fernandez M, Pennington CJ, Edwards DR, Overall CM, Lopez-Otin C. Collagenase-2 deficiency or inhibition impair experimental autoimmune encephalomyelitis in mice. J Biol Chem. 2008;283:9465–9474. doi: 10.1074/jbc.M709522200. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Zucker S, Gordon MK, Toole BP, Biswas C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J Biol Chem. 1997;272:24–27. [PubMed] [Google Scholar]
- Hartung H-P, Kieseier BC. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol. 2000;107:140–147. doi: 10.1016/s0165-5728(00)00225-3. [DOI] [PubMed] [Google Scholar]
- Igakura T, Kadomatsu K, Taguchi O, Muramatsu H, Kaname T, Miyauchi T, Yamamura K, Arimura K, Muramatsu T. Roles of basigin, a member of the immunoglobulin superfamily, in behavior as to an irritating odor, lymphocyte response, and blood-brain barrier. Biochem Biophys Res Commun. 1996;224:33–36. doi: 10.1006/bbrc.1996.0980. [DOI] [PubMed] [Google Scholar]
- Igakura T, Kadomatsu K, Kaname T, Muramatsu H, Fan QW, Miyauchi T, Toyama Y, Kuno N, Yuasa S, Takahashi M, Senda T, Taguchi O, Yamamura K, Arimura K, Muramatsu T. A null mutation in basigin, an immunoglobulin superfamily member, indicates its important roles in peri-implantation development and spermatogenesis. Dev Biol. 1998;194:152–165. doi: 10.1006/dbio.1997.8819. [DOI] [PubMed] [Google Scholar]
- Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, Black RA, Rossi JJ, Blobel CP, Dempsey PJ, Workman CJ, Vignali DA. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26:494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg RL, De Groot CJ, Montagne L, Freitag P, van der Valk P, Kappos L, Leppert D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain. 2001;124:1743–1753. doi: 10.1093/brain/124.9.1743. [DOI] [PubMed] [Google Scholar]
- Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- Naruhashi K, Kadomatsu K, Igakura T, Fan QW, Kuno N, Muramatsu H, Miyauchi T, Hasegawa T, Itoh A, Muramatsu T, Nabeshima T. Abnormalities of sensory and memory functions in mice lacking Bsg gene. Biochem Biophys Res Commun. 1997;236:733–737. doi: 10.1006/bbrc.1997.6993. [DOI] [PubMed] [Google Scholar]
- Newman TA, Woolley ST, Hughes PM, Sibson NR, Anthony DC, Perry VH. T-cell- and macrophage-mediated axon damage in the absence of a CNS-specific immune response: involvement of metalloproteinases. Brain. 2001;124:2203–2214. doi: 10.1093/brain/124.11.2203. [DOI] [PubMed] [Google Scholar]
- Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdenakker G, Nelissen I, Van Damme J. Functional roles and therapeutic targeting of gelatinase B and chemokines in multiple sclerosis. Lancet Neurol. 2003;2:747–756. doi: 10.1016/s1474-4422(03)00587-8. [DOI] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Renno T, Wilson A, Dunkel C, Coste I, Maisnier-Patin K, Benoit de Coignac A, Aubry JP, Lees RK, Bonnefoy JY, MacDonald HR, Gauchat JF. A role for CD147 in thymic development. J Immunol. 2002;168:4946–4950. doi: 10.4049/jimmunol.168.10.4946. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Stuve O, Dooley NP, Uhm JH, Antel JP, Francis GS, Williams G, Yong VW. Interferon beta-1b decreases the migration of T lymphocytes in vitro: effects on matrix metalloproteinase-9. Ann Neurol. 1996;40:853–863. doi: 10.1002/ana.410400607. [DOI] [PubMed] [Google Scholar]
- Tang Y, Kesavan P, Nakada MT, Yan L. Tumor-stroma interaction: positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol Cancer Res. 2004;2:73–80. [PubMed] [Google Scholar]
- Toft-Hansen H, Nuttall RK, Edwards DR, Owens T. Key metalloproteinases are expressed by specific cell types in experimental autoimmune encephalomyelitis. J Immunol. 2004;173:5209–5218. doi: 10.4049/jimmunol.173.8.5209. [DOI] [PubMed] [Google Scholar]
- Toft-Hansen H, Buist R, Sun XJ, Schellenberg A, Peeling J, Owens T. Metalloproteinases control brain inflammation induced by pertussis toxin in mice overexpressing the chemokine CCL2 in the central nervous system. J Immunol. 2006;177:7242–7249. doi: 10.4049/jimmunol.177.10.7242. [DOI] [PubMed] [Google Scholar]
- Weaver A, Goncalves da Silva A, Nuttall RK, Edwards DR, Shapiro SD, Rivest S, Yong VW. An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB J. 2005;19:1668–1670. doi: 10.1096/fj.04-2030fje. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Zabad RK, Agrawal S, Goncalves Dasilva A, Metz LM. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259:79–84. doi: 10.1016/j.jns.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Yurchenko V, Pushkarsky T, Li JH, Dai WW, Sherry B, Bukrinsky M. Regulation of CD147 cell surface expression: involvement of the proline residue in the CD147 transmembrane domain. J Biol Chem. 2005;280:17013–17019. doi: 10.1074/jbc.M412851200. [DOI] [PubMed] [Google Scholar]
- Zhou S, Zhou H, Walian PJ, Jap BK. CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer's disease amyloid beta-peptide production. Proc Natl Acad Sci U S A. 2005;102:7499–7504. doi: 10.1073/pnas.0502768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Khachi S, Hao Q, Shen F, Young WL, Yang GY, Chen Y. Upregulation of EMMPRIN after permanent focal cerebral ischemia. Neurochem Int. 2008;52:1086–1091. doi: 10.1016/j.neuint.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]